Embed Size (px)
Citation preview

Die Sauerstoff bestimmung in Metallen') Von Wolfgang Fischer
Forschungsinstitut fur Nichteisenmetalle, Freiberg/Snchscii
Die mechanischen und einige andere physikalische Eigenschaften der Metalle und Legiernngen werden z. T. in erheblichem MaBe durch die Anwesenheit ge- ringer Mengen nichtmetallischer Beimengungen beein- flufit. Dies gilt besonders fiir die Elemente, die unter normalen Bedingungen gasformig sjnd, also Wasser- stoff, Saucrstoff und Stickstoff. Die exakte analytische Bestimmung dieser Gase i n Metal len hat daher in den letzten Jahrzehnten immer mehr an Interesse geworinen.
Die erste Monographie iiber die Gasbestinimung in Metallen von 2. M . Turovcevu und L. L. Kunin er- schien 1959 in Moskau [I].
I n der vorliegenden Arbeit soll versucht werden, iiber die Sauerstoffbestimmung in Metallen einen einigermaaen vollstandigen ftberblick zu geben.
Die Frage, in welcher Form der Sauerstoff in Metnl- len vorliegt, bzw. woher der im Metall vorhandene Sauerstoff kommt, ist ebenfalls von Interease, wurde aher den Rahmen dieser Arbeit ubersteigen. Sauerstoff kann molekular (1. B. in Lunkern), atomar oder als Ion geliist im Metall, oder in einer Oxydbindung vor- kommen. Restimmend hierfur sind die Art des betref. fenden Metalls und die Bedingungen seiner Herstellung und Verarbcitung. Die Kenntnis der Sauerstoffliislich- keit eines Metalls und deren Temperaturabhangigkeit ist in jedem Falle fur den Analytiker von Nutzen.
Fur eine sinnvolle Probenahme sind diese kurz ge- etreiften Zusammenhiinge bereits wichtig. Eine all- gemeingiiltige Vorschrift fur eine richtige Probenahme zur Sauerstoffbestimmung in Metallen kann nicht ge- geben werden, jedoch sei auf einige wichtige, hierbei zu beachtende Punkte hingewiesen : Zur Ausschaltung von Oberflacheneffekten ist es vorteilhaft, kompalrte Proben zu verwenden, die eine moglichst kleine Ober- fliiche, bezopen auf ihre Masse, haben. Das bedeutet nicht, daB es sich um Kugeln handeln muB, aber kurze zylindrische oder quaderformige Proben eind .zu bevor- zugen.
Bei der Herstellung dieser Proben soll sich das Metallstiick moglichst wenig erwlrmen, aber auch nicht mit Schmicr- oder Kuhlmitteln in Beruhrung kommen. Haufig wird man kurz vor Beginn der Ana- lyse die gesamte Oberflache der Probe nochmals durch Irurzes Abfeilen oder Entfetten, z. B. mit CCl,, reini- gen. Im einzelnen hangt die Probenahme und -vor- bereitung jedoch auch von der gewahlten Sauerstoff - bestimmungsmethode ab.
Eine Besprechung der heute verfiigbaren Sauerstoff- bestimmungsmethoden wird erleichtert, wenn ahnliche Verfahren zu bestimmten Gruppen zusammengefaBt werden, etwa wie folgt:
1. HeiBextraktionsverfahren 1 .l. Vakuum-HeiBextraktionsverfahren
') Nach einem Vortraa, aehalten aul der Arbeitstflguns ,,Neue Anitlysenmethodeb in der NE-Metellurgie", veranstnltet von dcr ODRH gerneinasm mit der C'hemiachen GeaelLchatt in der DDR und den1 Porachnngainatitut fdr NE-Metalle, Vreibcrg, i r n Novcmher 1980 in Frcibew.
1.2. Tragergasverfahrcn 2 . Chrmische Verfahren 3. Physikalische Verfahren 4. Metallographische Verfahren.
1. Heilloxtraktionsvc.rfahrcri 1.1. Vakunm-HeiBoxtrnktiotisvcrPIlirrn
Das universellste und wahrscheinlich auch zuvcr- lassigste Verfahren zur Sauerstoffbestimmung in tlcn meisten handelsublichen Metallen ist z. Z. das sog. HeiBextraktionsverfahren. Das Prinzip dieser Methotle besteht darin, die moglichst lt~mpnlrt~e Untersurhungs- probe mit Graphit bei erhohten Trmperaturen im Vakuum zur Reaktion zu hiingen, etwa nach folgentlcr Gleiehung :
Das sich aus der Oxydphase hildende Kohlenmonoxyd wird abgepumpt, gesammelt und analysiert. Wichtig- ste Voraussetzung hierfiir ist, bedingt durch den an- gestrebten Mechanismus, daB diese Reaktion in einein moglichst sauberen Hochvakuumsystem durchgefuhrt wird. Um eine befriedigende Reaktionsgeschwintlig- keit zu erzielen, muB man meist die Probe bis zum Schmelzpunkt und daruber erhitzen (Vacuum-fusion- procedure).
Der Anwendung hoher Temperaturen im Vakuum sind Ieider Grenzen gesetzt, so da13 man z. B. fur die Untersuchung von Wolfram (Fp. -3380 "C), Moly1)- dan (Fp. N 2622OC) und Tantal (Fp. - 300OOC) nach anderen W e e n suchen muBte. Diese fand man einmal in der V a k h u m - F e s t - E x t r a k t i o n bei r t w n
,2000°C, zum an&en in der Anwendung einer Bacl- Techik. Bei der Festextraktion ist es notwendig, da0 die Probe gut vbn Fraphit umgeben ist und zumindent an der Grenzflache Metall-Kohlenstoff die Reaktion nach G1. (1) geniiqend schnell ahlauft. Die Voraus- setzungen fur dasl erfolgreiche Arbeiten mit einem Metallbad werden spater noch besonders besprochen.
1912 veroffentlichten Walker und Putrick 121 erst- mals Einzelheiten uber Versuche, mit einem HeiB- extraktionsverfahren zu brauchbaren Sauerstoffbe- stimmungen in Metallen zu kommen. In Deutschlancl beschrieben 1919 Oberhoffer und Mitarbeiter [3] eino HeiBextraktionsanlage. Bis zum Beginn des 2. Welt- krieges fanden in Deutschland diese Arbeiten eirien gewissen AbschluB mit einer von Thanheiser [4] und Mitarbeiter entwickelten Apparatur. Etwa zur gleichen Zeit erschienen von Sloman und Mitarbeiter [ 5 ] eine Reihe von grundlegenden Arbeiten. Seit Kriegsende sind eine Fiille von neuen Veroffentlichungen hinzu- gekommen, mit bedingt durch vollig neue Werkstoff - probleme. Man denke nur an das zeitweilig starke An- steigen der Titanproduktion und an das wachsende Interesse an Kcrnenergiewerkstoffen.
Me,O + C + Me,C + CO. (1)
1.1.1. Appwatives Fur den nicht naher mit der Thematik Vertrauten
sollen kurz einige Worte zum apparativen Aufwand
354 Z . Clrem., 1. Jg., Heft 14, 1961

fur die HeiBextraktion gesagt werden. Die aus den Arbeiten des ehem. Kaiser-Wilhelm-Instituts fur Eisenforschung (Oberhoffer, Thanheiser und Brauns u. a.) stammende Anordnung einer solchen Apparatur ist in Deut,schlancl vor rtllem auf dem Eisen- und 8tahl- gebiet rioch weit, verbreit,et,. Pirmcn, wie Strohleir i , Ilmenau untl Diisseldorf, uncl wie K ler s , Diisseldorf, vertreil)en diese Anlagen [(i], [TI. [HI. (her deren Auf- I)au kann man sich im ,,Handbuch fiir tlas EisenhiittJen- laboratorium" orientieren.
Fur die Untersuchung von NE-Metallen und -1,egie- rungen hat sich eine auf Arbeiten der National Research Corporation und des National Bureau of Standards euriiakgehende Apparatm BUR den z w m - ziger Jahren international mehr clurchgesetzt.
In Bild 1 ist schematisch eine solche Apparittur gezeigt, wie sie etwa z. B. Leybold in Koln unct Edwa,rds in England bauen. Die Apparatur best,eht, aus drei Hauptteilen: 1. dem Ofenteil, 2. dem Gas- forderteil und 3. tiem Analysatorteil. Der Hochtem- peratur-Vakuumofen ist induktionsheheizt. Man hrancht also keine Stronianschliisse ins Vakuum ein- zufuhren. Dtlr Ofen, der auf Arbeiten von Gddner und Beach [$I] zuriickgeht, erreicht bei einer HF-Gene- ratorleistung von etwa 2 1tW eine Temperatur von 2400 "C. Der Ofenmantel hest,eht nu r aus Pyrexglas und wird mit einem J,uftgelilase gekiihlt. Tm Ofen- innern ist. ein Q,uarzglastiegel an Platindriihten auf- gehiingt. Dieaer Tiegel cnthiilt, von Graphitpulver um- hiillt den cigentlichcn Graphittiegel fiir die Aufnahme der l\.letallprohen. Mc iilber den Glasmantel gcscho- hene Intluktionwpiilc wird mit Wasser gekiihlt. Am
Oberteil des Ofens sitzen die Anschlusse fiir den Gas- sbtransport, den Probeeinwurf, die pyrometrische Temperaturkontrolle und evtl. fur eine Deckelziehvor- richtung.
Eine moglichst kurze und geniigend weite Saug- leitung verbindet den Ofen mit einer Quecksilber- Diffusionspumpe, die die im Ofenteil frcigewordenen Gase moglichst rasch in den Analysatorteil saugt. Meist wird eine zwei- oder dreistufige Diffusionspumpe mit Saugleistungen his zu etwa 45 l/s hei 10'' Torr verwendet.
Die ahgesaugten Gase werden in ein vorher gut evakniertes Kreislaufsystem gepumpt, in dem eine I o wpr es su re - ana lys i s ausgefiihrt wird.
Die Qasanalyse bei vermindertem Druck hietet, vor allem bei kleinen Gasmengen viele Vorteile, z. B. ge- geniiher der klassischen Orsat-Gasanalyse. Gasmengen von 0,005 om3 konnen noch sicher ei-faBt werden. Vor dlem kann hierbei auf eine Sammlung des Gases bei Atmospharendruck verzichtet werden, die ziisiitzliche Rauelemente erfordert. Das Analysatorsystem hesteht wie bei einer 8tockschen Appnratur nur aus Glas und Quecksilher. Es hat also die Vorteile groBter Sauber- Ireit, Fettfreiheit usw. Deshalb kann man auch sehr ltleine Gasmengen gut damit erfassen. I n den Analy- satorltreislauf sind im wesentlichen eine Fdle mit speziell prapariertem Kupferoxyd, eine Magnssium- perchlorat-Falle und eine Kiihlfalle eingeschaltet. Das Umpumpen erfolgt mit einer Quecksilher-Diffusions- pumpe.
Die Messung der Gasmengen vor und nach den ein- zelnen Reaktions- und Kondensationsschritten der
Hiid I VakuumlieiUex truk - tionsapparatur mi t
HF-Induktions- heizung und Nieder-
druckanalysatnr (HIIR Symposium nn Titib-
ninm, ASTM Spec. Techn. Public. Nr. 204
(1951) 202) .
Blower, I3 = GeblUo zur Kiihlung: Furnacc, F = Ofen (HF-beheizt); Right-A4ngle Prism for Reading Temperature = 90°-Prismu zur Temperatnrmessung; Louding Arm = Probevorratsarnl ; Transfer Punlp (Two Stage) U P , = Hg-Diffusionspumpe (2-stUfig) zur Ciasextraktion: C;irculnting Pump (Three Stage) D P , = Umwklzpumpe (1-stofig); Exhaust Line = Hauptvakuumleitung; Ionisation :age = Ionisat,ionsmi~nouieter; M a i n Stopcock, S1, = Heupthahu: Muin Evacuating System Diffusion Pump A11 Metall DP. = II~tuptdiffusionspuni~,r iuis Metnll; To Mcchanicul Vacuuni Pump = zur Vnrpumpe; (:uOl,-J)rying Tube = CaC1,-Trockenrohr: Analytical System : AnHlysHtnr-Teil: l'oepler-Pump T (C: 3) = Tnepler-Pumpe; McLeod Gage = McLeod Manometer; SpeClal Mercury Cut off for Addition of Cia.scs = besonderer QuecksilberverschluB zur Gaszugabe: Expansion Volumes H I , H . = Ausdehnungs- KefliBe; CUO-T~RIJ U 7 Balk mit C ! u O : Mp(ClO,),-Trap, Z = Fnlle mit Mg(CIO,),; Cold Trap, J = Kuhlfnlle; Vacuum Manifold = VB- kiiiun-Sninnirllritiin~ mi' Hedienung der Hg-Versahliisse; Air Manifold = Luft-Hammelleitung zur Bedienuna der Hg-Verachlilsse;
(!, Iiis ( : , I ~~ ~uecksilber-Ver~rhliircRt.: S, biH S, 7 HIlhrie fiir Ha-VerschlBase
45' Z. (:hem., 1. JR., Heft. 12, Ill61 3%
Analyse erfolgt als Druckmessung in einem McLeod- Manometer. Zur Messung im McLeod muR allerdings jedesmal das Gas mit einer Toepler-Pumpe aus dem Kreislauf gezogen werden. Dieser Schritt ware sogar bei der Niederdruck- Analyso vermeidbar , wie in Arbeiten von Ransley u. a. [lo] gezeigt wurde. Jedoch bedeutet er kaum eine besondere Vedangerung der Analysenzeiten.
Die eigentliche Gasbestimmungsapparatur kann durch zusiitzliche Pumpen (z. B. 01-Diffusionspumpen hoher Leirtung) am Beginn einer Analysenreihe schnell und gut evakuiert werden.
1 .I .2. Durchfiihrung ekner Messung Fiir eine Messung, die etwa die Gasbestimmung in
5 bis 10 Proben umfabt, wird in den Ofen ein neuer Graphittiegel eingesetzt und der Probenvorratsarm iiber dem Ofen mit den Proben beschickt. Die Appa- ratur wird sofort nach dem Einsetzen der Proben ge- schlossen und mit moglichst leistungsfiihigen Pumpen evakuiert. Der Ofen, der vor allem durch seine Graphit- teile groRe Gasmengen gebunden enthiilt, wird nun oberhalb der gewunschten Analysentemperatur (Ex-
traktionstemperatur) ausgeheizt. Dies geschieht so lange, bis man nach Zuruckgehen auf Extraktionu- temperatur einen tragbaren Leerwert erhiilt.
Nach der Ermittlung des Leerwertes wird die erstc Probe in den Graphittiegel eingeschleust. Hierf iir wirtl allgemein die Ofentemperatur kurzzeitig erniedrigt (etwa 1200 bis 1400"C, je nach Art des Metalls). 1st die vorgesehene Arbeitstemperatur wieder erreicht, pumpt man so lange das entweichende Gas ab, bis durch Vakuummessung keine merkliche, ~ b c r den Leerwert hinausgehende Gasentwicklung festzustellen ist. Die Extraktion der ersten Probe ist damit beendet. Die Extraktionszeit liegt unter gdnstigen Bedingungen bei etwa 5 min, unter ungiinstigen dagegen bei 45 his (i0 min. Das extrahierte Gas besteht ini wesentlirlien aus Kohlenmonoxyd, Wasserstoff und Stickstoff, die in bekannter Weise analysiert werden.
Wahrend das Gas aus der ersten Probe analyqiert wird, lauft bereits die Extraktion der zweiten Probe nil
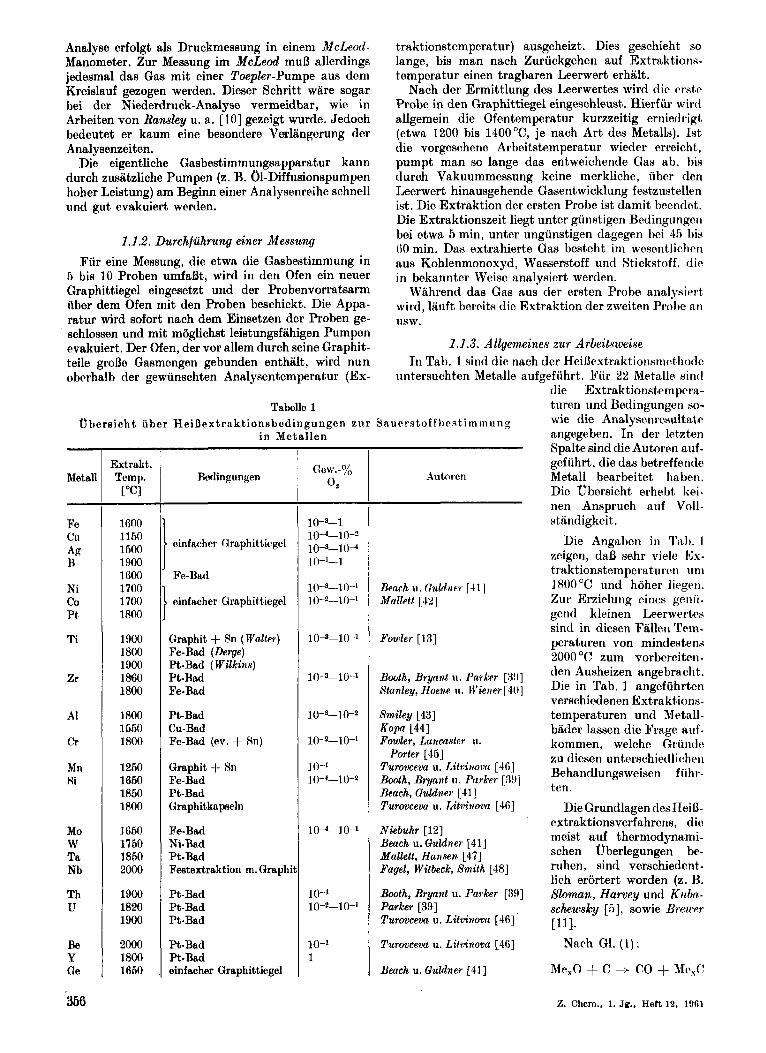
1.1.3. Allgemeines zur Arbeitsweise In Tab. 1 sind die nach der HeiBextraktiorisnictliotle
untersuchten Metalle aufgefiihrt. Fiir 22 Metalle xincl
IISW.
Tabelle 1 u he ra i c h t u b or H ei D e x t ra k t i on B bed in g u ng e n zii r 3 a u r r s t o f f 1) r SI t i m m 11 n g
in Metallen - FrletlEli
- Fe Ch Ag R
Ni co Pt
Ti
Zr
A1
Cr
Mn Hi
Mo W Ta Nb
Th U
Be Y a e
356
Extrakt,. Temp
LOCI
1600 1160 1600 1900 1600 1700 1700 1800
1900 1800 1900 1860 1800
1800 1660 1800
1260 1660 1860 1800
1660 1760 1860 2000
1900 1820 1900
2000 1800 1660
Bedinpingen
einfacher Oraphittiogel
Fe-Bad
einfacher Graphit,tiegel
Graphit + Pn (Wa,ller) Fe-Bad (Derge) Pt-Bad ( Wdkins) Pt-Bad Fe-Bad
Pt-Bad
Fe-Bad (ev. + Sn) &-Bad
Graphit + 8n Fe-Bad Pt-Bad Graphitkap~eln
Fe-Bad Ni-Bad Pt-Bad Festextrektion m. Graphj
Pt-Bad Pt-Bad Pt-Bad
Pt-Bad
einfacher Graphittiegel Pt-Bad
Autorrn
10-3-1 10-4--10--3
1 n-1-1
10-3--10-1
10-8-1 0 - 4
10-2-1 0-1
10-3-10-1
in- 3-10-1
10-9-104
10-*-10-'
10-1 10-~-10-2
10-4-1 0-1
10-1 10-*-10-'
10-1 1
Fouier [13]
Booth, Bryant 11. Parkrr [39 Rtanley, Hoene 11. Wienrr [do
SmiZey [43]
Fowler, La~icaster 11.
Turovceva u. Lituinova [40] Booth, Bryant ti. Parker [R!)] Beach, au2dner 1411 Turovceva u. Litvinova [4G]
Niebuhr [12] Beach u. Guldner [41 J dlallett, Hanaen [47] Fagel, Witbeck, Smith [48]
Booth, Bryant u. Parker [39] Parker [39] Turovceva u. Litvinova [46]
Turovceva u. Litvinova [4G]
Beach u. auldner [41]
KOP [441
Porter [46]
- die Extraktionstemprra- turen und Bedingungen so- wie die Analysenresultatr angegeben. In der leteten Spalte sind die Autoren auf- gefiihrt, die das betreffende Metall bearbeitet hnltwn. Die ubersicht erhebt kei- nen Anspruch auf Voll- stiindigkeit.
Die Angaben in Tab. 1 zeigen, da13 sehr vielc Ex- traktionstemperaturrn uni 1800°C und hoher liegeti. Zur Erzielung eines gwiri- gcnd kleinen Leerwertcv sind in diesen Fiillen Tern- peraturen von mindestens 2000 "C zum vorbrreiten- den Ausheizen angebracht. Die in Tab. 1 nngefuhrten verschiedenen Extraktions- temperaturen und Metall- bader lassen die Frage uiif- kommen, welchr Griintlc zu dieseri unterschiedliohen Behandlungsweiuen fii hr- ten.
Die Grundlagen dcs HeiR- extraktionsverfahrens, dirt meist auf thermodynami- schen Uberlegungen be- ruhen, sind verschiedent- lich erortert worden (z. R. Slomn, Harvey und Kicbo- schew&y [6], sowie R r e w r [ I l l -
Narh G1. (1) :
MexO + C --> CO + Mc,C
Z . Cham., 1. Jg.. Hett12. 1961

laat sich meist nur dadurch rrrcichen, daB man in Titan A 1,19 1,21 1,09 1.26 1916 1,18 f 0,064 Mchmelzfl~ssigem Zustand Tftm B 0,646 0,566 0,641 0,676 0,621 0,660 & 0,022
Titan C 0,158 0,143 0,152 0,130 0,146 0,148 f0,0076
aufgef uhrten Metallbader dienen daher dem Zweck, die Schmelztemperatur der zu untersuchenden Metalle herabzusetzen. AuSerdem fungieren diese Bader als Kohlenstoffiibefiriiger, indem sie entweder von Anfang an Kohlenstoff ent- halten (Stahlbad) oder beim Entgasen Kohlenstoff losen (Platinbad).
Fiir die Wahl einrs solchen Bades sind folgende Bedingungen ausschlaggebend : 1. es muS die Metall- proben leicht auflosen; 2. es mu13 wahrend der Ex- traktion flussig bleiben und darf bei der Arbeitstem- peratur kein festes Carbid bilden; 3. es sol1 bei der Arbeitstemperatur einen geringen Dampfdruck haben, an den kiilteren Stellen des Ofens nur wenig Beschlage bilden und dort ein schlechter Getter sein.
Dementsprechend hat man vor allem Fe, Ni, Co, Sn, 1% und Pd als Badmetalle verwendet. Zweck aller dieser Operationen ist jedoch, daD Reakt,ionsbedin- gutigen und Temperatur so gehalten werden, daB der Druck des entstehenden Kohlenmonoxyds groS genug ist, um den statischen Druck der Schmelee uberwinden zii konnen. Nur dann kann das CO abgepumpt und tler Analyse zugefiihrt werden.
Aus G1. (1) ergibt sich nach dcm MWG folgender CO-Druck :
pco = exp (- AG/RT) . (2) Im Bild 2 ist pco als Funktion der Temperatur gra-
phisch dargestellt. Fur einen speziellen Fall 1aSt sich hieraus - Carbidbildung des betreffenden Metalls vor- ausgesetzt - leicht die unbedingt notige Arbeitstem- peratur ablesen. Bei ihr mu13 der CO-Druck etwa 5 bis 10 Torr betragen (Niebuhr [12]).
Die quantitative Entwicklung des im CO gebun- clenen Sauerstoffs hiingt wesentlich von der Viskositit des verwendeten Metallbades ab. Durch Kohlenstoff- aufnahme aus dem Tiegel kann z.B. ein Stahlbad schon nach etwa einstundigem Arbeiten bei hoher Temperatur so ziih werden, da13 Gasblasen im Bad festgehalten werden. Das gleiche kann geschehen, wenn die Konzentration des ,,Gelosten" im ,,Lijsungs- mittel" ungunstig ist. Zur uberwindung dieser Schwie- rigkeiten sind die verschiedensten Vorschliige gemacht
f 594 f 3.9 f 6,l
Bild 2 Kohlenmon- oxyddruok ale Funktion der Temperrttur (nmh iVie6alrr I121)
lemperatur -
worden [MI, die hier nicht weiter diskutiert wcrdun konnen.
1.1.4. Qenauigkeit Jedes Analysenverfahren wird nach Reproduzier-
barkeit und Genauigkeit beurteilt. Fur die Sauerstoff- bostimmung nach dem HeiSextraktionsverfahren las- sen sich in dieser Hinsicht schwer allgemeingiiltige Aussagen machen. In Tab. 2 ist ein gunstiges Beispiel wiedergegeben. Bei den hochschmelzenden Metallen Mo, W, Ta sind die Ergebnisse wesentlich schlechter. Etwa 16% Varianz (relative Standardabweichung) gelten als gute Werte (Niebuhr [12]).
Bei solchen Ermittlungen wurden immer die Werte eines Laboratoriums, d. h. Werte nach einem ganz bestimmten Modus verglichen. Leider ist es oft so, daB bei Vergleichsanalysen versohiedener Laboratorien wesentlich groSere Unterschiede auftreten. Die Ur- sachen hierfiir konnen nur angedeutet werden. Die Probenahme und -vorbereitung bis zum Einbringen ins Vakuum sind sehr entscheidend. So zeigte z. B. Fowler [14] am Titan, daS bei Reinigung der Ober- fliiche durch Feilen der Sauerstoffgehalt einer Probc auf die Hiilfte sank.
Die Reduktion durch Kohlenstoff muB vollstiindig und das Ausbringen des gebildeten CO quantitativ sein. Letzteres ist besonders davon abhiingig, daB leistungsfiihige Pumpen zum schnellen Abtransport der Gase aus dem Ofenraum verwendet werden. An- dernfalls ist es moglich, d a B Reaktionen zwischen den Gasen und Metalldiimpfen und den unvermeidbaren Metallfilmen an kiilteren Stellen oberhalb der Heizeone eintreten (Gettern). Diese FehlergroDen haben Beach und W n e r [15] sehr exakt untersucht. Solche Fragen der Genauigkeit lieI3en sich wesentlich leichter bearbei- ten, wenn auch fiir die Gasbestimmung in Metallen Eichproben zur Verfugung stunden. Vorliiufig sind wir jedoch mehr oder weniger auf eine gute Zusammen- arbeit der interessierten Laboratorien angewieaep. Testversuche durch Zusatz definierter Mengen reiner Oxyde sind hiiufig ausgefuhrt worden, die eine gewisse uberprufung der ganzen Verfahrensweise gestatten.
1.2. Traigergseverfahren Smiky [ l G ] zeigte 1965, daD man nach dem Prinzip
der HeiSextraktion auch ohne Vakuum arbeiten.kann, indem man die Apparatur mit Inertgas (Argon) fiillt und spult. Die Vorteile sind hierbei Mar, weil der game Vakuumaufwand wegfiillt. AuSerdem hat man zum Teil Vorteile dadurch, daS im Inertgas weniger stii- rende Verdampf ungserscheinungen auftreten ah im Vakuum.
Die Analyse der im Triigergasstrom ausgetriebenen Gase erfolgt selbstverstiindlich auf anderem Wege sls bei der oben beschriebenen Methode. Abresch und Lemm [17] haben mit dieser Triigergastechnik Sauor-
2. Cham., 1. Jg., Haft 12, 1961 567

stoff im Stahl bestimmt. Bemerkenswert ist die von ihnen angegebene kurze Analysenzeit von 7 min/Probe. Auf die Bedeutung der Reinheit des verwendeten Argons sei noch hingewiesen.
1959 beschrieben Yanagisawa, Xeki und Watan.de [18] eine Apparatur zur Gasbestimmung in Metallen mit automatischem Gasanalysator, der nach dem Nie- derdruckprinzip arbeitet. SchlieBlich haben Fedchtinger und Mitarbeiter [19] 1959 gezeigt, wie durch Anwen- dung eines Gaschromatographen als Analysatorteil die HeiSextraktion verbessert und registrierend gestaltet
, werden kann. Fiir mehr kinetische Untersuchungen in der HeiBextraktionsapparatur ist die vorgeschlagenc ,,Gasevolographie" vielleicht sehr nutzlich. Auch hier wird versucht, mit einer Programmreglung der Tem- peratur und Driicke eine Automatisierung der Analyse zu erreichen. Eine Kombination von Vakuum-HeiB- extraktion und Triigergasverfahren mit Gaschromato- graphie stellt auch die neue Apparatur von Heriius, Hanau, dar, die von Qruber und Lesser [20] beschrieben wurde.
2. Chemische Verfahren Der Sauerstoffgehalt einiger Metalle liiBt sich nach
Reduktion der Oxydphase durch Wasserstoff ermit- teln, indem das gebildete Wasser bestimmt wircl. Die Reaktionsenthalpien einiger solcher Reduktionen zeigt Tab. 3. Eeriiber wird in ,,Analyse der Metalle" [21] ausfiihrlich berichtet.
Wegen der aufwendigen Hochreinigung und Hoch- trocknung des erforderlichen Wasserstoffs ist es bei kleinen Sauerstoffgehalten besser, mit ruhendem Wasserstoff a h mit stromendem zu arbeiten. Fur Pb, Cu, Bi, Mo und bestimmte Stiihle wurden auf diese Weise brauchbare Ergebnisse enielt. Genauigkeiten von &20% sind hierbei erreicht worden. Die vor- geschlagenen Ausfiihrungsformen einer solchen Be- stimmung unterscheiden sich meist nur in der Art der Wasserbestimmung (gravimetrisches und manome- trisches Verfahren).
Liegen im Metall oder einer Legierung leicht ver- dampfende Bestandteile, wie z. B. Phosphor oder Zink vor, so konnen erhebliche Storungen auftreten durch Reaktionen des Wassers mit den Kondensaten, die sich an kiilteren Stellen der Apparatur bilden.
Tabelle 3
&us Oxyden bei 26°C und 760 Torr (nach Rberiua und Kowalski [23])
Reaktionsenthalpien der Wasserstoff-Reduktion
- Nr. -
1 2 3 4 5 6 7 8 9
10 11
358
Oxyd R. Enth. [kcal/O]
- 52 41 36 26 20 21 10 I0 6 6 4
- Nr . -
12 13 14 16 16 17 18 19 20 21 22
~~
Oxyd R. Enth. [ kcal/O]
1 $ 3
4 7
21 28 47 64 69 7!J 90
Zinkoxyd kann init Wasserstoff nicht reduziert wer- den, weswegen von Hartwbann [22] die Reduktion rnit H,S eingefuhrt wurde, die nach der Gleichung
ZnO + H,S = ZnS + H,O (;v verliiuft. Auch hier wird das gebildete Wasser bc- stimmt.
Eine Neuerung in iihnlicher Richtung haben Eberius und Kowalski [23] vorgeschlagen. Sie liisen z. B. Zink- staub in HC1-Eisessig-Gemischen untl titrieren clas nach GI. (4 ) gebildetc Wasser init Karl-Fischer- Reagens.
Bie ermittelten so Gehalte von 3 ,is Y9,87% 15110 in Zn-StLuben.
Ein einfaches und schnelles, dabei doch sehr genaues Verfahren zur Sauerstoffbestimmung in Cu, Pb, Mo und W durch Reduktion im stromenden Wasserstoff und direkte Titration des Reduktionswassers rnit Karl- Fischer-Reagens geben Fischer und Mehlhorn [24] an.
Verschiedene, teils auch in , ,Analyse der Metalle" zitierte Autoren, haben die Umsetzung von Metall- oxydproben mit Schwefel oder Schwefelverbindungen vorgeschlagen, die zu einer Bestimmungsmoglichkeit des Sauerstoffs als SO, fuhrt. Meist wurde Schwefel- dampf im Stickstoffstrom uber die Probe gefiihrt und das gebildete SO, jodometrisch bestimmt. Untersucht wurden die Oxyde des Zn, Mo, W, Ni, Co, Mn, As, Sb, Pb, Cd, Cu. In den letzten Jahren hat ein Kreis sowjetischer Forscher das Verfahren zur Sauerstoff - bestimmung in Cu, Ni, Cr und Mo modifiziert.
Babko und Mitarbeiter [2b] fuhren die Reaktion niit Schwefel im Vakuum aus und bestimmen das gebildetc SO, kolorimetrisch mit Fuchsin-Formaldehyd. Kleiner und Mitarbeiter [26] behandeln die Metallproben bei 500 bis 1000°C mit Dischwefeldichlorid, das in einem Stickstoffstrom uber die Probe gefiihrt wird. DAS nach der Gleichung
( 3 ) gebildete SO, wird jodometrisch bestimmt. Bisher liegen Erfahrungen mit Cu, Ferrosilicium, Po, Zr, Cr- Legierungen und Ni-(3-Legierungen vor. Dic Ermitt- lung kleiner Sauerstoffgehalte in Metallen und Legie- rungen nach dem letztgenannten Verfahren diirfte erhebliche Schwierigkeiten bereiten. Vermutlich sind &us diesem Grund keine weiteren Arbeiten in dieser Richtung bekannt geworden. Vor allem fehlen ver- gleichende Bestimmungen nach anderen bewiihrten Verfahren .
Weiterhin sind die Ruckstandsverfahrcn zu erwah- nen, die teilweise in der Gruppe Isolationsverfahren in ,,Analyse der Metalle" aufgefiihrt sind. Eine Fulle von Arbeiten sind hieriiber bekannt geworden, die im Prinzip immer nene Varianten cles Grundproblems darstellen - exakte Trennung gcringer oxydischer Bestandteile vom Grundmetall. Der Ruckstand wird quantitativ gemessen. Leider besteht er moist nicht nur aus einem definierten Oxyd. Somit treten neue Probleme auf. Eine universelle Anwendung ist nicht moglich. Liegt der Sauerstoff in geloster Form vor, so kann diese Methode nicht angewendet werden.
Diese meist alteren Arbeiten haben fur die sog. inetallkundlichc Analyse (Klinger und Koch 1271) Bc- deutung erlangt, bei der ein cLifferenziertes anodisches
ZnO + 2HC1= ZnCI, + H,O. (4)
MeO, + 2S,CI, = MeC14 + SO, + 3S
%. CJIUIII., 1. Jg., HcFt 12, 1901

Aufloscn des Grunclmetalls in einem Elektrolyten mog- lich ist.
In Fortsetzung alterer Arbciten, die eine Oxydiso- licrung mit freiem Halogen oder Halogenwasserstoff - sauren versuchten, sind einige neuere Arbeiten zu nennen, die gleichzeitig rnit der Halogenierung des Metallanteils eine Kohlenstoffreduktion der Oxyde vornehmen. Dabei entsteht CO, das nach Oxydation zum CO, quantitativ .bestimmt wird. Auf diese Weise wird vor rrllem auch geloster Sauerstoff mit erfaBt. Elwell und Peake [28] chlorieren nach Mischen der Probe rnit Graphit im Argonstrom, wahrend Codell und Norwitz [29] im Heliumstrom bromieren. Sauer- stoffbestimmungen in Ti, Zr, Cr, V und Stahlen wurden nach diesen-Methoden ausgefuhrt und zum Teil mit Werten nach dem HeiBextraktionsverfahren vergli- chen. Die ubereinstimmung der Ergebnisse beider Ver- fahren ist gut. Besondere Schwierigkeiten konnen beim sog. Bromierungs-Kohlenstoff-Reduktions-Verfahren dadurch auftreten, daB die Proben zerkleinert werden miissen, zum Teil werden sie zerspant, zum Teil pul- verisiert. Hierbei darf keine meBbare Oxydation ein- treten. AuOerdem mu0 das verwendete Halogen ebenso wie die Apparatur moglichst ideal wasserfrei sein, damit keine zu hohen Blindwerte auftreten.
Fur die Sauerstoffbestimmung in Ti, TJ, Ge, Pb, V, Cr, Cu und W beschrieben 1963 Hoekstra und Kats [30] tin vollig neues Halogenierungsverfahren mit Brom- trifluorid. Entsprechend der Gleichung
3Me0, + 4BrF3 = 3MeF, + 2Br, + 3 0 , (6) rragiert Bromtrifluorid mit Metalloxyden unter Ent- wicklung von molekularem Sauerstoff. In gleicher Weise reagieren Metallnitride zu molekularem Stick- stoff. Die Reaktion wird in Teflon-GefaBen bei erhoh- ter Temperatur durchgefuhrt. Fluchtige Reaktions- produkte werden durch Kuhlfallen vom Sauerstoff und Stickstoff getrennt. Auf Metalle, die stabile Oxy- fluoride bilden, wie z. B. Mo, ist das Verfahren nicht anwendbar. Bei Sauerstoffbestimmungen in Titan und Titanlegierungen stellten Dupraw und O'Neill [31] eine gute ubereinstimmung mit den Werten nach dem Heiflextraktionsverfahren fest.
Dieser kurze AbriB des derzeitigen Standes der che- mischen Methoden zur Sauerstoffbestimmung in Metal- len zeigt schon deutlich, daB keine so universell an- wendbar ist wie die HeiBextraktionsmethode. In Labo- ratorien, in denen nur bestimmte Metalle oder Legie- rungen zur Untersuchung kommen, ist aus okonomi- schen Griinden hiinfig die kostspielige Anschaffung einer HeiBextraktionsanlage nicht moglich. Hier ist der Ein- satz von chemischen Methoden, wie z. B. der Wasser- stoffreduktion, sicher sehr sinnvoll. Natiirlich fiillt bei dicsen Methoden meist nur ein Sauerstoffwert bei einer Analyse an, wahrend die HeiBextraktion sozusagen nebenbei den Wasserstoff- und Stickstoffwcrt liefert.
3. Physikalische Varfahrcri Srit cinigen Jahren bemuht man sich, Sauerstoff-
bestimmungen uncl Gasbestimmungen uberhaupt spek- trographisoh durchzufuhren. Obwohl hierbei besondere Schwierigkeiten auftret'en, wird dieser Weg weiter ver- folgt, um zu kiirzeren Analysenzeiten zu kommen. Die Anregung muB hierbei unter vollstandigem Luft-
abschluB durch Verwendung von Vakuum oder einem. Edelgas erfolgen. Da die giirwtigen Spektrallinien doe Sauerstoffs im Bereich von 900 bis 1350 A, also im W liegen, mu13 man ungunstigere Linien unter Steigerung der Anregungsbedingungen verwenden, z. B. das Tri- plett bei 7771 L\.
Die Beschaffenheit der Probe und das Volumen- . element, in dem tatsachlich der Sauerstoff zur Anre. I
gung kommt, spielen eine grofle Rolle. Oberflachen- nahe Bezirke einer Probe lieferten teilweise vijllig irreale Analysenwerte. Um einen realen Probequer- schnitt zur Anregung zu bringen, werden verschiedene Wege eingeschlagen.
Rosen [32] schmilzt die Probe in einem als Katode geschalteten Graphittiegcl, der sich in eingm argon- gefiillten Entladungsrohr befindet. Das aus dem Oxyd gebildete Kohlenmonoxyd wird spektrographiert. Fassel und Mitarbeiter [33] bringen die Probe in einem Platinbad, das sich in einer Graphithohlelektrode be- findet, mit dem Gleichstrombogen zurn Schmelzen. Dies geschieht in einer Argonkammer, die vor der Argonfiillung evakuiert wird. Aus der Schmelze ent- weicht primiir Kohlenmonoxyd wie bei der HeiD- extraktionsanalyse, das aber im Bogen in Kohlenstoff und Sauerstoff zerfallt. Fassel hat rnit dieser Methode Sauerstoff in Ti, Zr, Nb, Ta, Th und in Stiihlen be- stimmt. Die Ergebnisse zeigten gute Ubereinstimmung mit Werten nach dem HeiBextraktionsverfahren und der Bromierungs-Kohlenstoff-Reduktionsmethode.
Wiihrend die beiden genannten Verfahren sozusagen Teile des HeiBextraktionsverfahrens iibernommen haben, gehen Koch und Mitarbeiter [34] etwas weiter und fuhren eine normale HeiBextraktion aus, nur da8 sie das abgepumpte Gas spektrochemisch analysieren. Von dem gesammelten Extraktionsgasgemisch wird cin Teil, etwa 0,15 cm3, in ein vorher evakuiertes Ent- ladungsrohr eingelassen und dort durch Hochfrequenz angeregt. Es wird also elektrodenlos gearbeitet ! Das emittierte Licht pamiert ein Quarzfenster, wird mono- chromatisiert und iiber einen SEV gemessen. Diese apparativ sehr anspruchsvolle Methode ist noch ent- wicklungsf ahig .
Prinzipiell kann zu allen genannten spektrometri- schen Methoden bemerkt werden, daB ihr Einsatz nur dann gerechtfertigt erscheint, wenn eine schon vgr- handene spektrometrische Einrichtung nur mit mog- lichst einfachen Mitteln erganzt zu werden braucht. Die von Fame1 vorgeschlagene Methode bringt wohl in dieser Richtung die meisten Vorteile.
Eine wesentliche Verkiirzung der Analysenzeiten ist aber im Vergleich zu modernen HeiBextraktionsan- lagen kaum zu erwarten. Auch bei spektrographischen Methoden mussen die Proben sorgfaltig vorbereitet in ein Vakuum- oder Inertgassystep eingebracht werden. Eine Verkurzung der Analysenzeit beim Ppek- trometrieren ist zum Beispiel gegeniiber dem J h s a t z eines Gaschromatographen nicht einmal garantiert. Moglicherweise ist aber cler Gaschromatograph billiger. uber diese Entwicklungstendenz wird die Zukunft ent-, scheiden.
Etwa seit 1953 sind Isotopenmethoden zur Sauer- stoffbestimmung in Fe, Cr, Cu, Ti und Zr bekannt geworden. Kirshenbaum und Mitarbeiter [35] haben cine Sauerstoffisotopenautauschmethode nach dem Vorbild von Hevesy und Paneth ausgearbeitet. Hierbei
Z. Uliom., 1. Jg., HcfL 12, 1961 359

wird die zu untersuchende Probe zusammen mit einer Vergleichsprobe, deren Verhaltnis la0 : l60 genau be- kannt ist, und Graphit im Vakuum weit oberhalb des Schmelzpunktes eingeschmolzen. Im Ofenraum stellt sich ein Gleichgewicht der Sauerstoffverbindungen ein, in dem das neue Verhaltnis l S 0 : la0 massenspektro- metrisch vermessen wird. Vorteilhnft bei dieser Arbeits- weise ist, daB keine quantitative Extraktion niitig ist. Dagegen mu13 aber die Temperatur so hoch gewiihlt werden, daD ein voUstZindiger statistischer Austausch dler Sauerstoffatome (Me0 ++ CO) gesichert ist. Die Methode ist apparativ sehr anspruchsvoll, so da13 bisher keine Routinebestimmungen ausgefiihrt wurden.
Neuerdings sind mehrere aktivierungsanalytische Methoden zur Sauerstoffbestimmung in Metallen be- kannt geworden. Osnwnd und Smales [36] entwickelten eine Methode, bei der die Metallprobe mit Lithium- fluorid gemischt im Reaktor mit Neutronen beschossen wird. Dabei spielen sich folgende Reaktionen ab:
(7)
Die so erzeugte Tritiumquelle setzt den Sauerstoff um nach
(8) n)
Das Fluor-18 ist bequem meBbar, da es unter Aus- sendung von Positronen (/?+-Strahler) mit einer Halb- wertszeit von 107 min zerfiillt.
Das Arbeiten mit Lithiurnfluorid ist fur kleine Sauerstoffgehalte nicht zu empfehlen, da im Lithium- fluorid Sauerstoff als Verunreinigung vorliegen kann. Born und Riehl [49] haben gezeigt, da13 durch Ver- wendung von Lithiummetall an Stelle des Fluorids diese Methode empfindlicher gemacht werden kann. uber eine neue direkte aktivierungsanalytische Sauer- stoffbestimmung in kompakten Berylliumproben be- richten Colemnn und Perkin [ 371. Gehalte von O,OOl% Sauerstoff sind damit noch bestimmbar. Hierbei wird die Probe direkt einem 14,6 MeV-NeutronenbeschuB ausgesetzt. Die Reaktion
160 18F.
1 6 0 16N (9)
liefert 16N, dessen /?-Zerfall bei einer Halbwertszeit von 7,4 s gemeasen werden mu& Der Gehalt an Verunreini- gungen, die bei Bestrahlung Produkte mit ebenfalls sehr kurzen Halbwertszeiten liefern, darf nur sehr niedrig sein.
Da alle diese Messungen meist nicht absolut durch- gefuhrt werden, mu13 man Proben mit bekannten Sauerstoffgehalten zur Eichung zur Verfugung haben. Hierin liegt ein Nachteil aller dieser Methoden. An anderen physikalischen Methoden, wie z. B. der Ront- gen-Fluoreszenzanalyse wird zur Zeit intensiv gearbei- tet. Mulvey [38] berichtete 1960 in London uber die apparativen Verbesserungen, die es ermoglichen, leichte Elemente, wie Sauerstoff, Stickstoff und Koh- lenstoff mit Rontgenlicht zu untersuchen.
4. Metallographische Verfahreii
SchlieDlich sei noch auf die metallographischen Ver- fahren zur abschiibzenden Ermittlung des Sauerstoff-
gehalts eines Metalls hingewiesen. Praktische Bedeu- t'ung hat die Bestimmung der Oxydanteile im Kupfer auf Grund der Schliffuntersuchungen erlangt (, ,Ana- lyse der Metalle").
Im Titan, aber nicht in seinen Legierungen, ist die Brinellhiirte ein Ma8 fur den Sauerstoff- und Stick- stoffgehalt. Bei Kenntnis des Stickstoffgehalts kann der Sauerstoffgehalt aus der Hiirte berechnet werden.
Welohe von den zahlreichen genannten Methoden zur Sauerstoffbestimmung in Metallen in Zukunft dau Feld behaupten wird, ist heute nicht vorauszusehen. Vorlaufig scheint die HeiBextraktion das universellste und bisher zuverlassigste Verfahren zu sein. Alle an- deren Verfahren eignen sich mehr oder minder fiir spezicllere Probleme, wobei die rein physikalischen Methoden stark an Interesse gewinnen, wahrend die meist alteren chemischen Verfahren an Bedeutung verlieren.
Literatnr Turovceua. %. M . und Kunin. L . L.: ,4ncl.l~se der Gase in Metallen, .Mosk%u/Leningrad, .1959. Walker, W. H. und Patrick, W. A . : Orig.-(;n~n. 8th (!onyr. Applied Chem. 21, 139 (1912). Oberhoiier. P . und Beulell, 9.: Stahleisen 89, 1584 (1919). Thanheher, G. und Brauns, E. : Arch. Eisenhtittenwes. 0, 435 (1936): Thanheiser, G. hnd Ploum, H.: Mitt. Kaiser- Wilhelm-Inst. Eisenfomch. 19, 105 (1937). Sloman, H. A. : J. Inst. Metala 71, 391 (1945); Slomaib,ll.d., Haruey, C. A . und Kubanchnaski, 0.: J. Inst. Metals 80, 391 (1951-52); Sloman, H. -4. und Rooney, T . E.: 4th Rep. of the Oxygen-Sub-committee on the Heterogeneity of Stecl Ingots 1944.
I6 1 Unger, If. D Schmik, K . G., Gerdonk, 6. H. und Schenck, 11.: Arch. Eisenlktenwes. 80, 411 (1959).
171 Lange 2). Stocmeier: Arch. Eisenhlittenwes. 29, 95 (1958). 181 Feichtinger, H. : Arch. Eisenhlittenwes. 26, 127 (1955); Berg-
und hiittenmlinn. Mh. montan. Hcchschule Leoben 100, 230 (1955).
I01 W n e r , W. C. und Beach, A . L.: Analytic. Cheni. $2, 366 (1950).
[lo1 Ramleu, C. E.: Analyst 72, 504 (1947). [111 Brewer, L.: U. S. Atom. Energy Comni. Report MDDC 366
(1946). Niebuhr, J . : 3. Plansee-Seminar, H. 313 (1958). Elwell, W . T . : The determin. of gases in metals, London 1960, 24, Spec. Report Nr. 68. Fowler, R. M . : Symposium on Titanium, ASTM Spec. Techn. Public. Nr. 204, 204 (1957). Beach, A . I,. und Uvldnor , I F r . C.: Analytir. Ghcni. 81, 1718 i i a m t ,'"""I.
1161 Smile$!, M - . ( f . : Analytic. Chem. $7, 1098 (1055): Synlp. of Determin. of Gases in Metals, ASTM Spec. Techu. Public.
[ I71 Abresch, K . und Lemm, H . : Arch. Eisenhiittenwes. 80, 1 i i a m t
mas, 25 (1957).
\A""",.
Yanagisawa, S., Seki, M . und Wdanabe, Y . : Wkrochim. Aota 1, (1059); siehe auch IUPAC-KongreP Liseabon 1956. Feichtinger, H . , Bhehthold, H . und Schuhknecht, W . : Schweiz. Arch. angew. WISE. Techn. 86, 426 (1959). Lesser, R. und Gruber, H.: Z. Metallkunde 51, 495 (1960). Analyst? der Metalle, II/2. Berlin 1953, 352. Hartmann, H. und Slr(ihZ, G . : Z. analyt. Chem. 144. 332 (1955); Harlmann, H. , Hotmaan, W . und Nchzclle-Schrepgiirn, K.-H.: Z. Metallkunde 48, 350 (1952). Eberiwr, E. und Kowakki, W . : Erzmetnll 7 , 229, 339 (1954). Fischer, W . und Mehlhorn, R.: Metall, ini Erscheinen. Babko, A. K . , Volkova, A . I . und B a k o , C'. F.: Zavod. Lab. 88, 136 m m ) . Kleiner, K . E. : YE~REHCKE& Xavnrecm* XypHan LUkrain. chem. J.1 $2, 809 (1956); Kleiner, K . E. und Markova, L. V . : Ebendn $8, 236 (1957). Klinger, P . und Koch, W . : Beitrwe znr metallkundlichen Anabse, Diisaeldorl: 1949. Elwell, W . T . und Peake, U . M . : Analyst 81, 734 (1957). Codell, M . und Norwitz, G . : Analytir. Chern. 97, 1083 (1955); 28, 2006 (1956); 80, 524 (1958). Hoekstra, N . R. und Katz, I . 1.: -4nalytic. (!hem. $6, 1608 (1953). Dupraw, M'. -4. und O'Neill, H . J.: Analytic. ('hem. 81, 1104 (1959). Rosen, B. : Rev. univ. Mines, MBttLllurg. Trav. publ., Sci Arts appl. Ind. 0, 445 (1953). Fassel, V . -4.. Gordon. W . A . und Tabeliag, R. W . : Synip. on Determin. of Owes in Metals, ASTM Spec. Techn. Publ. 228 (1957). Koch, W., Eckhard, S . und Stricker, F . : Arch. Mtsenhlitteu- wes. 80, 137 (1959). Kirehenbaum, A . D . , Monsmann, R. A . und Grossc, A. V. : Trans. Amer. SOC. Metals 46, 525 (1954). Osmond, R. G. D. und Rmalee, .+. -4.: Analytdcn cliini. Act11 10, 117 (1954). Coleman, R. F. und Perkin, J . L.: Analyst 84, 233 (195L)). Muluey, T.: Symp. on the Determin. cf Oases in Metals, London 1960, 255.
360 Z. Chem.. 1. Jg., Hett18, 1961

Booth, E. , Bvanl , F . J . und Parker, A . : Analyst 82, 50 (1957). Stanley, J . K., 2'. Iloenc, J . und Wieaer , 0.: Analytic. Chem. 28. 377 (1951). Behrh, A : I,. und Gi~ldner , I4.. (7. : ASTM Rpec. Techn. Public. 222, 15 (1957). Malleft, Jf. Jt'.: Trans. .4mrr. SOC. Metals 41, 870 (1949). Smiku , 11.. H.: ASTM Spec. Techn. PiibI. S?2, 25 (1957). K o p a , I d . : Hntiiick& Listy 14, 382 (1Y59). Fowler, H. A f . , Imicmter , T. ('. und Porler, U.: Uuctilc ~'liromliini, C'lrvrlnntl 1957, 121.
146 1 Turevreva, 2. M . und Lifvinova, N . F. : Proceed. of the Sth U. N. Internat. Conf. on the Peaceful Uaea of Atom Energy 1958, 28, 592.
[471 Mallell, M . W . und Hansen, W . R.: The Metal Molybdenum, Cleveland 1958, 365.
[481 FaoeZ, I . R., Witbeck, R. F . und Smith, H . A. : Analytic Chem. 81, 1115 (1959).
I491 Born, H . .I. und Riehl, A'.: A n ~ e w . ('hem. 72, 559 (1960).
ZCA 167 Eingegangen am 7. J u n i 1961
Elektronenresonanz-Spektren einiger Anthrasemichinone unter photochemischen, polarographischen und katalytischen Bedingungen
Von B. Elschner und R. Neubert
Physikdisches Institut der Friedrich-Schiller-Universitiit Jena (Direktor: Prof. Dr. U'. Schiilz)
und H. Berg und D. Tresselt Institut fiir Mikrobiologie und experirnentelle Therapie der DAW zu Berlin, Jena (Direktor: Prof. Dr. H . Knoll)
Eiiileituiig Die ubliche chemische Erzeugung von Semichinonen
ist vielfach mit Nebenreaktionen verbnnden, welche Elektronenresonanz-Messungen unter Umstanden er- schweren. Nur in wenigen Fallen [l] ist bisher die elektrolytische Darstellung mit anschlieaender Elek- tronenresonanz (EPR)-Messung versucht worden, je- doch konnte dabei die bereits bekannte Hyperfein- struktur (HfS) des Anthrasemichinons [2], [6] infolge cxperimenteller Unzulanglichkeiten nicht registriert werden.
Um die gunstigsten Herstellungsbedingungen unter AusschluB von Sauerstoff kennenzulernen, haben wir die photochemische, polarographische und kataly- tische Reduktion am 2,6-substituierten Anthrachinon durchgefuhrt und die dabei entstandenen, im Gleich- gewicht stabilisierten Anthrasemichinone in bezug auf ihre EPR-Spektren verglichen. Weitere Anthrachinon- clerivate hahen wir vergleichsweise nur photochemisch reduziert und danach ebenfalls EPR-Spektren auf- genommen.
1. Experimentelles 1.1. Elektronenresonanz-Apperetur
Der Mikrowellen-Aufbau der Apparatur ist in [3] beschrieben. Es handelt sich um eine Brucken-Anlage mit magischem T und Reflexionshohlraum mit stetig variabler Ankopplung. Die Klystronfrequenz und das Magnetfeld sind elektronisch stabilisiert. Die Regi- strierung des Signals erfolgt hinter einem 700 Hz ,,Lock-in"-Verstarker. Bei einer Zeitkonstante von 1 s wurde eine NachweisemDfindlichkeit von M l 014 angepaarte Spins Oe-Lhienbreite gemessen. Die verwendeten MeBrohr-
chen hatten einen Innendurchmesser von 2 mm. Das Magnetfeld ( H , = 3300 Oe) wurde mit einem
WeiPschen Elektromagneten (10 cm Polschuhdurch- messer) erzeugt. Die relative Inhomogenitat uber den Bereich der Probe betragt dabei - = 1 . Hfs- Abstande im EPR-Signal von 0,5 Oe konnen also noch gut aufgelost werden. Alle Messungen wurden bei Zimmertemperatur in verdunnten Losungen durch- gefuhrt. Als Losungsmittel diente in den meisten Pal- len ein Gemisch aus 40% Isopropylalkohol und 60% Wasner mit einem Gehalt an Natriumhydroxyd von 0,1 m. Der Isopropylalkohol war uber Na destilliert.
A H Ho
1.2. Die Bestrahlungseinrichtung Das MeBrohrchen oder die Kapillare der Zelle nach
Bild 1, unter LuftabschluB mit der entlufteten Chinon: losung gefullt, wird zunachst abgeschmolzen oder im anderen Falle mit Stopfen verschlossen. Danach konnte die Belichtung wegen der Stabilisierung des Radikalanions aul3erhalb des Hohlraumresonators mit dem ungefilterten Licht einer Hg-Hochstdrucklampe HBO 500 im Abstand von etwa 25 cm erfolgen. Die Dauer der Belichtung richtet sich nach der Photo- reduktionsgeschwindigkeit, ist von Probe zu Probe verschieden und schwankt zwischen 30 s und 3Qmin.
1.8. Die polerogrephische Apperatur Wenn auch an Stelle der klassischen Hg-Tropfelek-
trode eine stationare Hg-Katode zur Erzielung hohe- rer Ausbeute an Semichinonen Verwendung fend, so sind damit doch die Bedingungen der polarographi- schen Reduktion erfullt. Die Elektroreduktion wurde zunachst im Hohlraumresonator zwischen Quecksilber- katode und Platinanode durchgefuhrt. Spiiter wurde eine fur alle drei Reduktionsmethoden geeignete Zelle bevorzugt (Bild 1). Sie eignet sich zur Darstellyng von sauerstoffempfindlichen, jedoch sonst stabilen Radi- kallosungen. Im vorliegenden Fall wird unter Wasser- stoff-Ruhrung die Chinonlosung bei einer Stromstiirke von etwa 0,3mA 10 Minuten lang reduziert. Den Reduktionsverlauf kann man dabei prinzipiell ebenso wie bei den Verfahren mit der Tropfelektrode polaro- graphisch verfolgen, um die reduzierte Chinonmenge
Bild 1 . Zelle zur Darstellung von Radikalen. Daa ROhrchen nit Halin und Schliff dient zun&chst der Entliittung und dam naoh Hochsaugen der LOsung und VerschlieDen ah MeUrtbhrchen. Boden- queckailber dient 8.18 Katode, Platinblech hinter der Fritte im rech-
ten Srhenkel als Anode
46 Z. Chem., 1. Jg., Heft 12, 1961 361





























