Embed Size (px)
Citation preview

ETH Zürich
Abteilung für Biologie XA
Eine Calciumabhängige Proteinkinase aus
Lycopersicon esculentum:
Klonierung, Expression und Charakterisierung
Institut für Pflanzenwissenschaften ETH Zürich
Biochemie und Physiologie der Pflanzen
Prof. Dr. N. Amrhein
Diplomarbeit
vorgelegt von
Urs Gregor Stalder
Betreut durch:
Dr. Andreas Schaller
Zürich
April 2000

Inhaltsverzeichnis
- II -
Inhaltsverzeichnis
1. Einleitung 1
1.1 Pflanzenabwehr 1
1.2 Induzierte Abwehr 1
1.3 Die Rolle der Plasmamembran H+-ATPase 6
1.4 Aufgabenstellung 7
2. Material und Methoden 8
2.1 Material 8
2.1.1 Lösungen und Medien 8
2.1.2 Pflanzenmaterial 13
2.1.3 Bakterienstämme 13
2.2 Molekularbiologische Methoden 14
2.2.1 Klonierung des 3’-Endes der CDPK cDNA in den pBluescript SK(-) Vektor 14
2.2.2 RACE-PCR und Klonierung in den pCR®2.1-TOPO Vektor 14
2.2.3 Isolierung von Plasmid-DNA 15
2.2.4 DNA-Sequenzanalyse 15
2.2.5 Gesamt-RNA Extraktion 16
2.2.6 Northern-Blot Analyse 16
2.2.7 Isolierung der Genomischen DNA 17
2.2.8 Southern-Blot Analyse 18
2.2.9 Klonierung der CDPK cDNA in pGEX-G 19
2.2.10 Transformation von E. coli DH5α mit pGEX-G/CDPK-L/K 19
2.2.11 Transformation von E. coli BL21-(DE3)-RIL mit pGEX-G/CDPK-L/K 20
2.2.12 Anzucht von E. coli BL21-(DE3)-RIL Zellen 20
2.2.13 Aufschluss von E. coli BL21-(DE3)-RIL Zellen 21

Inhaltsverzeichnis
- III -
2.3 Protein-Analytische Methoden 21
2.3.1 SDS-PAGE / Western-Blot 21
2.3.2 Reinigung der GST-CDPK Fusionsproteine 21
2.3.3 Bindung von 45Ca2+ 22
2.3.4 Autophosphorylierung immobilisierter CDPK 22
2.3.5 Phosphoaminosäuretrennung mittels DC 23
2.3.6 Aktivitätstest 24
3. Resultate 26
3.1 Klonierung des 3’-Endes einer CDPK aus Tomate 26
3.2 Southern Blot Analyse 28
3.3 Northern Blot Analyse 29
3.4 Amplifizierung des 5’-Endes mittels RACE-PCR 31
3.5 Amplifizierung und Klonierung von CDPK-L und –K 38
3.6 Expression der CDPK in E. coli 40
3.7 Reinigung des Fusionsproteins 43
3.8 Bindung von 45Ca2+ 44
3.9 Autophosphorylierung der CDPK 45
3.10 Phosphoaminosäure Trennung 46
3.11 Aktivität gegenüber synthetischen Peptiden 47
4. Diskussion 49
5. Literaturverzeichnis 54
6. Abkürzungsverzeichnis 61
7. Anhang 63

Zusammenfassung
- I -
Zusammenfassung Im Rahmen der vorliegenden Arbeit wurde eine cDNA einer Calciumabhängigen-
Proteinkinase (CDPK) aus Tomate (Lycopersicon esculentum cv. Castelmart II), die der von
Yoon et al. (1999) beschriebenen CDPK aus Tabak homolog ist, unter Einsatz der RACE-
Methode kloniert, heterolog exprimiert und charakterisiert.
Das offene Leseraster kodiert ein Polypeptid von 553 Aminosäuren und weist zu der CDPK
aus Tabak eine Aminosäuresequenzidentität von 94% auf. Durch Southern Blot Analyse
wurde gezeigt, dass eine Sonde, die der 3’-untranslatierten Region der cDNA entspricht,
spezifisch mit nur einem Gen der CDPK Genfamilie hybridisiert. Eine Induktion des
entsprechenden CDPK-Transkriptes nach Verwundung liess sich mittels Northern Blot
Analyse nicht nachweisen. Es konnte aber festgestellt werden, dass Transkripte der CDPK ca.
4 Stunden nach Fusicoccin-Behandlung von Tomatenpflanzen akkumulierten. Das Protein
wurde in Fusion mit Glutathion-S-Transferase in E. coli exprimiert und aus bakteriellen
Extrakten gereinigt. Mit radioaktiv markiertem 45Ca2+ konnte gezeigt werden, dass die C-
terminale, aber nicht die katalytische Domäne, Calcium bindet und folglich alleinig für die
Calciumbindefähigkeit der CDPK und vermutlich für deren Ca2+-abhängige Aktivierung
verantwortlich ist. Das gereinigte Fusionsprotein wies eine Ca2+-abhängige Ser/Thr-
Kinaseaktivität auf und war zur Autophosphorylierung in der Lage. Für das synthetische
Oligopeptid Substrat Syntide-2 wurde ein apparenter KM-Wert zwischen 82 und 89 µM
ermittelt.

Einleitung
- 1 - 1
1. EINLEITUNG
1.1 Pflanzenabwehr
Die Abwehrmechanismen von Pflanzen sind ihrer sessilen Lebensweise angepasst und daher
kaum mit den entsprechenden Vorkehrungen bei tierischen Organismen zu vergleichen.
Pflanzen im Freiland unterliegen einer Vielzahl von Herausforderungen: infektiösen
Mikroorganismen, pflanzenfressenden Tieren und konkurrierenden Arten sowie
Schwankungen von Temperatur, Strahlung und Wasserversorgung. Daher überrascht es nicht,
dass sie komplexe chemische Anpassungen entwickelten, die ihr Überleben sichern. Hierin ist
sicherlich eine Ursache für die beeindruckende Strukturvielfalt der Sekundärverbindungen zu
suchen, welche wohl erst adäquates Reagieren auf die unterschiedlichsten Herausforderungen
ermöglicht.
Offensichtlich sind jene Sekundärverbindungen, welche als chemische Schutzstoffe agieren,
unter dem Druck einer sich stetig wandelnden Umwelt entstanden; ihre Abwehrfunktion
wurde weiterentwickelt. So haben sich bestimmte Alkaloide unter Selektionsdruck als
zweckmässig für die Arterhaltung erwiesen und ihre Synthesekapazität wurde im Laufe der
Evolution optimiert.
Zur Abwehr von Herbivoren, Mikroorganismen und Viren stehen den höheren Pflanzen im
wesentlichen vier Strategien zur Verfügung: Akkumulation toxischer Verbindungen im
Gewebe, Bildung selbiger aus nichttoxischen Vorstufen bei Gewebeverletzung, Bildung
mechanischer Barrieren und induzierte Bildung von Schutzstoffen bei Angriff.
1.2 Induzierte Abwehr
Während bei den beiden erst genannten Mechanismen die Abwehrstoffe permanent
einsatzbereit sind, werden sie bei letzteren erst als Reaktion auf eine Infektion bzw.
Verletzung gebildet: induzierte Abwehr. Sie beinhaltet u. a. die Verstärkung der Zellwände
durch Quervernetzung und Kalloseablagerung, die Synthese von Phytoalexinen,
antimikrobiell wirksamen Substanzen von geringer Spezifität. Ihre Bildung beschränkt sich
auf den Ort der Infektion, wo bei inkompatibler Wechselwirkung zwischen Pathogen und
pflanzlichem Gewebe, meist lokal begrenzt, Zellen und Angreifer absterben und eine

Einleitung
- 2 - 2
Abwehrnekrose hervorrufen. Diese Gewebeläsionen werden dadurch „abgedichtet“, dass die
Zellwände der umgebenden noch lebenden Zellen Lignifizierung, Verkorkung und/oder
Imprägnierung mit Polyphenolen erfahren (Greenberg et al., 1994; Somssich und Hahlbrock,
1998). Phytoalexine fehlen in der gesunden Pflanze oder liegen nur in geringen Quantitäten
vor. Ihre de-novo-Synthese wird generell durch spezifische Signalsubstanzen, die Elicitoren,
ausgelöst. Dabei handelt es sich oft um Oligosaccharide, welche am Infektionsort als
Abbauprodukte der Zellwände von Pilz oder Pflanzen anfallen (Yoshikawa et al. 1993).
Induktion von Abwehr ist auch über die Aktivierung von Genen möglich, deren Produkte
dann eine Schutzfunktion gegenüber dem Schädling übernehmen. Beispiel eines solchen
Mechanismus ist die wundinduzierte Produktion und Akkumulation von Proteinase-
Inhibitoren bei Vertretern der Familien Cucurbitaceae, Fabaceae, Salicaceae und Solanaceae
(Ryan, 1992 und dort angegebene Referenzen; Constabel et al., 1998). Die Wundantwort ist
systemisch, das heisst, die Expression der Abwehrproteine erfolgt nicht nur in unmittelbarer
Umgebung der Verletzung, sondern zeitlich versetzt auch in weiter entfernten Teilen der
Pflanze. Die ursprünglich gefundenen Inhibitor-Proteine der Tomate wurden zwei Familien,
Inhibitor I und II, zugeordnet (Bishop et al., 1984). Angriffsziele dieser Inhibitoren sind
Serin-Proteinasen, wie sie im tierischen Verdauungstrakt vorliegen. Über das gefressene
Blattmaterial kann der Inhibitor in das Verdauungssystem einer Raupe gelangen. Diese stellt
bald darauf ihr Wachstum ein, weil die essentiellen proteinogenen Aminosäuren nicht mehr
aus dem Abbau der aufgenommenen Pflanzenproteine zur Verfügung stehen und damit die
Synthese körpereigener Proteine behindert ist (Orozco-Cardenas et al., 1993). Später wurde
gezeigt, dass die systemische Wundantwort die Akkumulation einer Vielzahl weiterer
Proteine beinhaltet (Schaller et al., 1995; Bergey et al., 1996), die unter dem Begriff SWRPs
(systemic wound response proteins) zusammengefasst sind. Dabei handelt es sich nebst
Proteinaseinhibitoren um Polyphenoloxidase (Constabel et al., 1995), Proteasen (Hildmann et
al., 1992; Walker-Simmons und Ryan, 1977; Pautot et al., 1993; Schaller und Ryan, 1996),
sowie um Proteine, die möglicherweise eine wichtige Funktion bei der Transduktion des
Wundsignals einnehmen (McGurl et al., 1992; Heitz et al., 1997; Bergey et al., 1999).
Eine systemische Induktion von SWRPs setzt die Existenz einer Signaltransfer-Kette unter
Beteiligung von Signalmolekülen voraus. Die Suche nach Molekülen mit
Signalisierungseigenschaften, hat eine Vielzahl von Verbindungen hervorgebracht, wie
Pektin- (Bishop et al., 1981), Chitosan- und Chitin-Fragmente (Walker-Simmons und Ryan,
1984) aus pflanzlichen bzw. pilzlichen Zellwänden, die Hormone Ethylen (O’Donnell et al.,
1996), Abscisinsäure (Pẽna-Cortés et al., 1989) und Jasmonsäure (Farmer und Ryan, 1992),

Einleitung
- 3 - 3
sowie Methyljasmonat (Farmer und Ryan, 1990) und das Oligopeptid Systemin (Pearce et al.,
1991), die alle an der Übermittlung des Wundsignals beteiligt zu sein scheinen.
Von der Verwundung bis zur Akkumulation der Abwehrproteine führt ein komplexer
Signaltransduktionsweg (Abb. 1). Nach der gängigen Modellvorstellung von Farmer und
Ryan (1992) entsteht im geschädigten Gewebe aus Prosystemin, einem Vorstufen-Polypeptid
aus 200 Aminosäure-Resten, durch proteolytische Spaltung das 18 Aminosäure-Polypeptid
Systemin als eigentliches Peptidhormon, welches über das vaskuläre System der Pflanze
transloziert wird (Narváez-Vásquez et al., 1995). Die Transkription von Prosystemin-mRNA
ist auf tiefem Niveau konstitutiv und wird durch Verwundung systemisch markant erhöht
(McGurl et al., 1992). Die Perzeption von Systemin durch seinen Rezeptor im Plasmalemma
einer intakten Blattzelle (Scheer und Ryan, 1999) führt zur Aktivierung einer bestimmten
Lipase, welche die Freisetzung von Linolensäure aus Membranlipiden katalysiert (Farmer und
Ryan, 1992; Narváez-Vásquez et al., 1999). Über mehrere Reaktionsschritte des sogenannten
Octadecanoid-Weges, entsteht aus α-Linolensäure die Jasmonsäure (Vick und Zimmermann,
1984). Als Signalsubstanz aktiviert Jasmonsäure auf bisher unbekannte Weise die SWRP
Gene.
Als erste messbare Antwort auf die Interaktion von Systemin mit seinem Rezeptor kommt es
zu veränderten Ionenströmen über die Plasmamembran. In Mesophyllzellen von
Tomatenpflanzen bewirkt Systemin eine rasche Depolarisation des Plasmamembran-
Potentials und in Zellkulturen von L. peruvianum eine Alkanisierungsantwort (Moyen und
Johannes 1996; Felix und Boller 1995). Inhibierung der H+-ATPase führt die Akkumulation
der Transkripte wundinduzierbarer Proteine mit sich (Schaller und Oecking, 1999). Dagegen
bewirkt eine Hyperpolarisation des Plasmamembran-Potentials durch Aktivierung der H+-
ATPase eine Unterdrückung der Alkanisierungsantwort und der Expression von
wundinduzierbaren Abwehrgenen. Aufgrund dieser Daten besteht der Verdacht, dass die
Depolarisation des Plasmamembran-Potentials für die Auslösung der Wundantwort
notwendig ist. Dabei könnte die Inhibierung der H+-ATPase Aktivität eine Vermittlerrolle
spielen.
Die systemin-induzierte Alkanisierungsantwort in L. peruvianum erfordert den Einstrom von
Ca2+-Ionen und die Aktivität einer Proteinkinase (PK) (Schaller und Oecking, 1999). Somit
könnte ein Zusammenhang zwischen dem Calciumeinstrom und/oder der Proteinkinase und
der Inhibierung der H+-ATPase bestehen. Einen indirekten Beweis für die Beteiligung von
Ca2+ im Wundsignaltransduktionsweg erbrachten Bergey und Ryan (1999). Sie beobachteten
einen Anstieg der Calmodulin mRNA und Protein-Konzentration in den Blättern von

Einleitung
- 4 - 4
Tomaten nach Verwundung, sowie bei Fütterungsexperimenten mit Systemin,
Methyljasmonat und Linolensäure. Sie stellten weiter fest, dass die Induktion der
Calmodulinexpression zeitlich mit der von anderen Signalisierungskomponenten
zusammenfällt, sich jedoch von der späteren Expression der Abwehrgene unterscheidet.
Ob Calmodulin aber an der Regulation der Aktivität einer Proteinkinase beteiligt ist, ist noch
unklar. Wahrscheinlicher, weil erheblich rascher, ist die direkte Aktivierung einer Calcium-
abhängigen Proteinkinase durch erhöhten Spiegel cytosolischen Calciums, wie von Schaller
(1999) in einem Modell der systemin-induzierten Wundantwort vorgeschlagen (Abb. 1).
Nach diesem Modell bewirkt die Interaktion von Systemin mit seinem Rezeptor SR-160 einen
Anstieg der freien cytoplasmatischen Calciumionen-Konzentration, hervorgerufen durch das
Öffnen von plasma-membranständigen Ca2+ Kanälen, sowie das Entlassen von organell-
gebundenem Ca2+. Als Folge der erhöhten cytosolischen Ca2+-Konzentration kommt es zur
Inhibierung der H+-ATPase, wobei die Beteiligung einer Calcium-abhängigen-Proteinkinase
(CDPK) vermutet wird. Die Depolarisation des Plasmamembran-Potentials könnte den
Anstieg des Ca2+-Gehalts verstärken und so zur Aktivierung einer Phospholipase A2 (PLA2)
beitragen. Die Aktivität der PLA2 scheint weiterhin durch eine Proteinkinase aus der Familie
der Mitogen-aktivierten Proteinkinasen (MAPKs) kontrolliert zu sein (Stratmann und Ryan,
1997; Ryan und Pearce, 1998). Phospholipid-hydrolyse durch eine aktivierte Phospholipase
setzt Linolensäure als Ausgangssubstrat der durch den Oktadekanoid-Biosyntheseweg
gebildeten Jasmonsäure frei. Letztere aktiviert ihrerseits auf bisher unbekannte Weise die
Abwehrgene.

Einleitung
- 5 - 5
Abb. 1: Modell des durch Systemin ausgelösten Signalisierungsweges; nach Schaller (1999), modifiziert.

Einleitung
- 6 - 6
1.3 Die Rolle der Plasmamembran H+-ATPase
Die H+-ATPase der pflanzlichen Plasmamembran ist ein Enzym von etwa 100 kDa. Ihre
Funktion liegt in der Schaffung eines transmembranen H+-Gradienten. Das errichtete H+-
elektrochemische Potential nutzt die Zelle für den Antrieb vieler transmembranen
Transportvorgänge aber auch zur Regulation des intrazellulären pH-Wertes sowie des Zell-
Turgor (Serrano 1989, 1990; Michelet und Boutry 1995). In Anbetracht der wichtigen
Funktionen, die diesem Gradienten zukommen, und dem enormen Energieverbrauch der zu
dessen Aufrechterhaltung erforderlich und der bis zu einem Viertel des zellulär gebildeten
ATPs ausmachen kann, ist es nicht erstaunlich, dass die H+-ATPase komplexen
Regulationsvorgängen unterworfen ist. Ihre Aktivität wird in vivo über die Autoinhibitor-
Domäne am C-Terminus reguliert. Bei derer Entfernung stellt sich eine deutliche Steigerung
der Aktivität ein (Palmgren et al., 1991).
Das Pilzgift Fusicoccin von Fusicoccum amygdali Del. aktiviert die H+-ATPase durch
Interaktion mit ihrer Autoinhibitor-Domäne (Johansson et al., 1993). Es konnte gezeigt
werden, dass Fusicoccin einen Komplex aus 14-3-3 Proteinen mit dem C-Terminus der
ATPase stabilisiert (Jahn et al., 1997; Oecking et al., 1997; Baunsgaard et al., 1998).
Die Interaktion von 14-3-3 Proteinen mit dem C-Terminus der H+-ATPase hängt von dessen
Phosphorylierungsstatus ab. Nur wenn ein kritischer Threoninrest im C-Terminus der
Protonenpumpe phosphoryliert vorliegt, ist eine Bindung von 14-3-3 Proteinen und damit die
Aktivierung der ATPase möglich (Svennelid et al., 1999). Weiterhin wurde die
Phosphorylierung eines Serinrestes zwischen Transmembransegmenten 8 und 9 als wichtig
für die 14-3-3 Proteinbindung beschrieben (Marra et al., 2000). Bereits Schaller und Sussman
(1988) zeigten die Ca2+–abhängige Phosphorylierung an Serin- und Threonin-Resten der
Plasmamembran H+-ATPase. Phosphorylierungsereignisse, die die Bindung von 14-3-3
Proteinen ermöglichen würden zu einer Aktivierung der Protonenpumpe führen. In der Tat ist
eine Aktivierung der ATPase durch Phosphorylierung beobachtet worden (Bidway und
Takemoto, 1987; Suzuki et al., 1992). Desweitern scheint aber auch eine Inaktivierung durch
Phosphorylierung möglich zu sein (Vera-Estrella et al., 1994; Xing et al., 1996; Lino et al.,
1998, De Nisi et al., 1999).
Die für diese beiden Phosphorylierungsereignisse verantwortlichen Proteinkinasen sind
bislang nicht identifiziert worden. Es wird aber vermutet, dass daran calciumabhängige
Proteinkinasen (CDPKs) beteiligt sind (Camoni et al., 1998; De Nisi et al., 1999). CDPKs
weisen eine für sie charakeristische Struktur auf. Die N-terminale Kinase Domäne ist über ein

Einleitung
- 7 - 7
autoinhibitorisches Verbindungsstück mit der calmodulin-ähnlichen Domäne am C-Terminus
verbunden (Harper et al., 1994; Harmon et al., 1994). Einige dieser CDPKs fand man als
plasmamembranständige Enzyme in verschiedenen Spezies (Schaller et al., 1992; Verhey et
al., 1993).
Kürzlich wurde von Yoon et al. (1999) eine CDPK aus Tabak kloniert. Einige Hinweise
zeigen an, dass es sich bei dieser CDPK möglicherweise um die Kinase handelt, die für die
Inaktivierung der H+-ATPase nach Verwundung verantwortlich sein könnte. Denn zum einen
ist die von Yoon et al. (1999) beschriebene Kinase auf transkriptioneller Ebene
wundinduzierbar, zum anderen handelt es sich dabei um ein plasmamembranständiges
Enzym, dessen Substrat sich wahrscheinlich ebenfalls in der Plasmamembran befindet.
Zudem wird die Aktivität der CDPK durch Ca2+ erhöht. All dies sind Kriterien, die für die
Proteinkinase des Wundsignalwegs zu erwarten sind.
1.4 Aufgabenstellung
Im Rahmen der vorliegenden Arbeit sollte daher die Klonierung und Charkerisierung einer
CDPK aus Tomate (Lycopersicon esculentum cv. Castelmart II) versucht werden, die der von
Yoon et al. (1999) beschriebenen CDPK aus Tabak entspricht. Ein Datenbankvergleich mit
der CDPK aus Tabak wies auf die Existenz mehrerer ESTs der Tomate hin, die hohe
Sequenzübereinstimmung mit dem Tabakenzym aufwiesen. Unglücklicherweise umfasste der
EST mit der höchsten Sequenzübereinstimmung lediglich das 3’-Ende der cDNA, welches die
3’ untranslatierte Region und nur einen kleinen Teil der kodierenden Sequenz beinhaltete.
Ausgehend von dieser Sequenzinformation sollte sowohl das 3’-Ende als auch das 5’-Ende
der cDNA unter Einsatz der RACE-Methode kloniert werden. Die CDPK sollte anschliessend
heterolog exprimiert und charakterisiert werden.

Material und Methoden
- 8 - 8
2. MATERIAL UND METHODEN
2.1 Material
2.1.1 Lösungen und Medien
Die Chemikalien stammen, sofern nichts anderes angegeben ist, von FLUKA Chemie AG,
Buchs SG, Schweiz.
LB-Medium: 2.5% w/v LB Broth, Miller
(Difco Laboratories, Detroit, MI, USA)
2x YT Medium: 1.6% w/v Bactotrypton (Difco)
1.0% w/v Bactoyeastextrakt (Difco)
0.5% w/v NaCl
Plasmid DNA -, genom. DNA Isolierung
Mini-Prep Puffer: 25 mM Tris/HCl, pH 7.5
50 mM Glucose
10 mM EDTA
CTAB-Extraktionspuffer: 2% w/v CTAB
100 mM Tris/HCl, pH 8
20 mM EDTA, pH 8
1.4 M NaCl
CTAB-NaCl Puffer: 10% w/v CTAB
50 mM Tris/HCl, pH 8
10 mM EDTA, pH 8

Material und Methoden
- 9 - 9
CTAB/NaCl Fällungspuffer: 1% w/v CTAB
50 mM Tris/HCl, pH 8
10 mM EDTA, pH 8
Sequenzierung, Northern-, Southernblotting
20x SSC: 173.3 g NaCl
100 g Natriumcitrat x 3 H2O
aqua dest. ad 1 Liter
pH 7, autoklaviert
50x Denhardt’s Reagenz: 5 g Ficoll
5 g Polyvinylpyrrolidon
5 g Rinderserum Albumin
aqua dest. ad 500 ml
steril filtriert
1 M Kaliumphosphatpuffer: 1 M K2HPO4
1 M KH2PO4
pH 7
aqua dest. ad 1 Liter
10x STE: 0.1 M NaCl
10 mM Tris/HCl
1 mM EDTA
pH 8, autoklaviert
TE: 10 mM Tris/HCl
1 mM EDTA
pH 8, autoklaviert

Material und Methoden
- 10 - 10
1x TAE: 40 mM Tris/AcOH, pH 7.8
1 mM EDTA
autoklaviert
6x Ladepuffer: 0.25% w/v Bromphenolblau
0.25% w/v Xylencyanol
30% v/v Glycerin
10x Gelpuffer: 0.4 M MOPS
100 mM Natriumacetat
10 mM EDTA
pH 7, autoklaviert
Dentaturierungslösung: 1.5 M NaCl
0.5 M NaOH
Neutralisierungslösung: 1.5 M NaCl
0.5 M Tris/HCl
0.5 M EDTA
pH 8, autoklaviert
Hybridisierungslösung: 50% Formamid
5x SSC
50 mM Kaliumphosphatpuffer, pH 7
2x Denhardt’s
100 µg/ml Heringsspermien-DNA
0.5% w/v SDS
Sequenzierladepuffer: 80% Formamid
20% 25 mM EDTA
50 mg/ml Blue Dextran

Material und Methoden
- 11 - 11
1% Agarose Gel: 1 g Agarose
100 ml aqua dest.
1x TAE
10 µl EtBr (5 mg/ml)
Glutathion Sepharose 4B Affinitätschromatographie
Puffer A: 10 mM NaCl
1 mM EDTA
50 mM Tris/HCl, pH 8
1 mg/ml Lysozym
1 mM PMSF
Spatelspitze DNAse / pro 30 ml
Waschpuffer: 1% Nonidet 40 (NP 40) inPuffer A
Elutionspuffer: 5 mM red. Glutathion in 50 mM Tris/HCl,
pH 8
SDS-PAGE, Westernblotting
Trenngelpuffer: 1.5 M Tris/HCl, pH 8.8
0.4% SDS
25% Glycerin
Sammelgelpuffer: 0.5 M Tris/HCl, pH 6.8
0.4% SDS
30% Monomerengemisch: 29.2% Acrylamid
0.8% Bisacrylamid

Material und Methoden
- 12 - 12
2x Probenpuffer: 1.5 ml Sammelgelpuffer
1 ml 20% SDS
0.5 ml Glycerin
0.5 ml Mercaptoethanol
5 ml aqua dest.
Spatelspitze Bromphenolblau
10x Laufpuffer: 30 g Tris
144 g Glycin
10 g SDS
aqua dest. ad 1 Liter
Blottingpuffer: 25 mM Tris
192 mM Glycin
20% v/v Methanol
aqua dest. ad 1 Liter

Material und Methoden
- 13 - 13
2.1.2 Pflanzenmaterial
Für alle Untersuchungen wurde Lycopersicon esculentum cv. Castelmart II Tomatenpflanzen
eingesetzt. Ihre Anzucht erfolgte im Phytoschrank mit einem hell/dunkel Zyklus von 16.5 h
Licht (>140 µmolm-2s-1, 25°C) und 7.5 h Dunkel (18°C) bei 75% relativer Luftfeuchtigkeit.
Im Alter von 2-3 Wochen wurden sie zur Isolierung genomischer-DNA oder für
Fütterungsversuche mit Fusicoccin (3 µM) eingesetzt. In beiden Fällen wurde das gesamte
Sprossmaterial oberhalb der Kotyledonen verwendet.
2.1.3 Bakterienstämme
E. coli TOP10F’ F’{laclq Tn10 (TetR)} mcrA ∆(mrr-hsdRMS-
mcrBC)Φ80lacZ ∆M15 ∆lacX74 recA1 deoR
araD139 ∆(ara-leu)7697 galU galK rpsL(StrR)
endA1 nupG
E. coli DH5α supE44 hsdR17 recA1 endA1 gyrA96 thi-1
relA1
E. coli BL21-CodonPlus (DE3)-RIL F’ ampT hsdS (rB- mB
-) dcm+ Tetr galλ(DE3)
endA Hte [argU ileY leuW Camr]

Material und Methoden
- 14 - 14
2.2 Molekularbiologische Methoden 2.2.1 Klonierung des 3’-Endes der CDPK cDNA in den pBluescript SK(-) Vektor
Mit der Polymerase Kettenreaktion (PCR) wurde das 3’-Ende der CDPK cDNA aus cDNA-
Banken von der Tomatenblüte und dem Tomatenspross amplifiziert und mit
Restriktionsschnittstellen für die Enzyme EcoRI und XhoI versehen. Die PCR Reaktion
erfolgte in einem 50 µl Ansatz. Dieser enthielt 200 ng des Templats, 5 µl 10x M-MLV RT
Reaktionspuffer (Promega, Madison,WI, USA), 5 µl 5‘-Primer (10 µM), 5 µl 3‘-Primer (10
µM), 3 µl 25 mM MgCl2, 1 µl 10 mM dNTP’s und 26 µl nukleasefreies Wasser. Zudem
wurde der Mix mit einem TaqBeadTM (Heiss-Start-Polymerase, Promega) versetzt und mit 2
Tropfen Mineralöl überschichtet.
Das PCR-Programm umfasste 45 Zyklen (95 °C, 30 sec; 60 °C, 45 sec; 72 °C, 1 min). Die
DNA wurde durch eine Ethanolfällung aus dem Reaktionsansatz gereinigt, mit den Enzymen
EcoRI und XhoI restringiert, über ein 1 % (w/v) Agarose-Gel aufgetrennt und mit dem Biorad
Kit prep@gene aus dem Gel eluiert. Der pBluescript SK(-) Vektor wurde in gleicherweise
restringiert, mit CIAP (2 U Calf Intestine Alkaline Phosphatase) dephosphoryliert und durch
Agaroseelektrophorese gereinigt. Die Ligation mit T4-DNA Ligase wurde bei 16°C über
Nacht durchgeführt. Transformation von E. coli DH5α erfolgte auf gleiche Weise wie in
Abschnitt 2.2.10 beschrieben.
2.2.2 RACE-PCR und Klonierung in den pCR®2.1-TOPO Vektor
Die Amplifizierung des 5’ Endes der unvollständigen CDPK-cDNA erfolgte mit Hilfe des
„Smart™ RACE cDNA amplification Kit“ von Clontech (Palo Alto, Cal., USA). In einem
ersten Schritt erfolgte die Synthese der einzelsträngigen cDNA aus der Gesamt-RNA mittels
reverser Transkriptase. Dabei wurde das Protokoll des Herstellers unverändert übernommen
(im Manual Seite 8). Die auf diese Weise erhaltene einzelsträngige cDNA diente als Matrize
für die 5’RACE PCR. Abgesehen vom PCR-Programm wurde auch hier die Anleitung des
Herstellers (im Manual Seite 11) befolgt. Die dazu verwendeten Primer sind im Anhang
aufgeführt. Das leicht modifizierte „touch down“ PCR-Programm umfasste je 5 Zyklen
(94°C, 30 s; 74°C, 3 min), (94°C, 30 s; 72°C, 3 min), (94°C, 30 s; 70°C, 30 s; 72°C, 3 min)

Material und Methoden
- 15 - 15
sowie 35 Zyklen (94°C, 30 s; 68°C, 30 s; 72°C, 3 min). Die PCR-Produkte wurden durch
Agarosegelelektrophorese gereinigt und mit dem Biorad Kit prep@gene aus dem Gel eluiert.
Zur Klonierung wurde das TOPO TA Clonig® Kit von Invitrogen (Groningen, Nierderlande)
verwendet und gemäss Protokoll des Herstellers eingesetzt. Rekombinante Klone wurden auf
Vollmedium in Gegenwart von Ampicillin, IPTG und X-Gal selektioniert.
2.2.3 Isolierung von Plasmid-DNA
Plasmid-DNA wurde nach dem Prinzip der alkalischen Lyse isoliert (Birnboim und Doly,
1979). Die Anzucht der Bakterien erfolgte in 2 ml LB/Amp-Medium (100 ng Ampicillin/ml
LB-Medium) über Nacht bei 37°C und 220 rpm.
Die Übernacht-Kultur wurden bei 14'000 rpm für 2 min abzentrifugiert, der Überstand
verworfen und das Pellet in 100 µl Mini-Prep Puffer resuspendiert. Nach Zugabe von 150 µl
0.2 N NaOH, 1% (w/v) SDS und 5 min Inkubation auf Eis, wurde die Suspension ergänzt mit
150 µl 3 M Kaliumacetat und für weitere 5 min auf Eis gestellt. Durch Zentrifugation wurde
das Präzipitat entfernt und der Überstand wurde in ein neues Reaktionsgefäss mit 800 µl
vorgelegtem Ethanol transferiert. Es folgten weitere 15 min Inkubation auf Eis und 15 min
Zentrifugation der präzipitierten Plasmid-DNA, die anschliessend in 200 µl TE mit 1µl
pankreatischer RNAse (10 mg/ml) aufgenommen und 30 min bei 37°C inkubiert wurde.
Daran schloss sich je eine Extraktion mit Phenol und mit Phenol/Chloroform (1:1) an, gefolgt
von einer weiteren Ethanolfällung. Das Präzipitat wurde in 50 µl H2O aufgenommen und bei
–20°C gelagert.
2.2.4 DNA-Sequenzanalyse
Die DNA-Sequenzierung erfolgte nach der von Sanger et al. (1977) beschriebenen
Kettenabbruchmethode unter Einsatz von fluoreszenzmarkierten Dideoxynukleotiden. Diese
waren im Reaktionsmix enthalten (Perkin Elmer, Rotkreuz, Schweiz), ebenso die Taq-
Polymerase und der Puffer. In einem PCR-Ansatz von 10 µl wurden 250-500 ng Plasmid-
DNA, 1.6 pmol Primer und 4 µl Reaktionsmix eingesetzt. Es wurden 25 Reaktionszyklen wie
folgt durchgeführt: 95°C, 30 s; 50°C, 15 s; 60°C, 4 min. Die Reaktionsprodukte wurden mit

Material und Methoden
- 16 - 16
Ethanol gefällt, in 4 µl Ladepuffer aufgenommen und 2 min bei 95°C denaturiert. Die
Trennung der Nukleotidstränge erfolgte über ein 5% (w/v) Polyacrylamid-Gel in Gegenwart
von 8 M Harnstoff. Als Laufpuffer wurde 1x TBE verwendet. Die Elektrophorese dauerte bei
2500 V und 38 W 17 Stunden (ABI 373 DNA Sequencer, Perkin Elmer). Analysiert und
ausgewertet wurden die Daten mit Hilfe des Software-Paketes der University of Wisconsin
Genetics Computer Group.
2.2.5 Gesamt-RNA Extraktion
Die Gesamt-RNA wurde aus zwei Wochen alten Pflanzen L. esculentum cv. Castelmart II
gewonnen, die in flüssigem Stickstoff tiefgefroren und bis zur Verwendung bei –80°C
gelagert worden waren. Das gefrorene Pflanzenmaterial von jeweils 6 Pflanzen (ca. 0.8 g
Frischgewicht) wurde mit Mörser und Pistill in flüssigem Stickstoff zu einem feinem Pulver
zerrieben.
Die Extraktion erfolgte mit 300 µl Phenol, equilibriert in 100 mM Tris/HCl, pH 8, und 300 µl
Tris/HCl, pH 8. Die wässrige Phase wurde anschliessend vier weiteren Phenol- (equilibriert
in 100 mM Tris, pH 8), sowie einer Phenol/Chloroform (1:1) Extraktion unterzogen. An die
sich Ethanol-, Lithiumchlorid- und Ethanolfällungen anschlossen. Aufgenommen wurde das
Pellet in 50 µl TE. Die RNA-Konzentration wurde anhand der Absorption bei einer
Wellenlänge von 260 nm bestimmt und auf 1 µg/µl eingestellt.
2.2.6 Northern-Blot Analyse
Es wurden 5.5 µl RNA (1µg/µl) mit 1 µl 10x Formaldehydgelpuffer (Sambrook et al., 1989),
3.5 µl Formaldehyd und 10 µl Formamid gemischt, während 15 min bei 65°C denaturiert und
auf Eis abgekühlt. Nach Zugabe von 2 µl Formaldehyd-Ladepuffer wurde die Probe auf
einem 1.2% (w/v) Agarosegel in Gegenwart von 6.5% (v/v) Formaldehyd während 5 h bei
120 V elektrophoretisch aufgetrennt. Nach erfolgter Elektrophorese wurde das Gel 2x 20 min
in Wasser gewaschen. Die RNA wurde entweder mit Ethidiumbromid angefärbt oder mittels

Material und Methoden
- 17 - 17
Kapillartransfer auf eine Nitrocellulosemembran (BA 85, Schleier & Schuell, Dassel,
Deutschland) übertragen und mit einer radioaktiv markierten DNA-Sonde hybridisiert.
Für die Ethidiumbromidfärbung wurde das Gel 30 min in Wasser mit Ethidiumbromid (1
µg/ml) inkubiert und über Nacht in Wasser entfärbt. Die Nukleinsäurebanden wurden unter
UV-Licht (354 nm) sichtbar gemacht und dokumentiert (Eagle Eye, Stratagene, La Jolla, Cal.,
USA).
Der Kapillartransfer der RNA auf eine Nitrocellulosemembran erfolgte über Nacht in
Gegenwart von 10x SSC. Die Membran wurde danach für kurze Zeit in 2x SSC gewaschen
und im UV-Crosslinker (Stratagene) bestrahlt, um die RNA kovalent auf die
Membranoberfläche zu binden. Die Vorhybridisierung sowie die Hybridisierung wurden in 5x
SSC, 50 mM KPP, pH 7, 2x Denhardt’s, 100 µg/ml Heringssperma DNA und 0.5% SDS bei
42°C für 4 h bzw. über Nacht durchgeführt. Die radioaktive Markierung der Sonde erfolgte
nach der Methode von Feinberg und Vogelstein (1983). Dabei wurde ein kommerzielles
Produkt vorbereiteter Reagenzien (Prime-it) nach Angaben des Herstellers Stratagene
verwendet. Nichtinkorporierte Nukleotide wurden mittels Gelfiltration über Sephadex G50
(Amersham Pharmacia Biotech, Dübendorf, Schweiz) von der markierten Sonde getrennt. Der
Einbau radioaktiven dATP’s wurde im Scintillationszähler überprüft. Die sich an die
Hybridisierung anschliessenden Waschschritte erfolgten mit abnehmenden
Salzkonzentrationen. Sie wurden alle bei 60°C für je 30 min durchgeführt. Der erste
Waschschritt erfolgte mit einer 1x SSC, 0.5% (w/v) SDS Lösung. Auf diesen folgten zwei
weitere mit jeweils einer 0.2x SSC, 0.5% (w/v) SDS Lösung.
Die Membran wurde dann über Nacht auf einer PhosphorImager Kassette exponiert und
anschliessend am PhosphorImager unter Anwendung der entsprechenden Software (Image
Quant, Molecular Dynamics, Sunnyvale, Cal., USA) analysiert.
2.2.7 Isolierung der Genomischen DNA
Das in flüssigem Stickstoff schockgefrorene und bei –80°C gelagerte Pflanzenmaterial (10 g
Frischgewicht) wurde mit Mörser und Pistill in flüssigem Stickstoff zu einem feinen Pulver
zerrieben. Dieses wurde in 50 ml vorgewärmten (65°C) CTAB-Extraktionspuffer, welcher 2%
(v/v) 2-Mercaptoethanol enthielt, überführt. und für 30 min bei 65°C inkubiert. Während
dieser Zeit wurde der Ansatz mehrmals vorsichtig gemischt. Die anschliessende Extraktion
mit Chloroform wurde mit einem dem Ansatz entsprechenden Volumen durchgeführt. Bei

Material und Methoden
- 18 - 18
5000 xg wurde 5 min zentrifugiert. Dann wurde der zuvor abgenommenen wässrigen Phase
1/10 Volumenteil CTAB-NaCl Puffer zugesetzt und vorsichtig geschüttelt. Es schloss sich
eine weitere Chloroform-Extraktion an, worauf die wässrige Phase mit dem gleichen
Volumen CTAB-Fällungspuffer versetzt wurde. Da sich das Präzipitat nicht sogleich bildete,
wurde die Lösung für 30 min bei 65°C inkubiert. Nach einem zweiten Zentrifugationsschritt
bei 5000 xg für 5 min wurde das Pellet in 10 ml STE für 30 min bei 65°C resuspendiert und
anschliessend mit 6 ml Isopropanol überschichtet. Die ausgefallene DNA wurde mittels einer
Pasteurpipette aus der Interphase “gefischt” und in 80% (v/v) Ethanol gewaschen. Das
luftgetrocknete Pellet wurde in 2.5 ml TE aufgenommen und zur Elimination von RNA mit 10
µg/ml RNAse versehen. An die Inkubationszeit von 30 min bei 37°C folgte eine weitere
Präzipitation mit 800 µl 10 M Ammoniumacetat und 6.5 ml Ethanol. Das Präzipitat wurde bei
7500 xg und 4°C für 15 min sedimentiert, anschliessend in 80% (v/v) Ethanol gewaschen,
vacuumzentrifugiert (Savant) und in 0.5 ml TE aufgenommen. Die Konzentration der DNA-
Lösung wurde durch Messen der OD260 einer 1/50 Verdünnung bestimmt.
2.2.8 Southern-Blot Analyse
Die genomische DNA aus Sprossen von 20 Tage alten Tomaten wurde mit den Enzymen
DraI, EcoRI, HindIII und XbaI restringiert. Das Gesamtvolumen der einzelnen
Reaktionsansätze betrug 100 µl und beinhaltete 10 µg genom. DNA, 0.1 mg/ml BSA und 50
U der Restriktionsendonuclease im entsprechenden Puffer (MBI Fermentas, Vilnius,
Lithauen). Nach einer Reaktionszeit von 2 h bei 37°C wurden erneut 50 U des
Restriktionsenzyms zugegeben und für weitere 2 h inkubiert. Es folgten Hitzeinaktivierung
des Enzyms und Ethanolfällung der DNA. Die elektrophoretische Trennung der
Restriktionsfragmente wurde über Nacht bei einer Spannung von 40 V auf einem 0.8%
Agarose Gel durchgeführt. Als Elektrophoresepuffer wurde 1x TAE unter Zusatz von 0.25
µg/ml EtBr eingesetzt. Die im Gel immobilisierten DNA Fragmente wurden partiell
hydrolysiert (20 min in 500 ml 0.25 N HCl), denaturiert (30 min in 500 ml 1.5 M NaCl, 0.5 M
NaOH) und neutralisiert (2x 15 min in je 500 ml 1.5 M NaCl, 0.5 M Tris/HCl pH 8).
Der anschliessende Kapillartransfer der DNA Fragmente auf die Nitrocellulosemembran
erfolgte über Nacht in Gegenwart von 20x SSC. Alle weiteren Schritte wurden gemäss (2.2.6)
durchgeführt.

Material und Methoden
- 19 - 19
2.2.9 Klonierung der CDPK cDNA in pGEX-G
Beim pGEX-G Vektor (Schmid und Görlach, 1996) handelt es sich um ein Derivat des
pGEX-3X Vektors (Amersham Pharmacia Biotech). Der Vektor erlaubt die Expression eines
Proteins in N-terminaler Fusion mit Glutathion S-Transferase (GST) unter der Kontrolle des
mit IPTG induzierbaren tac-Promoters. Die Trennung des zu exprimierenden Proteins
(CDPK) von der GST Domäne ist durch proteolytische Spaltung mit Faktor Xa möglich.
Das offene Leseraster der CDPK cDNA wurde mittels PCR amplifiziert. Der eingesetzte 5’-
und 3’-Primer (im Anhang aufgeführt) war am 5’-Ende phosphoryliert, bzw. wies am Ende
eine Restriktionsschnittstelle für das Enzyme KpnI auf. Im Reaktionsansatz von 100 µl
Volumen wurden 500 ng cDNA, 0.5 µM beider Primer, 0.1 mM dNTPs und 2.5 U Pwo-
Polymerase im entsprechenden MgCl2 Puffer (Roche Diagnostics, Rotkreuz, Schweiz)
eingesetzt. Die PCR im DNA Thermal Cycler (Perkin Elmer Cetus) umfasste 25 Zyklen
(95°C, 15 s; 52°C, 30 s; 72°C, 2 min; nach dem zehnten Zyklus wurde die Reaktionszeit bei
72°C jeweils um 20 s gegenüber dem vorhergegangenen Zyklus verlängert. Die amplifizierten
PCR-Produkte wurden mit Phenol/Chloroform extrahiert, mit Ethanol präzipitiert und mit
KpnI restringiert. Die über ein Agarosegel aufgetrennten Produkte wurden mit dem Kit
prep@gene (BioRad) gereinigt. Entsprechend wurde der pGEX-G Vektor mit StuI und KpnI
restringiert und mit CIAP dephosphoryliert. Die Ligation erfolgte mit T4-DNA Ligase über
Nacht bei 16°C und resultierte in pGEX-G/CDPK-L. Auf gleiche Weise wurde eine C-
terminal um die calmodulin-ähnliche Domäne verkürzte Form der CDPK amplifiziert und
kloniert (Primer sind im Anhang aufgeführt). Der resultierende Expressionsvektor wurde mit
pGEX-G/CDPK-K bezeichnet.
2.2.10 Transformation von E. coli DH5α mit pGEX-G/CDPK-L/K
Für die Transformation wurden die Ligationsansätze aus 2.2.9 zu je 200 µl kompetenten E.
coli DH5α Zellen, die auf Eis aufgetaut wurden, zugegeben und 30 min auf Eis, 30 s bei 42°C
und 5 min auf Eis inkubiert. Der Ansatz wurde mit 800 µl LB-Medium ergänzt und für 45
min bei 37°C auf dem Schüttler inkubiert. Zum Ausplattieren wurden 50 µl bzw. 500 µl des
Ansatzes auf LB-Platten mit 100 mg/l Ampicillin gebracht und über Nacht bei 37°C inkubiert.
Mit den auf LB/Amp-Platten gewachsenen Kolonien wurden 2 ml LB/Amp-Flüssigkulturen,

Material und Methoden
- 20 - 20
(100 ng Ampicillin/ml LB-Medium) inokuliert. Die Flüssigkulturen wurden erneut über Nacht
bei 37°C und 220 rpm auf dem Schüttler inkubiert. Nach der Plasmid-Isolation (2.2.3) wurde
deren Identität anhand von Restriktions- und Sequenzanalyse (2.2.4) bestätigt.
2.2.11 Transformation von E. coli BL21-(DE3)-RIL mit pGEX-G/CDPK -L/K
Zur Transformation von E. coli BL21-(DE3)-RIL wurden Aliquots von 50 µl auftauender
kompetenter E. coli Zellen mit je 1 µl verdünntem β-Mercaptoethanol (1:10) versetzt und 10
min auf Eis gehalten. Während dieser Inkubationszeit wurden die Bakterien alle 2 min
vorsichtig gemischt. Nach der Zugabe von je 50 ng der pGEX-G/CDPK-L/K Plasmid-DNAs
(2.2.10) folgten 30 min Inkubation auf Eis, 20 s Hitzeschock bei 42°C und weitere 2 min auf
Eis. Die Ansätze wurden mit 450 µl LB-Medium ergänzt und für ca. 45 min im 37°C
Wasserbad inkubiert. Auf LB/Amp-Platten (100 mg/l Ampicillin) wurden jeweils 50 µl bzw.
200 µl des Transformationsansatzes ausplattiert und über Nacht bei 37°C inkubiert.
2.2.12 Anzucht von E. coli BL21-(DE3)-RIL Zellen
Die Anzucht der Bakterien erfolgte in 5 ml LB-Flüssigkulturen in Gegenwart von 100 mg/l
Ampicillin. Die über Nacht bei 37°C und 220 rpm gewachsenen Zellen wurden andern Tags
in 500 ml LB/Amp-Flüssigkulturen transferiert und wie zuvor bei 37°C und 220 rpm bis zu
einer optischen Dichte von 1.0 angezogen. Durch Zugabe von IPTG zu 1 mM wurde die
Expression des Fusionsproteins induziert. Daraufhin wurden die Schüttelkulturen bei
Raumtemperatur oder bei 30°C gehalten. Die Zellen wurden zu den angegebenen Zeitpunkten
abzentrifugiert (3 min, 2400 xg, 4°C) und das Sediment wurde bei -80 °C gelagert.

Material und Methoden
- 21 - 21
2.2.13 Aufschluss von E. coli BL21-(DE3)-RIL Zellen
Das Zellsediment einer 250 ml Kultur wurde mit Hilfe einer 10 ml Stabpipette in 30 ml Puffer
A (10 mM NaCl, 1 mM EDTA, 50 mM Tris/HCl, pH 8), 1 mg/ml Lysozym, 1 mM PMSF und
einer Spatelspitze DNAse resuspendiert. Die Lyse erfolgte während 20 min auf Eis. Der
Zellaufschluss erfolgte am Sonicator (W-380, Heat Systems, Ultrasonics). Dazu wurde die
Zellsuspension 5x 30 s mit Ultraschall behandelt. Zelltrümmer sowie unlösliche Proteine,
sogenannte „Inclusion Bodies“, wurden durch Zentrifugation (30 min, 2800 xg, 4°C) entfernt.
Der Überstand wurde über einen Faltenfilter filtriert und bis zu seiner Verwendung bei 4°C
gelagert. Zur Reinigung der unlöslichen Proteine wurde das Sediment unlöslicher Proteine mit
1% NP40 in 30 ml Puffer A gewaschen und erneut in 30 ml Puffer A resuspendiert. Aliquote
des Überstands als auch des Sediments wurden durch SDS-PAGE analysiert.
2.3 Protein-Analytische Methoden 2.3.1 SDS-PAGE / Western-Blot
Die Auftrennung des Proteingemisches erfolgte über ein 8% SDS-Polyacrylamidgel, in einem
diskontinuierlichen Gelsystem nach Lämmli (1970), welches sich aus einem 4.5%-igen
Sammelgel und einem 8%-igen Trenngel zusammensetzt. Vor dem Laden des Gels, wurde die
Proteinlösung mit SDS Lämmli Probenpuffer versehen und während 5 min bei 95°C
denaturiert. Die Elektrophorese dauerte 2.5 h bei 150 V. Zur Anfärbung der Proteinbanden
wurde Coomassie Brillant Blue R 250 eingesetzt. Der Transfer (Western Blot), der mittels
SDS-PAGE aufgetrennten Proteine auf eine Nitrocellulosemembran (Schleicher & Schuell),
erfolgte während 1 h bei 200 mA im Blottingpuffer (gemäss 2.1.1).
2.3.2 Reinigung der GST-CDPK Fusionproteine
Das Reinigungsprinzip der Glutathion Sepharose Affinitätschromatographie beruht auf der
Fähigkeit des GST Anteils des Fusionsproteins, an die Säulenmatrix zu binden.

Material und Methoden
- 22 - 22
Glutathion Sepharose 4B-Säulen von 2 ml Bettvolumen wurden bei 4°C mit Puffer A (aus
2.2.13) äquilibriert. Die Kapazität der Matrix beträgt 5 mg Fusionsprotein je 1 ml
Bettvolumen. Bei einer Flussrate von 4 ml/h wurde der Proteinextrakt (2.2.13) auf die Säule
aufgetragen und anschliessend bei erhöhter Flussrate (7 ml/h) mit 50 ml Puffer A gewaschen.
Nach einem weiteren Waschgang mit 15 ml 50 mM Tris/HCl, pH 8, wurden GST-CDPK
Fusionsproteine mit 5x 2 ml 10 mM reduziertem Glutathion in 50 mM Tris/HCl, pH 8, von
der Säule eluiert und bei 4°C oder -20°C gelagert. Die Bestimmung der Proteinkonzentration
der gesammelten Eluatfraktionen erfolgte nach Bradford (1976). Das Farbreagens (basierend
auf Coomassie Brilliant Blue G-250, Pierce Chem. Co.) wurde 1:10 (v/v) mit der zu
analysierenden Proteinlösung gemischt und anschliessend im Spektrophotometer bei einer
Wellenlänge von 595 nm gemessen. Die Proteinkonzentrationen wurde anhand einer BSA-
Eichgerade errechnet.
2.3.3 Bindung von 45Ca2+
Die mittels SDS-PAGE (2.3.1) aufgetrennten Proteine wurden elektrophoretisch auf eine
Nitrocellulosemembran übertragen (Western Blot, 2.3.1). Nach erfolgtem Transfer wurde die
Membran 3x 20 min in (60 mM KCl, 5 mM MgCl2, 10 mM Imidazol/HCl, pH 6.8)
gewaschen. Zum Test auf Bindung von 45Ca2+ wurde die Membran in 20 ml einer Lösung von
60 mM KCl, 5 mM MgCl2, 10 mM Imidazol/HCl, pH 6.8 und 20 µCi 45CaCl2 für 10 min im
Mini Hybridisierungsofen bei RT inkubiert. Anschliessend wurde die Membran in Wasser
gewaschen und für 12 h auf Kodak X-O-mat Röntgenfilm exponiert.
2.3.4 Autophosphorylierung immobilsierter CDPK
Die mittels SDS-PAGE (2.3.1) aufgetrennten Proteine wurden elektrophoretisch auf eine
Nitrocellulosemembran übertragen (Western Blot, 2.3.1; ohne Methanol im Transferpuffer).
Nach erfolgtem Transfer wurde die Membran zunächst in 30 mM HEPES mit 5% (w/v)
Magermilchpulver 30 min bei Raumtemperatur abgesättigt. Dann wurde die Membran mit
Denaturierungspuffer (7 M Guanidin Hydrochlorid, 50 mM Tris/HCl, pH 8.3, 50 mM DTT, 2
mM EDTA, 0.25% (w/v) Magermilchpulver) für 1 h bei RT inkubiert. Auf die Denaturierung

Material und Methoden
- 23 - 23
folgte eine Renaturierung. Der dazu eingesetzte Renaturierungspuffer (50 mM Tris/HCl, pH
7.5, 100 mM NaCl, 2 mM DTT, 2 mM EDTA, 0.1% Nonidet P-40, 0.25% (w/v)
Magermilchpulver) wurde während 16 h mehrmals ausgetauscht. Die Membran wurde danach
in 30 mM HEPES, pH 7.5 grüdlich gewaschen und anschliessend in calciumhaltigen - (50
mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 2 mM DTT, 1.1 mM CaCl2, 0.01µM
ATP, 0.0022 µM [γ-32P]-ATP (4500 Ci/mmol)) bzw. calciumfreiem Kinase Puffer (50 mM
HEPES, pH 7.5, 10 mM MgCl2, 5 mM EGTA, 2 mM DTT, 0.01µM ATP, 0.0022 µM [γ-32P]-
ATP (4500 Ci/mmol)) während 30 min bei RT im Mini-Hybridisierungsofen inkubiert. Es
folgten Waschschritte mit 30 mM HEPES, pH 7.5, gefolgt von 1 N HCl (1 h, 40°C), gefolgt
von 30 mM HEPES, pH 7.5. Daraufhin wurde die Membran 24 h auf einer PhosphorImager-
Kassette (Molecular Dynamics) exponiert. Die Auswertung erfolgte mit der Software Image
Quant (Molecular Dynamics).
2.3.5 Phosphoaminosäuretrennung mittels DC
Die Analyse von Phosphoaminosäuren wurde nach der Methode von Shi et al (1999) in leicht
abgeänderter Form übernommen. Das nach (2.3.2) gereinigte GST-CDPK-L Protein (5 µg)
wurde zur Autophosphorylierung in einem Reaktionsvolumen von 500 µl mit 50 mM HEPES,
pH 7.5, 10 mM MgCl2, 1 mM EGTA, 1.1 mM CaCl2, 2 mM DTT, 0.025 µM ATP und 0.022
µM [γ-32P]-ATP (4500 Ci/mmol) für 30 min bei 30°C inkubiert. Zur Präzipitation des
Proteins mit 10% (w/v) Trichloressigsäure wurde der Ansatz 10 min auf Eis gehalten und
anschliessend 10 min zentrifugiert. Daran schlossen sich drei Waschschritte mit je 5% TCA
und ein weiterer mit 95% (w/v) Ethanol an. Das Protein wurde in 200 µl 6 N HCl bei 110°C
für 1 h hydrolysiert. Das Hydrolysat wurde in vacuo eingetrocknet, dann in 6 µl einer
wässrigen Lösung nichtradioaktiver Phosphoaminosäuren (je 2 mg/ml P-Thr, P-Tyr und P-
Ser) resuspendiert und auf eine cellulosebeschichtete Dünnschichtplatte (E. Merck,
Darmstadt, Deutschland) aufgetragen. Die zweidimensionale Auftrennung der hydrolysierten
Aminosäuren erfolgte in erster Dimension während 10 h in Isobuttersäure:0.5 M NH4OH (5:3,
v/v). In zweiter Dimension erfolgte die Trennung während 7 h in Propionsäure:1 M
NH4OH:2-Propanol (45:17.5:17.5, v/v/v). Die Platte wurde getrocknet und zur
Aminosäureanfärbung mit einer 0.25% Ninhydrin/Aceton Lösung besprüht und 10 min bei
60°C inkubiert. Zur Autoradiographie wurde die Platte für 20 min auf Kodak X-O-mat

Material und Methoden
- 24 - 24
Röntgenfilm exponiert. Durch Vergleich des Röntgenfilms und der gefärbten Platte konnten
die in der CDPK autophosphorylierten Aminosäuren identifiziert werden.
2.3.6 Aktivitätstest
Kinetische Analysen der CDPK Aktivität mit dem Oligopeptid Syntide-2
(PLARTLSVAGLPGKK) als Substrat wurden nach Harmon et al. (1994) durchgeführt. In
einem 50 µl Reaktionsansatz wurden 30 nM CDPK mit unterschiedlich hohen
Substratkonzentrationen (10 µM, 20 µM, 30µM, 40 µM, 50 µM, 100 µM 150 µM) und dem
Kinase Puffer (50 mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 1.1 mM CaCl2, 2 mM
DTT, 0.1 mg/ml BSA) zusammengebracht. Nach einer Inkubationszeit von 5 min bei 30°C,
wurde die Reaktion durch Zugabe von 60 µM [γ-32P]-ATP (500 cpm/pmol) gestartet und für
weitere 15 min bei 30°C inkubiert. Abgestoppt wurde die Reaktion durch Auftragen von 10 µl
des Reaktionsansatzes auf einen Streifen (1 cm x 2 cm) Phosphocellulosepapiers (Whatman
P81). Die Streifen wurden anschliessend gründlich in 150 mM H3PO4 gewaschen und zum
Trocknen mit Aceton gespült. Die luftgetrockneten Proben wurden mit 4 ml
Scintillationslösung versetzt und am Scintillationszähler gemessen.

Resultate
- 26 - 26
3. RESULTATE Mittels eines Datenbankvergleichs wurde ein EST der Tomate (tomest: expressed sequence
tag der Tomate) identifiziert, der hohe Sequenzierähnlichkeit mit der von Yoon et al. (1999)
beschriebenen Calcium-abhängigen Proteinkinase aus Tabak aufwies. Ein volllänge cDNA
Klon dieser CDPK aus Tomate sollte mittels PCR kloniert werden.
3.1 Klonierung des 3’Endes einer Calciumabhängigen Proteinkinase (CDPK) aus Tomate
Zur Klonierung des 3’-Endes dieser CDPK cDNA wurden cDNA-Banken von der
Tomatenblüte und dem Tomatenspross, die in den pBluescript SK(-) Vektor konstruiert
worden waren (angelegt von Andreas Schaller), als Matrize eingesetzt. Die Amplifikation der
CDPK cDNA erfolgte mittels PCR. Als 5’-Primer wurde ein Oligonukleotid eingesetzt (vgl.
Abb. 2), dessen Sequenz von dem EST (tomest) abgeleitet worden war. Der T7 Primer des
pBluescript Vektors diente als 3’-Primer.
Von den 22 Klonen, die der Sequenzierung zugeführt wurden, wiesen alle 22 Klone dieselbe
Sequenz auf. Lediglich die Länge der 3’-untranslatierten Regionen unterschied sich in den
cDNAs die aus der Spross-, bzw. Blüten-Bank isoliert worden waren. Die erhaltene
Nukleotidsequenz des 3’-Endes der CDPK cDNA stimmte vollständig mit dem EST der
Datenbank überein (Abb. 2). Die cDNA wies eine singuläre HindIII-Schnittstelle auf. Durch
Restriktion mit HindIII und mit XhoI im Bereich des „Polylinkers“ des Vektors konnte ein
Fragment generiert werden, welches der 3’-untranslatierten Region der mRNA entspricht.
Dieses Fragment wurde als Sonde bei Northern- und Southern Blot Analysen eingesetzt.

Resultate
- 27 - 27
1 50
klon25 .......... .......... .......... GGTATGGGTG ATGAGGCCAC tomest GATGAATTGG AAAACGCTAT GAAGGAATAT GGTATGGGTG ATGAGGCCAC 51 100 klon25 CATCAAGGAG ATAATCGCAG AGGTGGGCAC TGACAATGAC GGTAGGATCA tomest ATTACGAGGA GTTCTGTGCT ATGATGAGAA GTGGAACAAC ACAACCACAA 151 200 klon25 CAAAAGCTTT TCTAGTGCAT CGAGAGCAAA TTACCCTTGC AACGAGCAGA tomest CAAAAGCTTT TCTAGTGCAT CGAGAGCAAA TTACCCTTGC AACGAGCAGA
201 250 klon25 GAACGTTCAC CAGCCAGCAC GATGGAGAGA TGTCTATCAC GAGAGGATCA tomest GAACGTTCAC CAGCCAGCAC GATGGAGAGA TGTCTATCAC GAGAGGATCA 251 300 klon25 GTAACTTAAC ATACTTCCAT ATTTCTTCAA TCCTTCAGCT ATCTTCAAAA tomest AGCACTGGAG TACATGAGAC CTTTTTCGTG TAGGTTGAGT TCGCGTGGAA 351 400 klon25 AATGTGTGTA TAGCTAGTTA ATGGTATATT TCGAGTTTCT TGTAATGTAT tomest AATGTGTGTA TAGCTAGTTA ATGGTATATT TCGAGTTTCT TGTAATGTAT 401 450 klon25 TTGATCCCCC TTCGTAATTT TGTGTGGATC GCGTTGTGAA GTTGACCTCC tomest TTGATCCCCC TTCGTAATTT TGTGTGGATC GCGTTGTGAA NNAAA..... 451 500 klon25 CGTAGAGATT ATAACAAGAC AATCTGAATA TAGCTTTATG CTTGAAAAAA tomest .......... .......... .......... .......... ..........
Abb. 2: Nukleotidsequenz des 3’-Endes der CDPK cDNA, isoliert aus der Blüten-Bank. Die kursiv gedruckten
Nukleotide bilden die Schnittstelle für HindIII. Der PCR Primer (LePK32) ist hervorgehoben. Die cDNAs,
welche aus dem Spross isoliert wurden, waren um die unterstrichene Nukleotidsequenz kürzer.
3.2 Southern Blot Analyse
LePK52
Resultate
- 28 - 28
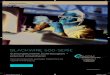
Genomische DNA der Tomatenpflanze wurde mit verschiedenen Restriktionsenzymen (DraI,
EcoRI, HindIII und XbaI) restringiert und nach Trennung über ein Agarosegel (Abb. 3A) auf
eine Nitrocellulosemembran transferiert. Die Hybridisierung der Membran erfolgte mit dem
HindIII/XhoI-Fragment der bislang erhaltenen CDPK-cDNA (s. 3.1), welches der 3’-
untranslatierten Region des Transkriptes entspricht. DNA-Fragmente, welche mit der Sonde
hybridisierten, weisen im Southern Blot eine Markierung auf (Abb. 3B). In jeder Spur konnte
nur ein Fragment nachgewiesen werden. Offensichtlich ist die eingesetzte Sonde
genspezifisch, d.h. sie hybridisiert mit nur einem Gen der CDPK-Genfamilie. Die Sonde ist
demnach geeignet, in weiterführenden Experimenten die Expression dieser CDPK-Isoform
auf Northern Blots zu untersuchen.
Abb. 3: Southern-Analyse genomischer DNA aus Lycopersicon esculentum cv. Castelmart II, die mit XbaI,
HindIII, EcoRI und DraI geschnitten wurde. Es wurden jeweils 10 µg genom. DNA verwendet,
elektrophoretisch aufgetrennt, auf eine Nitrocellulosemembran transferiert und dort mit einer CDPK-Sonde
(entsprechend dem 3’untranslatierten Bereich der mRNA) hybridisiert. Die Lage und Grösse bekannter DNA
Fragmente (Gibco/BRL, Basel, Schweiz) sind links der Abbildungen angegeben. Sowohl das EtBr- gefärbte Gel
(A), als auch der davon erhaltene Gel-Blot (B) sind gezeigt.
3.3 Northern Blot Analyse
XbaI
HindIII
DraI
EcoR
I
XbaI
HindIII
DraI
EcoR
I
0.5
1
1.6
3
5
12
kb
0.5
1
1.6
3
5
12
kb
A B
XbaI
HindIII
DraI
EcoR
I
XbaI
HindIII
DraI
EcoR
I
0.5
1
1.6
3
5
12
kb
0.5
1
1.6
3
5
12
kb
A B
0.5
1
1.6
3
5
12
kb
0.5
1
1.6
3
5
12
kb
A B
Resultate
- 29 - 29
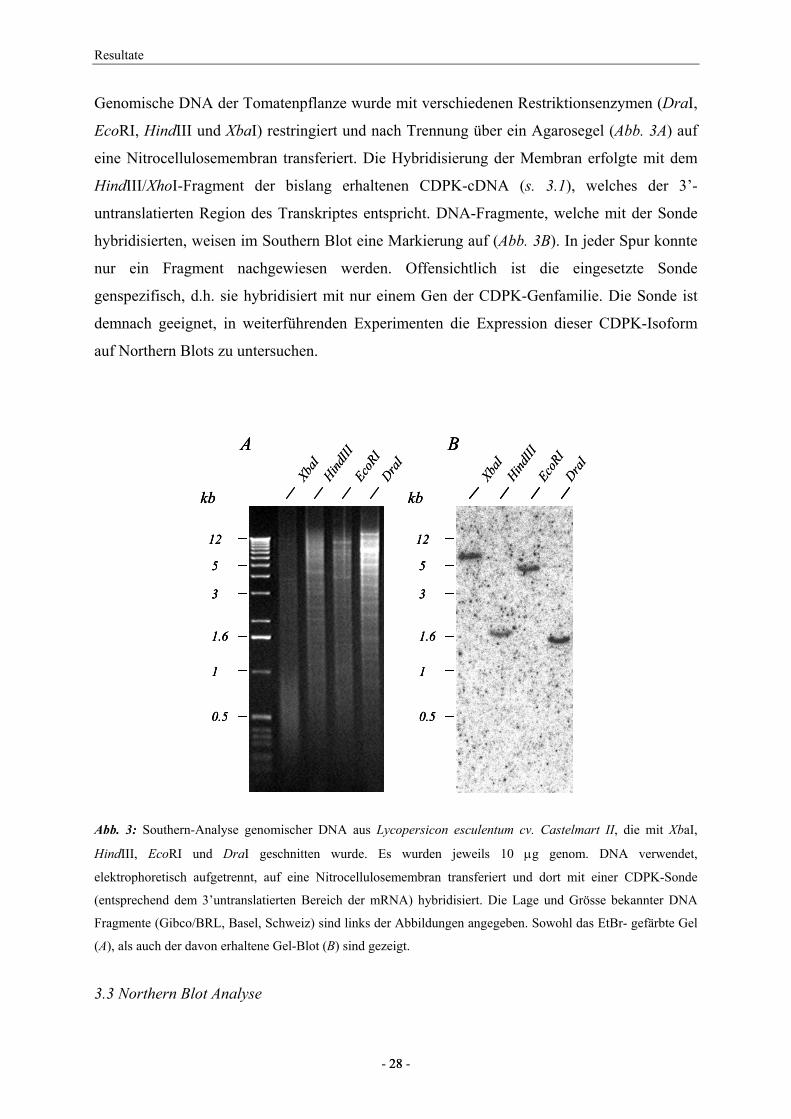
Zunächst wurden die organspezifische Expression in Tomatenpflanzen untersucht. Dazu
wurde die Gesamt-RNA aus Kotyledonen, Blättern, Wurzeln, Blüten und aus in Suspension
kultivierten Zellen isoliert, aufgetrennt und zur Sichtung der entsprechenden mRNAs mit der
unter 3.2 beschriebenen Sonde hybridisiert. Es zeigte sich, dass die CDPK in allen hier
untersuchten Organen exprimiert ist. Die Transkriptspiegel waren jedoch in Kotyledonen und
Blättern wesentlich geringer als in Wurzeln, Blüten oder undifferenzierten Zellen (Abb. 4).
Abb. 4: Northern Blot Analyse der CDPK Expression in verschiedenen Organen der Tomate. Jeweils 4.5 µg
Gesamt-RNA aus Kotyledonen (1), Blättern (2), Wurzeln (3), Blüten (4) und Zellkulturen (5) wurden der RNA
Gelblot Analyse unterzogen. Die Blots wurden mit einer spezifischen CDPK- und einer Ubiquitin Sonde
hybridisiert und mittels des PhosphorImagers analysiert.
Weiterhin wurde eine mögliche Induzierbarkeit durch Verwundung oder Fusicoccin
untersucht. Während sich die Transkriptspiegel der CDPK in Blättern verwundeter
Tomatenpflanzen nicht änderten (Daten nicht gezeigt), war eine deutliche Akkumulation des
CDPK
UBQ
1 2 3 4 5
CDPK
UBQ
1 2 3 4 5

Resultate
- 30 - 30
Transkripts in Blättern Fusicoccin behandelter Pflanzen zu beobachten (Abb. 5). Die
Induktion war 4 h nach Gabe des Toxins maximal, danach nahm der „steady state“ Gehalt des
CDPK Transkriptes wieder ab. Die Induktion der CDPK Expression durch Fusicoccin ist
daher schneller als die von typischen „Pathogenesis related“ (PR) Proteinen (Abb. 5). Das in
Abbildung 5 gezeigte, mit Ethidiumbromid gefärbte Gel ist mit jenem identisch, von dem die
RNA auf eine Membran transferiert wurde. Es zeigt, dass die Gesamt-RNA-Konzentration in
den entsprechenden Spuren gleich war.
Abb. 5: Northern Blot Analyse Fusicoccin gefütterter Tomatenpflanzen. Zur Analyse wurden jeweils 4.5 µg
Gesamt-RNA eingesetzt. Die RNA-Gel Blots wurden mit radioaktiv markierten Sonden der CDPK-cDNA (dem
HindIII/XhoI-Fragment) und zur Kontrolle mit einer sauren Chitinase hybridisiert. Ein weiteres Gel wurde mit
Ethidiumbromid gefärbt. 0 – 8 h: Zeitangabe nach Beginn der Fusicoccin-Behandlung.
3.4 Amplifizierung des 5’-Endes mittels RACE-PCR
0 h 0.5 h
1 h 1.5 h4 h 6 h 8 h2 h
CDPK
Saure Chitinase
Ethidiumbromidgel25s rRNA
18s rRNA
0 h 0.5 h
1 h 1.5 h4 h 6 h 8 h2 h
CDPK
Saure Chitinase
Ethidiumbromidgel25s rRNA25s rRNA
18s rRNA18s rRNA

Resultate
- 31 - 31
Die vorausgegangene Northern Blot Analyse (3.3) zeigte, dass der Anteil des CDPK
Transkriptes an der Gesamt-RNA 4 Stunden nach Behandlung mit Fusicoccin relativ hoch
war. Aufgrund dieser neu gewonnenen Erkenntnis diente die RNA aus 4 Stunden mit
Fusicoccin behandelten Pflanzen als Ausgangsmaterial für eine Amplifikation des CDPK 5’-
Endes durch RACE (rapid amplification of cDNA ends)-PCR. Wie zuvor wurden auch hier
zwei zu tomest komplementäre Oligonukleotide als 3’-Primer eingesetzt (PKGSP1, LePK32;
s. Anhang). Die generierten RACE Produkte wurden anschliessend gelelektrophoretisch
aufgetrennt (Abb. 6).
Abb. 6: Amplifikation der CDPK cDNA (5’Bereich) mittels RACE PCR. Als Templat diente die Gesamt-RNA
aus Fusicoccin-behandelten (4 h) Tomatenpflanzen. Die RACE Produkte wurden über ein EtBr-gefärbtes
Agarosegel getrennt. In den Spalten 1 & 2 wurde PKGSP1, in 4 & 5 LePK32 als 3’ PCR Primer benutzt. Die zu
erwartende Produktegrösse beträgt ungefähr 2 kb.
Die amplifizierten Produkte von etwa 2 kb zeigten eine von verwandten CDPKs zu
erwartende Grösse. Die in dem Agarosegel identifizierten Banden wurden isoliert und in den
1 2 M 4 5
0.5
23
1.6
1
kb1 2 M 4 5
0.5
23
1.6
1
kb

Resultate
- 32 - 32
TOPO Vektor kloniert. Nach Isolierung des Plasmids wurde durch Restriktionsverdau mit
EcoRI das Vorhandensein der entsprechenden Insertion überprüft (Abb. 7). Die cDNA ist im
Vektor beiderseits von einer EcoRI Schnittstelle flankiert. Zudem scheint die inserierte CDPK
cDNA über eine interne EcoRI Schnittstelle zu verfügen (Abb. 7). Die Grösse der beiden
beobachteten Restriktionsfragmente (1.2 und 0.8 kb) entspricht in Summe der zu erwartenden
Grösse von 2 kb.
Abb. 7: EcoRI-Restriktionsverdau von 9 unabhängigen Klonen der CDPK cDNA im TOPO Vektor. Die
restringierte Plasmind-DNA wurde im Vergleich mit einem Grössenmarker (1 kb DNA-Ladder)
gelelektrophoretisch aufgetrennt.
In lediglich 2 von 9 Fällen (Spuren 1 und 5) ist eine cDNA Insertion zu beobachten und
bestätigt das Vorhandensein des gewünschten Inserts. Der leichte Unterschied in
Fragmentgrössen zwischen Klon 1 und 5 ist auf die unterschiedlichen Primer (PKGSP1 bzw.
LePK32) zurückzuführen.
Für die Sequenzbestimmung wurde die cDNA zunächst von den Enden her ansequenziert.
Dazu wurden als Primer die Oligonukleotide aus der RACE-PCR sowie 2 Primer
flankierender Vektorsequenzen benutzt. Die so gewonnene Sequenzinformation erlaubte es,
Oligonukleotide auszuwählen, die dann als Primer in weiteren Sequenzierreaktionen
eingesetzt wurden. Auf diese Art und Weise konnten beide Stränge der cDNA vollständig
durchsequenziert werden. Ihre Sequenz, sowie die damit überlappende Sequenz des zuvor
klonierten 3’-Endes der cDNA (vgl. Abb. 2, Klon 25) und die davon abgeleitete
Aminosäuresequenz sind in Abbildung 9 dargestellt. Wie aus der Sequenz zu entnehmen ist,
erstreckt sich die cDNA über 2229 Nukleotide. An der Position 219 beginnt mit dem ATG
Codon das offene Leseraster (ORF), welches 1659 bp umfasst und für ein Protein von 553
Aminosäuren kodiert. Ein zusätzliches Stopcodon im selben Leseraster 5’-aufwärts lässt
1 2 3 4 5 6 7 8 9 M
1 kb
1 2 3 4 5 6 7 8 9 M
1 kb

Resultate
- 33 - 33
vermuten, dass es sich hierbei um eine Volllänge-cDNA handelt. Die vom ORF abgeleitete
Aminosäuresequenz weist Ähnlichkeit zu bekannten pflanzlichen CDPKs auf. Die
Sequenzähnlichkeit mit der von Yoon et al. (1999) beschriebenen Tabak CDPK ist dabei
besonders ausgeprägt (94% identische Aminosäuren). Durch den Sequenzvergleich lassen
sich leicht die putative katalytische Domäne und die C-terminale Calcium-binde Domäne mit
vier charakteristischen EF-Hand Motiven ausmachen (Abb. 8).
Abb. 8: Aminosäuresequenzvergleich der CDPK aus Tomate mit der aus Tabak (NtCDPK1). Die Katalytische
Domäne, das autoinhibitorische, verbindende Peptid und die Calcium-binde Domäne mit den EF-Hand Motiven
sind gekennzeichnet. Horizontale Striche (-) weisen auf identische Aminosäuren hin, während Punkte (.) Lücken
symbolisieren, um eine maximale Sequenzidentität zu erlangen. Der Sequenzvergleich wurde mit dem
Programm „gap“ des GCG-Programmpaketes erstellt.
GGCAAGTGGGGAAAAAGCCTAGAAAAATAACAAGGGCAAAAGCTGGCAAAGAAAGTGTGT 1 ---------+---------+---------+---------+---------+---------+ 60 CCGTTCACCCCTTTTTCGGATCTTTTTATTGTTCCCGTTTTCGACCGTTTCTTTCACACA
LeCDPK MGGCFSKKYT QQDAN.GHRA GRR.VNQAYQ KPPQPQPERP YQPQPQQERP 48 NtCDPK1 ---------R –EG--G-Y-- T--NA--E-- ----H----- ----...... 44 LeCDPK YQPPPQPAYQ PPPQPKPQPQ PHPVPVTVQS GQPQDQMQGP HMNNILGKPF 98 NtCDPK1 ....---..- -Q---R---- A-..T--X-P X-T------- -L-------F 86 LeCDPK EEIRKLYTLG KELGRGQFGV TYYCTENSTG NPYACKSILK RKLVSKNDRE 148 NtCDPK1 -D---Q---- ---------- --H------- ---------- ----R----- 136 LeCDPK DMKREIQIMQ HLSGQPNIVE FKGAYEDRQS VHLVMELCAG GELFDRIIAR 198 NtCDPK1 ---------- ---------- --------H- ---------- ---------- 186 LeCDPK GYYSEKDAAE IIRQIVNVVN ICHFMGVMHR DLKPENFLLT SKDENAMLKA 248 NtCDPK1 ---------- -----A---- ---------- ---------- ------K--- 236 LeCDPK TDFGLSVFIE EGKVYRDIVG SAYYVAPEVL RRSYGKEADV WSAGVILYIL 298 NtCDPK1 ---------- ---------- ---------- ---------- ---------- 286 LeCDPK LSGVPPFWAE TEKGIFNTIL KGEIDFQSDP WPSISNSAKD LIRKMLTQEP 348 NtCDPK1 ---------- -------A-- ---------- ---------- ---------- 336 LeCDPK RKRITSAQVL EHPWLRLGEA SDKPIDSAVL SRMKQFRAMN KLKKLALKVI 398 NtCDPK1 ---M------ ------M--- ---------- ---------- ---------- 386 LeCDPK AENLSEEEIK GLKAMFHNID TDNSGTITYE ELKSGLARLG SKLTETEVKQ 448 NtCDPK1 ---------- ------A--- ---------- ---------- ---------- 436 LeCDPK LMEAADVDGN GSIDYIEFIT ATMHRHRLER DEHLFKAFQH FDKDHSGFIT 498 NtCDPK1 ---------- -T-------- ---------- ---------Y ---------- 486 LeCDPK RDELENAMKE YGMGDEATIK EIIAEVGTDN DGRINYEEFC AMMRSGTTQP 548 NtCDPK1 -----S---- ---------- ------D--- ---------- ------.--- 535 LeCDPK QQKLF* 554 NtCDPK1 -P---* 540
EF-Hand1
EF-Hand1
EF-Hand1
EF-Hand1
Autoinhibitorische Domäne
Katalytische Domäne
Variable Domäne LeCDPK MGGCFSKKYT QQDAN.GHRA GRR.VNQAYQ KPPQPQPERP YQPQPQQERP 48 NtCDPK1 ---------R –EG--G-Y-- T--NA--E-- ----H----- ----...... 44 LeCDPK YQPPPQPAYQ PPPQPKPQPQ PHPVPVTVQS GQPQDQMQGP HMNNILGKPF 98 NtCDPK1 ....---..- -Q---R---- A-..T--X-P X-T------- -L-------F 86 LeCDPK EEIRKLYTLG KELGRGQFGV TYYCTENSTG NPYACKSILK RKLVSKNDRE 148 NtCDPK1 -D---Q---- ---------- --H------- ---------- ----R----- 136 LeCDPK DMKREIQIMQ HLSGQPNIVE FKGAYEDRQS VHLVMELCAG GELFDRIIAR 198 NtCDPK1 ---------- ---------- --------H- ---------- ---------- 186 LeCDPK GYYSEKDAAE IIRQIVNVVN ICHFMGVMHR DLKPENFLLT SKDENAMLKA 248 NtCDPK1 ---------- -----A---- ---------- ---------- ------K--- 236 LeCDPK TDFGLSVFIE EGKVYRDIVG SAYYVAPEVL RRSYGKEADV WSAGVILYIL 298 NtCDPK1 ---------- ---------- ---------- ---------- ---------- 286 LeCDPK LSGVPPFWAE TEKGIFNTIL KGEIDFQSDP WPSISNSAKD LIRKMLTQEP 348 NtCDPK1 ---------- -------A-- ---------- ---------- ---------- 336 LeCDPK RKRITSAQVL EHPWLRLGEA SDKPIDSAVL SRMKQFRAMN KLKKLALKVI 398 NtCDPK1 ---M------ ------M--- ---------- ---------- ---------- 386 LeCDPK AENLSEEEIK GLKAMFHNID TDNSGTITYE ELKSGLARLG SKLTETEVKQ 448 NtCDPK1 ---------- ------A--- ---------- ---------- ---------- 436 LeCDPK LMEAADVDGN GSIDYIEFIT ATMHRHRLER DEHLFKAFQH FDKDHSGFIT 498 NtCDPK1 ---------- -T-------- ---------- ---------Y ---------- 486 LeCDPK RDELENAMKE YGMGDEATIK EIIAEVGTDN DGRINYEEFC AMMRSGTTQP 548 NtCDPK1 -----S---- ---------- ------D--- ---------- ------.--- 535 LeCDPK QQKLF* 554 NtCDPK1 -P---* 540
EF-Hand1
EF-Hand1
EF-Hand1
EF-Hand1
Autoinhibitorische Domäne
Katalytische Domäne
Variable Domäne

Resultate
- 34 - 34
AGCATTTTCATGGAGTTTTTTCCCATGAAGTCAATGAAGAGGGTTCAAAACGTTGGCATT 61 ---------+---------+---------+---------+---------+---------+ 120 TCGTAAAAGTACCTCAAAAAAGGGTACTTCAGTTACTTCTCCCAAGTTTTGCAACCGTAA TTCTGATCTGTGGAATATCTTTTAGGTTGTTGTTTTTTTTGTAACTTGTTGAGGAATTGA 121 ---------+---------+---------+---------+---------+---------+ 180 AAGACTAGACACCTTATAGAAAATCCAACAACAAAAAAAACATTGAACAACTCCTTAACT AGTATCCAAAGTTCAATCTTGTTGAATAGACTGTAGAAATGGGTGGTTGTTTTAGCAAGA 181 ---------+---------+---------+---------+---------+---------+ 240 TCATAGGTTTCAAGTTAGAACAACTTATCTGACATCTTTACCCACCAACAAAATCGTTCT * M G G C F S K K - AGTATACCCAACAAGATGCTAATGGGCATAGGGCAGGAAGAAGAGTTAATCAAGCATATC 241 ---------+---------+---------+---------+---------+---------+ 300 TCATATGGGTTGTTCTACGATTACCCGTATCCCGTCCTTCTTCTCAATTAGTTCGTATAG Y T Q Q D A N G H R A G R R V N Q A Y Q - AAAAACCACCACAACCCCAGCCAGAAAGGCCATATCAGCCTCAGCCCCAGCAGGAAAGGC 301 ---------+---------+---------+---------+---------+---------+ 360 TTTTTGGTGGTGTTGGGGTCGGTCTTTCCGGTATAGTCGGAGTCGGGGTCGTCCTTTCCG K P P Q P Q P E R P Y Q P Q P Q Q E R P - CATATCAGCCACCGCCACAGCCAGCATATCAGCCACCGCCACAGCCAAAGCCACAACCTC 361 ---------+---------+---------+---------+---------+---------+ 420 GTATAGTCGGTGGCGGTGTCGGTCGTATAGTCGGTGGCGGTGTCGGTTTCGGTGTTGGAG Y Q P P P Q P A Y Q P P P Q P K P Q P Q - AGCCCCACCCTGTTCCTGTTACTGTGCAGTCTGGACAGCCCCAAGACCAAATGCAAGGAC 421 ---------+---------+---------+---------+---------+---------+ 480 TCGGGGTGGGACAAGGACAATGACACGTCAGACCTGTCGGGGTTCTGGTTTACGTTCCTG P H P V P V T V Q S G Q P Q D Q M Q G P - CCCATATGAATAACATATTGGGAAAGCCTTTTGAGGAAATTAGAAAGCTCTATACACTTG 481 ---------+---------+---------+---------+---------+---------+ 540 GGGTATACTTATTGTATAACCCTTTCGGAAAACTCCTTTAATCTTTCGAGATATGTGAAC H M N N I L G K P F E E I R K L Y T L G - GGAAAGAATTGGGTAGGGGTCAGTTTGGAGTGACTTACTATTGTACTGAGAACTCAACTG 541 ---------+---------+---------+---------+---------+---------+ 600 CCTTTCTTAACCCATCCCCAGTCAAACCTCACTGAATGATAACATGACTCTTGAGTTGAC K E L G R G Q F G V T Y Y C T E N S T G - GGAACCCTTATGCTTGCAAGTCCATACTTAAGAGGAAGCTTGTAAGCAAGAATGATAGGG 601 ---------+---------+---------+---------+---------+---------+ 660 CCTTGGGAATACGAACGTTCAGGTATGAATTCTCCTTCGAACATTCGTTCTTACTATCCC N P Y A C K S I L K R K L V S K N D R E - AGGATATGAAGAGGGAAATTCAGATTATGCAGCATTTGAGTGGGCAGCCAAACATTGTGG 661 ---------+---------+---------+---------+---------+---------+ 720 TCCTATACTTCTCCCTTTAAGTCTAATACGTCGTAAACTCACCCGTCGGTTTGTAACACC D M K R E I Q I M Q H L S G Q P N I V E -
PKNT1
PK52
PK33

Resultate
- 35 - 35
AATTCAAGGGTGCTTATGAAGATAGGCAGTCAGTACACCTTGTGATGGAACTTTGTGCTG 721 ---------+---------+---------+---------+---------+---------+ 780 TTAAGTTCCCACGAATACTTCTATCCGTCAGTCATGTGGAACACTACCTTGAAACACGAC F K G A Y E D R Q S V H L V M E L C A G - GAGGAGAGTTGTTTGACAGGATTATCGCCCGTGGATATTACTCAGAGAAGGATGCTGCTG 781 ---------+---------+---------+---------+---------+---------+ 840 CTCCTCTCAACAAACTGTCCTAATAGCGGGCACCTATAATGAGTCTCTTCCTACGACGAC G E L F D R I I A R G Y Y S E K D A A E - AGATTATTAGACAGATTGTAAATGTTGTTAACATTTGCCATTTCATGGGTGTCATGCATA 841 ---------+---------+---------+---------+---------+---------+ 900 TCTAATAATCTGTCTAACATTTACAACAATTGTAAACGGTAAAGTACCCACAGTACGTAT I I R Q I V N V V N I C H F M G V M H R - GGGATCTCAAGCCAGAGAATTTCTTACTGACTAGTAAGGATGAAAATGCTATGTTGAAGG 901 ---------+---------+---------+---------+---------+---------+ 960 CCCTAGAGTTCGGTCTCTTAAAGAATGACTGATCATTCCTACTTTTACGATACAACTTCC D L K P E N F L L T S K D E N A M L K A - CAACTGATTTTGGACTTTCTGTCTTCATTGAAGAAGGGAAGGTGTACCGTGATATAGTTG 961 ---------+---------+---------+---------+---------+---------+ 1020 GTTGACTAAAACCTGAAAGACAGAAGTAACTTCTTCCCTTCCACATGGCACTATATCAAC T D F G L S V F I E E G K V Y R D I V G - GTAGTGCATATTATGTTGCCCCTGAAGTGTTGCGGCGTAGTTATGGGAAGGAAGCAGATG 1021 ---------+---------+---------+---------+---------+---------+ 1080 CATCACGTATAATACAACGGGGACTTCACAACGCCGCATCAATACCCTTCCTTCGTCTAC S A Y Y V A P E V L R R S Y G K E A D V - TATGGAGTGCAGGTGTTATTTTGTATATTCTGCTCAGTGGTGTACCTCCATTTTGGGCTG 1081 ---------+---------+---------+---------+---------+---------+ 1140 ATACCTCACGTCCACAATAAAACATATAAGACGAGTCACCACATGGAGGTAAAACCCGAC W S A G V I L Y I L L S G V P P F W A E - AAACTGAAAAGGGAATATTTAATACCATACTAAAAGGAGAAATTGACTTCCAAAGTGATC 1141 ---------+---------+---------+---------+---------+---------+ 1200 TTTGACTTTTCCCTTATAAATTATGGTATGATTTTCCTCTTTAACTGAAGGTTTCACTAG T E K G I F N T I L K G E I D F Q S D P - CATGGCCATCAATATCAAATAGTGCCAAGGACCTTATTCGGAAAATGCTAACGCAGGAGC 1201 ---------+---------+---------+---------+---------+---------+ 1260 GTACCGGTAGTTATAGTTTATCACGGTTCCTGGAATAAGCCTTTTACGATTGCGTCCTCG W P S I S N S A K D L I R K M L T Q E P - CAAGGAAGAGAATTACTTCCGCGCAAGTTCTTGAACATCCATGGCTTCGACTTGGAGAAG 1261 ---------+---------+---------+---------+---------+---------+ 1320 GTTCCTTCTCTTAATGAAGGCGCGTTCAAGAACTTGTAGGTACCGAAGCTGAACCTCTTC R K R I T S A Q V L E H P W L R L G E A - CATCAGATAAGCCAATAGACAGTGCAGTCCTGTCCAGAATGAAGCAGTTCAGAGCAATGA 1321 ---------+---------+---------+---------+---------+---------+ 1380 GTAGTCTATTCGGTTATCTGTCACGTCAGGACAGGTCTTACTTCGTCAAGTCTCGTTACT S D K P I D S A V L S R M K Q F R A M N -
PK34
PK54
PK53
PK32
PKCT2

Resultate
- 36 - 36
ACAAGCTCAAAAAACTCGCATTGAAGGTCATTGCGGAAAATTTATCTGAAGAAGAAATCA 1381 ---------+---------+---------+---------+---------+---------+ 1440 TGTTCGAGTTTTTTGAGCGTAACTTCCAGTAACGCCTTTTAAATAGACTTCTTCTTTAGT K L K K L A L K V I A E N L S E E E I K - AGGGTCTGAAAGCTATGTTTCACAACATTGATACGGACAATAGTGGCACAATCACTTATG 1441 ---------+---------+---------+---------+---------+---------+ 1500 TCCCAGACTTTCGATACAAAGTGTTGTAACTATGCCTGTTATCACCGTGTTAGTGAATAC G L K A M F H N I D T D N S G T I T Y E - AAGAACTGAAGTCGGGATTGGCTCGGCTTGGATCAAAGCTAACGGAGACTGAAGTCAAGC 1501 ---------+---------+---------+---------+---------+---------+ 1560 TTCTTGACTTCAGCCCTAACCGAGCCGAACCTAGTTTCGATTGCCTCTGACTTCAGTTCG E L K S G L A R L G S K L T E T E V K Q - AACTCATGGAAGCTGCTGATGTGGATGGAAATGGATCAATTGATTACATTGAGTTTATTA 1561 ---------+---------+---------+---------+---------+---------+ 1620 TTGAGTACCTTCGACGACTACACCTACCTTTACCTAGTTAACTAATGTAACTCAAATAAT L M E A A D V D G N G S I D Y I E F I T - CTGCAACGATGCATAGACATCGGCTAGAAAGGGATGAGCATCTTTTTAAAGCTTTTCAGC 1621 ---------+---------+---------+---------+---------+---------+ 1680 GACGTTGCTACGTATCTGTAGCCGATCTTTCCCTACTCGTAGAAAAATTTCGAAAAGTCG A T M H R H R L E R D E H L F K A F Q H - ATTTTGACAAGGACCATAGTGGATTCATCACGCGAGATGAATTGGAAAACGCTATGAAGG 1681 ---------+---------+---------+---------+---------+---------+ 1740 TAAAACTGTTCCTGGTATCACCTAAGTAGTGCGCTCTACTTAACCTTTTGCGATACTTCC F D K D H S G F I T R D E L E N A M K E –
AATATGGTATGGGTGATGAGGCCACCATCAAGGAGATAATCGCAGAGGTGGGCACTGACA 1741 ---------+---------+---------+---------+---------+---------+ 1800 TTATACCATACCCACTACTCCGGTGGTAGTTCCTCTATTAGCGTCTCCACCCGTGACTGT Y G M G D E A T I K E I I A E V G T D N - ATGACGGTAGGATCAATTACGAGGAGTTCTGTGCTATGATGAGAAGTGGAACAACACAAC 1801 ---------+---------+---------+---------+---------+---------+ 1860 TACTGCCATCCTAGTTAATGCTCCTCAAGACACGATACTACTCTTCACCTTGTTGTGTTG D G R I N Y E E F C A M M R S G T T Q P - CACAACAAAAGCTTTTCTAGTGCATCGAGAGCAAATTACCCTTGCAACGAGCAGAGAACG 1861 ---------+---------+---------+---------+---------+---------+ 1920 GTGTTGTTTTCGAAAAGATCACGTAGCTCTCGTTTAATGGGAACGTTGCTCGTCTCTTGC Q Q K L F * TTCACCAGCCAGCACGATGGAGAGATGTCTATCACGAGAGGATCAGTAACTTAACATACT 1921 ---------+---------+---------+---------+---------+---------+ 1980 AAGTGGTCGGTCGTGCTACCTCTCTACAGATAGTGCTCTCCTAGTCATTGAATTGTATGA TCCATATTTCTTCAATCCTTCAGCTATCTTCAAAAAGCACTGGAGTACATGAGACCTTTT 1981 ---------+---------+---------+---------+---------+---------+ 2040 AGGTATAAAGAAGTTAGGAAGTCGATAGAAGTTTTTCGTGACCTCATGTACTCTGGAAAA
PKCT1 LePK32
LePK52
PKGSP1

Resultate
- 37 - 37
TCGTGTAGGTTGAGTTCGCGTGGAAAATGTGTGTATAGCTAGTTAATGGTATATTTCGAG 2041 ---------+---------+---------+---------+---------+---------+ 2100 AGCACATCCAACTCAAGCGCACCTTTTACACACATATCGATCAATTACCATATAAAGCTC TTTCTTGTAATGTATTTGATCCCCCTTCGTAATTTTGTGTGGATCGCGTTGTGAAGTTGA 2101 ---------+---------+---------+---------+---------+---------+ 2160 AAAGAACATTACATAAACTAGGGGGAAGCATTAAAACACACCTAGCGCAACACTTCAACT CCTCCCGTAGAGATTATAACAAGACAATCTGAATATAGCTTTATGCTTGAAAAAAAAAAA 2161 ---------+---------+---------+---------+---------+---------+ 2220 GGAGGGCATCTCTAATATTGTTCTGTTAGACTTATATCGAAATACGAACTTTTTTTTTTT AAAAAAAAA 2221 --------- 2229 TTTTTTTTT
Abb. 9: Sequenz der CDPK cDNA aus Lycopersicon esculentum cv. Castelmart II und die davon abgeleitete
Primärstruktur. Das ORF beginnt mit dem Startcodon für Methionin bei Position 219 und endet mit dem
Stoppcodon in Position 1880. Die zur Sequenzaufklärung verwendeten Primer sind eingezeichnet und benannt.
Auch die zur RACE-PCR eingesetzten Primersequenzen (PKGSP1 und LePK32) sind hervorgehoben.
Dargestellt sind die vereinigten Sequenzen des 5’-RACE Produktes und des Klon 25 (vgl. 3.1) der dem 3’-Ende
der cDNA entspricht. markiert das 5’-Ende des Klon 25 und markiert das 3’-Ende des RACE Produktes.
3.5 Amplifizierung und Klonierung von CDPK-L und –K
Voraussetzung für weiterführende biochemische Untersuchungen ist die Expression des
offenen Leserasters der CDPK cDNA in Bakterien. Zu diesem Zweck musste zuerst das zu
exprimierende ORF in einen geeigneten Expressionsvektor gebracht werden.
In dem von Görlach und Schmid (1996) entwickelten pGEX-G Vektor haben wir ein ideales
Klonierungssystem gefunden. Dieser Vektor erlaubt die Expression eines Proteins in Fusion
mit Glutathion-S-Transferase in E. coli. Unter der Kontrolle des tac-Promoters kann mit IPTG
die Proteinexpression induziert werden. Zudem ermöglicht die Erkennungssequenz für die
Protease Faktor Xa eine allfällige Trennung des Fusionproteins.
Die Integration der zu exprimierenden cDNA-Konstrukte in den pGEX-G Vektor erfolgte
über die Restriktionsschnittstellen StuI und KpnI in der „multiple cloning site“ (MCS). Die
schematische Darstellung (Abb. 10) gibt die Klonierungsstrategie wieder.

Resultate
- 38 - 38
Abb. 10: Schematische Darstellung des pGEX-G Expressionsvektors. Die CDPK-L/K cDNAs wurden in die
StuI/KpnI–Schnittstellen der MCS des Vektors ligiert. Das Fusionsprotein GST-CDPK wird über den tac-
Promoter induziert und kann durch Faktor Xa gespalten werden.
Das offene Leseraster der CDPK cDNA wurde für die Klonierung in den pGEX-G Vektor
mittels PCR amplifiziert. Dazu wurden die Oligonukleotide PKNT1 und PKCT1 (s. Anhang
und Abb. 9) als 5’- und 3’-Primer eingesetzt. Als Templat diente die CDPK cDNA im TOPO-
Vektor (i.e. das 5’-RACE Produkt). Auf gleiche Weise wurde mit Hilfe des Primers PKCT2
eine C-terminal um die calmodulin-ähnliche Domäne verkürzte Form der CDPK amplifiziert
und kloniert. Die so erhaltenen Klone wurden der Sequenzanalyse zugeführt, mit der
Ausgangssequenz verglichen und für identisch befunden.
Sequenzier-Fragmente von pGEX-G/CDPK-L pGEX 5' <----------------+ PKNT1 <---------------+ PK52 <------------+ PK54 <----------------+ PK53 <---------------+ pGEX 3' +--------------> 5’RACE +------------------------------------------------------------------> Produkt |------|------|------|------|------|------|------|------|------|------| 0 200 400 600 800 1000 1200 1400 1600 1800
GSTtac mcs
pGEX-G(~4950 bp )
Faktor Xa
KpnIStuICDPK
GSTtac mcs
pGEX-G(~4950 bp )
Faktor Xa
KpnIStuICDPK
KpnIStuICDPK

Resultate
- 39 - 39
Sequenzier-Fragmente von pGEX-G/CDPK-K pGEX 5' <---------------+ PKNT1 <-------------+ PK52 <--------------+ PK54 <------------+ pGEX 3' +----------------> 5’RACE +------------------------------------------------------------------> Produkt |------|------|------|------|------|------|------|------|------|------| 0 200 400 600 800 1000 1200 1400 1600 1800
Abb. 11: Sequenzierstrategie der in den pGEX-G Vektor klonierten CDPK-L/K. Die Pfeile geben Länge sowie
Orientierung der in einzelnen Analysen ermittelten Sequenzen wieder. Sie sind im Vergleich zu der Sequenz des
als Templat eingesetzten 5’-RACE Produktes dargestellt. Alle Primer-Sequenzen sind im Anhang aufgeführt.
3.6 Expression der CDPK in E. coli
Für die Expression in Escherichia coli BL-21(DE3)-RIL Zellen wurden vier 250 ml
Flüssigkulturen bei 37°C bis zu einer optischen Dichte von ungefähr 1 angezogen und durch
Zusatz von 1 mM IPTG induziert. Daraufhin wurde von jedem Klon eine Kultur bei
Raumtemperatur und eine zweite bei 30°C gehalten. Die Expression des Fusionsproteins in
Abhängigkeit der Zeit wurde durch Entnehmen von 1 ml Aliquots in regelmässigen
Zeitabständen analysiert. Für die Kontrolle wurden Ansätze mitgeführt, die nicht durch IPTG
induziert wurden.
Die Auftrennung der zuvor mit SDS Probenpuffer aufgeschlossenen Zellen erfolgte über ein
8% SDS-Polyacrylamidgel, welches zur Sichtbarmachung der Proteine mit Coomassie
Brillant Blue angefärbt wurde. Den Expressionsverlauf in Abhängigkeit von Zeit und
Temperatur gibt Abbildung 11 wieder. Sie zeigt eine massive Akkumulation der beiden GST-
Fusionsproteine, sowohl bei 30°C als auch bei RT. Aufgrund der Primärstruktur wurde für die
GST-CDPK Konstrukte eine molekulare Masse von 71.2 kDa bzw. 86.9 kDa errechnet. Mit
einer apparenten molekularen Masse von 66 kDa zeigt sich für CDPK-K eine geringe
Abweichung vom errechneten Wert. Für beide Proteine wurde nach 5 Stunden Inkubation bei

Resultate
- 40 - 40
Raumtemperatur die stärkste Akkumulation ausgemacht. Bemerkenswert ist der grosse
prozentuale Anteil von CDPK-L/K am Gesamtprotein.
Die Expression heterologer Proteine in E. coli führt oft zur Bildung von sogenannten
"Inclusion Bodies", die eine unlösliche Form des zu exprimierenden Proteins darstellen. Um
zu klären, ob GST-CDPK-L/K in löslicher Form oder aber als "Inclusion Bodies" exprimiert
werden, wurden wie beim zuvor beschriebenen Experiment bakterielle Extrakte in
Abhängigkeit der Inkubationsdauer und Temperatur erstellt, die durch Zentrifugation in eine
lösliche und eine unlösliche Fraktion getrennt wurden. Die SDS-PAGE Analyse der
gewonnenen Fraktionen zeigte, dass der Grossteil des exprimierten Proteins unlöslich ist
(Abb. 12). In Anbetracht des sehr hohen Expressionsspiegels ist der Anteil des löslichen
Proteins für weiterführende Experimente aber immer noch ausreichend. Auch die Expression
an löslichem Protein ist nach 5 Stunden Inkubationszeit bei Raumtemperatur maximal.

Resultate
- 41 - 41
Abb. 11: SDS-PAGE Analyse der Zeitabhängigkeit der GST-CDPK Expression in E. coli BL-21(DE3)-RIL
Zellen. Für GST-CDPK-K und GST-CDPK-L wurden je zwei Ansätze bei unterschiedlichen Temperaturen
angezogen und zu den angegebenen Zeitpunkten nach der IPTG Zugabe jeweils Aliquots von 1 ml entnommen.
In den Kontrollspuren ist der Zellextrakt von nicht IPTG-induzierten E. coli Zellen gezeigt. Es wurden gleiche
Volumina Zellsuspension auf 8% SDS-PAGE Gelen analysiert. Die Massen der Markerproteine (BioRad) sind in
kDa angegeben. Sowohl das SDS-Gel der GST-CDPK-L (A), als auch jenes der GST- CDPK-K (B) sind gezeigt.
3h1h 2h 4h 5h 1h 1.5h
2h 3h Kon
trolle
3h
Kon
trolle
0h
Kon
trolle
5h
Kon
trolle
0h
Mar
ker
kDa3h1h 2h 4h 5h 1h 1.5h
2h 3h Kon
trolle
3h
Kon
trolle
0h
Kon
trolle
5h
Kon
trolle
0h
Mar
ker
kDa
200
11697
66CDPK-K
200
11697
66
4545
B
97
200116
66
45
CDPK-L
RT 30°RT 30°
97
200116
66
45
97
200116
66
45
200116
66
45
A
3h1h 2h 4h 5h 1h 1.5h
2h 3h Kon
trolle
3h
Kon
trolle
0h
Kon
trolle
5h
Kon
trolle
0h
Mar
ker
kDa3h1h 2h 4h 5h 1h 1.5h
2h 3h Kon
trolle
3h
Kon
trolle
0h
Kon
trolle
5h
Kon
trolle
0h
Mar
ker
kDa3h1h 2h 4h 5h 1h 1.5h
2h 3h Kon
trolle
3h
Kon
trolle
0h
Kon
trolle
5h
Kon
trolle
0h
Mar
ker
kDa3h1h 2h 4h 5h 1h 1.5h
2h 3h Kon
trolle
3h
Kon
trolle
0h
Kon
trolle
5h
Kon
trolle
0h
Mar
ker
kDa
200
11697
66CDPK-K
200
11697
66
4545
B 200
11697
66CDPK-K
200
11697
66
4545
B
97
200116
66
45
CDPK-L
RT 30°RT 30°RT 30°RT 30°
97
200116
66
45
97
200116
66
45
200116
66
45
A

Resultate
- 42 - 42
Abb. 12: SDS-PAGE Analyse von löslichen sowie unlöslichen Anteilen der GST-CDPK-L/K. E. coli BL-
21(DE3)-RIL Zellen wurden bei Raumtemperatur und 30°C angezogen. Zu den angegebenen Zeitpunkten nach
Induktion mit IPTG wurden Aliquots entnommen, bakterielle Extrakte erstellt, durch Zentrifugation in lösliche
und unlösliche Fraktionen getrennt und durch SDS-PAGE analysiert. Es wurden gleiche Volumenanteile des
Gesamtextraktes aufgetragen, die denen in Abb. 11 entsprechen. Zu sehen sind die beiden mit Coomassie
Brillant Blue angefärbt Polyacrylamidgele von GST-CDPK-L (A) und GST-CDPK-K (B). Die grösste
Akkumulation an löslichem Protein wurde bei RT nach 5 Stunden erreicht.
Mar
ker
5h2h 2h 5h3h 1.5h
3h1.5h
30° RT
u nlö
slic
h
unlö
slic
h
lösl
ich
lösl
ich
kDa
A
B
97
200
66
45
116
97
200
66
45
116
CDPK-L
CDPK-K
Mar
ker
5h2h 2h 5h3h 1.5h
3h1.5h
30° RT
u nlö
slic
h
unlö
slic
h
lösl
ich
lösl
ich
kDa
A
B
97
200
66
45
11697
200
66
45
11697
200
66
45
116
200
66
45
116
97
200
66
45
116
CDPK-L
CDPK-K

Resultate
- 43 - 43
3.7 Reinigung des Fusionsproteins
Die nachfolgende Reinigung der GST-CDPKs erfolgte mittels Affinitätschromatographie.
Dank dieser Methode lassen sich Fusionsproteine in einem Schritt reinigen. Das auf eine
Glutathion Sepharose 4B Säule aufgetragene Proteingemisch wird über den GST-Teil des
Fusionsproteins an die Matrix gebunden und durch Elution mit reduziertem Glutathion von
der Säule gewaschen.
In Abbildung 13 sind im einzelnen die Reinigungsschritte, begonnen mit dem Zellextrakt bis
hin zum gereinigten Fusionsprotein, dokumentiert. Die im GST-CDPK-L Eluat zu
beobachtende Proteinbande weist eine apparente molekulare Masse von 87 kDa auf, was der
molekularen Masse von 86903 Da sehr nahe kommt. Dafür ist eine gewisse Abweichung
zwischen apparenter und errechneter Masse bei der GST-CDPK-K festzustellen. Die
erwartete Masse beträgt 71173 Da, die apparente Masse aber ist 66 kDa.
Die Proteinkonzentration in den Eluaten wurde nach Bradford (1976) bestimmt. Es ergab sich
eine Ausbeute von 9 mg GST-CDPK-L und 6.5 mg GST-CDPK-K pro 500 ml
Bakterienkultur. Es ist auffällig, dass der Anteil der in unlöslicher Form exprimierten
Fusionsproteine hier wesentlich geringer ist als in den unter 3.6 beschriebenen Vorversuchen
(vgl. Abb. 11).
Abb. 13: Reinigung der rekombinanten GST-CDPK-L (A) und GST-CDPK-K (B) aus E. coli. Je eine 0.5 Liter E.
coli Kultur für GST-CDPK-L (A) und GST-CDPK-K (B) wurden durch IPTG induziert und die Zellen wurden
nach 5 h Inkubation bei Raumtemperatur abzentrifugiert. Die Zellen wurden aufgeschlossen und der Rohextrakt
(Spur 1, 1/106 des Gesamtvolumens) wurde durch Zentrifugation in eine unlösliche (Spur 2, 1/106 des
Gesamtvolumens) und eine lösliche Fraktion (Spur 3, 1/106 des Gesamtvolumens) getrennt. Der Extrakt wurde
auf eine GST-Sepharose-Säule appliziert. Der Durchlauf (1/106) ist in den Spuren 4 gezeigt. Gebundenes Protein
wurde mit red. Glutathion (10 mM) eluiert (Spur 5, 0.8 bzw. 0.4 µg). Die Abbildung zeigt mit Coomassie
Brilliant Blue gefärbte SDS-PAGE Gele. Als Molekulargewichtsmarker wurde der „broad molecular weight
standards“ (BioRad) verwendet.
B
66 kDa
1 2 3 4 5A
87 kDa
1 2 3 4 5B
66 kDa
1 2 3 4 5A
87 kDa
1 2 3 4 5

Resultate
- 44 - 44
3.8 Bindung von 45Ca 2+
Ähnlich zu den tierischen Ca2+/Calmodulin Kinasen soll die Aktivierung der CDPK über eine
Konformationsänderung der C-terminalen Domäne nach Calciumbindung erfolgen. Die von
uns klonierte und exprimierte CDPK-L weist im Gegensatz zu der verkürzten CDPK-K eine
C-terminale Domäne auf, mit den vier charakteristischen Ca2+-bindenden EF-Hand Motiven.
Für die Beweiserbringung der aufgrund der Sequenz gemachten Aussagen (vgl. 3.4 und Abb.
8) wurde die Calciumbindefähigkeit mit radioaktiv markiertem 45Ca2+ überprüft. Zu diesem
Zweck wurden die mittels SDS-PAGE aufgetrennten Proteine (0.5 µg) elektrophoretisch auf
eine Nitrocellulosemembran transferiert und in einer 45CaCl2 (1.7 mM, 1 mCi L-1) Lösung
inkubiert.
Wie die Autoradiographie (Abb. 14) bestätigt, verfügt demnach nur die GST-CDPK-L über
Calciumbindestellen, nicht so die GST-CDPK-K. Dies zeigt, dass die C-terminale, aber nicht
die katalytische Domäne, Calcium bindet und folglich alleinig für die Calciumbindefähigkeit
der CDPK und vermutlich für deren calciumabhängige Aktivierung verantwortlich ist. Die
Calciumbindefähigkeit der GST-CDPK-L bleibt auch nach Einfrieren und Lagerung bei
−20°C erhalten (Abb. 14).
Abb. 14: Bindung von 45Ca2+ durch GST-CDPK-L. Je 0.5 µg der gereinigten GST-CDPK-L (Spur 1 + 4) und
GST-CDPK-K (Spur 2 + 5) wurden über ein 8% SDS Polyacrylamidgel getrennt, elektrophoretisch auf eine
Nitrocellulosemembran übertragen und durch Amidoblack angefärbt (A). Anschliessend wurden die
membrangebundenen Proteine mit 45Ca2+ inkubiert (B). In der mittleren Spur (3) ist der
Molekulargewichtsmarker „broad molecular weight standards“ (BioRad) zu sehen. Die Proteinproben in Spuren
1 + 2 waren bei 4°C, jene in Spuren 4 + 5 bei –20°C gelagert worden.
A
B
1 2 3 4 5
11697
66
kDa
87 kDa
A
B
1 2 3 4 5
A
B
1 2 3 4 5
11697
66
kDa
87 kDa

Resultate
- 45 - 45
3.9 Autophosphorylierung der CDPK
Hier sollte die Fähigkeit der GST-CDPK zur autokatalytischen Phosphorylierung untersucht
werden. Dazu wurden die Fusionsproteine wie zuvor (3.8) durch SDS-PAGE aufgetrennt und
elektrophoretisch auf eine Nitrocellulosemembran übertragen. Die so immobilisierten
Proteine wurden zunächst denaturiert und dann auf der Mmebran renaturiert. Durch
Inkubieren des Blots in einem Ca2+- oder EGTA-haltigen Puffer, wurde die
Autophosphorylierungsreaktion in Gegenwart von [γ-32P]-ATP ausgeführt.
Die radioaktivmarkierten Banden in Abbildung 15B repräsentieren die autophosphorylierten
Proteine. Wie die Autoradiographie zeigt, ist nur die GST-CDPK-L zur autokatalytischen
Inkorporation radioaktiven Phosphats in der Lage. Diese Phosphat-Inkorporation wird in
Gegenwart des Calciumchelators EGTA (5 mM) vollständig inhibiert. Dieser Befund deutet
darauf hin, dass es sich bei der Autophosphorylierung um eine Ca2+-abhängige Reaktion
handelt. Wie die Calciumbindefähigkeit bleibt auch die autokatalytische Aktivität nach
Einfrieren und Lagern bei –20°C erhalten (Abb. 15).
Abb. 15: Ca2+-abhängige Autophosphorylierung der rekombinanten CDPK. Aliquots von 0.5 µg der gereinigten
GST-CDPK-L und GST-CDPK-K wurden durch Western Blot auf eine Nitrocellulosemembran transferiert und
mit [γ-32P]-ATP- haltigem Puffer in Gegenwrt von EGTA oder Ca2+ inkubiert. Das inkorporierte [32P] Phosphat
wurde autoradiographisch nachgewiesen (B). Zum Vergleich dient das in Abbildung (A) abgebildete 8% SDS
Polyacrylamidgel. Das Ladeschema sowie die Proteinmengen und Laufbedingungen waren zu jenem des
Western Blots identisch.
CDPK
-L /
4°C
CDPK
-L /
-20°
C
CDPK
-K /
4°C
CDPK
-K /
-20°
C
EGTA(5mM)
Ca 2+
(1.1mM)
A
B
87 kDa
66 kDa
87 kDa
CDPK
-L /
4°C
CDPK
-L /
-20°
C
CDPK
-K /
4°C
CDPK
-K /
-20°
C
CDPK
-L /
4°C
CDPK
-L /
-20°
C
CDPK
-K /
4°C
CDPK
-K /
-20°
C
CDPK
-L /
4°C
CDPK
-L /
-20°
C
CDPK
-K /
4°C
CDPK
-K /
-20°
C
EGTA(5mM)
Ca 2+
(1.1mM)
A
B
87 kDa
66 kDa
87 kDa
CDPK
-L /
4°C
CDPK
-L /
-20°
C
CDPK
-K /
4°C
CDPK
-K /
-20°
C
CDPK
-L /
4°C
CDPK
-L /
-20°
C
CDPK
-K /
4°C
CDPK
-K /
-20°
C
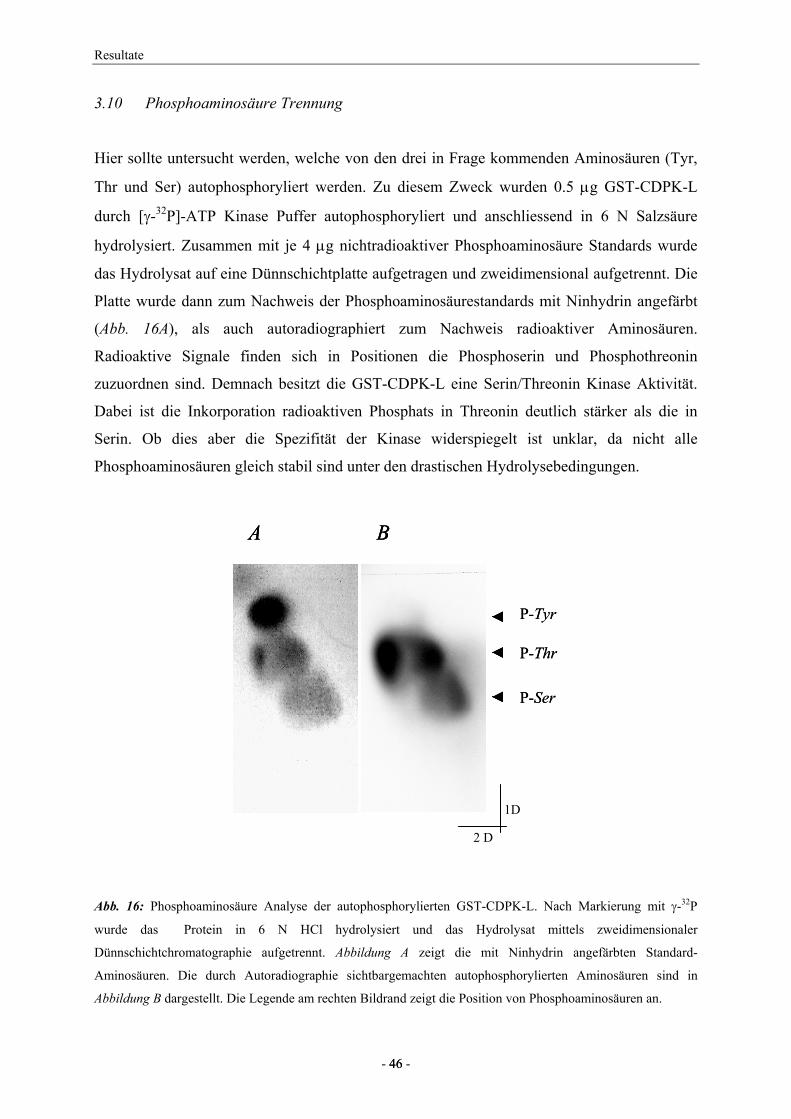
Resultate
- 46 - 46
3.10 Phosphoaminosäure Trennung
Hier sollte untersucht werden, welche von den drei in Frage kommenden Aminosäuren (Tyr,
Thr und Ser) autophosphoryliert werden. Zu diesem Zweck wurden 0.5 µg GST-CDPK-L
durch [γ-32P]-ATP Kinase Puffer autophosphoryliert und anschliessend in 6 N Salzsäure
hydrolysiert. Zusammen mit je 4 µg nichtradioaktiver Phosphoaminosäure Standards wurde
das Hydrolysat auf eine Dünnschichtplatte aufgetragen und zweidimensional aufgetrennt. Die
Platte wurde dann zum Nachweis der Phosphoaminosäurestandards mit Ninhydrin angefärbt
(Abb. 16A), als auch autoradiographiert zum Nachweis radioaktiver Aminosäuren.
Radioaktive Signale finden sich in Positionen die Phosphoserin und Phosphothreonin
zuzuordnen sind. Demnach besitzt die GST-CDPK-L eine Serin/Threonin Kinase Aktivität.
Dabei ist die Inkorporation radioaktiven Phosphats in Threonin deutlich stärker als die in
Serin. Ob dies aber die Spezifität der Kinase widerspiegelt ist unklar, da nicht alle
Phosphoaminosäuren gleich stabil sind unter den drastischen Hydrolysebedingungen.
Abb. 16: Phosphoaminosäure Analyse der autophosphorylierten GST-CDPK-L. Nach Markierung mit γ-32P
wurde das Protein in 6 N HCl hydrolysiert und das Hydrolysat mittels zweidimensionaler
Dünnschichtchromatographie aufgetrennt. Abbildung A zeigt die mit Ninhydrin angefärbten Standard-
Aminosäuren. Die durch Autoradiographie sichtbargemachten autophosphorylierten Aminosäuren sind in
Abbildung B dargestellt. Die Legende am rechten Bildrand zeigt die Position von Phosphoaminosäuren an.
P-Tyr
A B
P-Thr
P-Ser
P-Tyr
A BA B
P-Thr
P-Ser
1D
2 D

Resultate
- 47 - 47
Welche Serin- und Threoninreste der CDPK durch Autophosphorylierung modifiziert werden
konnte noch nicht ermittelt werden. Erste Versuche, diese Reste durch einen
massenspektrometrischen Vergleich tryptischer Peptide der phosphorylierten bzw.
unphosphorylierten CDPK zu identifizieren, führten bislang zu keinem Ergebnis.
3.11 Aktivität gegenüber synthetischen Peptiden
Roberts und Harmon (1992) zeigten, dass sich synthetische Peptide, welche über die Sequenz
eine K/R-X-X-Ser/Thr Sequenz verfügen, als Substrat von CDPKs eignen. Aufgrund dieses
Befundes viel unsere Substratwahl für die Aktivitätsmessungen auf das Oligopeptid Syntide-2
(PLARTLSVAGLPGKK; Phosphorylierungsmotiv ist unterstrichen).
Geeignete Reaktionsbedingungen (Enzymkonzentration, Reaktionsdauer), welche einen
linearen Reaktionsverlauf gewährleisten wurden in Vorversuchen ermittelt. Die kinetischen
Untersuchungen wurden darum in Ansätzen von 50 µl 30 nM GST-CDP-L und variierenden
Substratkonzentrationen in einem calcium- und [γ-32P]-ATP-haltigen Puffer durchgeführt.
Nach einer Reaktionszeit von 15 min wurde die Reaktion durch Entnahme von 10 µl und
Auftragen auf Phosphocellulosepapier abgestoppt. Die Papierstreifen mit dem ionisch
gebundenen Reaktionsprodukt wurden anschliessend gewaschen und im Scintillationszähler
gemessen.
Die gemessenen Aktivitäten wurden nun gegen die jeweilige Substratkonzentration
aufgetragen. Die in Abbildung 17A dargestellte Kurve zeigt den charakteristischen Verlauf
einer Michaelis Menten Sättigungskurve. Die nach Lineweaver und Burk linearisierten Daten
sind in Abbildung 17B gezeigt. In vier unabhängigen Experimenten wurden KM-Werte für
Syntide-2 von 82 bis 89 µM ermittelt. Es bleibt darauf hinzuweisen, dass sich der
Aktivitätsverlauf bei höheren Substratkonzentrationen nicht mehr durch die Michaelis-
Menten Gleichung beschreiben liess. Die Ursache dafür mag darin liegen, dass sich mit dem
Threonin in Position 5 von Syntide-2 eine zweite potentielle Phosphorylierungsstelle im
Substrat befindet. Die Frage bleibt letztlich aber ungeklärt. Zwei weitere Peptide, nämlich
Systemin (AKQSKPPSKRDPPKMQTD) und Myelin-basic Protein (QKRPSQRSKYL)
wurden als mögliche Substrate getestet, wurden aber durch GST-CDPK-L nicht
phosphoryliert.

Resultate
- 48 - 48
Abb. 17A: Aktivität von GST-CDPK-L gegenüber Syntide-2. Die Figur zeigt das repräsentative Ergebnis eines
von vier Experimenten. Jeder Punkt stellt den Mittelwert einer vierfach Bestimmungen dar.
Abb. 17B: Linearisierung der in Abb. 17A gezeigten Daten nach Lineweaver-Burk.
0
100
200
300
400
500
600
700
0 20 40 60 80 100 120
Akt
ivitä
t (cp
m)
Syntide-2 Konzentration (µM)
A
0
100
200
300
400
500
600
700
0 20 40 60 80 100 120
Akt
ivitä
t (cp
m)
Syntide-2 Konzentration (µM)
A
1/Syntide-2 (1/µM)
1/A
ktiv
ität (
1/cp
m)
B
y = 0.0709x + 0.0008R2 = 0.9912
0
0.001
0.002
0.003
0.004
0.005
0 0.01 0.02 0.03 0.04 0.05 0.06
1/Syntide-2 (1/µM)
1/A
ktiv
ität (
1/cp
m)
B
y = 0.0709x + 0.0008R2 = 0.9912
0
0.001
0.002
0.003
0.004
0.005
0 0.01 0.02 0.03 0.04 0.05 0.06

Diskussion
- 49 -
4. Diskussion
Während viele der biochemische Mechanismen zur Signaltransduktion zwischen Pflanzen und
Tieren konserviert zu sein scheinen – so weist etwa der Wundsignalweg der Pflanzen grosse
Ähnlichkeiten auf zur Eicosanoid Signalübermittlung in Tieren (Ryan und Pearce, 1998) –
gibt es auch deutliche Unterschiede, die ihre Ursache in der unterschiedlichen Lebensart und
Antwort auf Umwelteränderungen finden mögen (Stone und Walker, 1995; Satterlee und
Sussman, 1998). In Pflanzen wie in Tieren sind einzigartige Signaltransduktionskomponenten
anzutreffen. Dazu zählt auch eine neue Klasse von Proteinkinasen, die den Namen Calcium-
abhängige (Calmodulin-unabhängige) Proteinkinase (CDPKs) trägt. Vertreter dieser Klasse
sind nur im Pflanzenreich anzutreffen (Roberts und Harmon, 1992). CDPKs sind überwiegend
Ser/Thr Proteinkinasen (Stone und Walker, 1995). Wie der Name schon verrät, wird die
Kinaseaktivität direkt durch Bindung von Calcium an eine Calmodulin-ähnliche Domäne
reguliert. Die Primärstruktur der CDPKs umfasst eine N-terminale Kinase Domäne, die über
ein autoinhibitorsches Verbindungspeptid mit der calmodulin-ähnlichen Domäne am C-
Terminus verbunden ist. Kennzeichnend für die C-terminale Domäne sind vier Ca2+-bindende
EF-Hand Motive. In Abwesenheit von Calcium kommt es zur Inhibierung der Kinaseaktivität
durch Interaktion der autoinhibitorischen (Pseudosubstrat) – mit der katalytischen Domäne
(Binder et al., 1994; Harmon et al., 1994 ; Harper et al., 1994 ; Huang et al., 1996 ; Yoo und
Harmon, 1996). Obwohl CDPKs stark konserviert sind, so sind doch bei den verschiedenen
Vertretern der CDPK Familie unterschiedliche Eigenschaften zu beobachten. So kann die
Zahl der charakteristischen Ca2+-bindenden EF-Hand Motive zwischen 1 und 4 variieren, was
wahrscheinlich eine unterschiedliche Kinetik der Enzym-Aktivität in Abhängigkeit von
Calcium zur Folge hat (Zhao et al., 1994). Ausserdem verfügen CDPKs über eine stark
variierende N-terminale Region, die oft eine Erkennungssequenz zur N-Myristoylierung
aufweist.
Seit der ersten Charakterisierung einer CDPK (Harmon et al., 1987) wurden weitere
Isoformen gefunden und die entsprechenden Gene bzw. cDNAs kloniert. Es zeigte sich, dass
CDPKs einer grossen Gen-Familie angehören: bis zu 20 verschiedene Isoformen werden in
Arabidopsis thaliana vermutet (Hrabak et al., 1996), neun bzw. drei Isoformen wurden in
Mais (Saijo et al., 1997) und Reis (Kawasaki et al., 1993; Breviario et al., 1995) identifiziert.
Strukturelle Unterschiede zwischen den Isoformen sind überwiegend in der Anzahl der EF-
Hand Motive sowie in der Länge und Zusammensetzung des N-terminalen Endes

Diskussion
- 50 -
auszumachen. Wenn auch CDPKs in letzter Zeit viel Aufmerksamkeit auf sich zogen, sind
ihre biologischen Funktionen weiterhin grösstenteils ungeklärt.
Im Rahmen dieser Arbeit konnte mittels RACE-PCR die cDNA einer CDPK aus Tomate
amplifiziert werden. Das offene Leseraster der cDNA kodiert ein Polypeptid von 553
Aminosäuren. Die vom ORF abgeleitete Aminosäuresequenz weist alle charakteristischen
Merkmale einer CDPK auf. Durch den Sequenzvergleich lassen sich die putative katalytische
Domäne sowie die C-terminale Ca2+-binde Domäne mit den vier EF-Hand Motiven aus
machen. Die Sequenzähnlichkeit mit der von Yoon et al. (1999) beschriebenen Tabak CDPK
ist dabei besonders ausgeprägt. Es zeigte sich eine Aminosäuresequenzidentität von 94%.
Zudem verfügt die CDPK ebenfalls über die Pro-Gln-reiche Sequenz am N-terminus, die ein
Charakteristikum dieser Isoform zu sein scheint (Yoon et al. 1999).
Southern-Analyse genomischer DNA mit dem 3’-untranslatierten Fragment der CDPK-cDNA
zeigte, dass die Sonde spezifisch mit nur einem Gen der CDPK Genfamilie hybridisierte.
Daher ist die Sonde geeignet, die Expression der hier interessierenden CDPK auf Northern
Blots zu untersuchen. Während sich anders als bei der Tabak CDPK (Yoon) keine Induktion
nach Verwundung nachweisen lies, ergab die Northern Blot Analyse, dass Transkripte der
CDPK nach Fusicoccin-Behandlung von Tomatenpflanzen akkumulierten. Die Menge des
Transkriptes war bereits 1.5 Stunden nach Gabe des Toxins erhöht und wies nach 4 h das
Maximum auf. Da Fusicoccin zu einer Aktivierung der H+-ATPase führt (Serrano, 1990;
Palmgren, 1991), ist die erhöhte Expression der CDPK in Antwort auf FC-Gabe
möglicherweise ein Indiz, dass die CDPK ebenfalls an der Regulation der ATPase beteiligt
ist. Es ist denkbar, dass die Zelle einer erhöhten Aktivität der H+-ATPase nach FC-
Behandlung entgegenzuwirken bestrebt ist, und daher verstärkt eine CDPK exprimiert, die in
der Lage ist, die ATPase zu inaktivieren. Damit wäre trotz der ausbleibenden
Wundinduzierbarkeit eine Rolle für die hier untersuchte CDPK in der Regulation der
Aktivität der H+-ATPase denkbar.
In weiterführenden Experimenten wird zu klären sein, ob die H+-ATPase Substrat der CDPK
ist, in vitro und in vivo. Auch wird zu untersuchen sein, ob eine mögliche Phosphorylierung
der H+-ATPase durch die CDPK eine Aktivitätsänderung zur Folge hat.

Diskussion
- 51 -
Ca2+-abhängige Autophosphorylierung
Wie das Autophosphorylierungs-Experiment (3.9) zeigte, verfügt nur die GST-CDPK-L über
autokatalytische Fähigkeiten, nicht so die um die C-terminale Domäne verkürzte GST-CDPK-
K. Dieser Befund deutet darauf hin, dass es sich bei der autokatalytischen Phosphorylierung
um eine calciumabhängige Reaktion handelt. Er steht in Einklang mit zwei unabhängigen
Studien (Harmon et al., 1994; Harper et al., 1994), die die autoinhibitorischen Eigenschaften
der Verbindungsdomäne von CDPKs untersuchten: Die Verbindungsdomäne ist in der Lage
in Abhängigkeit von Ca2+ Calmodulin wie auch die C-terminale Calmodulin-ähnliche
Domäne zu binden. Das deutet an, dass die Autoinhibierung der CDPK ähnlich wie bei
Calmodulin-abhängigen Proteinkinasen aufgehoben wird. Bei der Myosin-light-chain Kinase
sowie der Calmodulin-abhängigen Proteinkinase vom Typ II (CaMKII) bewirkt die Bindung
von Calmodulin an ihre autoinhibitorische Domäne eine Stimulierung der Kinase Aktivität
(Bagchi et al., 1992; Kemp und Pearson, 1991; Soderling, 1990). Obwohl das Phänomen der
Autophosphorylierung in zahlreichen CDPKs (Roberts and Harmon, 1992) beobachtet werden
konnte, ist bislang weitgehend unklar ob und inwieweit die Autophosphorylierung die Ca2+-
Abhängigkeit der Aktivierung oder die Kinase-Aktivität der CDPKs beeinflusst. Bogre et al.
(1988) und Saha und Singh (1995) beobachteten eine Stimulierung bzw. eine Hemmung der
Kinase-Aktivität nach Autophosphorylierung. Ähnlich wurde auch für die Phosphorylase
Kinase eine Stimulierung durch Autophosphorylierung beobachtet (King et al., 1983)
während sie bei CaMKII reversibel die Ca2+/Calmodulin-Abhängigkeit zu regulieren scheint
(Lai et al., 1993). Denkbar wäre aber auch eine Veränderung in der Interaktion mit
Effektorproteinen in Abhängigkeit vom Phosphorylierungsstatus. Eine mögliche Funktion der
Autophosphorylierung für die hier charakterisierte CDPK bleibt ungeklärt.

Diskussion
- 52 -
Membran-assozierte CDPK
Verschiedene plasmamembran-assozierte Proteine sind durch Myristat, einer C14 gesättigten
Fettsäure, modifiziert. Die kovalente Verkettung von Myristat erfolgt über eine Peptidbindung
zum Glycin der N-terminalen Domäne mit der Konsensus-Sequenz Met-Gly-x-x-x-[Ser/Thr].
Katalysiert wird die Reaktion von der N-Myristyl-Transferase und verläuft co-translationell
nach Abspaltung des Start-Methionins durch M-aminopeptidase (Russell et al., 1994).
Obgleich die Modifizierung stabil ist, ist die daraus resultierende Hydrophobizität eines
myristoylierten Peptids nicht ausreichend für eine stabile Verankerung des Proteins in der
Plasmamembran. Dies lässt vermuten, dass bei der Myristoylierung zusätzliche Mechanismen
zum Tragen kommen, die der Stabilität des plasmamembran-assozierten Proteins dienen.
Häufig ist beispielsweise eine zusätzliche Palmitoylierung an einem Cystein-Rest nahe dem
myristoylierten Glycin. Auch eine Anhäufung positiver Ladungen, die eine ionische
Wechselwirkung mit den Phospholipiden der Plasmamembran erlauben, wird beobachtet
(Yalovsky et al., 1999).
Die N-terminale Aminosäuresequenz zahlreicher CDPKs weisen die Konsensus-Sequenz für
eine N-Myristoylierung auf. Vorausgegangene Arbeiten zeigten, dass einige der
Calciumabhängigen-Proteinkinasen membran-assoziert vorliegen. So konnten zum Beispiel
CDPKs von Gerste (Klimczak und Hind, 1990) und Hafer (Schaller et al., 1992) von der
Membran isoliert und gereinigt werden. Darüber hinaus gelang die Identifizierung
membranständiger Substrate, die von CDPKs phosphoryliert werden; so das Protein Nodulin
26 (Roberts und Harmon, 1992), die Plamamembran H+-ATPase (Schaller und Sussman,
1998), TIP, ein vakuoläres Wasserkanal Protein (Johnson und Chrispeels, 1992), Chlorid-
Kanal in Vakuolen (Pei et al., 1996), sowie ein Kaliumkanal der Schliesszellen (Li et al.,
1995). Darüber hinaus ist für eine CDPK aus Cucurbita pepo L. die N-terminale
Myristoylierung in vitro nachgewiesen worden (Ellard-Ivey et al., 1999).
Die Vereinigung von CDPK und Plasmamembran könnte durch Ca2+ reguliert sein (Yuasa et
al., 1995). Recoverin, ein Ca2+-bindendes Photorezeptorprotein von Wirbeltieren, bindet
durch eine N-terminal verknüpfte Myristoyl-Gruppe an die Membran. Dabei wird der
Assoziations-Prozess von gebundenem Calcium reguliert. Es wird vermutet, dass durch Ca2+-
Bindung der hydrophobe NH2-Terminus von Recoverin exponiert wird und so mit der
Membran interagieren kann. Nach diesem Model könnte die kovalent gebundene Myristoyl-
Gruppe der CDPK an die Plasmamembran binden (Yalovsky et al., 1999).

Diskussion
- 53 -
Die abgeleitete Aminosäuresequnz der identifizierten CDPK aus Tomate weist die besagte N-
terminale Konsensus-Sequenz Met-Gly-x-x-x-[Ser/Thr] auf, wie sie bereits schon bei anderen
CDPKs beobatet worden ist. Um einen Einblick in die biologische Funktion der CDPK zu
erhalten, ist es von grossem Interesse ihre Funktionalität zu überprüfen und die Lokalisation
der CDPK zu untersuchen. Denkbar wäre etwa eine Zerstörung des Konsensus-Motivs durch
„site-directed mutagenesis“ und ein Vergleich der in vitro Myristoylierung des Wildtyp und
mutierten Proteins. Weiterhin wäre eine Bestimmung der Lokalisation in vivo und eine
Untersuchung der Ca2+-Abhängigkeit der Membranassoziation durch transiente Expression
von CDPK-GFP (green fluorescent protein)-Fusionsproteinen in Pflanzenzellen möglich.
Diese Untersuchung sollen in weiterführenden Arbeiten vorgenommen werden.

Literaturverzeichnis
- 54 -
5. LITERATURVERZEICHNIS
Bagchi, I.C., Huang, Q., Means, A.R. (1992). Identification of amino acids essential for calmodulin binding and activation of smooth muscle myosin light chain kinase. J. Biol. Chem. 267: 3024-3029. Baunsgaard, L., Fuglsang, A.T., Jahn, T., Korthout, H.A.A.J., de Boer, A.H., Palmgren, M.G. (1998). The 14-3-3 protein associates with the plasma membrane H+-ATPase to generate a fusicoccin binding complex and a fusicoccin responsive system. Plant J. 13: 661-671. Bergey, D.R., Howe, G.A., Ryan, C.A. (1996). Polypeptide signaling for plant defensive genes exhibits analogies to defense signaling in animals. Proc. Natl. Acad. Sci. USA 92: 8675-8679. Bergey, D.R., Orozco-Cardenas, M., de Moura, D.S., Ryan, C.A. (1999). A wound- and systemin-inducible polygalacturonase in tomato leaves. Proc. Natl. Acad. Sci. USA 96: 1756-1760. Bergy, D.R., Ryan, C.A. (1999). Wound- and systemin–inducible calmodulin gene expression in tomato leaves. Plant Mol. Biol. 40: 815-823. Bidway, A.P., Takemoto, J.Y. (1987). Bacterial phytotoxin, syringomycin, induces a protein-kinase mediated phosphorylation of red beet plasma membrane polypeptides. Proc. Natl. Acad. Sci. USA 84: 6755-6759. Binder, B.M., Harper, J.F., Sussman, M.R. (1994). Characterisation of an Arabidopsis calmodulin-like domain protein kinase purified from Escherichia coli using an affinity sandwich technique. Biochemistry 33: 2033- 2041. Bishop, P.D., Makus, J.K., Pearce, G., Ryan, C.A. (1981). Proteinase inhibitor inducing factor activity in tomato leaves resides in oligosaccharides enzymically released from cell walls. Proc. Natl. Acad. Sci. USA 78: 3536-3540. Bishop, P.D., Pearce, G., Bryant, J.E., Ryan, C.A. (1984). Isolation and characterisation of the proteinase inhibitor-inducing factor from tomato leaves. Identity and activity of the poly- and oligogalacturonide fragments. J. Biol. Chem. 259: 13172-13177. Birnboim, H., Doly, J. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucl. Acids Res. 7: 1513-1523. Bogre, L., Olah, Z., Dudits, D. (1988). Calcium dependent protein kinase from alfalfa (Medicago varia): partial purification and autophosphorylation. Plant Sci. 58: 135-144. Bradford, M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72: 248-254.

Literaturverzeichnis
- 55 -
Breviario, D., Morello, L., Giani, S. (1995). Molecular cloning of two novel rice cDNA sequences encoding putative calcium-dependent protein kinases. Plant Mol. Biol. 27: 953-967. Camoni, L., Fullone, R.F., Marra, M., Aducci, P. (1998). The plasma membrane H+-ATPase from maize roots is phosphorylated in the C-terminal domain by a calcium-dependent protein kinase. Plant Physiol. 104: 549-555. Constabel P.C., Bergey D.R., Ryan C.A. (1995). Systemin activates synthesis of wound inducible tomato leaf polyphenol oxidase via the octadecanoid defense signalling pathway. Proc. Natl. Acad. Sci. USA 92: 407-411. Constabel, P.C., Yip, L., Ryan, C.A. (1998). Prosystemin from potato, black night-shade, and bell pepper: primary structure and biological activity of predicted systemin polypeptides. Plant Mol. Biol. 36: 55-62. De Nisi, P., Dell’Orto, M., Pirovano, L., Zocchi, G. (1999). Calcium-dependent phosphorylation regulates the plasma-membrane H+-ATPase activity of maize (Zea mays L.) roots. Planta 209: 187-194. Ellard-Ivey, M., Hopkins, R.B., White, T.J., Lomax, T.L. (1999). Cloning, expression and N-terminal myristoylation of CpCPK1, a calcium-dependent protein kinase from zucchini (Cucurbita pepo L.). Plant Mol. Biol. 39: 199-208. Farmer, E.E., Ryan, C.A. (1990). Interplant communication: airborne methyl jasmonate induces synthesis of proteinase inhibitors in plant leaves. Proc. Natl. Acad. Sci. USA 87: 7713-7716. Farmer, E.E., Ryan, C.A. (1992). Octadecanoid precursors of jasmonic acid activate the synthesis of wound-induible proteinase inhibitors. Plant Cell 4: 129-134. Farmer, E.E., Ryan, C.A. (1992). Octadecanoid-derived signals in plants. Trends Cell Biol. 2: 236-241. Feinberg, A.P., Vogelstein, B. (1983). A technique for radioactive labeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132: 6’13. Felix, G., Boller, T. (1995). Systemin induces rapid ion fluxes and ethylene biosynthesis in Lycopersicon peruvianum cells. Plant J. 7: 381-389. Greenberg, J.T., Guo, A., Klessig, D.F., Ausubel, F..M. (1994). Programmed cell death in plants: a pathogen-triggered response activated coordinately with multiple defense. Cell 77: 551-563. Hrabak, E.M., Dickmann, I.J., Satterlee, J.S., Sussman, M.R. (1996). Characterisation of eight new members of the calmodulin-like domain protein kinase gene family from Arabidopsis thaliana. Plant Mol. Biol. 31: 405-412. Harmon, A.C., Putnam-Evans, C., Cormier, M.J. (1987). A calcium-dependent but calmodulin-independent protein kinase from soybean. Plant Physiol. 83: 830-837.

Literaturverzeichnis
- 56 -
Harmon, A.C., Yoo, B.C., McCaffery, C. (1994). Pseudosubstrate inhibition of CDPK, a protein kinase with a calmodulin-like domain. Biochemistry 33: 7278-7287. Harper, J.F., Huang, J.F., Lloyd, S.J. (1994). Genetic identification of an autoinhibitor in CDPK, a protein kinase with a calmodulin-like domain. Biochemistry 33: 7267-7277. Harper, J.F., Sussman, M.R., Schaller, G.E. Putnam-Evans, C., Charbonneau, H., Harmon, A.C. (1991). A calcium-dependent protein kinase with a regulatory domain similar to calmodulin. Science 252: 951-954. Heitz, T., Bergey, D.R., Ryan, C.A. (1997). A gene encoding a chloroplast-targeted lipoxygenase in tomato leaves is transiently induced by wounding, systemin, and methyl jasmonate. Plant Physiol. 114: 1085-1093. Hildmann, T., Ebneth, M., Pẽna-Cortés, H., Sanchez-Serrano, J., Willmitzer, L., Prat, S. (1992). General roles of abscisic and jasmonic acids in gene activation as a result of mechanical wounding. Plant Cell 4: 1157-1170. Huang, J.F., Teyton, L., Harper, J.F. (1996). Activation of a Ca2+-dependent protein kinase involves intramolecular binding of a calmodulin-like regulatory domain. Biochemistry 35: 13222-13230. Jahn, T., Fuglsang, A.T., Olsson, A., Brüntrup, I.M., Collinge, D.B., Volkmann, D., Sommarin, M., Palmgren, M.G., Larsson, C. (1997). The 14-3-3 protein interacts directly with the C-terminal region of the plant plasma membrane H+-ATPase. Plant Cell 9: 1805-1814. Johansson, F., Sommarin, M., Larsson, C. (1993). Fusicoccin activates the plasma membrane H+-ATPase by a mechanism involving the C-terminal inhibitory domain. Plant Cell 5: 321-327. Johnson, K.D., Chrispeels M.J. (1992). Tonoplast-bound protein kinase phosphorylates tonoplast intrinsic protein. Plant Physiol 100: 1787-1795. Kawasaki, T., Hayashida, N., Baba, T., Shinozaki, K., Shimada, H. (1993). The gene encoding a calcium-dependent protein kinase located near the sbeI gene encoding starch branching enzyme I is specifically expressed in developing rice seeds. Gene 129: 183-189. Kemp, B.E., Pearson, R.B. (1991). Protein kinase phosphorylation site sequences and consensus specificity motifs: tabulations. Methods Enzymol. 200: 62-81. King, M.W., Fitzgerald, T.J., Carlson, G.M. (1983). Characterisation of initial autophosphorylation events in rabbit skeletal muscle phosphorylase kinase. J. Biol. Chem. 263: 9925-9930. Klimczak, L.J., Hind, G. (1990). Biochemical similarities between soluble and membrane-bound calcium-dependent protein kinases of barley. Plant Physiol 92: 919-923. Lai, Y., Nairn, A.C., Greengard, P. (1986). Autophosphorylation reversibly regulates the Ca2+ /calmodulin dependence of Ca 2+/calmodulin-dependent protein kinase II. Proc. Natl. Acad. Sci. USA 83: 4253-4257.

Literaturverzeichnis
- 57 -
Lämmli, U.K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. Li, J., Lee, Y.R.J., Assmann, S.M. (1998). Guard cells possess a calcium-dependent protein kinase that phosphorylates the KAT1 potassium channel. Plant Physiol. 116: 785-795. Lino, B., Baizabal-Aguirre, V.M., Gonzáles de la Vara, L.E. (1998). The plasma-membrane H+-ATPase from beet root is inhibited by a calcium-dependent phosphorylation. Planta 204: 352-359. Marra, M., Olivari, C., Visconti, S., Albumi, C., Aducci, P., De Michelis, M.I. (2000). A phosphopeptide corresponding to the cytosolic stretch connecting transmembrane segments 8 and 9 of the plasma membrane H+-ATPase binds 14-3-3 proteins and inhibits Fusicoccin-induced activation of the H+-ATPase. Plant Biol. 2: 11-16. McGurl, B., Pearce, G., Orozco-Cardenas, M., Ryan, C.A. (1992). Structure, expression, and antisense inhibition of the systemin precursor gene. Science 255: 1570-1573. Michelet, B., Boutry, M. (1995). The plasma membrane H+-ATPase. A highly regulated enzyme with multiple physiological functions. Plant Physiol. 108: 1-6. Moyen, C., Johannes, E. (1996). Systemin transiently depolarises the tomato mesophyll cell membrane and antagonises fusicoccin-induced extracellular acidification of mesophyll tissue. Plant Cell Environ. 19: 464-470. Narváez-Vásquez, J., Florin-Christensen, J., Ryan, C.A. (1999). Positional specificity of a phospholipase A activity induced by wounding, systemin, and oligosaccharide elicitors in tomato leaves. Plant Cell 11: 2249-2260. Narvaez-Vasquez, J., Pearce, G., Orozco-Cardenas, M.L., Franceschi, V.R., Ryan, C.A. (1995). Autoradiographic and biochemical evidence for the systemic translocation of systemin in tomato plants. Planta 195: 593-600. O’Donnell, P.J., Calvert, C.M., Atzorn, R., Wasternack, C., Leyser, H.M.O., Bowles, D.J. (1996). Ethylene as a signal mediating the wound response of tomato plants. Science 274: 1914-1917. Oecking, C., Piotrowski, M., Hagemeier, J., Hagemann, K. (1997). Topology and target interaction of the fusicoccin-binding 14-3-3 homologs of Commelina communis. Plant J. 12: 441-453. Orozco-Cardenas, M., McGurl, B., Ryan, C.A. (1993). Expression of an antisense prosystemin gene in tomato plants reduces resistance toward Manduca sexta larvae. Proc. Natl. Acad. Sci. USA 90: 8273-8276. Palmgren, M.G. (1991). Regulation of plant plasma membrane H+-ATPase activity. Physiol. Plant. 83: 314-323. Pautot, V., Holzer, F.M., Reisch, B., Walling, L. (1993). Leucine aminopeptidase: an inducible component of the defense response in Lycopersicon esculentum (tomato). Proc. Natl. Acad. Sci. USA 90: 9906-9910.

Literaturverzeichnis
- 58 -
Pearce, G., Strydom, D., Johnson, S., Ryan, C.A. (1991). A polypeptide from tomato leaves induces the synthesis of wound-inducible proteinase inhibitor proteins. Science 253: 895-898. Pei, Z.M., Ward, J.M., Harper, J.F., Schroeder, J.I. (1996). Anovel chloride channel in Vicia faba guard cell vacuoles activated by the serine/threonine kinase, CDPK. EMBO J. 15: 6564-6574. Pẽna-Cortés, H., Sanchez-Serrano, J., Mertens, R., Willmitzer, L., Prat, S. (1989). Abscisic acid is involved in the wound-induced expression of the proteinase inhibitor II gene in potato and tomato. Proc. Natl. Acad. Sci. USA 86: 9851-9855. Roberts, D.M., Harmon, A.C. (1992). Targets of intracellular calcium signals in higher plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 43: 375-414. Russell, J.D., Bhatnager, R.S., Knoll, L.J., Gordon, J.I. (1994). Genetic and biochemical studies of protein N-myristoylation. Annu. Rev. Biochem. 63: 869-914. Ryan, C.A. (1992). The search for the proteinase inhibitor-inducing factor, PIIF. Plant Mol. Biol. 19: 123-133. Ryan, C.A., Pearce, G. (1998). Systemin: a polypeptide signal for plant defensive genes. Annu. Rev. Cell Dev. Biol. 14: 1-17. Saha, P., Singh, M. (1995). Characterisation of a winged bean (Psophocarpus tetragonolobus) protein kinase with a calmodulin-like domain: regulation by autophosphorylation. Biochem. J. 305: 205-210. Saijo, Y., Hata, S., Sheen, J., Izui, K. (1997). cDNA cloning and prokaryotic expression of maize calcium-dependent protein kinase. Biochim. Biophys. Acta. 1350: 109-114. Sambrook, J., Fritsch, E.F., Maniatis, T. (1989). Molecular cloning. A laboratory manual. New York, Cold Spring Harbor Laboratory Press. Sanger, F., Nicklen, S., Coulsen, A.R. (1977). DNA sequencing with chain-termination inhibitors. Proc. Natl. Acad. Sci. USA 74: 5463-5467. Satterlee, J.S., Sussman, M.R. (1998). Unusual membrane-associated protein kinases in higher plants. J. Membrane Biol. 164: 205-213. Schaller, A. (1999). Oligopeptide signalling and the action of systemin. Plant Mol. Biol. 40: 763-769. Schaller, A., Bergey, D.R., Ryan, C.A. (1995). Induction of wound response genes in tomato leaves by bestatin, an inhibitor of aminopeptidases. Plant Cell 7: 1893-1898. Schaller, A., Oecking, C. (1999). Modulation of plasma membrane H+-ATPase activity differentially activates wound and pathogen defense responses in tomato plants. Plant Cell 11: 263-272.

Literaturverzeichnis
- 59 -
Schaller, A., Ryan, C.A. (1996). Molecular cloning of tomato leaf cDNA encoding an aspartic protease, a systemic wound response protein. Plant Mol. Biol. 31: 1073-1077. Schaller, G.E., Harmon, A.C., Sussman, M.R. (1992). Characterization of a calcium- and lipid-dependent protein kinase associated with the plasma membrane of oat. Biochemistry 31: 1721-1727. Schaller, G.E., Sussman, M.R. (1988). Phosphorylation of the plasma membrane H+-ATPase of oat roots by a calcium-stimulated protein kinase. Planta 173: 509-518. Scheer, J.M., Ryan, C.A. (1999). A 160 kDa systemin receptor on the cell surface of Lycopersicon peruvianum suspension cultured cells: kinetic analyses, induction by jasmonate and photoaffinity labelling. Plant Cell 11: 1525-1535. Schmid, J., Görlach, J. (1996). Introducing StuI sites improves vectors for the expression of fusion proteins with factor Xa cleavage sites. Gene 170: 145-146. Serrano, R. (1989). Structure and function of plasma membrane ATPase. Annu. Rev. Plant Physiol. Plant Mol. Biol. 40: 61-94. Serrano, R. (1990). Plasma membrane ATPase. In: Larsson, C., Moller, I.M. (eds). The Plant Plasma Membrane, structure, function and molecular biology. Springer-Verlag pp. 127-153. Shi, J., Kim, K., Ritz, O., Albrecht, V., Gupta, R., Harter, K., Luan, S., Kudla, J. (1999). Novel protein kinases associated with calcineurin B-like calcium sensors in Arabidopsis. Plant Cell 11(12): 2393-2406. Soderling, T.R. (1990). Protein kinases. Regulation by autoinhibitory domains. J. Biol. Chem. 265: 1823-1826. Somssich, I.E., Hahlbrock, K. (1998). Pathogen defense in plants - a paradigm of biological complexity. Trends Plant Sci. 3: 86-90. Stone, J.M., Walker, J.C. (1995). Plant protein kinase and stress signal transduction in plants. Science 274: 1900-1902. Stratmann, J.W., Ryan, C.A., (1997). Myelin basic protein kinase activity in tomato leaves is induced systemically by wounding and increases in response to systemin and oligosaccharide elicitors. Proc. Natl. Acad. Sci. USA 94: 11085-11089. Suzuki, Y.S., Wang, Y., Takemoto, J.Y. (1992). Syringomycin-stimulated phosphorylation of the plasma membrane H+-ATPase from red beet storage tissue. Plant Physiol. 99: 1314-1320. Svennelid, F., Olsson, A., Piotrowski, M., Rosenquist, M., Ottman, C., Larsson, C., Oecking, C., Sommarin, M. (1999). Phosphorylation of Thr-948 at the C terminus of the plasma membrane H+-ATPase creates a binding site for the regulatory 14-3-3 protein. Plant Cell 11: 2379-2392. Vera-Estrella, R., Barkla, B.J., Higgins, V.J., Blumwald, E. (1994). Plant defense to fungal pathogens. Activation of host-plasma membrane H+-ATPase by elicitor-induced enzyme dephosphorylation. Plant Physiol. 104: 209-215.

Literaturverzeichnis
- 60 -
Verhey, S.D., Gaiser, J.C., Lomax, T.L. (1993). Protein kinase from the zucchini. Characterisation of calcium-requiring plasma-membrane kinases. Plant Physiol 103: 413-419. Vick, B.A., Zimmerman, D.C. (1984). Biosynthesis of jasmonic acid by several plant species. Plant Physiol. 75: 458-461. Walker-Simmons, M., Ryan, C.A. (1977). Wound-induced peptidase activity in tomato leaves. Biochem. Biophys. Res. Comm. 74: 411-416. Walker-Simmons, M., Ryan, C.A. (1984). Proteinase inhibitor synthesis in tomato leaves. Induction by chitosan oligomers and chemically modified chitosan and chitin. Plant Physiol. 76: 787-790. Xing, T., Higgins, V.J., Blumwald, E. (1996). Regulation of plant defense response to fungal pathogens: two types of protein kinases in the reversible phosphorylation of the host plasma membrane H+-ATPase. Plant Cell 8: 555-564. Yalovsky, S., Rodríguez-Concepción, M., Gruissem, W. (1999). Lipid modifications of proteins - slipping in and out of membranes. Trends Plant SCI. 4: 439-445. Yoo, B.C., Harmon, A.C. (1996). Intramolecular binding contributes to the activation of CDPK, a protein kinase with a calmodulin-like domain. Biochemistry 35: 12029-12037. Yoon, G.M., Cho, H.S., Ha, H.J., Liu, J.R., Lee, H.-s.P. (1999). Characterisation of NtCDPK1, a calcium-dependent protein kinase gene in Nicotiana tabacum, and the activity of its encoded protein. Plant Mol. Biol. 39: 991-1001. Yoshikawa, M., Yamaoka, N., Takenchi, Y. (1993). Elicitors: Their significance and primary modes of action in the induction of plant defense reactions. Plant Cell Physiol. 34: 1163-1173. Yuasa, T., Takahashi, K., Muto, S. (1995). Purification and characterization of a Ca2+-dependent protein kinase from the halotolerant green alga Dunaliella tertioecta. Plant Cell Physiol. 36: 699-708. Zhao, Y., Pokutta, S., Maurer, P., Lindt, M., Franklin, R.M., Kappers, B. (1994). Calcium-binding properties of a calcium-dependent protein kinase from Plasmodium falciparum and the significance of individual calcium-binding sites for kinase activation. Biochemistry 33: 3714-3721.

Abkürzungsverzeichnis
- 61 -
6. ABKÜRZUNGEN
ATP Adenosintriphosphat
bp Basenpaare
BSA Bovine serum albumin
cDNA Complementary DNA
CDPK Calciumabhängige Proteinkinase
CDPK-K Teillänge-CDPK
CDPK-L Volllänge-CDPK
CIAP Calf intestine alkaline phosphatase
cpm Counts per minute
CTAB Cetyltrimethylammoniumbromid
dATP Desoxyriboadenosintriphosphat
DC Dünnschichtchromatographie
DMSO Dimethylsulfoxid
DNA Desoxyribonucleic acid
dNTP Desoxyribonukleotidtriphosphat
DTT 1,4-Dithio-DL-threitol
E. coli Escherichia coli
EDTA Ethylendiamin-N,N,N‘,N‘-tetraessigsäure
EGTA Ethylenglycol-Bis-(2-Aminoethyl)-Tetraessigsäure
EST Expressed sequence tag
EtBr Ethidiumbromid
FC Fusicoccin
GST Glutathion-S-Transferase
HEPES 4-(2-Hydroxymethyl)-Piperazin-1-Ethansulfonsäure
IPTG Isopropylthio-β-Galaktopyranosid
kb Kilobase
kDa Kilodalton
KM Michaeliskonstante
KPP Kaliumphosphatpuffer
LB-Medium Luria-Bertani Medium
mcs Multiple Cloning Site

Abkürzungsverzeichnis
- 62 -
MOPS 3-(N-morpholino) propansulfonsäure
mRNA Messenger RNA
OD Optische Dichte
ORF Open reading frame
PAGE Polyacrylamide gel electrophoresis
PCR Polymerase chain reaction
PK Protein Kinase
PMSF Phenylmethylsulfonylfluorid
PR Pathogenesis related
Pwo-Polymerase Polymerase aus Pyrococcus woesei
RACE Rapid amplification of cDNA ends
RNA Ribonucleic acid
RNAse Ribonuklease
rpm Revolutions per minute
RT Raumtemperatur
SDS Sodium dodecyl sulfate
SWRPs Systemic wound response proteins
Taq-Polymerase Polymerase aus Thermus aquaticus
TCA Trichloroacetic acid
Tris Tris-(hydroxymethyl)-aminomethan
v/v Volume per volume
w/v Weight per volume
X-Gal [(5-Brom-4-chlor-3-indolyl)-β-D-galactosid]

Anhang
- 63 -
7. ANHANG
7.1 Primer Sequenzen Die verwendeten Oligonukleotide sind von Microsynth, Balgach, Schweiz. PCR Primer: LePK52: 5 ´-GGGAATTCGGTATGGGTGATGAGGCCAC-3 ´ unterstrichen: eingeführte EcoRI-Schnittstelle RACE-PCR Primer: LePK32: 3 ´-GGGAATTCGCTCTCGATGCACTAGAAAAG-5 ´ PKGSP1: 3 ´-CGTGCTGGCTGGTGAACGTTCTCTGCTC-5 ´ Sequenzierprimer: PK52: 5 ´-TGGGAAAGAATTGGGTAGGG-3 ´ PK53: 5 ´-AGTGATCCATGGCCATCAAT-3 ´ PK54: 5 ´-CTTCATTGAAGAAGGGAAGG-3 ´ PK32: 3 ´-GTTCAAGAACTTGCGCGGAAG-5 ´ PK33: 3 ´-ACAATGTTTGGCTGCCCACT-5 ´ PK34: 3 ´-GACACCCATGAAATGGCAAA-5 ´

Anhang
- 64 -
Pwo-PCR Primer: PKNT1: 5 ´-ATGGGTGGTTGTTTTAGCAAGAAGT-3 ´ PKCT1: 3 ´-GGGGTACCCTAGAAAAGCTTTTGTTGTGGTTG-5 ´ PKCT2: 3 ´-GGGGTACCCTATTACAGACCCTTGATTTCTTCTTCA-5 ´ unterstrichen: eingeführte KpnI-Schnittstellen fett gedruckt: Startcodon fett, kursiv: eingeführte Stoppcodons

Danksagung
Prof. Dr. N. Amrhein hat mir ermöglicht, in seiner Professur die Diplomarbeit in der Gruppe
für Biochemie und Physiologie der Pflanzen auszuführen.
Dr. Andreas Schaller hat mich in seine Arbeitsgruppe aufgenommen und mich fachlich wie
menschlich hervorragend betreut.
David Frasson, Ingar Janzik, Jochen Strassner, Werner Schmidt und Felix Hauser standen
mir, wenn immer nötig mit Rat und Tat hilfsbereit zur Seite.
Ihnen und allen nicht namentlich erwähnten Mitarbeitern der Gruppe Amrhein sei an dieser
Stelle ganz herzlich gedankt.