Embed Size (px)
Citation preview

1
R.E.Scharf 2010
Hämophilie A / B und
von-Willebrand-Syndrom
BDT- und DGTI-Weiterbildungsseminar, Bielefeld, 06. bis 11. Mai 2012
Rüdiger E. Scharf Institut für Hämostaseologie, Hämotherapie und
TransfusionsmedizinHemophilia Comprehensive Care Center
Universitätsklinikum Düsseldorf und
Biologisch-Medizinisches Forschungszentrum
Heinrich Heine Universität Düsseldorf
R.E.Scharf 2010
Hämostatisches Gleichgewicht
Hämostasefördernd
Hämostasehemmend
PlättchenadhäsionPlättchenaggregationFibrinbildung
FibrinolyseAntiaggregatorische MechanismenInhibitoren der Fibrinbildung
R.E.Scharf 2010
Hämostase
Primäre Hämostase Sekundäre HämostasePlättchenadhäsionPlättchenaktivierungPlättchenaggregation„hemostatic plug“
GerinnungsaktivierungThrombingenerierungFibrinbildungPlättchen-Fibrin-Thrombus

2
R.E.Scharf 2010
R.E.Scharf 2010
Warum bluten Hämophilie-Patienten ?
R.E.Scharf 2010
FX FXa
Prothrombin (FII) Thrombin (FIIa)
TF / FVIIa
FVII Tissue factor (TF)
Fibrinogen Fibrin
RE. Scharf2004
FIXFVIIIa FVIII / vWF
FIXa
FVII
FXIII
FXIIIa
FVa FV
XIIa
XIa
3
R.E.Scharf 2010
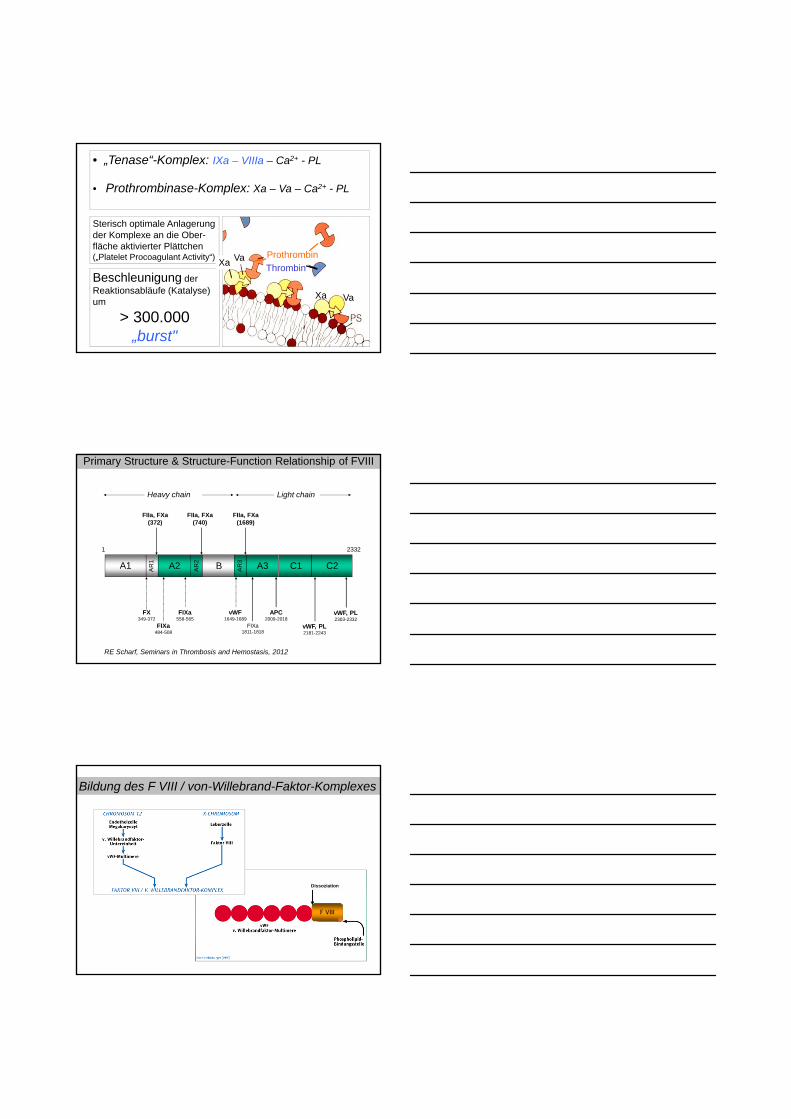
• „Tenase“-Komplex: IXa – VIIIa – Ca2+ - PL
• Prothrombinase-Komplex: Xa – Va – Ca2+ - PL
Sterisch optimale Anlagerungder Komplexe an die Ober-fläche aktivierter Plättchen(„Platelet Procoagulant Activity“)
Beschleunigung der Reaktionsabläufe (Katalyse) um
> 300.000„burst"
Thrombin
ProthrombinXa Va
VaXa
R.E.Scharf 2010
A1 A2 A3 C1 C2B
1 2332
AR
2
AR
1
AR
3
vWF, PL2181-2243
APC2009-2018
FIXa1811-1818
vWF1649-1689
FX349-372
FIXa484-508
FIXa558-565
FIIa, FXa (372)
FIIa, FXa (740)
FIIa, FXa (1689)
vWF, PL2303-2332
Heavy chain Light chain
Primary Structure & Structure-Function Relationship of FVIII
RE Scharf, Seminars in Thrombosis and Hemostasis, 2012
R.E.Scharf 2010
Faktor VIII / vWF-Komplex
Bildung des F VIII / von-Willebrand-Faktor-Komplexes
Faktor VIII / vWF-Komplex
F VIII
Dissoziation

4
R.E.Scharf 2010
BA2 C1 C2A1
HGR
50 43
45 25
90 kDa 80 kDa
80 kDa
73 kDa
73 kDa
200 kDa
1313
740
372
562336
1689FIIa FIIa
FIIa
NH2 COOHA3
APC 3. Inactivation(APC, FXa)
1. Secretion(Golgi)
2. Activation(FIIa)
Domain Structure and Processing of Factor VIII
Heavy-chain Light-chain
N Heimburger, 1999; P Fay, 2000 (modified)
R.E.Scharf 2010
A1
C1 C2A3
BA2
Cu2+
GPIbIIb3
Resting Platelet
A3 C1
A1Cu2+
C2
APC or XaCleavage
Inactivation
A3 C1
A1Cu2+
C2
A2 Dissociation
A2
A3 C1
A1Cu2+
C2
B
IIaActivation
A3 C1
A2A1
Cu2+C2
GPIbIIb3
Activated Platelet
Activation
ComplexAssembly
Model of Activation and Inactivation of Factor VIII
R.E.Scharf 2010
Angeborene Gerinnungsstörungen
1:5.000

5
R.E.Scharf 2010
Severe hemophilia A
missense15%
unknown4%
inversions42%
large deletion
8%
nonsense14%
frameshift17%
Mild-Moderate hemophilia A
missense86%
unknown10%
frameshift2%slicing
2%
Factor VIII Gene Defects in Severe andMild to Moderate Hemophilia A
(Data from RJ Kaufman and PJ Fay, 2000)
R.E.Scharf 2010
Missense Mutationsin the Factor VIII
Gene
Structural domainsof the protein
Data from Kaufman & Fay, 2000
R.E.Scharf 2010
Angeborene Gerinnungsstörungen
1:30.000

6
R.E.Scharf 2010
Historisches zur Hämophilie
5.Jh. - Erwähnung im babylonischen Talmud: „Wenn zweiv. Chr. Jungen bei der Beschneidung verblutet sind, keine
Beschneidung aller weiteren Söhne der Mutter, ihrerTöchter und ihrer Schwestern“.
1803 - Erbgang der Bluterkrankheit1828 - Begriff „Hämophilie“ (Diss. eines Schoenlein-Schülers)1926 - Erstbeschreibung des von-Willebrand-Syndroms
„Pseudohämophilie“1937 - Hämophilie als Faktor VIII-Mangel identifiziert
„antihämophiles Globulin“1952 - Abgrenzung der Hämophilie B (Biggs)1965 - Therapie der Hämophilie A mit Kryopräzipitaten1967 - Therapie der Hämophilie B mit PPSB1968 - Erstes plasmatisches Faktor VIII-Konzentrat1984 - AIDS-Katastrophe
R.E.Scharf 2010
Vererbung Hämophilie A / B
R.E.Scharf 2010
Beugekontraktur

7
R.E.Scharf 2010
Vererbung Hämophilie A / B
R.E.Scharf 2010
Vererbung Hämophilie A / B
R.E.Scharf 2010
Knieversteifung, AtrophieDeformation, Destruktionund Immobilisation

8
R.E.Scharf 2010
Schicksal des Hämophilie-Krankenin der Vergangenheit – die vier Vs:
• verkrüppelt
• vereinsamt
• verarmt
• verblutet
Kniegelenkblutung, Hämophilie A, schwere Verlaufsform
R.E.Scharf 2010
Hämophilie A / B Hämostaseologische Diagnostik
• Suchtest: aPTT
• verlängert bei plasmatischen Defekten des endogenen
(= intrinsischen) Systems (z.B. FVIII:C- bzw. FIX:C-Mangel)
• Einzelfaktor-Analyse (Aktivität, ggf. Plasma-Misch-Versuch)
• Differentialdiagnose der F VIII:C-Aktivitätsminderung
von-Willebrand-Syndrom
R.E.Scharf 2010
(XII)XI
IXVIII VII
X
V
II
aktiviertepartielle Thrombo-plastinzeit (APTT)
Prothrombin-zeit n. Quick
(INR)
Thrombinzeit
Fibrinogen
Welcher Test wofür ?
Intrinsisches System
ExtrinsischesSystem
anti-Faktor Xa-Aktivität

9
R.E.Scharf 2010
Thrombus/Oberflächeaktivierter Thrombozyten
Müller & Renné Curr Opin Hematol 2008; 15: 516-521
R.E.Scharf 2010
Blutung Hämostase
Thrombose
FXII
Rolle von FXII in einer revidierten Hämostase-Balance
Th. Renné 2009
Take home message:FXII hat spezifische Funktion für die Thrombusbildung, aber keineBedeutung für die Blutstillung
FVII/TF
Plättchen
R.E.Scharf 2010
Hämophilie A / B
Klinisches Leitsymptom: manifeste Blutung
Blutungsneigung
Bezug zur Restaktivität von Faktor VIII:C bzw. Faktor IX:C
Verlaufsformen der Hämophilie A / B:
• Schwere H. A / B Aktivität: < 2% • Mittelschwere H. A / B Aktivität: > 2 - <5%• Leichte H. A / B Aktivität: > 5 - <15%• Subhämophilie A / B Aktivität: >15 - < 50%
Konsensus-Empfehlung zur Hämophilie-Behandlung in Deutschland (2003)

10
R.E.Scharf 2010
Klinik und spezifische Pathogenese der Blutungsmanifestationen
• Gelenkblutungen• Muskel- und Weichteilblutungen• Pseudotumorbildung• intrakranielle Blutungen• Organblutungen (Nieren, GI-Trakt)
R.E.Scharf 2010
C.F. Sch., 64 J.
R.E.Scharf 2010
11
R.E.Scharf 2010
R.E.Scharf 2010
vorherA.M., 29 J.
nach offener Kürettageund Transplantation
R.E.Scharf 2010
Hämophile Arthropathie
• Bei schwerer Hämophilie A / B (ca. 45% der Patienten)
• ausgeprägte Blutungstendenz,
hohe Neigung zu Spontanblutungen (F-Restaktivität < 3%)
• 85% der hämorrhagischen Ereignisse sind Gelenk-Einblutungen:
- 80% Knie-, Sprung - und Ellbogengelenk
(Anatomie: ± muskuläre Gelenkführung; lange Hebel-
verhältnisse; Überstrapazierung M.-Band-Apparat)
- Hüft - und Schultergelenk seltener betroffen

12
R.E.Scharf 2010
Warum bluten Patienten mit schwerer Hämophilie bevorzugt in die Extremitätengelenke ?
Warum Gefahr eines „Zielgelenks“ ?
R.E.Scharf 2010
Ausbildung eines Targetgelenks
R.E.Scharf 2010
Entwicklung der hämophilen Arthropathie
• Mikrotraumata im alltäglichen Bewegungsablauf • Aura: bewußte Wahrnehmung des Gelenks vor
klinisch manifester Einblutung• 2-Zeitigkeit: erst rel. blande, dann massive Blutung• Synovitis: akute chron., hyperplastisch-vulnerabel,
Mikroblutungen, Hämosiderineinlagerungen• Reaktive Knorpel- u. Knochenläsionen, Deformation• Muskelatrophie, Gelenkstabilität, Koordination,Blutungsbereitschaft u. Blutungsfrequenz
• Hämotherapie, Physiotherapie, orthopäd. Mitbetreung• Integration, Langzeitprognose, Lebensqualität

13
R.E.Scharf 2010
Hämophilen Arthropathie
R.E.Scharf 2010
Gelenkblutung
DeformierungInstabilität
Synovitis
Distension derGelenkkapsel
reflektorischeMuskelhemmung
Arthropathie derStreckmuskulatur
BeugekontrakturSubluxation
Bewegungseinschränkung
Zerstörung desGelenkknorpels
Entwicklung der hämophilen Arthropathie
R.E.Scharf 2010
Prophylaxe
Vermeidung vonBlutungsfolgen
Verbesserte bzw.optimierte Koordination
Entwicklung eines gesunden
Bewegungsapparates
Vermeidung vonBlutungen
Prävention der hämophilen Arthropathie

14
R.E.Scharf 2010
Weichteilblutungen bei schwerer Hämophilie (ca. 13%)
Cave: Kompartment-Syndrom
R.E.Scharf 2010
Hämatom des re. M. iliopsoas
R.E.Scharf 2010
Infiziertes Weichteilhämatom li. SchulterP.W., 34 J., HIV-positiv gekammerter Abzess

15
R.E.Scharf 2010
Pseudotumor re. Oberschenkel
vor nach FVIII-Hämotherapie
R.E.Scharf 2010
Ziele der Hämophiliebehandlung
R.E.Scharf 2010
Hemophilia Comprehensive Care Center
Kennzeichen des HCCC:
Das Hämophilie-Zentrum ist in ein NETZWERK
zur fachübergreifendenDiagnostik und Therapie
eingebunden.

16
R.E.Scharf 2010
Hemophilia Comprehensive Care Center
Das Behandlungsteam des Zentrumskümmert sich um alle• medizinischen• psychosozialen • praktischenBelange des Bluterpatienten
R.E.Scharf 2010
Hemophilia Comprehensive Care Center
Comprehensive care is total care
Die „Amis“ bringen‘s auf den Punkt:
R.E.Scharf 2010
Hämotherapie mit Gerinnungsfaktor-Konzentraten Konzepte und Substitutionsregime
Zwei Behandlungsprinzipien:
1. Vorbeugende Behandlung (Prophylaxe)- Dauertherapie (kontrollierte Selbstbehandlung)(Kinder, rez. Blutungen, Stress, präoperativ)
2. Ereignisorientierte Bedarfsbehandlung („on demand“)- Symptombezogene Therapie
Konsensus-Empfehlung zur Hämophilie-Behandlung in Deutschland (2003)

17
R.E.Scharf 2010
Kriterien, welche die Hämophilie-Therapie beeinflussen
Ziel: Vermeidung von Blutungen bzw. der Blutungsfolgen Festlegung eines individuellen Substitutionsregimes
1. Patientenkollektiv- Lebensalter (bei Säuglingen u. Kleinkindern höhere Dosis/kg/KG)- Schweregrad der Hämophilie- Vorgeschichte- Hemmkörperbildung- individuell unterschiedliche Recovery und Halbwertszeit
2. Klinische Situation- Häufigkeit und Lokalisation vorausgegangener Blutungen- Gelenkzustand (orthopädischer und radiologischer Score)- Begleiterkrankungen (Hepatitis, HIV)- Behandlungsanlass (anstehende Operation, akute Einblutung)
3. Soziale Situation, PatientenwilleKonsensus-Empfehlung zur Hämophilie-Behandlung in Deutschland (2003)
R.E.Scharf 2010
Langzeitbehandlung mit Faktor VIII-Konzentrat (Erwachsener)
Mittlere Dosis: 20-30 E/kg/KG mindestens dreimal / Woche(Halbwertszeit FVIII: 8 - 12 h)
Hämophilie B: weniger Injektionen / Woche (Halbwertszeit FIX: 18 - 24 h)
Individuelle Anpassung nach klinischer Situation
Konsensus-Empfehlung zur Hämophilie-Behandlung in Deutschland (2003)
R.E.Scharf 2010
Phamakokinetik von Faktor IX nach Gabe vonBerinin HS bei einem Patienten mit Hämophilie B
18
R.E.Scharf 2010
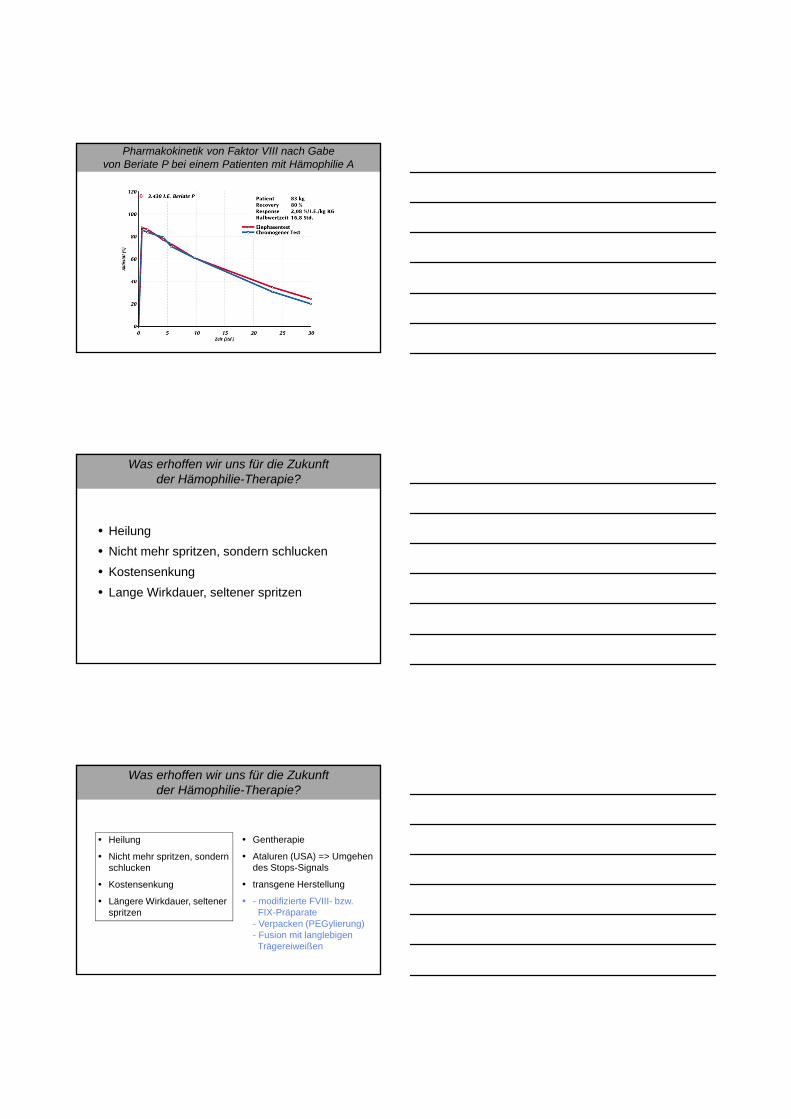
Pharmakokinetik von Faktor VIII nach Gabevon Beriate P bei einem Patienten mit Hämophilie A
R.E.Scharf 2010
Was erhoffen wir uns für die Zukunft der Hämophilie-Therapie?
• Heilung
• Nicht mehr spritzen, sondern schlucken
• Kostensenkung
• Lange Wirkdauer, seltener spritzen
R.E.Scharf 2010
Was erhoffen wir uns für die Zukunft der Hämophilie-Therapie?
• Heilung
• Nicht mehr spritzen, sondern schlucken
• Kostensenkung
• Längere Wirkdauer, seltener spritzen
• Gentherapie
• Ataluren (USA) => Umgehen des Stops-Signals
• transgene Herstellung
• - modifizierte FVIII- bzw.FIX-Präparate
- Verpacken (PEGylierung)- Fusion mit langlebigenTrägereiweißen
19
R.E.Scharf 2010
55
Tag2 3 4 5 6 7
20
40
100
80
60
10
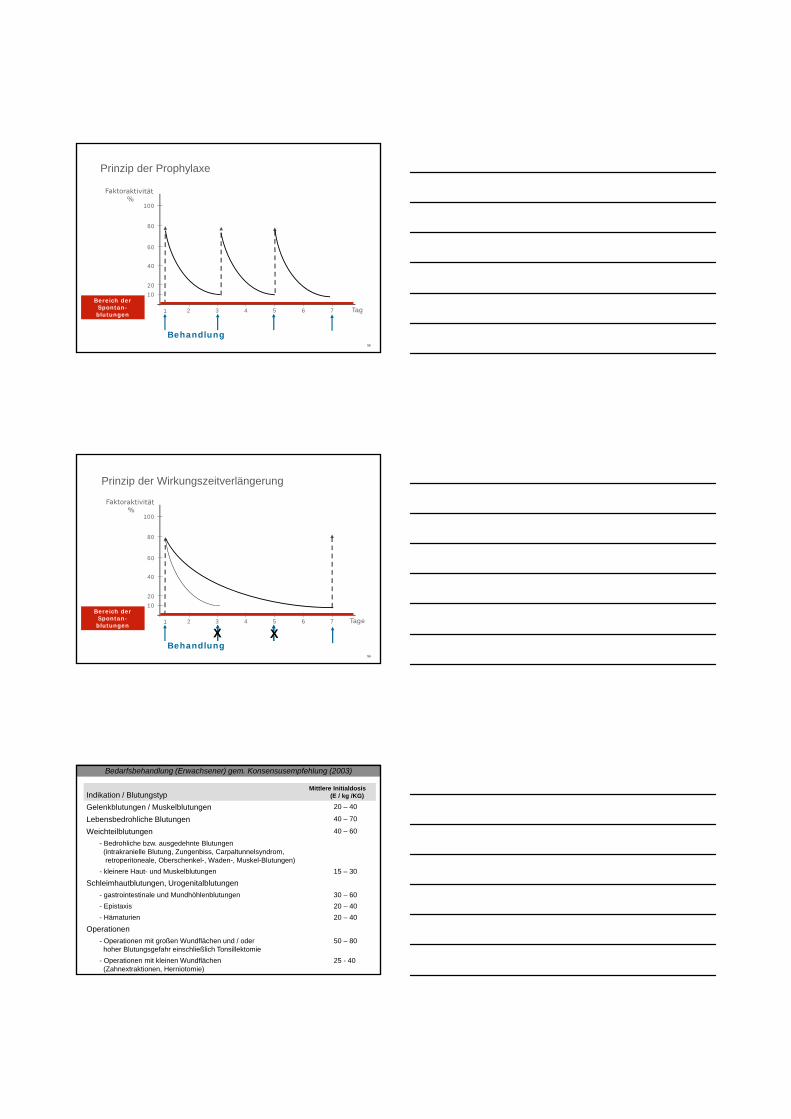
Prinzip der Prophylaxe
Behandlung
1
Bereich der Spontan-blutungen
R.E.Scharf 2010
56
Tage2 3 4 5 6 7
20
40
100
80
60
10
Prinzip der Wirkungszeitverlängerung
Behandlung
1
XX
Bereich der Spontan-blutungen
R.E.Scharf 2010
Bedarfsbehandlung (Erwachsener) gem. Konsensusempfehlung (2003)
Indikation / BlutungstypMittlere Initialdosis
(E / kg /KG)
Gelenkblutungen / Muskelblutungen 20 – 40
Lebensbedrohliche Blutungen 40 – 70
Weichteilblutungen 40 – 60
- Bedrohliche bzw. ausgedehnte Blutungen(intrakranielle Blutung, Zungenbiss, Carpaltunnelsyndrom,retroperitoneale, Oberschenkel-, Waden-, Muskel-Blutungen)
- kleinere Haut- und Muskelblutungen 15 – 30
Schleimhautblutungen, Urogenitalblutungen
- gastrointestinale und Mundhöhlenblutungen 30 – 60
- Epistaxis 20 – 40
- Hämaturien 20 – 40
Operationen
- Operationen mit großen Wundflächen und / oder hoher Blutungsgefahr einschließlich Tonsillektomie
50 – 80
- Operationen mit kleinen Wundflächen (Zahnextraktionen, Herniotomie)
25 - 40

20
R.E.Scharf 2010
Hemmkörper - Spezifische Inhibitoren -
Alloantikörper
• bei Patienten mit angeborenen Gerinnungsdefektengegen die substituierten Gerinnungsfaktoren
• Refrakterität der Hämotherapie (Faktorenkonzentrate) • Hemmkörperhämophilie A bzw. B
- erhöhtes Blutungsrisiko
Autoantikörper
• gegen körpereigene Gerinnungsproteine gerichtet(meist FVIII:C bzw. FIX)
• spontan bei zuvor gesunden Individuen• erworbene Hemmkörperhämophilie
- erhöhtes Blutungsrisiko !!
R.E.Scharf 2010
Acquired Hemophilia: Results of the Düsseldorf StudyHEMOPHILIA
CCCHHUMC
Acquired Hemophilia A (AHA) - Clinical phenotype
R.E.Scharf 2010
Epidemiologie: erworbene Hemmkörperhämophilie
erworbener Faktor VIII:C-Autoantikörper:- Inzidenz: 0.2-1 /1 Mio. Einwohner/Jahr
- gleich häufig bei Frauen und Männern - Alter < 50 Jahre: überwiegend weibliches Geschlecht
(postpartale Hemmkörper)- Alter > 50 Jahre: überwiegend männliches Geschlecht
- Häufigkeitsverteilung 2 Altersgipfel: - zwischen 20. - 30. und 60. - 80. Lebensjahr; Median ~ 60 J.
erworbene Faktor IX-Autoantikörper:- seltener als FVIII:C-Autoantikörper, - keine Inzidenzschätzungen - nur Einzelfälle beschrieben

21
R.E.Scharf 2010
Akute Blutstillungstherapie bei Autoantikörpern gegen FVIII:C bzw. IX
Patienten Behandlungsoptionen
- Low Responder FVIII:C- bzw. FIX-Konzentrat (plasmatisch od rekombinant)
HK-Titer < 5 BE DDAVP (Minirin)
- High Responder Präparate, die den Inhibitor umgehen (FVIII „bypassing activity“)
HK-Titer > 5 BE - rekombinanter FVIIa (NovoSeven)
- Prothrombin-Komplex (PPSB)
- aktivierter Prothrombin-Komplex (FEIBA)
Präparat mit „anderer Antigenität“
(wird nicht oder nur teilweise von den FVIII-Ak neutralisiert)
- Porciner FVIII (Hyate C)
Immunadsorption
- Staphylokokkenprotein A
- Anti-Ig-Antikörper
R.E.Scharf 2010
FVII FVII Tissue factor (TF)
FIXFVIIIa FVIII / vWF
FIXa
FXIII
FXIIIa
FVa FVFX FXa
Prothrombin (FII)
Fibrinogen Fibrin
Thrombin (FIIa)
FVIIaaPCC
rFVIIa FIIa, FVIIa,FIXa, FXa
inhibitory autoantibody
amplification of thrombin generation by FVIII:C by-passing agents
TF / FVIIa
Therapieprinzip bei Hemmkörper-Hämophilie
R.E.Scharf 2010
Green & Lechner 1981(n=215)
Kessler et. al. 1991(n=65)
keine Grundkrankheit 46.1 % 52.5 %
Autoimmunerkrankung
(incl. Hauterkrankungen)22.9 % 19.0 %
Post partum 7.3 % 11.0 %
Solide Tumoren 3.2 % 12.0 %
Lymphoproliferative Erkrankungen2.3 % 1.5 %
medikamentös 5.6 % 3.0 %
sonstige 11.8 % 1.5 %
Assoziierte Grunderkrankungen der erworbenen Hemmkörperhämophilie

22
R.E.Scharf 2010
Underlying disorders in patients with acquired hemophilia A
* Monocenter study, § multicenter studies. The underlying disease was recorded in # 150 of 172 or ## 178 of 215 patients, respectively. Abbreviations: SLE: systemic lupus erythematosus, COPD: chronic obstructive pulmonary disease.
Disorder Number of patients (%)
Düsseldorf Study*Gheisari et al. [10]
n = 32
Heidelberg Study *Huth-Kühne [12]
n=42
UK Study §Collins et al. [5]
n = 150 #
Kessler & Ludlam § [14]n = 65
Green & Lechner § [9]n = 178 ##
None 12 (37.5) 20 (47.6) 95 (63.3) 34 (52.3) 82 (46.1)
Autoimmune disease 7 (21.9) 9 (21.4) 25 (16.7) 11 (17.0) 32 (18.0)
- Rheumatoid arthritis 3 ( 9.4) not available 9 ( 6.0) not available 14 ( 7.9)
- SLE none not available 3 ( 2.0) not available 10 ( 5.6)
- Others 4 (12.5) not available 16 (10.7) not available 8 ( 4.5)
Malignancy 3 ( 9.4) 7 (16.7) 22 (14.7) 9 (13.9) 12 ( 6.7)
Dermatologic disorders none none 5 ( 3.3) 1 ( 2.0) 8 ( 4.5)
Respiratory disease 9 (28.1) none none 1 ( 2.0) 7 ( 3.9)
- Asthma 1 ( 3.1) 5 ( 2.8)
- COPD 8 (25.0)
Pregnancy-related orpost-partum
1 ( 3.1) 6 (14.3) 3 ( 2.0) 7 (10.8) 13 ( 7.3)
RE Scharf, Seminars in Thrombosis and Hemostasis, 2012
R.E.Scharf 2010
not time & temp. dependent
Negative personal & famiily history of bleeding disorder
Isolatedprolonged APTT
Suspect coagulation factor deficiency, vWD, or LA
Exclude heparin contamination
Confirm APTT
Consider anticoagulant influence
Occult bleeding
Acute bleeding
No bleeding
Mixing test Incubation of patient and pooled normal plasma mixed in a ratio of 1:1 (vol/vol) at 37°C for 2 hrs
APTT correction Weak or no APTT correction
Time & temperature . dependent
Suspect coagulation factor or vWF deficiency Suspect acquired hemophilia A or LA
Measure FVIII & inhibitor Testing for LA
Acquired hemophilia A
Consult HCCC
negative positive
Lupus anticoagulant (LA)
Measure FVIII, IX, XI, XII & vWF
Single factor deficiency or vWD
RE Scharf, Seminars in Thrombosis and Hemostasis, 2012
Diagnostic algorithm in patients with isolated prolonged aPTT
R.E.Scharf 2010
Reduktions- bzw. Eliminationstherapien
Nach der Therapie der akuten Blutung weiterhin hohes Blutungsrisiko bei erworbener Hemmkörperhämophilie
Behandlungsoptionen:
- Immunsuppressive Therapie mit - Steroiden- Cyclophosphamid- Retuximab
- Immunadsorption bzw. Plasmaseparation
- Immuntoleranztherapie (ITT)
23
R.E.Scharf 2010
2nd line:
&
irrespective of residual FVIII:C activity & inhibitor titer
Clinical findings &
AHA plusactive bleeding
Acute anti-hemor-rhagic treatment
Synopsis of clinical, laboratory &
imaging findings
&
Immediate immunosuppression
if no underlying disease detectable
if life-threatening bleeding persists
High-dose rFVIIa + activated PCC
Prednisolone + cyclophosphamide
Rituxibab (375 mg/m2/week)
1st line:
Inhibitor elimination
Immunotoleranceregimens
Immunoadsorption or exchange plasmapheresis
Diagnosis of any underlying disease related to AHA?
Imaging findings
Synopsis of clinical & laboratory
findings
Diagnosis of AHA
Laboratory findings
RE Scharf, Seminars in Thromb and Hemost, 2012
R.E.Scharf 2010
Substitution
IgG-Gabe
Endoxan
Urbason
ohne SubstitutionFVIII:C
0
30
60
90
120
150
180
210
240
FVIII:C
HK-Titer
0 7 19 93 Tage
~ 22 BE
% FVIII:C
Immuntoleranzbehandlung Grete J. *1925
ImmunadsorptionBeginn
R.E.Scharf 2010
Acquired Hemophilia: Results of the Düsseldorf StudyHEMOPHILIA
CCCHHUMC
Acquired Hemophilia A - Monocenter Study
complete remission (CR) n=26 patients (81 %)
(2 pts with relapse* CR)
partial remission (PR) n= 3 patients (9.5 %)
early deaths n= 3 patients (9.5 %)
*when prednison was discontinued
Results 3: Clinical outcome of AHA (n=32)
early deaths:
1 of acute MI (CR of AHA, no association with rFVIIa)
1 of sepsis (active AHA, while on immunosuppression)
1 of acute lung bleeding (in association with pulmonary sarcoidosis

24
R.E.Scharf 2010
R.E.Scharf 2010
R.E.Scharf 2010
Beziehung zwischen Blutungszeit und peripherer Thrombozytenkonzentration (Harker, 1972)
Bleeding Time (min)

25
R.E.Scharf 2010
Klassische Hämophilie Pseudohämophilie
BlutungstypGelenk- und
WeichteilblutungenSchleimhautblutungen
Gerinnungszeit hochgradig verlängert normal
Blutungszeitnormal hochgradig verlängert
Erbgang X-chromosomal-rezessivautosomal
(dominant/rezessiv)
R.E.Scharf 2010
Historisches zur Hämotherapiedes von-Willebrand-Syndroms (vWS)
1953 Berichte über Erniedrigung des FVIII bei vWS, also des antihämophilen Gerinnungsfaktors, der bei Hämophilie A fehlt
1956 Plasmafraktionierung zur Hämotherapie (Cohn-Fraktion-I-0)
1957 Transfusion der Cohn-Fraktion I-0 führt zu FVIII-Anstieg bei Hämophilie A und Korrektur der Blutungszeit bei vWS
1963 Beobachtung, dass Transfusionen von Hämophilie A-Plasma (!)den Hämostasedefekt bei vWS vorübergehend beheben kann
1966 Kryopräzipitate zur Therapie der Hämophilie und des vWS
R.E.Scharf 2010
EndoplasmicReticulum
Golgi Apparatus /Weibel-Palade Bodies
Pro-vWF Dimer
FreeSignal peptides
Pre-Pro-vWF Pro-vWF Monomer
1.
2.3.
4.
FreePropolypeptides
Pro-vWF Multimer
vWF Multimer
Signal peptide
Propeptide
vWFSubunit
RE. Scharf2004
N N
N N NN N N N
N N N N
C C C C C CC C
C C C C C CC CC
S-S
S-S S-S S-S
S-SS-SS-S
S-SS-S
S-S S-S

26
R.E.Scharf 2010
von-Willebrand-Faktor
Subunit Dimer
MW: 550-20.000 kDaMultimer
Hochmolekulare Multimere => hohe Kapazität f. Blutstillung
R.E.Scharf 2010
JE Sadler: N Engl J Med 2003; 349: 323-25
R.E.Scharf 2010
Multimer Size
Opt
ical
Den
sity
S M L
1
510

27
R.E.Scharf 2010
- FVIII:C / vWF Complex -
CollagensGlycosaminoglycans
- Aggregation -- Adhesion -
GPIb-IX GPIb-IX
GPIb-IX GPIIb-IIIa
GPIIb-IIIa GPIIb-IIIa
Endothelial cells
Platelet
PlateletPlatelet
vWF multimers
von Willebrand factor von Willebrand factor
FVIII:C
R.E.Scharf 2010
Single Platelets Adherent onto Thrombogenic Matrices
R.E.Scharf 2010
Platelet Aggregate Adherent onto Thrombogenic Matrices

28
R.E.Scharf 2010
ThrombinADP
KollagenAdrenalin
aktiviertesPlättchen
TxA2, ADPSerotonin (5-HT)
VasokonstriktionPlättchenaktivierung
Plättchenin Ruhe
nach GV Born (1962), modifiziert
Principle of Platelet AggregometryLight trans-mittance
Light source
Light trans-mittance
Light source
ADPKollagenAdrenalin
Platelet
GPIIb-IIIaFg
R.E.Scharf 2010
1 min
Ligh
t Tra
nsm
issi
on
Collagen (6.25 g/ml)Epinephrine (50 M)ADP (5 M)
C
P
P
C
P
P
C
R.E.Scharf 2010
Immobilized Fg
Subendothelium
vWFCollagen ECEC
Scharf et al., 2004
Recruitment and Activation of Platelets
GPIb-IX-V
RestingPlatelet
GPIa-IIa GPIIb-IIIa restingGPVI
vWF Plasma Fg
ActivatedPlatelet
GPIIb-IIIa activated
GPIIb-IIIa activated
Outside-in signal
Thrombin ADP, Serotonin
p-Selectin
TXA2

29
R.E.Scharf 2010
vWF
Immobilized Fg
Subendothelium
vWFCollagen ECEC
GPIb-IX-V
Resting Platelet
GPIa-IIa GPIIb-IIIa restingGPVI
Plasma Fg
ActivatedPlatelet
GPIIb-IIIa activated
GPIIb-IIIa activated
Outside-in signal
Thrombin ADP, Serotonin
p-Selectin
TXA2
1
2
3
4
5
6
7Sequence of events
(from bottom to top)
7. Procoagulant activity6. Secretion and release5. Platelet aggregation4. Platelet activation3. Signal transduction2. Platelet adhesion1. Vascular lesion
R.E.Scharf 2010
von-Willebrand-Syndrom (vWS)
• quantitativer oder qualitativer Defekt des
von-Willebrand-Faktors
• häufigste angeborene Hämostasestörung
• Prävalenz: 10.000 pro 1 Million
• erworbenes vWS bei Herzvitien, Neoplasien,
• Autoimmunerkrankungen, Hypothyreose und
• Therapie mit Valproinsäure
R.E.Scharf 2010
Klassifikation des von-Willebrand-Syndroms
1. Quantitative StörungenTyp 1, Typ 3
2. Qualitative StörungenTyp 2 mit zahlreichen Varianten

30
R.E.Scharf 2010
Hämophilievon-Willebrand-
Syndrom
BlutungstypGelenk- und
WeichteilblutungenSchleimhautblutungen
Gerinnungszeit hochgradig verlängert normal
Blutungszeitnormal hochgradig verlängert
Erbgang X-chromosomal-rezessivautosomal
(dominant/rezessiv)
R.E.Scharf 2010
Ursachen des angeborenen vWS
1. Mangel:in unzureichender Menge synthetisierter, funktionell intakter vWF
2. Defekt:strukturell abnormer, deshalb funktionelldefekter vWF
3. Kombination quantitativer und qualitativer Störungen
R.E.Scharf 2010
Art und Häufigkeit von Blutungen bei Patientenmit von-Willebrand-Syndrom

31
R.E.Scharf 2010
Severe von Willebrand Disease Type 1 and 2Definition
• Lifelong history of bleeding and
at least one of the following abnormalities
• Bleeding time longer than 15 min• Risticetin cofactor activity < 10%• Factor VIII:C activity < 20%
AB Federici et al. Blood 103: 2032-2038 (2004)
R.E.Scharf 2010
Differentialtherapie des vWS
erworbenes von Willebrand-Syndrom:Behandlung der Grunderkrankung
angeborenes von Willebrand-Syndrom:spezielle Maßnahmen je nach Typ(1, 2A, 2B, 2N, Pseudo-vWS, 3)
R.E.Scharf 2010
Ziel der (Hämo)-Therapie beim vWS
1. Anstieg des von-Willebrand-Faktors im PlasmaNormalisierung der FVIII:C-AktivitätNormalisierung der PlättchenfunktionKorrektur der verlängerten Blutungszeit
2. Stillung akuter Blutungen
3. Prävention von Blutungskomplikationen

32
R.E.Scharf 2010
Klassifikation & Therapie des von Willebrand-Syndroms
Parameter Typ 1 Typ 2A Typ 2B Pseudo-vWS Typ 3
Häufigkeit 70 - 80 % 10 - 15 % 3 - 5 % ca. 1 % 1 - 3 %
vWF-Ag fehlt
vWF-Akt fehlt
RIPA normal normal fehlt
Therapie DDAVP vWF-Konz. vWF-Konz. Thrombozyten vWF-Konz.
Multimere
normal
fehlen
R.E.Scharf 2010
Running gel
Dye frontN P1 N P2 P3 P4
vWDType 2A
R.E.Scharf 2010
von-Willebrand-Syndrom:Therapiemodalitäten
Induktion einer Synthesesteigerung des vWF (Östrogene)
Induktion einer erhöhten Freisetzung des vWF (DDAVP)
exogene Zufuhr (Hämotherapie mit vWF-Konzentraten)

33
R.E.Scharf 2010
Phe3
Tyr2
Cys1
Gln4
Asn5
Cys6 Pro7 Gly9 NH2S-S
Phe3
Tyr2
Cys1
Gln4
Asn5
Cys6 Pro7 Gly9 NH2S-S
Vasopressin(Arginin-Vasopressin, AVP)
Desmopressin(1-Desamino-8-D-Arginin-Vasopressin, DDAVP)
L-Arg8H2N
D-Arg8H
R.E.Scharf 2010
Per
cen
tag
e o
f N
orm
al C
on
tro
l Pla
sma
Ble
edin
g T
ime,
BT
(m
in)
Effect of DDAVP on vWF and FVIII:C levels and BT in vivo
R.E.Scharf 2010
Prinzipien der Hämotherapie
1. Gezielte, bedarfsgerechte und risikoadaptierte Substitution
("so viel wie nötig, so wenig wie möglich")
2. Minimierung transfusionsassoziierter Risiken (z.B. virusinaktivierte Produkte)
3. Qualitätskontrolle des Präparates
4. Prüfung des Behandlungseffektes durch therapiebegleitende
hämostaseologische Untersuchungen
34
R.E.Scharf 2010
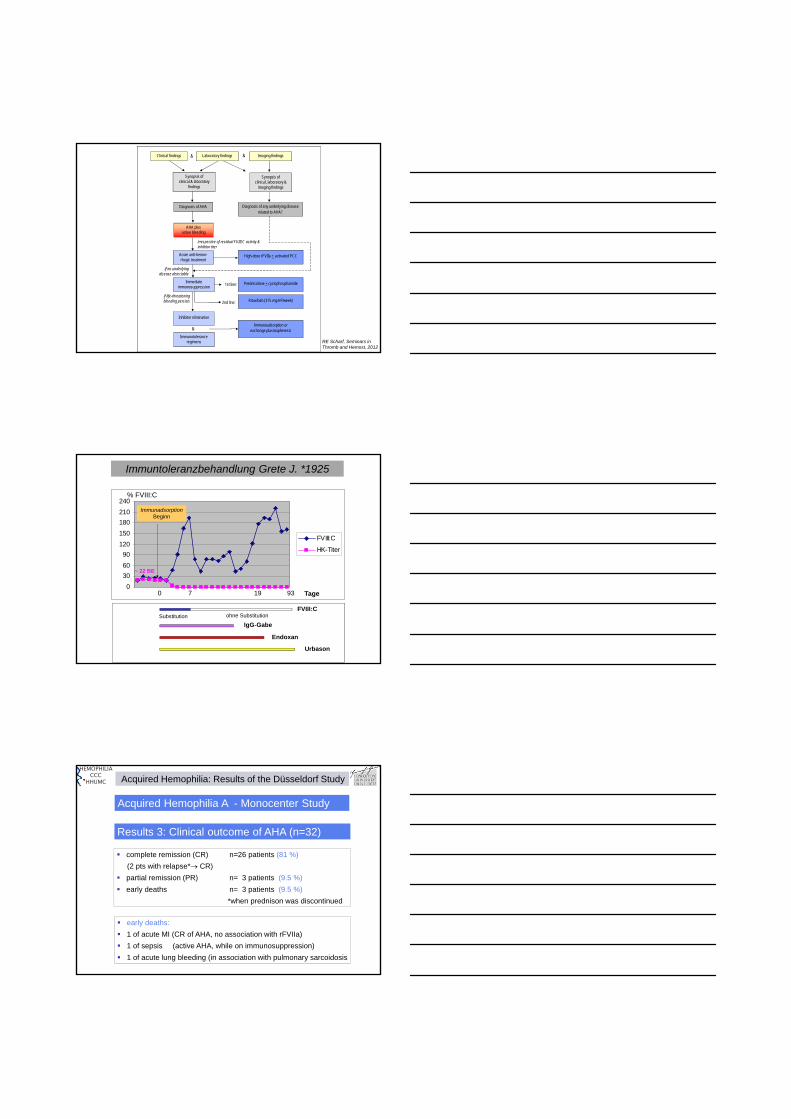
NP P CR CO CO CO CR12 11 21
Industrial
R.E.Scharf 2010
Multimerenanalyse verschiedener Faktor VIII-Konzentratein Gel mit mittlerer Auflösung (1.6 % LGT-Agarose)
Haemate P
R.E.Scharf 2010

35
R.E.Scharf 2010
Antworten zum Bestehen der Facharzt-Prüfung
1. FVIII oder Faktor IX-Mangel (Hämophilie A oder B)
2. FXII-Mangel (häufige Ursache!!)
3. FXI-Mangel
4. Mangel an Präkallikrein oder HMW-Kininogen (selten)
5. Antikörper gegen FVIII o.FIX (Hemmkörper-Hämophilie)
Differentialdiagnose der aPTT-Verlängerung I
6. von-Willebrand-Syndrom (Typ 2 und 3, inkonstant bei Typ 1)
7. Therapie mit UFH, LMWH (gering) und Hirudinen
8. Therapie mit neuen oralen Antikoagulanzien
(direkte Thrombininhibitoren z.B. Dabigatran = Pradaxa
oder FXa-Inhibitoren z.B. Rivaroxaban = Xarelto)
9. Aktiviertes Protein C (APC) – Spitzfindigkeit!
R.E.Scharf 2010
Antworten zum Bestehen der Facharzt-Prüfung
10. Erniedrigte Aktivitäten von FII, FV und/oder FX
(dann auch Prothrombinzeit n. Quick erniedrigt !)
11. Dysfibrinogenämien (selten)
12. Extrem erniedriegte Fibrinogenspiegel (< 50 mg/dl)
Differentialdiagnose der aPTT-Verlängerung II
Klinische Bedeutung der aPTT-Verlängerung bei FXII-Mangel?





























