Embed Size (px)
Citation preview

Modellierung und Charakterisierung starrer
knäuelförmiger Makromoleküle:
Polynorbornene
Dissertation
zur
Erlangung des Doktorgrades der Naturwissenschaften
(Dr. rer. nat.)
dem
Fachbereich Chemie
der Philipps-Universität Marburg
vorgelegt von
Stefan Krügel
aus Rotenburg/Fulda
Marburg/Lahn 1998

-II-
Vom Fachbereich Chemie der Philipps-Universität Marburg als
Dissertation am 20. Juli 1998 angenommen.
Erstgutachter: Prof. Dr. J. H. Wendorff
Zweitgutachter: Prof. Dr. W. Heitz
Tag der mündlichen Prüfung: 21. Juli 1998

-III-
Inhaltsverzeichnis
1 EINLEITUNG...............................................................................................................1
2 GRUNDLAGEN ...........................................................................................................8
2.1 Kettenkonformation und Flexibilität....................................................................8
2.2 Atomistische Simulationsverfahren....................................................................112.2.1 Quantenmechanische Verfahren ......................................................................12
2.2.1.1 Ab initio-Verfahren ...................................................................................122.2.1.2 Dichte-Funktional-Verfahren.....................................................................142.2.1.3 Identifikation von Minimumstrukturen .....................................................15
2.3 Kraftfeld-Methoden .............................................................................................152.3.1 Ensembles ........................................................................................................152.3.2 Ergoden-Hypothese..........................................................................................16
2.4 Molekularmechanik-Simulationen .....................................................................162.4.1 Methoden zur Lokalisierung von Potentialminima..........................................17
2.4.1.1 Minimierungsalgorithmen .........................................................................172.4.1.2 Simulated Annealing .................................................................................18
2.5 Molekulardynamik-Simulationen.......................................................................192.5.1 Verwendete Kraftfelder....................................................................................202.5.2 Molekulardynamik-Simulationen bei konstanter Temperatur .........................232.5.3 Modellierung des kondensierten Zustands.......................................................242.5.4 Behandlung der atomaren Partialladungen ......................................................25
2.5.4.1 Partielle Kompensation der Orbitalelektronegativität ...............................252.5.4.2 Charge-Equilibration-Schema ...................................................................26
2.6 Auswahl der Modellsysteme................................................................................26
2.7 Charakterisierung des Packungsverhaltens ......................................................27
2.8 Experimentelle Untersuchungsmethoden ..........................................................272.8.1 Röntgen-Weitwinkel-Streuung (WAXS).........................................................272.8.2 Röntgen-Kleinwinkel-Streuung (SAXS) .........................................................282.8.3 Differentialkalorimetrie (DSC) ........................................................................282.8.4 Thermisch-Mechanische Analyse (TMA)........................................................282.8.5 Kontaktwinkelmessungen ................................................................................28

-IV-
3 KETTENKONFORMATION STARRER KNÄUEL...........................................29
3.1 Simulationen zur Kettenkonformation ..............................................................293.1.1 Rotationspotentiale ..........................................................................................293.1.2 Eindimensionale Rotationspotentiale...............................................................30
3.1.2.1 Eindimensionales Rotationspotential für Butan ........................................313.1.2.2 Eindimensionales Rotationspotential für Biphenyl ...................................333.1.2.3 Eindimensionales Rotationspotential dimeres Norbornan.........................34
3.1.3 Zweidimensionale Rotationspotentiale ............................................................383.1.3.1 2D-Rotationspotential Pentan....................................................................393.1.3.2 2D-Rotationspotential Paraphenylen .........................................................403.1.3.3 2D-Rotationspotential Norbornan .............................................................42
3.2 Computersimulationen zur Konformation der Einzelkette im Vakuum .......453.2.1 Molekulardynamik-Simulationen im Vakuum ................................................453.2.2 Monte Carlo-Simulationen zur Kettenkonformation .......................................49
3.3 Molekulardynamik-Simulationen im kondensierten Zustand........................51
3.4 Experimentelle Untersuchungen zur Kettenkonformation ..............................543.4.1 Kettenkonformation aus Lichtstreu-Experimenten ..........................................543.4.2 Kettenkonformation aus viskosimetrischen Untersuchungen..........................55
3.5 Zusammenfassung Kapitel 3 ...............................................................................58
4 LOKALE KETTENDYNAMIK STARRER KNÄUEL .........................................59
4.1 Lokale Kettendynamik starrer Knäuel im Vakuum.........................................594.1.1 Kettengestalt und Fluktuationen (12er-Kette)..................................................61
4.1.1.1 Polyvinylchlorid (PVC) .............................................................................614.1.1.2 Polyparaphenylen (PPP) ............................................................................634.1.1.3 Polynorbornen (PN)...................................................................................65
4.2 Kettendynamik starrer Knäuel im kondensierten Zustand .............................684.2.1 Lokale Kettendynamik der 12er-Kette im kondensierten Zustand ..................684.2.2 Auswertung der Torsionswinkel-Fluktuationen...............................................704.2.3 Molekulardynamik-Simulationen zur Glasumwandlung .................................744.2.4 Experimentelle Bestimmung der Glasumwandlung ........................................76
4.3 Glasumwandlung starrer Knäuel .......................................................................774.3.1 Mechanismus der Glasumwandlung bei flexiblen Polymeren.........................784.3.2 Mechanismus der Glasumwandlung bei starren Knäueln................................80
4.4 Makroskopische Beweglichkeit...........................................................................82
4.5 Zusammenfassung Kapitel 4 ...............................................................................85

-V-
5 DER KONDENSIERTE ZUSTAND STARRER KNÄUEL.................................86
5.1 Statische Eigenschaften im kondensierten Zustand........................................865.1.1 Strukturelle Eigenschaften im kondensierten Zustand.....................................87
5.1.1.1 Röntgenweitwinkelstreuung (WAXS).......................................................875.1.1.2 Röntgenkleinwinkelstreuung (SAXS) .......................................................90
5.2 Thermodynamische Eigenschaften im kondensierten Zustand .......................915.2.1 Löslichkeitsparameter ......................................................................................92
5.2.1.1 Oberflächenspannung ................................................................................95
5.3 Mechanische Eigenschaften.................................................................................985.3.1 Computersimulationen.....................................................................................985.3.2 Experimentelle Untersuchung der mechanischen Eigenschaften...................101
5.4 Permeationseigenschaften..................................................................................1035.4.1 Molekulardynamik-Simulationen zum Permeationsverhalten.......................103
5.5 Zusammenfassung Kapitel 5 .............................................................................106
6 MODIFIKATION STARRER KNÄUEL DURCH KATALYSATORWAHL ..108
6.1 Polymerisation von Olefinen mit Metallocenen.............................................109
6.2 Modifikation des Kraftfeldes.............................................................................110
6.3 Computersimulationen zur Polymerisation von Poly-bicylo[2.2.1]hept-2-en(Polynorbornen)........................................................................................................114
6.3.1 Mechanismus der Polymerisation von Polynorbornen...................................1146.3.2 Polymerisation mit Pd(II)-Komplexen...........................................................1156.3.3 Polymerisation mit Cr(III)-Komplexen..........................................................1166.3.4 Sterischer Anspruch des Katalysators ............................................................116
6.4 Molekularmechanik-Simulationen zur Polymerisation von Polynorbornen1176.4.1 Allgemeines zu den Molekularmechanik-Simulationen ................................1186.4.2 Polymerisation mit Pd(II)tetrakisacetonitril-bis-tetrafluoroborat ..................1206.4.3 Polymerisation mit Cylopentadienyl-methyl-Cr(III)......................................1226.4.4 Polymerisation mit Cp*-methyl-Cr(III) .........................................................124
6.5 Experimentelle Charakterisierung der Polynorbornene................................125
6.6 Zusammenfassung Kapitel 6 .............................................................................126

-VI-
7 MODIFIKATION DER ROTATIONSBEHINDERUNG STARRERKNÄUEL.................................................................................................................128
7.1 Modifikation durch Einführung lateraler Substituenten.............................1297.1.1 Polynorbornencarbonsäuremethylester (PNme).............................................130
7.1.1.1 1D-Rotationspotential..............................................................................1307.1.1.2 2D-Rotationspotential trimerer Norbornen-2-carbonsäuremethylester ...1327.1.1.3 Lokale Kettendynamik Polynorbornencarbonsäuremethylester (12mer) 1337.1.1.4 Monte Carlo-Simulation Polynorbornencarbonsäuremethylester............1367.1.1.5 Experimentelle Charakterisierung ...........................................................136
7.1.2 Polynorbornenimid (PNim)............................................................................1387.1.2.1 1D-Rotationspotential..............................................................................1387.1.2.2 2D-Rotationspotential des trimeren Norbornen-imids ............................1407.1.2.3 Lokale Kettendynamik 12er-Kette Polynorbornenimid...........................1427.1.2.4 Molekulardynamik-Simulation Polynorbornenimid 50er-Kette..............1437.1.2.5 Experimentelle Charakterisierung ...........................................................146
7.2 Modifikation des Polymerbackbones durch Heteroatome .............................1497.2.1 Polynorbornenamide (PNam).........................................................................149
7.2.1.1 1D-Rotationspotential..............................................................................1507.2.1.2 2D-Rotationspotential Polynorbornenamid(PNam) ................................1527.2.1.3 Lokale Kettendynamik 12er.....................................................................1537.2.1.4 Molekulardynamik-Simulation von Polynorbornenamid (PNam)...........1557.2.1.5 Experimentelle Charakterisierung ...........................................................157
7.2.2 Polynorbornenoxide (PNox) ..........................................................................1597.2.2.1 1D-Rotationspotential Polynorbornenoxid..............................................1607.2.2.2 2D-Rotationspotential Polynorbornenoxid..............................................1617.2.2.3 Lokale Kettendynamik 12er.....................................................................1627.2.2.4 Molekulardynamik-Simulationen an Polynorbornenoxid (PNox) ...........1637.2.2.5 Experimentelle Charakterisierung ...........................................................164
7.3 Zusammenfassung Kapitel 7 .............................................................................165
8 ZUSAMMENFASSUNG..........................................................................................167
ANHANG......................................................................................................................172

Einleitung 1
1 Einleitung
Physikalische Eigenschaften polymerer Materialien hängen sowohl von ihrer
makroskopischen als auch von ihrer mikroskopischen Struktur ab [1,2]. Konformation
und Beweglichkeit der Polymerketten kontrollieren die thermodynamischen,
strukturellen, rheologischen und mechanischen Eigenschaften im kondensierten
Zustand. Synthetische Polymere lassen sich aufgrund ihrer Flexibilität und ihrer Gestalt
entweder den flexiblen Knäueln oder den starren stäbchenförmigen Polymeren
zuordnen.
Flexible Polymerketten liegen in ihrer thermodynamisch günstigsten Form als
statistische Knäuel vor. Dimension und Gestalt der Polymerkette hängen im
wesentlichen von den Bindungswinkeln und den Rotationspotentialen zwischen
benachbarten Kettensegmenten ab. Typischerweise zeichnen sich flexible Polymere, wie
z.B. Polyethylen (PE) oder Polystyrol (PS) (Abb. 1.1) durch das Auftreten von
Entanglements und durch viskoelastische Eigenschaften aus.
**
n
Polyethylen (PE)
C6H5C6H5C6H5
**
n
Polystyrol (PS)
Abb. 1.1: Beispiele für flexible Polymere
Flexible Polymere sind entweder amorph oder teilkristallin, wobei sie im teilkristallinen
Zustand Lamellenkristalle ausbilden. Externe mechanische Belastungen führen zu
Konformationsänderungen, die sich auf mikroskopischer Ebene durch die Veränderung
von Torsionswinkeln äußert. Eine Polymer-Einzelkette sollte entropieelastische
Eigenschaften besitzen. Flexible Polymerketten zeigen aufgrund ihrer Knäuelgestalt

Einleitung 2
eine gute Löslichkeit. Die hohe Flexibilität der Polymerketten führt in der Regel zu
niedrigen Glastemperaturen, die eine Verarbeitung dieser Materialien aus der Schmelze
ermöglichen. Amorphe Polymere besitzen aufgrund der hohen Beweglichkeit und des
im amorphen Festkörper eingeschlossenen freien Volumens nur eine bedingte
Verwendbarkeit als Barrierematerial für Verpackungs- oder Isolationszwecke. Dieses
freie Volumen kann für die Diffusion kleinerer Moleküle, wie z.B. Sauerstoff oder
Wasserdampf, ausreichend sein.
Starre stäbchenförmige Polymere wie z.B. das Polyparaphenylterephthalamid (PPTA,
Kevlar) oder Polyparaphenylen (PPP) (Abb. 1.2) zeichnen sich durch einen
flüssigkristallinen Charakter aus.
CO CONH
NH* *n
Polyparaphenylterephthalamid (PPTA; Kevlar)
** n
Polyparaphenylen (PPP)
Abb. 1.2: Beispiele für starre Stäbchen
Als charakteristisches Merkmal tritt neben der Orientierungsordnung noch die
Krümmungselastizität auf. Flüssigkristalle werden als lyotrop bezeichnet, wenn der
flüssigkristalline Charakter der Polymere in Lösung in Erscheinung tritt und nach der
Entfernung des Lösemittels erhalten bleibt. Bei thermotropen Flüssigkristallen wird die
Ausbildung der flüssigkristallinen Eigenschaften thermisch induziert [3]. Die starre
stäbchenförmige Gestalt dieser Polymerketten führt zusammen mit intermolekularen
Wechselwirkungen, wie z.B. Wasserstoffbrückenbindungen, zu einer schlechten
Löslichkeit. Durch die hohen Schmelztemperaturen starrer stäbchenförmiger Polymere
wird eine thermoplastische Verarbeitung in der Regel unmöglich. In den meisten Fällen
erfolgt bereits vor dem Erreichen der Verarbeitungstemperatur die chemische
Zersetzung.

Einleitung 3
Die vorliegende Arbeit befaßt sich mit dem Konzept der starren Knäuel, einer neuen
Polymerklasse, deren Eigenschaften zwischen denen der flexiblen knäuelförmigen
Polymere und denen der starren Stäbchen liegen sollten.
Dieses Konzept basiert auf den folgenden Grundgedanken:
Um zu einer starren knäuelförmigen Polymerkette zu gelangen, sollten die
Bindungswinkel zwischen den Kettensegmenten ungleich 180° betragen. Dies ist eine
Voraussetzung für die knäuelförmige Gestalt des Polymers. Im Rotationspotential
sollten die Potentialminima so angeordnet sein, daß eine lange Polymerkette die Gestalt
eines statistischen Knäuels besitzt. Weiterhin sollten die Potentialbarrieren so hoch sein,
daß der Übergang von einem Potentialminimum in ein anderes Gebiet niedriger Energie
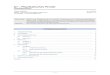
nur sehr selten oder gar nicht auftreten kann. In Abbildung 1.3 sind die Anforderungen
an das Rotationspotential eines starren Knäuels schematisch dargestellt.
0 30 60 90 120 150 180 210 240 270 300 330 3600,0
0,1
0,2
0,3
0,4
0,5
0,6
RT
rel.
Ene
rgie
[a.
u.]
Torsionswinkel φ [°]
Abb. 1.3: Anforderungen an das Rotationspotential starrer Knäuel

Einleitung 4
Um eine Äquipartition der Potentialminima zu vermeiden, sollten sich die einzelnen
Minima energetisch deutlich voneinander unterscheiden.
Starre knäuelförmige Makromoleküle sollten aufgrund dieser Kriterien bezüglich des
Rotationspotentials eine Reihe ungewöhnlicher Eigenschaften besitzen:
• Die Kettenkonformation der starren Knäuel sollte bereits während der Polymerisation
festgelegt sein. Aufgrund der hohen Rotationsbarrieren sollte die Einstellung der
Kettenkonformation bei der Polymerisation ein einmaliger und unveränderbarer
Vorgang sein.
• Derartig konformationsbehinderte Polymerketten sollten aufgrund der
eingeschränkten Beweglichkeit eine hohe Glastemperatur besitzen. Falls die
Glasumwandlung starrer Knäuel mit Konformationsänderungen verbunden wäre,
müßten diese bei sehr hohen Temperaturen zu erwarten sein.
• Die Starrheit der Polymerketten sollten keinerlei Reptationsprozesse zulassen. Dies
würde sich vor allem auf das Fließverhalten auswirken. Durch die
Rotationsbehinderung und die stark eingeschränkte Beweglichkeit der Ketten sollten
Fließprozesse erst bei stark erhöhten Temperaturen auftreten. Es ist zu erwarten, daß
Diffusionsprozesse im kondensierten Zustand stark behindert sind. Kleine Moleküle
wären in dem statischen, freien Volumen eingeschlossen und damit immobilisiert.
• Die Kettenkonformation der starren Knäuelmoleküle sollte unabhängig von der Art
des verwendeten Lösemittels sein. Lösemittel könnten keinerlei Einfluß auf die
Gestalt der Polymerkette ausüben, da die Kettenkonformation nicht geändert werden
kann. Aufgrund dieser Überlegungen ist zu erwarten, daß alle Lösemittel für die
starren Knäuel gleich gut sind, also kein Quellen und kein Kollabieren auftritt.
Kettenmoleküle, die der Polymerklasse der starren Knäuel zuzuordnen sind, müssen auf
mikroskopischem Niveau bestimmte Anforderungen an das Rotationspotential bzw.

Einleitung 5
dessen Potentialbarrieren erfüllen. Sobald die Eigenschaften der Potentialbarrieren
starrer knäuelförmiger Polymere den oben beschriebenen Kriterien genügen, sind
ungewöhnliche dynamische Eigenschaften zu erwarten. Die Anzahl von
Konformationsübergängen wird, auf die Lebensdauer der Kette bezogen, sehr gering
sein. Der Zeitraum für das Auftreten eines Konformationsübergangs wird unter diesen
Bedingungen länger sein als der experimentelle Beobachtungszeitraum. Diese Tatsache
würde dem Einfrieren einer einzelnen Kette entsprechen. De Gennes hat für diese
Situation den Begriff des „Einzelkettenglases“ geprägt [4]. Es ist zu erwarten, daß diese
eingefrorene Kettenkonformation bis zum Auftreten von Konformationsübergängen
erhalten bleibt, was nicht notwendigerweise mit der Glasumwandlung verknüpft sein
muß. Das mechanische Verhalten der starren Knäuel sollte aufgrund der stark
eingeschränkten Beweglichkeit mit dem Begriff der Energieelastizität charakterisierbar
sein.
Zur Konstruktion einer rotationsbehinderten Polymerkette lassen sich prinzipiell die
folgenden beiden Möglichkeiten anführen:
• Verwendung eines sterisch anspruchsvollen Monomers direkt im Backbone
• Einführung sterisch anspruchsvoller Substituenten an ein flexibles Polymerbackbone
Als typischen Vertreter eines sterisch anspruchsvollen Monomers, das zur Realisierung
einer rotationsbehinderten Polymerkette verwendet werden kann, ist das Norbornen zu
nennen. Die vorliegende Arbeit wird das Konzept der starren Knäuel mit Hilfe des Poly-
bicyclo[2.2.1]hept-2-ens bzw. Polynorbornen (PN) (Abb. 1.4) als Prototyp dieser neuen
Polymerklasse vorstellen.

Einleitung 6
Abb. 1.4: Polynorbornen (PN)
Mittels einer Kombination aus Computersimulationen an Modellsystemen und
experimentellen Untersuchungen soll das Konzept der starren Knäuel im Rahmen dieser
Arbeit überprüft werden. Weiterhin soll die vorliegende Arbeit zeigen, ob sich mit Hilfe
von computergestützten Simulationsverfahren Struktur-Eigenschaftsbeziehungen für
neuartige polymere Systeme aufklären und vorhersagen lassen. Die Kombination aus
Computersimulationen und Experimenten ist die Voraussetzung für die Entwicklung
neuer Polymer-Werkstoffe.
Die vorliegende Arbeit wird sich mit Untersuchungen zur Kettenkonformation und den
besonderen Eigenschaften des Rotationspotentials der Polynorbornene beschäftigen. Der
Vergleich mit bekannten flexiblen und starren stäbchenförmigen Polymeren soll es
ermöglichen, die Eigenschaften der Polynorbornene zu bewerten. Die n-dimensionalen
Potentialenergie-Hyperflächen verschiedener Modellsysteme werden mittels
Molekularmechanik- und Molekulardynamik-Simulationen untersucht. Ergebnisse aus
Computersimulationen zur Kettenkonformation der Polynorbornene werden mit
experimentellen Ergebnissen aus Lichtstreu-Experimenten und viskosimetrischen
Untersuchungen überprüft.
Durch Molekulardynamik-Simulationen im Vakuum und im kondensierten Zustand
wird der Einfluß verschiedener Umgebungen auf die Kettengestalt und die lokale
Kettendynamik des Polynorbornens untersucht. Die Betrachtung von Torsionswinkel-
Fluktuationen bzw. der Halbwertsbreiten der Torsionswinkel-Verteilung wird
Rückschlüsse auf den Potentialverlauf der n-dimensionalen Potentialenergie-

Einleitung 7
Hyperfläche bei unterschiedlichen Temperaturen erlauben. Mit Hilfe von
Molekulardynamik-Simulationen zur Glasumwandlung des Polynorbornens wird die
thermische Ausdehnung eines dreidimensionalen Modellsystem als Funktion der
Temperatur modelliert. In diesem Zusammenhang werden weiterhin verschiedene
Mechanismen für die Glasumwandlung diskutiert.
Im darauf folgenden Abschnitt werden die strukturellen Eigenschaften der
Polynorbornene durch Streuexperimente charakterisiert. Mittels Computersimulationen
erfolgt eine Abschätzung des Hildebrandschen Löslichkeitsparameters, des
Permeationsverhaltens gegenüber Sauerstoff sowie der mechanischen Eigenschaften des
Polynorbornens. Die mechanischen Eigenschaften und die Oberflächeneigenschaften der
Polynorbornene werden mit den Ergebnissen aus experimentellen Untersuchungen
verglichen.
Durch Molekularmechanik-Simulationen wird weiterhin untersucht, ob die
Konformation der Polymerkette tatsächlich bereits während der Polymerisation
endgültig festgelegt wird und ob eine Beziehung zwischen der Struktur des Katalysators
bzw. dessen Ligandensphäre und der resultierenden Mikrostruktur des Polymers besteht.
Am Ende dieser Arbeit steht die Untersuchung, wie sich verschiedene chemische
Modifikationen der Polymerkette auf die Eigenschaften der Polynorbornene auswirkt.
Dies wird unter dem Gesichtspunkt der beiden bereits erwähnten Möglichkeiten zur
Modifikation der Polymerkette erfolgen. Am Beispiel von jeweils zwei modifizierten
Polynorbornenen sollen die Struktur-Eigenschaftsbeziehungen für die laterale
Substitution und die Modifikation des Polymerbackbones aufgezeigt werden. Für diesen
Vergleich werden sowohl Computersimulationen als auch ausgewählte experimentelle
Ergebnisse verwendet.

Grundlagen 8
2 Grundlagen
In diesem Kapitel werden die Grundlagen zur Beschreibung der Konformation von
Polymerketten vorgestellt und die Formalismen beschrieben. Weiterhin werden die in
der vorliegenden Arbeit verwendeten Simulationsverfahren erläutert.
2.1 Kettenkonformation und Flexibilität
Die makroskopischen Eigenschaften polymerer Werkstoffe werden vor allem durch die
Kettenkonformation beeinflußt. Diese wird wiederum durch die Rotationsfreiheitsgrade
der Polymerketten bestimmt. Mit dem Übergang von flexiblen, knäuelförmigen Ketten
zu starren stäbchenförmigen Polymeren erfolgt der Übergang vom amorphen bzw.
teilkristallinen Zustand zum flüssigkristallinen Zustand. Aus der abnehmenden
Flexibilität der Polymerketten resultiert aus thermodynamischer Sicht eine Erhöhung der
Schmelz- bzw. Glastemperaturen sowie eine Verschlechterung der Löslichkeit. In Bezug
auf die mechanischen Eigenschaften erfolgt ein Übergang vom duktilen zum spröden
Verhalten der Materialien. Die Untersuchung der mikroskopischen Kettenkonformation
bzw. der Kettenflexibilität erlaubt eine Abschätzung der makroskopischen
Eigenschaften neuer polymerer Materialien.
Die Konformation kleiner Moleküle ist weitgehend durch feste Bindungslängen und
-winkel festgelegt. Rotationen um einzelne chemische Bindungen sind sowohl in
flexiblen als auch in starren Polymerketten möglich. Rotationen um Bindungen erfolgen
im allgemeinen leichter als die Deformationen von Bindungslängen oder
Bindungswinkeln, was sich in den großen energetischen Unterschieden zwischen
Bindungsenergien und den Potentialbarrieren dokumentiert. Die Beschreibung der
Konformation einzelner Polymerketten muß aus diesem Grund durch die Untersuchung

Grundlagen 9
der Beweglichkeit und damit der Rotationsfreiheitsgrade der einzelnen Bindungen in
den Makromolekülen erfolgen.
Bei Polymeren resultiert aufgrund der großen Anzahl der internen Freiheitsgrade eine
ebenso große Anzahl individueller Kettenkonformationen. Bei flexiblen Polymerketten
kann aufgrund der weitgehend unabhängigen Abfolge der Bindungsrotationen keine
definierten Kettenkonformationen mehr angeben werden. Zur Charakterisierung der
Kettenkonformation verwendet man deshalb Größen, die die Kettengestalt beschreiben,
wie z.B. den Kettenendenabstand und den Gyrations- bzw. Trägheitsradius. Die
Beschreibung dieser gemittelten Konfigurationen erfolgt mit Hilfe der statistischen
Mechanik der Kettenmoleküle von P. J. Flory [1].
Flory verwendet in diesem Zusammenhang eine vereinfachte Darstellung des
Kettenrückgrates anstelle der exakten chemischen Struktur. Die Polymerkette wird auf
ein Skelett von Atomen Ai reduziert, die durch die Bindungsvektoren →l
imiteinander
verbunden sind. Zur Beschreibung der Polymerkette müssen nicht alle Atome Ai explizit
berücksichtigt werden. Längere starre Bereiche einer Kette lassen sich in sinnvoller
Weise durch Segmente mit einer virtuellen Bindungslänge repräsentieren, um die
Darstellung der Kettenkonformation zu vereinfachen. Die hieraus resultierenden
Bindungsvektoren →l
i besitzen demnach kein chemisches Äquivalent, d.h. sie stellen
keine realen Bindungen dar. Diese vergröbernde Darstellung von Makromolekülen
basiert auf der Verwendung modellhafter Bindungsvektoren und virtueller Atome. Die
Längen dieser Vektoren →li
und die Winkel (φ) zwischen ihnen, sowie die Rotationen
um die Bindungsvektoren besitzen je nach dem verwendeten Modell dieselbe
Bedeutung wie die entsprechenden Größen der realen Polymerkette. In diesem Kontext
wird die Polymerkette „renormiert“, so daß die Ketteneigenschaften mit der universellen
Theorie der Kettenstatistik beschrieben werden können. Dieser Renormierungsfaktor
kann hierbei schon für sich allein ein Maß für die Länge einzelner linearer
Kettensegmente und somit für die Kettenflexibilität sein.
Eine zentrale Größe zur Beschreibung der Kettenkonformation eines Polymers ist der
mittlere quadratische Kettenendenabstand ⟨RE2⟩. Dieser Wert entspricht dem

Grundlagen 10
statistischen Mittelwert. Alternativ zu diesem Kettenendenabstand kann der mittlere
quadratische Trägheitsradius (Gyrationsradius) ⟨RG2⟩ betrachtet werden. Bei einem
hinreichend hohem Polymerisationsgrad n gilt für beide Größen die folgende
Beziehung:
R RG E2 21
6= (2.1)
Bei der einfachsten Darstellung einer flexiblen Polymerkette geht man davon aus, daß
alle Bindungswinkel und Torsionen mit der gleichen Wahrscheinlichkeit auftreten,
wobei die Bindungslängen li als konstant angenommen werden (freely jointed chain,
fjc). Für den Kettenendenabstand einer solchen Kette gilt dann:
R l nlE fjc ii
n2 2
1
2==∑ =
(2.2)
Die Konformation einer realen Kette wird dagegen im allgemeinen durch sterische und
elektronische Wechselwirkungen zwischen den einzelnen Kettensegmenten beeinflußt.
Dies bedeutet für den Kettenendenabstand:
R l l lE real ii
i ji j n
2 2
0
= +∑ ∑< ≤ ≤
(2.3)
Das Verhältnis zwischen dem quadratischen Kettenendenabstand der realen Kette und
dem Kettenendenabstand des Modells der freely jointed chain, ⟨RE2⟩real / ⟨RE
2⟩fjc wird als
charakteristisches Verhältnis Cn bezeichnet. Eine wichtige Kenngröße für die
Beschreibung der Kettengestalt ist der Grenzfall unendlich langer Ketten, sie wird als C∞
bezeichnet. Die Summation läuft über alle Bindungsvektoren.
Das Modell der Rotational Isomeric State (RIS)-Theorie [1,5] liefert die Grundlage für
die Berechnung des charakteristischen Verhältnisses C∞ durch die folgende Beziehung:

Grundlagen 11
Cr
nl∞ =2
2 (2.4)
In Gleichung 2.4 bedeutet ⟨r2⟩ den mittleren quadratischen Kettenendenvektor, n den
Polymerisationsgrad der Polymerkette und l die Segmentlänge des Monomers.
Neben diesen für die Kettengestalt von Polymeren charakteristischen Größen, wird in
der Literatur auch der dimensionslose Korrelationskoeffizient ρ durch die Bildung des
Verhältnisses von ⟨RE2⟩ / ⟨RG
2⟩ zur Charakterisierung der Kettengestalt herangezogen
[1,6].
2.2 Atomistische Simulationsverfahren
Die theoretische Beschreibung mikroskopischer Größen von Makromolekülen erfordert
eine mehr oder weniger starke Abstraktion mikroskopischer Eigenschaften [7-11]. Die
Behandlung molekularer Systeme mit quantenmechanischen Methoden ist aufgrund des
Rechenaufwandes nur bis zu einer relativ kleinen Systemgröße möglich. Die diesem
Verfahren zugrundeliegende Schrödinger-Gleichung ist nur für Systeme mit maximal
drei Teilchen exakt lösbar [12]. Zur Beschreibung größerer molekularer Systeme ist die
Verwendung geeigneter Näherungsverfahren erforderlich. Die Grundlage der
quantenmechanischen Simulationsmethoden ist die Born-Oppenheimer-Näherung [13],
die die Korrelation zwischen Atomkernen und Elektronen aufhebt. Mit Hilfe dieser
Näherung wird die Schrödinger-Gleichung zu einer rein elektronischen Schrödinger-
Gleichung, was die Grundlage aller Dichte-Funktional-, ab initio und semiempirischen
Rechenverfahren bildet [14-16].
Der Übergang von der Quantenmechanik zur klassischen Mechanik kann durch den
Übergang von der Schrödinger-Gleichung zur Lagrange-Gleichung vollzogen werden.
Auf der letzten Näherung beruhen alle Molekulardynamik-Simulationsverfahren.

Grundlagen 12
2.2.1 Quantenmechanische Verfahren
2.2.1.1 Ab initio-Verfahren
Die Berechnung molekularer Eigenschaften erfolgt durch die Lösung der N-Elektronen-
Schrödinger-Gleichung:
H(N) Ψ = E (N) Ψ (2.5)
In Gleichung 2.5 steht H(N) für den Viel-Elektronen-Hamilton-Operator, Ψ für die
Wellenfunktion und E für den entsprechenden Erwartungswert der Energie. Die exakte
Lösung dieser Gleichung ist nur für einige wenige Systeme möglich. Um diese
Gleichung für größere Systeme lösen zu können, wurde mit Hilfe der Molekülorbital-
Theorie (MO-Theorie) ein Verfahren zur näherungsweisen Lösung dieser Gleichung
entwickelt. Dieses Konzept beruht auf der Näherung, daß die Bewegung jedes der N
Elektronen im Molekül auf einem mittleren Potential beruht, das durch die übrigen
Elektronen und die Kerne des Moleküls erzeugt wird. Für jedes Elektron kann somit ein
effektiver Hamilton-Operator Heff(i) aufgestellt werden. Der N-Elektronen-Hamilton-
Operator H(N) läßt sich in dieser Näherung als Summe von N effektiven Ein-
Elektronen-Hamilton-Operatoren Heff(i) formulieren:
( ) ( )H N H ieffi
N
==∑
1
(2.6)
In ähnlicher Weise kann die N-Elektronen-Schrödinger-Gleichung durch N Ein-
Elektronen-Schrödinger-Gleichungen substituiert werden:
H i i i ieff ( ) ( ) ( ) ( )ϕ ε ϕ= (2.7)

Grundlagen 13
Im Rahmen der LCAO-Methode (Linear Combination of Atomic Orbitals) lassen sich
die Molekülorbitale des zu betrachtenden Systems durch eine lineare Kombination der
einzelnen Atomorbitale beschreiben:
ϕ χ( )i bij jj
N
==
∑1
(2.8)
Bei den Wellenfunktionen χi verwendet man als Orbitalfunktionen aus
rechentechnischen Gründen sog. Slater-Type Orbitals (STO). Diese Funktionen fallen
radial mit einem Expansionskoeffizienten exponentiell ab. In der Praxis werden diese
STO durch eine Kombination mathematisch einfacherer Gaussian-Type Orbitals (GTO)
angenähert.
Zur Durchführung der ab initio- bzw. DFT-Rechnungen wurden für diese Arbeit die
Programme Gaussian 94 [17] Revision C.2. und D implementiert auf dem System IBM
RS/6000 Scalable POWERparallel SP Marburg sowie auf dem Hessischen
Höchstleistungsrechner des Typs Fujitsu/SNI VPP300/6, Technische Hochschule
Darmstadt und GAMESS [18], Version 4.0, auf DEC Alpha Workstations verwendet.
Zur Bezeichnung der mit quantenmechanischen Verfahren berechneten Geometrien wird
die Terminologie aus [19] übernommen.
Bei den Berechnungen molekularer Eigenschaften wurden mit dem Programmpaket
Gaussian 94 die Basissätze STO-3G [20,21], 3-21G [22], 6-31G(d) [23,24] von Pople
sowie spezielle Basissätze, wie z.B. effektive Core-Potentiale (ECP) von Hay und Wadt
[25-27] verwendet. Bei Verwendung der Software GAMESS wurden die Mini- und
Midi-Basissätze von Huzinaga [28], sowie Split-Valence-Basissätze von Dunning-Hay
[29], Binning-Curtiss [30] und Wachters [31,32] eingesetzt.

Grundlagen 14
2.2.1.2 Dichte-Funktional-Verfahren
Die Dichte-Funktional-Theorie-Verfahren (DFT) basieren auf der Behandlung der
Elektronenkorrelation über allgemeine Funktionale der Elektronendichte [33,34]. Diese
Methoden haben ihren Ursprung im Hohenberg-Kohn-Theorem, daß die Existenz eines
speziellen Funktionals beweist, welches sowohl den energetischen Grundzustand als
auch die Elektronendichte exakt beschreibt [35,36]. Dieses Theorem liefert jedoch keine
Aussagen über die genaue Form dieses Funktionals. Im Gegensatz zu den ab initio-
Verfahren erfolgt die Beschreibung des molekularen Systems durch die physikalische
Observable der Elektronendichte im Gegensatz zur Wellenfunktion. Im Sinne von Kohn
und Sham wird die elektronische Energie durch die angenäherten Funktionale aktueller
Dichte-Funktional-Theorie-Methoden folgenderweise aufgeteilt:
E E E E ET V J XC= + + + (2.9)
In Gleichung 2.9 bedeuten ET die kinetische Energie, EV die potentielle Energie der
Kern-Elektron-Anziehung und der Abstoßung zwischen den Kernen, EJ die Elektron-
Elektron-Abstoßung (Coulomb-Wechselwirkung der Elektronendichte) und EXC der
Austausch-Korrelationsterm, der die verbleibenden Korrelationswechselwirkungen
berücksichtigt. Bei den verwendeten Austausch-Funktionalen unterscheidet man im
wesentlichen lokale (LDA, Local Density Approximation) [37] und non-lokale bzw.
gradienten-korrigierte (GDA) Funktionale [38].
Für die DFT-Rechnungen in Kapitel 6 wurde das Becke 3-Parameter-Hybrid-
Funktional, B3LYP, verwendet [39]. Bei diesem Gradienten-korrigierten Funktional
wurden sowohl die Werte als auch die Gradienten der Elektronenspin-Dichte
berücksichtigt.

Grundlagen 15
2.2.1.3 Identifikation von Minimumstrukturen
Durch die Berechnung der 2. Ableitung der Energie nach den Kernkoordinaten wurden
die berechneten Molekülgeometrien als energetische Minima identifiziert. Dieses
Vorgehen ist äquivalent mit der Berechnung der Kraftkonstanten bzw. der
Schwingungsfrequenzen in der harmonischen Näherung nach Gleichung 2.10 [40]:
FE
X Xiji j
= ∂∂ ∂
2
0
(2.10)
Die berechnete Struktur kann einem globalen Minimum zugeordnet werden, wenn alle
berechneten Schwingungsfrequenzen positive Vorzeichen besitzen. Übergangszustände,
wie z.B. Sattelpunkte oder lokale Minima, besitzen mindestens eine negative
Eigenfrequenz [41,42].
2.3 Kraftfeld-Methoden
2.3.1 Ensembles
Bei der Durchführung atomistischer Computersimulationen ist das Konzept des
Phasenraums von fundamentaler Bedeutung [7,43]. Für ein Molekül aus N Atomen
besteht der klassische Phasenraum Γ(r
r ,r
p ) aus 6N Dimensionen, wobei jeweils 3N auf
die Kernkoordinaten (r
r ) und 3N auf die jeweiligen Impulse (r
p ) entfallen. Der Zustand
eines molekularen Systems zu einem Zeitpunkt t kann vollständig durch diese 6N
allgemeinen Koordinaten erfolgen, die einen Punkt im Phasenraum erzeugen.
Molekulardynamik-Simulationen erzeugen eine Sequenz von Punkten im Phasenraum,
die zeitlich verknüpft sind. Die zeitliche Veränderung dieses Punktes wird durch die
Bewegungsgleichungen auf einer Trajektorie durch den Phasenraum bestimmt. Diese

Grundlagen 16
Gleichungen hängen von der Art des zu betrachtenden Ensembles ab und bilden somit
das Interface zwischen den makroskopischen Eigenschaften und dem Phasenraum, der
für das Modellsystem zugänglich ist. Ein Ensemble kann als Menge von Punkten im
Phasenraum interpretiert werden, die bestimmten Randbedingungen, wie z.B. einem
konstanten Volumen oder einer konstanten Temperatur, unterliegen.
2.3.2 Ergoden-Hypothese
Die Ergoden-Hypothese ist für die Generierung molekularer Ensembles durch Monte
Carlo- und Molekulardynamik-Simulationen von zentraler Bedeutung [44,45]. Diese
Hypothese postuliert, daß unterschiedliche Startstrukturen sowohl mit Monte Carlo- als
auch mit Molekulardynamik-Verfahren unter identischen Randbedingungen zu
denselben Punkten im Phasenraum führen sollten. Durch die Ergoden-Hypothese ist es
möglich, das Zahlenmittel bzw. Ortsmittel und das Zeitmittel gleichwertig zu
verwenden. Exakte Mittelwerte können streng genommen, nur durch unendlich lange
Trajektorien erhalten werden, die über alle relevanten Bereiche des Phasenraums
samplen.
2.4 Molekularmechanik-Simulationen
Molekularmechanik-Verfahren (MM) verwenden das Konzept expliziter Bindungen
zwischen den Atomen und stellen auf diese Weise eine Grundlage für die Modellierung
von Potentialenergie-Hyperflächen (PES) dar [46]. Molekularmechanik-Programme
verwenden zur Berechnung molekularer Eigenschaften approximierte Potentialenergie-
Oberflächen. Die berechneten Eigenschaften entsprechen denen molekularer Strukturen
im Gleichgewicht. Die Potentialenergie-Hyperfläche läßt sich durch geeignete
Kraftfelder generieren. Diese Simulationsverfahren sind in der Regel lediglich auf
molekulare Systeme im Bereich des Gleichgewichtszustandes anwendbar [11]. Die

Grundlagen 17
Durchmusterung des Konfigurationsraums bzw. das Auffinden des globalen Minimums
auf der Potentialenergie-Oberfläche ist in entscheidender Weise von der
Leistungsfähigkeit der verwendeten Minimierungsalgorithmen abhängig. Die aus den
Molekularmechanik-Simulationen abgeleiteten mikroskopischen Eigenschaften
beziehen sich auf eine Temperatur von 0 Kelvin.
2.4.1 Methoden zur Lokalisierung von Potentialminima
2.4.1.1 Minimierungsalgorithmen
Die Potentialenergie U eines molekularen Systems aus N Atomen, deren Gradient sowie
deren zweite Ableitung, die Hesssche Matrix, spannen als Funktion der
Atomkoordinaten eine sehr komplexe Potentialenergie-Hyperfläche in 3N-5
Dimensionen auf. Den Potentialminima auf dieser Potentialenergie-Hyperfläche kommt
hierbei eine besondere Bedeutung zu, da sich das molekulare System mit sehr hoher
Wahrscheinlichkeit in den Konformationen niedriger Energie befindet. Aus diesem
Grund sind die zur Verwendung stehenden numerischen Verfahren zum Auffinden der
Minima auf der Potentialenergie-Hyperfläche von großer Bedeutung.
Minimierungsalgorithmen, die zum Auffinden der Potentialminima verwendet werden,
basieren auf der Berechnung der ersten bzw. zweiten Ableitung der Potentialenergie U
nach den Kernkoordinaten. In der vorliegenden Arbeit wurden Algorithmen wie z.B.
Steepest Descent und Conjugate Gradient [47] verwendet, die jeweils die erste
Ableitung verwenden. Die Anwendung dieser Methoden führt in der Regel zu einer
schnellen Konvergenz bei der Minimierung ausgedehnter Systeme. Da diese Verfahren
ausschließlich das nächste Minimum finden können, ist es notwendig, bei Systemen mit
einer sehr rauhen Potentialenergie-Hyperfläche eine andere Methode anzuwenden. Zur
Exploration des Konfigurationsraums hochdimensionaler Systeme wie z.B. Polymere,
ist es deshalb erforderlich, ein anderes Verfahren einzusetzen, um zu Konformationen
mit niedriger Energie zu gelangen. Die Wahrscheinlichkeit Pi einer Konformation i

Grundlagen 18
hängt über die Boltzmann-Beziehung in Gleichung 2.11 exponentiell von dessen
Energie Ei ab.
P ei
E
kTi
=−
(2.11)
Typischerweise sind für ein hochmolekulares Ensemble nur wenige energetisch niedrig
liegende Konformationen von Bedeutung, um das gesamte molekulare System zu
charakterisieren.
2.4.1.2 Simulated Annealing
Das Simulated Annealing [48] erlaubt die Durchmusterung eines wesentlich größeren
Konformationsraums, als dies mit den in Abschnitt 2.4.1.1. vorgestellten Minimierungs-
Algorithmen möglich ist. Hierzu werden Monte Carlo- oder Molekulardynamik-
Simulationen bei sehr hohen, physikalisch unrealistischen Temperaturen durchgeführt,
die es ermöglichen, Potentialbarrieren zu überwinden. Die hohe kinetische Energie
ermöglicht es auf diese Weise, einen weitaus größeren Teil der Potentialenergie-
Hyperfläche zu erreichen. Durch dieses Vorgehen kann vermieden werden, daß das
System in einem kleinen Bereich niedriger Potentialenergie eingeschlossen bleibt.
Aufgrund des Boltzmann-Kriteriums in Gleichung 2.11 besitzen die durch das
Abkühlen des Systems erhaltenen energetisch niedrig liegenden Konformationen eine
sehr hohe Wahrscheinlichkeit.
Ein verfahrenstechnisches Analogon zum Simulated Annealing findet sich bei der
Produktion von Silizium-Einkristallen. Hierbei wird die Siliziumschmelze langsam
abgekühlt, um zu einem Einkristall zu gelangen, dessen Struktur einem globalen
Minimum der Freien Energie entspricht. Simulated Annealing ist demnach ein
Simulationsverfahren, das diesen Prozeß zum Auffinden der optimalen Lösung aus einer
Reihe potentieller Lösungen nachbildet.

Grundlagen 19
2.5 Molekulardynamik-Simulationen
Die molekulardynamischen Simulationsmethoden (MD) bilden im Bereich der
klassischen Mechanik ein dem quantenmechanischen Verfahren analoges Verfahren.
Der Abstraktionsgrad der Molekulardynamik-Verfahren steht zwischen dem der Monte
Carlo-Methoden und dem der quantenmechanischen Verfahren. Das Verfahren
ermöglicht durch atomistische Simulationen die Behandlung molekularer Systeme mit
mehreren tausend Atomen. Bei der Durchführung von Molekulardynamik-Simulationen
werden zur Erzeugung von Konfigurationsensembles die natürlichen Bewegungsgesetze
auf alle Atome des molekularen Systems angewandt. Der Vorteil dieses Verfahrens
besteht darin, daß auch dynamische Informationen über das Modellsystem erhalten
werden.
Die mathematischen Grundlage des Molekulardynamik-Verfahrens liefert die Lagrange-
Gleichung [7,9-11]. Das dynamische Verhalten eines molekularen Systems kann durch
einen klassischen Hamiltonian in Kombination mit speziellen Randbedingungen, wie
z.B. konstantem Druck oder konstanter Temperatur, beschrieben werden. Die zeitliche
Entwicklung des molekularen Systems wird durch die gleichzeitige Integration der
Newtonschen Bewegungsgleichungen (Gleichungen 2.12 und 2.13) aller Atome
berechnet. Bei der Durchführung des Molekulardynamik-Verfahrens erhält man als
Ergebnis die Trajektorie des Modellsystems.
( )d r t
dtm Fi
i i
2
21
r
r
= − (2.12)
( )r
r r
rF
V r r
riN
i
=−∂
∂1 ,...,
(2.13)
In Gleichung 2.12 wird die auf das Atom i wirkende Kraft mit r
Fi und die Zeit mit t
bezeichnet. Die Durchführung von Molekulardynamik-Simulationen erfordert die
Berechnung des Gradienten der potentiellen Energie V(r) in Gleichung 2.13, die eine

Grundlagen 20
differenzierbare Funktion der Atomkoordinaten r
rN sein muß. Die Integration von
Gleichung 2.13 erfolgt über kleine Zeitintervalle ∆t, die typischerweise 0,25 fs bei
united-atom- und 0,1 fs bei all-atom-Modellsystemen betragen. Statische
Gleichgewichtseigenschaften lassen sich durch Mittelung über die Trajektorie erhalten.
Die Trajektorie sollte hinreichend lang sein, um ein für den Systemzustand
repräsentatives Ensemble darzustellen.
Die elektronischen Eigenschaften der Atome werden bei Molekulardynamik-
Simulationen durch räumlich isotrop wirkende Ladungsverteilungen und durch die Van-
der-Waals-Wechselwirkungen beschrieben. Kovalente Bindungen werden im
einfachsten Fall durch Federn repräsentiert, deren Charakteristik durch harmonische
Potentiale, oder realistischer durch Morse-Potentiale beschrieben wird. Die
Parametrisierung dieser Charakteristika wird als Kraftfeld bezeichnet und bildet die
Grundlage vieler kommerzieller Molekulardynamik-Simulations-Programme. Die
Parametrisierung erfolgt in der Regel für bestimmte Materialklassen.
2.5.1 Verwendete Kraftfelder
Im Rahmen dieser Arbeit wurden die Molekularmechanik bzw. Molekulardynamik-
Simulationen mit der Software Cerius2, Version 2.0 von Molecular Simulations Inc.
[49] auf IBM RS/6000-Workstations durchgeführt. Die verwendeten Kraftfelder wurden
in der in der Software Cerius2 implementierten Version eingesetzt.
Hierfür wurden für die Molekularmechanik-Simulationen an den Übergangsmetall-
Komplexen das Universal Force Field 1.01 [50] und für die Molekulardynamik-
Simulationen der Polymeren, für die Einzelketten und für die periodischen Systeme, das
Dreiding 2.21-Kraftfeld [51] verwendet. Die funktionalen Formen der beiden
verwendeten Kraftfelder werden in den Gleichungen 2.14 (Dreiding 2.21) und 2.15
(Universal Force Field 1.01) beschrieben:

Grundlagen 21
Dreiding 2.21
( ) [ ] ( )[ ]{ }
( ) [ ] ( )
E k R R C V n
C DQ Q
R
e e ijk ijk j ij jk jk
l li j
ij
= − + − + − −
+ − + − +
− −
1
2
1
2
1
21
1
22 322 0637
2 0 2 0
0 2
012 6
cos cos cos
cos cos ,
Θ Θ ς ς
ψ ψ ρ ρε
(2.14)
Im Potentialausdruck in Gleichung 2.14 stellen der erste, zweite und vierte Term jeweils
einfache harmonische Potentiale für die Deformation von Bindungslängen,
Bindungswinkeln und Inversions-Wechselwirkungen, der dritte Term eine Cosinus-
Funktion für die Beschreibung des Torsionspotentials dar. Die letzten beiden Terme
beschreiben die funktionale Form der nicht-bindenden Wechselwirkungen in Form eines
kurzreichweitigen Lennard-Jones-[12-6]-Potentials und eines Coulomb-Terms für die
langreichweitigen elektrostatischen Wechselwirkungen.
Der wesentliche Unterschied des Dreiding 2.21-Kraftfeldes in Gleichung 2.14 zum
Universal Force Field 1.01 in Gleichung 2.15 besteht darin, daß in letzterem die
Potentialausdrücke für die Bindungswinkel und die Torsionswinkel als Fourier-Reihe
entwickelt werden.
Universal Force Field (UFF) 1.01
(2.15)
( )
( ) ( )
E kij r r K C n K C n
K C C C Dx
x
x
x
Q Q
R
ij ijk nn
m
ijkl n ijkln
m
ijkl ijkl ijkl IJij ij i j
ij
= − + +
+ + + + −
+
+
= =∑ ∑1
2
2 2 322 0637
2
0 0
0 1 2
6 12
cos cos
cos cos ,
θ φ
ω ωε

Grundlagen 22
Die Berechnung der Bindungslängen erfolgt, wie in Gleichung 2.16 beschrieben, durch
Summation über der Bindungsradien der einzelnen Atome, ri und rj, sowie durch zwei
zusätzliche Korrekturterme zur Berücksichtigung der Bindungsordnung, rBO, und der
Elektronegativität, rEN.
r r r r rij i j BO EN= + + + (2.16)
Bei den allgemein verwendeten Kraftfeldern teilt man in sogenannte Klasse I-
Kraftfelder und Klasse II-Kraftfelder ein, wobei erstere auf empirischen Daten basieren.
Die Parametrisierung von Klasse II-Kraftfeldern beruht ausschließlich auf den
Ergebnissen quantenmechanischer Rechnungen [52].
Zur Durchführung der Molekularmechanik-Simulationen wurden die Module
Conformer Search, Conformer Analysis, Minimizer sowie Charges verwendet, bei der
Durchführung der Molekulardynamik-Simulationen die Module Polymer Builder,
Minimizer, Dynamics Simulation, Dynamics Analysis sowie Charges. Alle
Molekulardynamik-Simulationen wurden als all atom-Simulationen, d.h. unter
expliziter Berücksichtigung aller Atome, mit einem Zeitschritt von 0,1 fs durchgeführt.

Grundlagen 23
2.5.2 Molekulardynamik-Simulationen bei konstanter Temperatur
Bei der Integration der Newtonschen Bewegungsgleichungen 2.12 und 2.13 bleibt die
Gesamtenergie des Modellsystems erhalten. Die Simulation dieses adiabatischen
Systems mit konstantem Volumen führt zu einem mikrokanonischen Ensemble. Zur
Berechnung der meisten molekularen Eigenschaften ist dieses Ensemble jedoch nicht
sinnvoll, da es sich meist um die Untersuchung temperatur- oder druckabhängiger
Prozesse handelt.
Die Durchführung von Molekulardynamik-Simulationen bei konstanter Temperatur ist
prinzipiell durch mehrere Verfahren realisierbar. Die Methoden reichen von der ad hoc
erfolgenden Reskalierung der Atomgeschwindigkeiten [53], um die Temperatur zu
regulieren, bis zur Formulierung modifizierter Lagrangescher Bewegungsgleichungen,
die die Durchführung der Molekulardynamik-Simulation innnerhalb eines definierten
Temperaturintervalls erzwingt [54-56].
Im Rahmen dieser Arbeit wurden die Molekulardynamik-Simulationen bei konstanter
Temperatur ausschließlich unter Verwendung des Berendsen-Thermostats durchgeführt
[57]. Mit diesem Verfahren wird das zu untersuchende System an ein externes
Wärmebad gekoppelt, das die geforderte Temperatur besitzt. Dieses Wärmebad dient als
Quelle thermischer Energie, die dem System Wärme zuführt oder entzieht. Die
Geschwindigkeiten der Atome werden nach jedem Rechenschritt reskaliert, so daß die
thermischen Fluktuationen proportional der Temperaturdifferenz zwischen dem System
und dem Wärmebad sind. Die Anbindung des Systems an das Wärmebad erfolgt durch
den Kopplungsparameter τ in Gleichung 2.17:
( ) ( )( )dT t
dtT T tBad System= −1
τ (2.17)
Bei den Molekulardynamik-Simulationen in dieser Arbeit wurde für den
Kopplungsparameter ein Wert von τ = 0,1 ps verwendet.

Grundlagen 24
2.5.3 Modellierung des kondensierten Zustands
Mit den in Abschnitt 2.5.1. vorgestellten Kraftfeldern ist es möglich, Kettenmoleküle im
Vakuum zu behandeln, nicht aber im festen bzw. flüssigen Zustand oder in der
Schmelze. Hierdurch ist es nicht möglich, Aussagen über Bulkeigenschaften des
Modellsystems zu erhalten. Eine elegante Lösung dieses Problems liefert die
Anwendung sogenannter periodischer Randbedingungen, die es erlaubt, die
Bulkeigenschaften molekularer Systeme im kondensierten Zustand anhand einer relativ
geringen Anzahl von Atomen zu modellieren [7,11]. Das betrachtete Modellsystem
befindet sich hierbei in einer virtuellen Zelle mit parallelen Seitenwänden, die in alle
drei Raumrichtungen repliziert wird. Die Wände dieser Zellen sind für alle Teilchen
permeabel, in der Art, daß Teilchen, die auf einer Seite aus der Zelle heraustreten auf
der gegenüberliegenden wieder in die Zelle eintreten. Durch diese Bedingungen bleibt
die Teilchenzahl in der Zelle konstant. Prinzipiell sind fünf Zellformen denkbar, die
durch Translation der zentralen Zelle, den Raum vollständig erfüllen können. Hierzu
gehören das Parallelepiped, das hexagonale Prisma, eine modifzierte Oktaederzelle, der
rhombische Oktaeder und der elongierte Dodekaeder [58]. In der vorliegenden Arbeit
wurde bei den Molekulardynamik-Simulationen ausschließlich eine kubische Zelle
verwendet, die einen Sonderfall des Parallelepipeds darstellt. Die Verwendung
periodischer Randbedingungen führt in Abhängigkeit von den verwendeten
Zelldimensionen dazu, daß die Wellenlängen von Fluktuationen nicht größer sein
können als die verwendete Zelle [7].
Die Anwendung periodischer Randbedingungen führt zu einem virtuell unendlichen
System. Dieses Modell wäre mit einer unendlichen Anzahl von Wechselwirkungen
zwischen der zentralen Zelle und deren Repliken verbunden, die nichtbindenden
Wechselwirkungen wären explizit nicht zu behandeln. Verschiedene Methoden, wie
z.B. die Beschreibung der Van-der-Waals-Wechselwirkungen durch einen Cutoff in
Kombination mit einem Korrekturterm [59], das Cell Multipole Expansions-Verfahren
[60-62] oder die Reaction Field-Methode [63-65] erlauben eine physikalisch sinnvolle
Behandlung der nichtbindenden Wechselwirkungen.

Grundlagen 25
2.5.4 Behandlung der atomaren Partialladungen
2.5.4.1 Partielle Kompensation der Orbitalelektronegativität
Zur Berechnung der Partialladungen in den rein organischen Modellsystemen wurde der
Gasteiger-Marsili-Ansatz verwendet, der auf dem Konzept der partiellen Kompensation
der Orbitalelektronegativität basiert [66].
Dieses Konzept beruht auf der Annahme, daß Elektronen von den elektropositiveren
Elementen zu den elektronegativeren fließen. Dieser Ladungsfluß führt zu einer
positiven Ladung der elektropositiven Elemente bzw. einer negativen Ladung der
elektronegativen Elemente. Ursache für diesen Ladungsfluß ist das Streben des Systems,
die unterschiedlichen Elektronegativitäten auszugleichen. Dieser Effekt wird im
Rahmen des Gasteiger-Marsili-Ansatzes durch ein iteratives Verfahren beschrieben. Bei
jedem Schritt des Iterations-Verfahrens wird immer weniger Ladung zwischen den
gebundenen Atomen transferiert. Die elektronische Ladung, die während der k-ten
Iteration von einem Atom A zu einem Atom B transferiert wird, ist durch die Gleichung
2.18 gegeben:
Q k Bk
Ak
A
k( )( ) ( )
=−
+
χ χχ
α (2.18)
In Gleichung (2.18) beschreibt QK( )
die transferierte elektronische Ladung, χ Bk( ) und
χ Ak( ) die Elektronegativitäten der Atome A und B, χ A
+ die Elektronegativität des
Kations des elektropositiveren Atoms und α den Dämpfungsfaktor in der k-ten Potenz.
Gasteiger und Marsili nahmen für den Dämpfungsfaktor α den Wert 0,5 an. Der
Dämpfungsfaktor αk reduziert den Einfluß stärker elektronegativer Atome.

Grundlagen 26
2.5.4.2 Charge-Equilibration-Schema
Das Charge-Equilibration-Schema von Rappé et al. [67] verwendet zur Bestimmung der
Ladungsverteilung bei Molekulardynamik-Simulationen eine Reihenentwicklung für das
Atompotential, das aus atomaren Ionisationspotentialen, Elektronenaffinitäten und
Atomradien generiert wird, in Kombination mit einer zusätzlichen Abschirmungs-
Wechselwirkung zwischen allen im System vorhandenen Ladungen.
Die Vorteile dieser Methode sind eine gute Übereinstimmung mit experimentellen
Dipolmomenten und den atomaren Partialladungen, die aus ab initio-Rechnungen
erhalten werden. Wesentlicher Nachteil bei der Verwendung des Charge-Equilibration-
Schemas ist der hohe Rechenaufwand.
2.6 Auswahl der Modellsysteme
Bei der Polymerisation von Norbornen und dessen Derivaten sind aufgrund der
bicyclischen Struktur prinzipiell verschiedene Verknüpfungen möglich. Experimentelle
Untersuchungen zur Kettenstruktur durch Kernmagnetische Resonanzspektroskopie
(NMR) haben gezeigt, daß die Monomere in den untersuchten Polynorbornen
ausschließlich eine cis-exo-Verknüpfung zeigen [68,69].

Grundlagen 27
2.7 Charakterisierung des Packungsverhaltens
Das Packungsverhalten von Polymeren kann nach van Krevelen [70] über das
Verhältnis von molarem Volumen Vm und Van-der-Waals-Volumen Vw charakterisiert
werden. Das molare Volumen Vm hängt über die Dichte ρ vom Molekulargewicht des
untersuchten Polymers ab. Die Berechnung des Van-der-Waals-Volumen Vw erfolgt
über die Bestimmung der Connolly-Oberfläche mit Hilfe eines geeigneten Algorithmus
[71]. Die Connolly-Oberfläche ergibt sich in der Näherung interpenetrierender
Kugelschalen durch deren Abtastung mit einer Sonde mit infinitesimalen Radius.
Das Verhältnis von molarem Volumen Vm und Van-der-Waals-Volumen Vw besitzt für
glasbildende Polymere, mit einer Glastemperatur oberhalb von 25°C, im Mittel einen
Wert von 1,600 ± 0,045. Für kristalline Polymere wird ein Verhältnis von 1,435 ± 0,045
angegeben.
2.8 Experimentelle Untersuchungsmethoden
2.8.1 Röntgen-Weitwinkel-Streuung (WAXS)
Zur Untersuchung der räumlichen Anordnung im kondensierten Zustand wurden
Röntgen-Weitwinkeluntersuchungen (WAXS) an Pulver- und Filmproben mit einem
Goniometer (Siemens D5000) im 2Θ/Θ-Betrieb durchgeführt. Für die Untersuchungen
wurde Ni-gefilterte Cu(Kα)-Strahlung der Wellenlänge λ = 1,54 Å verwendet.

Grundlagen 28
2.8.2 Röntgen-Kleinwinkel-Streuung (SAXS)
Röntgen-Kleinwinkeluntersuchungen (SAXS) wurden an Filmproben mit einer Kratky-
Kleinwinkelkamera durchgeführt. Für die Untersuchungen wurde Ni-gefilterte Cu(Kα)-
Strahlung der Wellenlänge λ = 1.54 Å verwendet.
2.8.3 Differentialkalorimetrie (DSC)
Die differentialkalorimetrischen Messungen erfolgten mit einer Meßzelle der Firma
Mettler vom Typ DSC 30 in Kombination mit einem TA-Prozessor Mettler TA 3000.
Die pulverförmigen Proben wurden unter Schutzgasatmosphäre gemessen. Die
Messungen wurden mit verschiedenen Aufheizraten durchgeführt.
2.8.4 Thermisch-Mechanische Analyse (TMA)
Für die thermisch-mechanischen Untersuchungen wurden mit dem Gerät TMA7 von
Perkin Elmer durchgeführt. Die Messung der Penetration einer Wolframnadel mit einer
Auflagefläche von 0,28 mm2 erfolgte bei einer Aufheizrate von 10 K/min. Die
Auflagekraft der Nadel betrug 100 mN.
2.8.5 Kontaktwinkelmessungen
Die Bestimmung der Oberflächenenergien wurde an Polymerfilmen, die durch Spin
Coating hergestellt wurden, mit Hilfe eines Kontaktwinkelmeßgeräts der Firma Krüss
durchgeführt.

Kettenkonformation starrer Knäuel 29
3 Kettenkonformation starrer Knäuel
Dieses Kapitel wird sich mit der Charakterisierung und der Einordnung der
Kettenkonformation starrer Knäuel befassen. Für die Modellsysteme Polynorbornen
(PN, starres Knäuel), Polyethylen (PE, flexibles Knäuel) und Polyparaphenylen (PPP,
starres stäbchenförmiges Polymer) sollen die entsprechenden ein-, zwei- und n-
dimensionalen Potentialenergie-Hyperflächen charakterisiert werden. Das Ziel dieses
Kapitels ist es, die charakteristischen Eigenschaften der Polymerketten, wie z.B.
Kettenendenabstand und Gyrationsradius aber auch die Neigung zum Kettenkollaps im
Vakuum von Polynorbornen mit denen bekannter Polymere zu vergleichen. Durch
diesen Vergleich sollen die konformationsabhängigen Eigenschaften starrer Knäuel
quantitativ erfaßt und eingeordnet werden.
3.1 Simulationen zur Kettenkonformation
3.1.1 Rotationspotentiale
Die Konformation einer einzelnen Polymerkette im Raum wird im wesentlichen durch
die Verteilung der Torsionswinkel φi um die einzelnen chemischen Bindungen bestimmt
[1,72]. Im allgemeinen sind durch sterische und elektronische Wechselwirkungen nicht
alle Torsionswinkel energetisch äquivalent. Aus diesem Grund ist es sinnvoll, die
Wahrscheinlichkeiten für das Auftreten energetisch bevorzugter Konformationen durch
die Berechnung des Rotationspotentials zu untersuchen. Bei der Berechnung des
Rotationspotentials für ein polymeres bzw. ein oligomeres System erhält man die
Potentialenergie als Funktion des Torsionswinkels zwischen zwei benachbarten
Monomeren. Kennt man das Rotationspotential, d.h. den Verlauf der Potentialenergie

Kettenkonformation starrer Knäuel 30
bei der Drehung um eine oder mehrere Bindungen in Oligomeren, kann die
Wahrscheinlichkeit für das Auftreten bestimmter Torsionswinkel angegeben werden.
In Abbildung 3.1 ist die Verknüpfung zwischen den Monomeren in Polyethylen (PE),
Polynorbornen (PN) und Polyparaphenylen (PPP) dargestellt.
Polyethylen (PE) Polynorbornen (PN) Polyparaphenylen (PPP)
Abb. 3.1: Verknüpfung und Bindungswinkel zwischen den Monomeren
Bei Polyethylen und Polynorbornen beträgt der Bindungswinkel zwischen den
Monomeren 120°, was der Projektion des sp3-hybridisierten Kohlenstoffs entspricht.
Beim Polyparaphenylen zeigt sich zwischen den Monomereinheiten ein Bindungswinkel
von 180°, der zu einer vollkommen gestreckten Polymerkette führt.
3.1.2 Eindimensionale Rotationspotentiale
Bei der Berechnung des Rotationspotentials für die ausgewählten Modellsysteme erhält
man den Potentialverlauf als Funktion des Torsionswinkels. Jedes Rotationspotential
wird sowohl durch die Lage der Potentialminima als auch durch die Barrierenhöhe
charakterisiert. Um den Einfluß sowohl sterischer als auch elektronischer Effekte auf die
potentielle Energie zu untersuchen, lassen sich diese Rechnungen mit Hilfe
quantenmechanischer Verfahren durchführen [73,74]. Bei diesen Rechnungen wurde die

Kettenkonformation starrer Knäuel 31
gesamte Struktur zunächst ohne Einführung von äußeren Beschränkungen hinsichtlich
der Energie minimiert. Durch die Berechnung des Schwingungspektrums, d.h. die
Berechnung der zweiten Ableitung der Energie nach den Kernkoordinaten, konnte die
minimierte Struktur einem globalen Minimum zugeordnet werden.
3.1.2.1 Eindimensionales Rotationspotential für Butan
Das Rotationspotential für das Butan wurde durch schrittweise Drehung des in
Abbildung 3.2 definierten Torsionswinkel φ um jeweils 1° berechnet. Die Rechnungen
wurde mittels Molekularmechanik-Simulationen unter Verwendung des Dreiding 2.21-
Kraftfeldes durchgeführt. Die Ergebnisse quantenmechanischer Rechnungen (MP2/6-
311G**//MP2/6-311G**) finden sich in [75].
12
34
Abb. 3.2: Definition des Torsionswinkels φ über drei Bindungen für Butan
In Abbildung 3.3 ist der Potentialverlauf des eindimensionalen Rotationspotentials von
Butan dargestellt.

Kettenkonformation starrer Knäuel 32
0 30 60 90 120 150 180 210 240 270 300 330 3600
5
10
15
20
25
30
35
40
45
50
55
60
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 3.3: Eindimensionales Rotationspotential von Butan
Das dreizählig symmetrische Rotationspotential vom Butan in Abbildung 3.3 zeigt drei
relative breite Potentialminima bei 60° (lokales Minimum, gauche-), 180° (globales
Minimum, trans) sowie 290° (lokales Minimum, gauche+). Der Übergang von der
trans-Form in die gauche-Form, d.h. gauche+- oder gauche--Form, erfordert mit rund
15,0 kJ/mol eine niedrigere Energie als der direkte Übergang der gauche+- in die
gauche--Form mit 25,0 kJ/mol. Der energetische Unterschied zwischen dem globalen
Minimum und den lokalen Minima beträgt lediglich rund 2,5 kJ/mol. Bei
Raumtemperatur sind alle drei Potentialminima mit einer Fluktuation von jeweils ± 20°
besetzt. Eine Trennung der verschiedenen Konformere ist aufgrund der hohen
Interkonversionsrate im Bereich von 1010 s-1 nicht möglich [1].

Kettenkonformation starrer Knäuel 33
3.1.2.2 Eindimensionales Rotationspotential für Biphenyl
Das Rotationspotential für das Biphenyl wurde durch schrittweise Drehung des in
Abbildung 3.4 definierten Torsionswinkel φ um jeweils 10° berechnet. Die Rechnungen
wurden mittels quantenmechanischer Rechnungen unter Verwendung des 6-31G(d)-
Basissatzes von Pople et al. [23,24] durchgeführt.
1
2
3
4
Abb. 3.4: Definition des Torsionswinkels φ über drei Bindungen für Biphenyl
Der Potentialverlauf in Abbildung 3.5 zeigt ein symmetrisches Rotationspotential, daß
durch vier äquienergetische Minima charakterisiert ist. Die niedrigen Potentialbarrieren
zwischen den Minima bei 45° und 130° sowie denen zwischen 230° und 315° besitzen
eine Barrierenhöhe von rund 5,0 kJ/mol. Aufgrund dieser Barrierenhöhe ist zu erwarten,
daß die Übergänge zwischen den o.g. Minima bei Raumtemperatur auftreten. Die trans-
und die cis-Konformation bilden als energetisch hochliegende Konformationen die
Potentialbarrieren im eindimensionalen Rotationspotential des Biphenyls.

Kettenkonformation starrer Knäuel 34
0 30 60 90 120 150 180 210 240 270 300 330 3600
5
10
15
20
25
30
35
40
45
50
55
60
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 3.5: Eindimensionales Rotationspotential von Biphenyl (RHF/6-31G(d))
Die äquienergetischen Potentialminima in Abbildung 3.5 sollten bei Raumtemperatur
aufgrund der niedrigen Potentialbarrieren gleich stark besetzt sein [1].
3.1.2.3 Eindimensionales Rotationspotential dimeres Norbornan
Das Rotationspotential für das dimere Norbornan wurde durch schrittweise Drehung des
in Abbildung 3.6 definierten Torsionswinkel φ um jeweils 10° berechnet.

Kettenkonformation starrer Knäuel 35
1 2 3 4
Abb. 3.6: Definition des Torsionswinkels φ über drei Bindungen
Für die quantenmechanische Berechnung des eindimensionalen Rotationspotentials für
das dimere Norbornan wurde der 3-21G-Split-Valenz-Basissatz von Pople et al. [22]
verwendet.
0 30 60 90 120 150 180 210 240 270 300 330 3600
5
10
15
20
25
30
35
40
45
50
55
60
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 3.7: Rotationspotential für dimeres Norbornan (RHF/3-21G)

Kettenkonformation starrer Knäuel 36
Das Rotationspotential in Abbildung 3.7 zeigt ein globales Minimum bei 60° und zwei
lokale Minima bei 180° bzw. 310°. Die Potentialenergien der beiden lokalen Minima
liegen um 17,0 bzw. 18,0 kJ/mol oberhalb des globalen Minimums, ein Sprung aus dem
Minimum bei 60° muß über eine Barriere von etwa 25,0 kJ/mol erfolgen.
Um die Konformationseigenschaften der Norbornan-Oligomere besser beschreiben und
vergleichen zu können, erscheint es sinnvoll, mit Tabelle 3.1 eine dem trans-gauche-
Formalismus analoge Bezeichnung der Minima für das Polynorbornen einzuführen. Als
Bezugssystem soll das Butan gelten, da in diesem System ebenso wie im Norbornan die
sp3-hybridisierten Kohlenstoffatome im Polymerbackbone für sich ein dreizähliges
Rotationspotential hervorrufen.
Tabelle 3.1:
Nomenklatur für Konformationseigenschaften von Butan und dimerem Norbornan
Butan dimeres Norbornan
Torsionswinkel φ Bezeichnung Torsionswinkel φ Bezeichnung
180° trans, t, antiperiplanar 60° n_min
300° gauche +, g+, synclinal 180° n_min +
60° gauche -, g-., synclinal 310° n_min-
Im Falle des dimeren Norbornens ist für den Übergang von n_min nach n_min+ ein
Energiebetrag von 25,0 kJ/mol, für den Übergang von n_min nach n_min- mehr als 30,0
kJ/mol notwendig, während für den Übergang zwischen den beiden lokalen Minima
rund 55,0 kJ/mol erforderlich wären.
Der Vergleich mit dem Potentialverlauf der eindimensionalen Rotationspotentiale von
Polyethylen (Modellsystem Butan) bzw. von Polyparaphenylen (Modell Biphenyl) zeigt
kaum Unterschiede in den Höhen der entsprechenden Potentialbarrieren. Polynorbornen
kann also aufgrund des eindimensionalen Rotationspotentials mit einer Barrierenhöhe

Kettenkonformation starrer Knäuel 37
von 25 kJ/mol nicht als absolut starres Knäuel angesehen werden. Es erscheint aber
sinnvoll, daß die Steifigkeit des Polynorbornens durch Nachbargruppeneffekte
kontrolliert wird. Aus diesem Grund wird im folgenden Abschnitt das zweidimensionale
Rotationspotential betrachtet.
Über die Boltzmann-Beziehung kann weiterhin abgeschätzt werden, daß das Verhältnis
der Besetzungszahlen für das lokale Minimum bei 180° und das absolute Minimum bei
60° rund 1 / 25000 beträgt.
Die Energien, die für eine freie Rotation um die zentrale Bindung im Butan, Biphenyl
und dimeren Norbornan mindestens aufgewendet werden müssen, sind in Tabelle 3.2
angeführt:
Tabelle 3.2:
Energien für eine freie Rotation um die zentrale Bindung aus eindimensionalen
Rotationspotentialen
Dimer Potentialbarriere [kJ/mol]
Butan 22,0
Biphenyl 25,0
dimeres Norbornan 55,0
Abbildung 3.8 zeigt den Vergleich des Potentialverlaufs des eindimensionalen
Rotationspotentials für das dimere Norbornan im globalen Minimum mit dem
Potentialverlauf eines harmonischen Oszillators. Dieser Vergleich zeigt, daß die
Torsionswechselwirkungen in einem sehr großen Torsionswinkelbereich von ± 50° um
das Minimum mit einem harmonischen Potential beschrieben werden können.

Kettenkonformation starrer Knäuel 38
-60 -50 -40 -30 -20 -10 0 10 20 30 40 50 600
2
4
6
8
10
12
14
16
18
20
22
24
26
Harmonisches Potential
Ausschnitt Rotationspotentialdimeres Norbornan
rel.
Ene
rgie
[kJ
/mol
]
relativer Torsionswinkel φ [°]
Abb. 3.8: Vergleich Potentialverlauf im globalen Minimum von PN und harm. Oszillator
Dieser Vergleich wird in Kapitel 4 noch einmal in die Betrachtungen einbezogen.
3.1.3 Zweidimensionale Rotationspotentiale
Durch die Berechnung zweidimensionaler Rotationspotentiale läßt sich ein größerer
Konformationsraum durchsuchen, der die Verhältnisse in einer langen Polymerkette
besser beschreibt als das eindimensionale Rotationspotential. Durch das Vorhandensein
eines dritten Norbornan-Monomeren werden Nachbargruppeneffekte, die durch die
dreidimensionale Gestalt des Norbornans verursacht werden, berücksichtigt. Aufgrund
der Systemgröße wurde die Berechnung des zweidimensionalen Rotationspotentials
mittels Molekularmechanik-Simulationen durchgeführt. Für diese Rechnungen wurde
das Dreiding 2.21-Kraftfeld in Kombination mit dem Charge-Equilibration-Schema von
Gasteiger und Marsili [66] verwendet.

Kettenkonformation starrer Knäuel 39
3.1.3.1 2D-Rotationspotential Pentan
Zu der Berechnung der zweidimensionalen Rotationspotentiale geht man von einem
trimeren System aus. Die Potentialenergie des Trimers erhält man als Funktion zweier
fixierter Torsionswinkel durch die Minimierung der Gesamtstruktur hinsichtlich der
Energie. Die folgenden Potentialenergie-Hyperflächen (PES) wurden durch jeweils rund
1400 einzelne Rechnungen erhalten.
Für das Pentan, als Modellsystem für das Polyethylen, findet man ein symmetrisches
zweidimensionales Rotationspotential. Die Farbskalierung in Abbildung 3.9 zeigt, daß
sich alle Potentialminima für dieses System durch Kombinationen der trans- und der
gauche+- bzw. gauche--Konformation erklären lassen. Weiterhin erkennt man aus der
Potentialenergie-Hyperfläche, daß die niedrigsten Potentialbarrieren für die Übergänge
zwischen den trans-gauche+-, trans-gauche--, gauche+-trans- bzw. gauche--trans-
Konformationen über die trans-trans-Konformation mit rund 17,0 kJ/mol bei
Raumtemperatur übersprungen werden können. Dieser Wert liegt nur um rund 2,0
kJ/mol über den Rotationsbarrieren des eindimensionalen Rotationspotentials
(Abbildung 3.3.) zwischen der trans- und der gauche-- bzw. der gauche+-
Konformation. Aus Abbildung 3.9. ist zu erkennen, daß diese Potentialenergie-
Hyperfläche fünf annähernd äquienergetische Potentialminima aufweist. Bei
Raumtemperatur sollten mindestens diese fünf Potentialminima besetzt sein [1].

Kettenkonformation starrer Knäuel 40
0 60 120 180 240 300 360
0
60
120
180
240
300
360
Torsionswinkel 1 [°]
Tor
sion
swin
kel 2
[°]
25.00 -- 26.00 24.00 -- 25.00 23.00 -- 24.00 22.00 -- 23.00 21.00 -- 22.00 20.00 -- 21.00 19.00 -- 20.00 18.00 -- 19.00 17.00 -- 18.00 16.00 -- 17.00 15.00 -- 16.00 14.00 -- 15.00 13.00 -- 14.00 12.00 -- 13.00 11.00 -- 12.00 10.00 -- 11.00 9.000 -- 10.00 8.000 -- 9.000 7.000 -- 8.000 6.000 -- 7.000 5.000 -- 6.000 4.000 -- 5.000 3.000 -- 4.000 2.000 -- 3.000 1.000 -- 2.000 0 -- 1.000
Abb. 3.9: Zweidimensionales Rotationspotential für Pentan
(Energien in kJ/mol)
Anhand der Äquipotentiallinien in Abbildung 3.9 kann ein Minimumspfad durch die
Potentialenergie-Hyperfläche geführt werden, so daß für eine vollständige Rotation
einer Bindung mindestens 25,0 kJ/mol aufzuwenden sind. Dieser Wert zeigt im
Vergleich zur Rotationsbarriere des eindimensionalen Rotationspotentials des Butans
denselben Wert.
3.1.3.2 2D-Rotationspotential Paraphenylen
Die Berechnung des zweidimensionalen Rotationspotentials des para-Terphenyls mit
Hilfe von Molekularmechanik-Simulationen führt zu der in Abbildung 3.10

Kettenkonformation starrer Knäuel 41
dargestellten Potentialenergie-Hyperfläche. Die Ergebnisse von Molekularmechanik-
Rechnungen mit dem Softwarepaket SYBYL [76] zu Polyparaphenylen-Oligomeren
finden sich in [77].
0 60 120 180 240 300 3600
60
120
180
240
300
360
Torsionswinkel 1 [°]
Tor
sion
swin
kel 2
[°]
21.00 -- 22.00 20.00 -- 21.00 19.00 -- 20.00 18.00 -- 19.00 17.00 -- 18.00 16.00 -- 17.00 15.00 -- 16.00 14.00 -- 15.00 13.00 -- 14.00 12.00 -- 13.00 11.00 -- 12.00 10.00 -- 11.00 9.000 -- 10.00 8.000 -- 9.000 7.000 -- 8.000 6.000 -- 7.000 5.000 -- 6.000 4.000 -- 5.000 3.000 -- 4.000 2.000 -- 3.000 1.000 -- 2.000 0 -- 1.000
Abb. 3.10: Zweidimensionales Rotationspotential für para-Terphenyl
(Energien in kJ/mol)
Aus dieser symmetrischen Potentialenergie-Hyperfläche ist sehr gut zu erkennen, daß
die koplanare Anordnung der Phenylringe ein Energiemaximum, d.h. eine
Potentialbarriere, darstellt. Weiterhin ist aus den Äquienergieflächen ersichtlich, daß die
einzelnen Bindungen zwischen den Monomeren über einen sehr weiten Bereich von
120° näherungsweise frei rotieren können. Die Potentialbarriere in diesem System
entsteht durch sterische und starke elektronische Wechselwirkungen der Phenylringe.
Die minimal aufzuwendende Energie für eine vollständige Rotation um eine Bindung
zwischen zwei Phenylringen beträgt, ähnlich wie im Fall des Pentans, rund 22,0 kJ/mol.

Kettenkonformation starrer Knäuel 42
Ein wesentlicher Unterschied ist jedoch die Tatsache, daß beide Bindungen im para-
Terphenyl aufgrund des Bindungswinkels von 180° unabhängig voneinander sind.
3.1.3.3 2D-Rotationspotential Norbornan
Berechnet man das zweidimensionale Rotationspotential für das trimere Norbornan,
findet man eine asymmetrische Potentialenergie-Hyperfläche (Abb. 3.11), die durch
einige wenige energetisch bevorzugte Minima charakterisiert ist.
0 60 120 180 240 300 3600
60
120
180
240
300
360
Torsionswinkel 1 [°]
Tor
sion
swin
kel 2
[°]
98.00 -- 101.5 94.50 -- 98.00 91.00 -- 94.50 87.50 -- 91.00 84.00 -- 87.50 80.50 -- 84.00 77.00 -- 80.50 73.50 -- 77.00 70.00 -- 73.50 66.50 -- 70.00 63.00 -- 66.50 59.50 -- 63.00 56.00 -- 59.50 52.50 -- 56.00 49.00 -- 52.50 45.50 -- 49.00 42.00 -- 45.50 38.50 -- 42.00 35.00 -- 38.50 31.50 -- 35.00 28.00 -- 31.50 24.50 -- 28.00 21.00 -- 24.50 17.50 -- 21.00 14.00 -- 17.50 10.50 -- 14.00 7.000 -- 10.50 3.500 -- 7.000 0 -- 3.500
Abb. 3.11: Zweidimensionales Rotationspotential für trimers Norbornan
(Energien in kJ/mol)

Kettenkonformation starrer Knäuel 43
Das zweidimensionale Rotationspotential des trimeren Norbornans zeigt, daß sich zwei
der Potentialminima dieses Modellsystems durch die Kombination aus den
Konformationen n_min und n_min+ generieren lassen. Weiterhin treten vier zusätzliche
Nebenminima auf, die sich nicht durch eine einfache Kombination der Minima des
eindimensionalen Rotationspotentials erklären lassen. Der relative Energieunterschied
zwischen den beiden niedrigsten Hauptminima und den vier Nebenminima beträgt 8,0
kJ/mol. Vielmehr führt der sterische Anspruch der Norbornan-Monomere zu
zusätzlichen, energetisch bevorzugten Konformationen, was bei einer Vergrößerung des
Systems zu einer komplexen mehrdimensionalen Potentialenergie-Hyperfläche führt. In
der Abbildung 3.11 erkennt man, daß sich alle Potentialminima auf der
Potentialenergie-Hyperfläche in einem relativ kleinen, quadratischen Bereich mit einer
Größe von jeweils 180° befinden. Die hohen Potentialbarrieren von mehr als 100,0
kJ/mol schränken die Beweglichkeit des trimeren Systems auf diesen schmalen Bereich
ein. Die Potentialbarriere für einen Konformationsübergang durch vollständige Rotation
ist beim Übergang vom eindimensionalen zum zweidimensionalen Rotationspotential
von rund 50,0 kJ/mol auf über 100,0 kJ/mol angestiegen.
Ein wesentliches Ergebnis dieser Betrachtungen ist, daß im trimeren Norbornan
Nachbargruppeneffekte auftreten. Diese Nachbargruppeneffekte mit den jeweils
übernächsten Monomeren verursachen im zweidimensionalen Rotationspotential hohe
Potentialbarrieren. Es ist zu erwarten, daß die hohen Potentialbarrieren auf der n-
dimensionalen Potentialenergie-Hyperfläche zu einem starren knäuelförmigen
Kettengestalt führen.
Abbildung 3.12 zeigt einen schematischen Vergleich der Potentialenergie-Hyperflächen
des Pentans und para-Terphenyls mit der des trimeren Norbornans. Bei der Darstellung
wurde die Potentialenergie-Hyperfläche des trimeren Norbornens bei 26,0 kJ/mol
geschnitten. Dies entspricht derjenigen Energie, die erforderlich ist, um beim Pentan
und para-Terphenyl eine vollständige Rotation auszuführen.

Kettenkonformation starrer Knäuel 44
0 60 120 180 240 300 3600
60
120
180
240
300
360
Torsionswinkel 1 [°]
Tor
sion
swin
kel 2
[°]
25.00 -- 26.00 24.00 -- 25.00 23.00 -- 24.00 22.00 -- 23.00 21.00 -- 22.00 20.00 -- 21.00 19.00 -- 20.00 18.00 -- 19.00 17.00 -- 18.00 16.00 -- 17.00 15.00 -- 16.00 14.00 -- 15.00 13.00 -- 14.00 12.00 -- 13.00 11.00 -- 12.00 10.00 -- 11.00 9.000 -- 10.00 8.000 -- 9.000 7.000 -- 8.000 6.000 -- 7.000 5.000 -- 6.000 4.000 -- 5.000 3.000 -- 4.000 2.000 -- 3.000 1.000 -- 2.000 0 -- 1.000
Abb. 3.12: Ausschnitt der Potentialenergie-Hyperfläche des trim. Norbornans mit Energien ≤ 26,0 kJ/mol
(Energien in kJ/mol)
Abbildung 3.12 stellt demnach denjenigen Bereich der Potentialenergie-Hyperfläche des
trimeren Norbornans dar, der ausgehend vom globalen Potentialminimum mit Energien
von rund 26,0 kJ/mol erreicht werden kann. Dieser Bereich beträgt schätzungsweise 10
bis 15% der gesamten Potentialenergie-Hyperfläche des trimeren Norbornans.

Kettenkonformation starrer Knäuel 45
3.2 Computersimulationen zur Konformation der Einzelkette im
Vakuum
3.2.1 Molekulardynamik-Simulationen im Vakuum
Um den in Abschnitt 3.2.3.3 beobachteten Einfluß der Nachbargruppeneffekte auf die
Gestalt einer langen Polymerkette zu untersuchen, wurden Molekulardynamik-
Simulationen an Einzelketten bei unterschiedlichen Temperaturen durchgeführt. Die
verwendeten Polynorbornen-Ketten aus cis-exo verknüpften Monomeren besaßen einen
Polymerisationsgrad 100.
Vor der Durchführung der eigentlichen Molekulardynamik-Simulationen wurden die
Polymerketten mittels einer Kombination aus Simulated Annealing und Anwendung
eines Minimierungsverfahrens hinsichtlich der potentiellen Energie optimiert. Das
Verfahren des Simulated Annealing erlaubt es, durch wiederholtes Aufheizen und
Abkühlen und anschließendem Minimieren einen weitaus größeren Teil des
Konfigurationsraumes zu durchmustern als dies durch einfaches Minimieren zu
erreichen wäre. Zur Minimierung der Polymerstruktur wurde ein Conjugate Gradient-
Algorithmus verwendet. Die zunächst durchgeführte Molekularmechanik-Simulation
zum Erreichen des Minimums der potentiellen Energie entspricht einer Temperatur von
0 K. Die Abnahme der o.g. Kettenparameter zu Beginn der Molekulardynamik-
Simulation wird durch die zusätzlich kinetische Energie verursacht. Die bei der
Simulation verwendete Temperatur von 300 K führt dazu, daß ein größerer Teil der n-
dimensionalen Potentialenergie-Hyperfläche zugänglich wird.
Die Molekulardynamik-Simulationen an den Polynorbornenketten wurden im Vakuum
durchgeführt, um ein schlechtes Lösemittel mit einer sehr niedrigen Dichte und einer
sehr niedrigen Viskosität zu simulieren [78,79]. Durch die Simulationen unter
Berücksichtigung dieser Randbedingungen ist es möglich, innerhalb relativ kurzer
Simulationszeit Bewegungen großer Kettenfragmente zu beobachten.

Kettenkonformation starrer Knäuel 46
Die Auswertung der Molekulardynamik-Simulation zeigt, daß die Polynorbornenkette
nicht kollabiert. Dies erkennt man in Abbildung 3.13 daran, daß der Kettenenden-
Abstand RE und der Gyrationsradius RG zwar 100 ps lang abnehmen, anschließend aber
um Mittelwerte von rund 78,0 Å bzw. 33,0 Å fluktuieren.
0 50 100 150 200 250 300 350 4000
20
40
60
80
100
120
140
160
180
200
Gyrationsradius
Kettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 3.13: Molekulardynamik-Simulation Polynorbornen bei 300 K (Einzelkette)
Untersucht man die Fluktuationen der beiden Kettenparameter in dem Intervall
zwischen 150 und 350 ps, so erhält man für den Kettenendenabstand RE 78,0 Å ± 2,6 Å
und für den Gyrationsradius RG 33,0 Å ± 2,6 Å. Das Verhältnis von ⟨RE2⟩ / ⟨RG
2⟩ liefert
einen Wert von 5,5. Das charakteristische Verhältnis C100 ergibt einen Wert von 8,2.
Wertet man eine Molekulardynamik-Simulation einer Polynorbornen-Einzelkette mit
einem Polymerisationsgrad 100 aus, die bei 600 K durchgeführt wurde, findet man die

Kettenkonformation starrer Knäuel 47
folgende zeitliche Entwicklung des Kettenendenabstandes RE und Gyrationsradius RG,
die in Abbildung 3.14 dargestellt ist.
660 680 700 720 740 760 780 800 820 8400
20
40
60
80
100
120
140
160
180
200
GyrationsradiusKettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 3.14: Molekulardynamik-Simulation Polynorbornen bei 600 K (Einzelkette)
Das charakteristische Ergebnis dieser Molekulardynamik-Simulation ist, daß die
Polynorbornenkette selbst bei höheren Temperaturen, wie etwa 600 K, nicht kollabiert,
wohl aber enger wird. Die Untersuchung der zeitlichen Entwicklung des
Kettenendenabstandes RE und des Gyrationsradius RG im Intervall zwischen 650 bis 850
ps in Abbildung 3.10. liefern 54,5 Å ± 7,5 Å bzw. 25,9 Å ± 2,5 Å. Das dimensionslose
Verhältnis ⟨RE2⟩ / ⟨RG
2⟩ besitzt einen Wert von 4,6. Das charakteristische Verhältnis C100
berechnet sich zu einem Wert von 4,0.
Der Vergleich mit Ergebnissen von Molekulardynamik-Simulationen an flexiblen
Polymerketten zeigt deutliche Unterschiede. Die Simulation einer Polyvinylchloridkette
(PVC) von Mattice et al. [78] bei 300 K zeigt bereits nach 50 ps ein deutliches

Kettenkonformation starrer Knäuel 48
Kollabieren der Polymerkette. Der Kettenendenabstand des Polymers mit einem
Polymerisationsgrad von 100 fluktuiert um ungefähr 21 Å, der Gyrationsradius um rund
13 Å. Das dimensionslose Verhältnis ⟨RE2⟩ / ⟨RG
2⟩ besitzt einen Wert von 2,6.
Für die untersuchten Modellsysteme erhält man für die Korrelationen aus dem
quadratischen Kettenendenabstand ⟨RE2⟩ und quadratischen Gyrationsradius ⟨RG
2⟩ die in
Tabelle 3.3. aufgeführten Werte:
Tabelle 3.3. :
Korrelationen für das Verhältnis von ⟨RE2⟩ / ⟨RG
2⟩
Polymer ⟨RE2⟩ / ⟨RG
2⟩
Polyvinylchlorid (PVC), 100mer bei 300 K [69] 2,6
Polynorbornen (PN), 100mer bei 300 K 5,5
Polynorbornen (PN), 100mer bei 600 K 4,4
Die Werte für ideale Geometrien belaufen sich 2,0 für kollabierte Ketten, 6,0 für Gauß-
Knäuel und 12,0 für starre Stäbchen [78].
Der Vergleich der Verhältnisse von ⟨RE2⟩ / ⟨RG
2⟩ mit denen von flexiblen Ketten zeigt
deutlich, daß Polynorbornene bei Raumtemperatur als relativ starre Knäuel vorliegen
und daß bei Temperaturerhöhung nur ein schwacher Übergang zu einer stärker
kollabierten Struktur zu beobachten ist.
Die zeitliche Entwicklung der Kettenkonformation, die aus der Molekulardynamik-
Simulation von Polynorbornen erhalten wird, erscheint auf den ersten Blick
ungewöhnlich, da die ursprüngliche Kettengestalt erhalten bleibt und ein Kollaps dieser
Polymerkette nicht stattfindet. Um die Ergebnisse hinsichtlich Kettenkonformation und

Kettenkonformation starrer Knäuel 49
Kettengestalt des Polynorbornens zu überprüfen, wurden Monte Carlo-Simulationen zur
Kettenkonformation eingesetzt.
3.2.2 Monte Carlo-Simulationen zur Kettenkonformation
Die Anwendung des Monte Carlo-Verfahrens zur Untersuchung der Konformation der
Polynorbornenkette ermöglicht es, innerhalb kurzer Zeit ein relativ großes Ensemble
von Polymerketten zu generieren. Im Gegensatz zum Molekulardynamik-Verfahren, das
auf die Erzeugung des zeitlichen Mittels abzielt, ist mit der Monte Carlo-Methode das
Zahlenmittel zugänglich [80,81].
In der Rotational Isomeric State (RIS)-Theorie wird jeder Bindung eines Moleküls ein
diskreter Rotationszustand zugewiesen [1]. Die einzelnen Zustände werden aus den
Potentialminima des Rotationspotentials abgeleitet. Die Theorie der rotationsisomeren
Zustände ersetzt das kontinuierliche Rotationspotential durch eine Verteilung diskreter
Rotationszustände. Die Grundlage für die Definition dieser diskreten Rotationszustände
ist das Vorliegen von Rotationsbarrieren größer RT, bzw. 2,48 kJ/mol. Durch die
Definition rotationsisomerer Zustände kann für das Polynorbornen eine Tabelle
gekoppelter Rotationszustände aufgestellt werden, die es ermöglicht, hinreichend
realistische Polymerketten aufzubauen. Durch dieses Vorgehen ist es möglich, die
energetischen Unterschiede der einzelnen Minima aus dem zweidimensionalen
Rotationspotential beim Aufbau der Polymerketten zu berücksichtigen. Mit Hilfe der
Boltzmann-Beziehung werden die einzelnen Rotationszustände gewichtet und die
statischen Potentialmimima durch Fluktuationsbreiten an die berechneten
Potentialverläufe aus ein- bzw. zweidimensionalen Rotationspotentialen angepaßt. Die
Tabelle der gekoppelten rotationsisomeren Zustände berücksichtigt auf diese Weise die
für das Polynorbornen charakteristischen Nachbargruppeneffekte.
Generiert man z.B. ein Ensemble aus 1000 Polynorbornenketten mit einem
Molekulargewicht von 20.000 g/mol, so findet man bei Raumtemperatur am häufigsten

Kettenkonformation starrer Knäuel 50
Polymerketten mit einem Gyrationsradius von rund 60 Å, wie aus Abbildung 3.15 zu
erkennen ist [82].
30 35 40 45 50 55 60 650
10
20
30
40
50H
äufi
gkei
t
Gyrationsradius [Å]
Abb. 3.15: Häufigkeitsverteilung des Gyrationsradius von Polynorbornen (MC)
Aus der in Abbildung 3.15 dargestellte Häufigkeitsverteilung erhält man ein
charakteristisches Verhältnis C200 von 12,1, was charakteristisch für semiflexible
Polymerketten ist.
Der Vergleich der charakteristischen Verhältnisse aus Molekulardynamik- und Monte
Carlo-Simulationen zeigen Werte von 8,2 bzw. 12,1. Die zahlenmäßigen Unterschiede
sind auf das Versagen der Ergoden-Hypothese zurückzuführen. Dies ist für ein
Einzelkettenglas im Sinne von de Gennes [4] durchaus verständlich, entspricht dies
doch einem Nicht-Gleichgewichtszustand. Das mit Hilfe der Molekulardynamik-
Simulationen erzeugte Ensemble besitzt andere Eigenschaften als das mit Monte Carlo-
Verfahren generierte Ensemble, d.h. Orts- bzw. Zahlenmittel können nicht mit dem
Zeitmittel verglichen werden.

Kettenkonformation starrer Knäuel 51
3.3 Molekulardynamik-Simulationen im kondensierten Zustand
Die räumliche Gestalt einer Polymerkette wird sowohl durch kurzreichweitige als auch
durch langreichweitige Wechselwirkungen beeinflußt. Die kurzreichweitigen
Wechselwirkungen entstehen zwischen Monomeren, die nur durch wenige Bindungen
voneinander getrennt sind. Die langreichweitigen Volumeneffekte nehmen mit
wachsender Kettenlänge stark zu, da es sich hierbei um Wechselwirkungen zwischen
weiter entfernten Kettensegmenten handelt. Die langreichweitigen Wechselwirkungen
werden durch die Wechselwirkungen mit dem Lösemittel bzw. der Umgebung um die
Kette ebenfalls stark beeinflußt [83]. Zur Beschreibung dieser Wechselwirkungen dient
das sogenannte Ausschlußvolumen, was den Unterschied zwischen der idealen und der
realen Kettengestalt charakterisiert. Polymerketten nehmen im sog. Theta-Zustand eine
ideale Konformation ein, da das ausgeschlossenes Volumen in diesem ausgezeichneten
Zustand Null wird. In diesem Zustand kompensieren sich gerade die repulsiven und
attraktiven Wechselwirkungen. Polymerketten können diesen Zustand sowohl in
geeigneten Lösemitteln als auch in ihren Schmelzen annehmen [1].
Mittels geeigneter Molekulardynamik-Simulationsverfahren läßt sich der kondensierte
Zustand von Polymeren durch Modellsysteme mit periodischen Randbedingungen
nachbilden [84-88]. Bei diesen Computersimulationen wird eine sog. Polynorbornen-
Parent-Chain von deren identischen Abbildern in allen Raumrichtungen umgeben.
Für diese Simulation wurde eine Polymerkette aus 100 Monomeren eingesetzt. Für die
Zelle mit periodischen Randbedingungen wurde die experimentelle Dichte von 1,094
g/cm3 [89] verwendet. Die Simulationszeit betrug 0,9 ns. Die Simulationsbedingungen
mit konstantem Druck und konstanter Temperatur (NPT-Ensemble) wurden durch den
Berendsen-Thermostat (sog. T-Damping) [57] realisiert. Bei dieser Simulation wurde
ein externer Korrekturdruck zur Kompensation des Abschneidens der langreichweitigen
Wechselwirkungen durch die Verwendung eines Cut-Offs eingesetzt [59].

Kettenkonformation starrer Knäuel 52
100 110 120 130 140 150 160 170 180 190 20020
30
40
50
60
70
80
90
100
Gyrationsradius
Kettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 3.16: Molekulardynamik-Simulation Polynorbornen im kondensierten Zustand (300 K)
Abbildung 3.16 zeigt die zeitliche Entwicklung des Kettenendenabstandes RE und des
Gyrationsradius RG bei 300 K. Die Auswertung der Molekulardynamik-Simulation führt
zu einem Kettenendenabstand von 79,4 Å ± 2,5 Å bzw. zu einem Gyrationsradius von
37,7 Å ± 1,2 Å. Das Verhältnis aus ⟨RE2⟩ / ⟨RG
2⟩ liefert einen Wert von 4,5. Aus dieser
Molekulardynamik-Simulation erhält man ein charakteristisches Verhältnis C100 von
8,4. Allgemein ist aus den Abbildungen 3.13 und 3.16 zu erkennen, daß die
Fluktuationen der Polynorbornenkette im kondensierten Zustand wesentlich gedämpfter
sind als bei der Molekulardynamik-Simulation der Einzelkette im Vakuum.
Führt man dieselbe Molekulardynamik-Simulationen im kondensierten Zustand bei 600
K durch, findet man die in Abbildung 3.17 dargestellte zeitliche Entwicklung des
Kettenendenabstandes RE und des Gyrationsradius RG.

Kettenkonformation starrer Knäuel 53
100 120 140 160 180 200 220 240 260 280 30020
30
40
50
60
70
80
90
100
Gyrationsradius
Kettenendenabstand
Abs
tand
[ Å
]
Simulationszeit [ps]
Abb. 3.17: Molekulardynamik-Simulation Polynorbornen im kondensierten Zustand (600 K)
Die Auswertung der Molekulardynamik-Simulation in Abbildung 3.17 liefert ein
charakteristische Verhältnis C100 von 4,1. Die Korrelation von ⟨RE2⟩ / ⟨RG
2⟩ liefert einen
Wert von 3,9. Die Polymerkette zeigt infolge der höheren Temperatur im kondensierten
Zustand wesentlich stärkere Fluktuationen als bei 300 K. Der Vergleich mit der
Molekulardynamik-Simulation der Polynorbornen-Einzelkette bei 600 K im Vakuum (s.
Abb. 3.14) zeigt deutlich geringere Fluktuationen des Kettenendenabstands RE und des
Gyrationsradius RG. Das charakteristische Verhältnis C100 besitzt mit 4,1
näherungsweise denselben Wert wie für die Einzelkette im Vakuum bei 600 K. Das
dimensionslose Verhältnis ⟨RE2⟩ / ⟨RG
2⟩ ist im Vergleich zwischen der isolierten Kette
und der Polymerkette in der Schmelze von 4,6 auf 3,9 abgesunken.

Kettenkonformation starrer Knäuel 54
3.4 Experimentelle Untersuchungen zur Kettenkonformation
Die Computersimulationen zur Kettenkonformation des Polynorbornens haben gezeigt,
daß sowohl Theta-Bedingungen (Simulation der Polymer-Schmelze) als auch ein
schlechtes Lösemittel (Simulation im Vakuum) bei unterschiedliche Temperaturen zu
ähnlichen Kettenkonformationen führen. Zur Überprüfung dieser Ergebnisse aus den
Computersimulationen zur Kettenkonformation wurde ein Vergleich mit
experimentellen Ergebnissen vorgenommen. Die Bestimmung der Kettendimensionen
erfolgte mittels Lichtstreu-Experimenten und viskosimetrischen Untersuchungen in sehr
verdünnter Lösung.
3.4.1 Kettenkonformation aus Lichtstreu-Experimenten
Durch Lichtstreu-Experimente lassen sich Strukturen bis zur Größenordnung der halben
Wellenlänge der verwendeten Strahlung bestimmen [90-92]. Die Bestimmung der
Kettendimension von Makromolekülen basiert auf der Winkelabhängigkeit der
Streustrahlung, die von den gelösten Makromolekülen hervorgerufen wird. Untersucht
man die Teilchengeometrie der Polynorbornene mit Hilfe von statischen und
dynamischen Lichtstreu-Experimenten, findet man die in Tabelle 3.4 aufgeführten
Verhältnisse von Gyrationsradius RG und hydrodynamischen Radius RH.
Tabelle 3.4:
Ergebnisse der Lichtstreu-Experimente von Polynorbornenen [93]
Molekulargewicht Mw [g/mol] RG [nm] Rh[nm] σ = RG/RH
65.400 12,1 7,2 1,68
255.000 24,0 16,3 1,47
351.000 25,3 16,9 1,50
433.000 27,5 18,7 1,47

Kettenkonformation starrer Knäuel 55
Aus dem Vergleich der gefundenen Werte für das Verhältnis von Gyrationsradius RG
und hydrodynamischen Radius RH mit denen für ideale Teilchengeometrien in Tabelle
3.5 ist zu erkennen, daß die Polynorbornene in Lösung als Gauß-Knäuel vorliegen.
Tabelle 3.5:
Verhältnis von Gyrationsradius RG und hydrodynamischen Radius RH für ideale
Teilchengeometrien
Teilchengeometrie σ = RG / RH
Kugel 0,75
Gauß-Knäuel 1,50
Stäbchen 2,00
3.4.2 Kettenkonformation aus viskosimetrischen Untersuchungen
Die Untersuchung der Kettengestalt mit Hilfe viskosimetrischer Messungen basiert auf
der Bestimmung der intrinsischen Viskosität [η] definiert durch [83,91]:
[ ] ( )η
η=
→−
lim
c crel
0
1 (3.1)
In Gleichung 3.1 bedeuten ηrel die relative Viskosität, d.h. das Verhältnis der Viskosität
der Lösung zu der des reinen Lösemittels, und c die Konzentration in Masse je
Volumeneinheit (g/dl). Für hinreichend lange Kettenmoleküle in stark verdünnten
Lösungen läßt sich diese Gleichung näherungsweise durch die Mark-Houwink-
Gleichung beschreiben.

Kettenkonformation starrer Knäuel 56
[ ]η α= ⋅K M v (3.2)
In Gleichung 3.2 steht α für den Mark-Houwink-Parameter. Die Ergebnisse der
viskosimetrischen Untersuchungen zur Bestimmung der Kettendimensionen in stark
verdünnten Lösungen von Polynorbornenen sind in Tabelle 3.6 zusammengefaßt:
Tabelle 3.6:
Ergebnisse der viskosimetrischen Untersuchungen an Polynorbornenen [93]
Polymer Lösemittel Konz. in g/dl αMH
Polynorbornen Chlorbenzol 5,97⋅10-4 0,560
Polynorbornen Cyclohexan 9,58⋅10-4 0,510
Der Vergleich mit theoretischen Mark-Houwink-Parametern idealer Teilchengeometrien
in Tabelle 3.7 zeigt, daß Polynorbornene in verdünnter Lösung als Gauß-Knäuel
vorliegen.
Tabelle 3.7:
Theoretische Mark-Houwink-Parameter idealer Teilchengeometrien
Teilchengeometrie αMN
Kugel 0
Gauß-Knäuel 0,5
Stäbchen 2,0

Kettenkonformation starrer Knäuel 57
Ein weiteres wichtiges Ergebnis aus den viskosimetrischen Untersuchungen ist die
Tatsache, daß die Kettengestalt der Polynorbornene offensichtlich unabhängig von der
Art des verwendeten Lösemittels ist. Bei den verwendeten Lösemitteln handelt es sich
um Verbindungen mit signifikant unterschiedlichen Löslichkeitsparametern, wie aus
Tabelle 3.8 zu erkennen ist.
Tabelle 3.8:
Löslichkeitsparameter der verwendeten Lösemittel [94]
Lösemittel Löslichkeitsparameter δ [(cal/cm3)0.5]
Chlorbenzol 9,5
Cyclohexan 8,2
Diese Ergebnisse zeigen, daß sich Polynorbornen als Prototyp starrer Knäuel auch in
Lösemitteln löst, deren Löslichkeitsparameter sich deutlich voneinander unterscheiden.
Normalerweise wäre zu erwarten, daß die Polymerkette in einem der beiden
verwendeten Lösemittel kollabiert bzw. expandiert. Die unterschiedlichen
Löslichkeitsparameter der verwendeten Lösemittel signalisieren deutlich verschiedene
Wechselwirkungen für die gelösten Polynorbornenketten. Durch diese Betrachtungen
wird deutlich, daß die Kettenkonformation durch die individuelle Struktur der
Polynorbornene nicht aber durch die Umgebung beeinflußt wird.

Kettenkonformation starrer Knäuel 58
3.5 Zusammenfassung Kapitel 3
Das Norbornan erfüllt die in der Einleitung geforderten Kriterien an das Monomer
hinsichtlich einer geeigneten Lage der Potentialminima im Rotationspotential und
hinreichend hoher Rotationsbarrieren. Die Untersuchung der Kettenkonformation der
Polynorbornene zeigt eine Reihe unerwarteter Ergebnisse.
Starre Knäuel bzw. das Polynorbornen besitzen aufgrund der hohen Potentialbarrieren
im zweidimensionalen Rotationspotential eine sehr rauhe n-dimensionale
Potentialenergie-Hyperfläche. Der dazugehörige Konfigurationsraum ist infolge der
hohen Rotationsbarrieren mit den Verfahren der Molekularmechanik nur schwer zu
durchmustern. Die Computersimulationen haben gezeigt, daß das Polynorbornen als
Prototyp der starren Knäuel gelten kann. Die Kettengestalt wird von der Temperatur und
der mikroskopischen Umgebung im Sinne von „guten“ und „schlechten“ Lösemitteln
kaum beeinflußt. Monte Carlo-Simulationen haben gezeigt, daß Polynorbornene im
Vakuum ein Knäuel bilden, aber im Gegensatz zu den flexiblen Ketten nicht kollabieren
können.
Experimentelle Untersuchungen zur Kettengestalt zeigen eine sehr gute
Übereinstimmung mit den Ergebnissen der Computersimulationen. Polynorbornene
besitzen in Lösemitteln mit deutlich unterschiedlichen Löslichkeitsparametern immer
dieselbe Gestalt eines Gauß-Knäuels: Die Kettengestalt der Polynorbornene ist
unabhängig von der Art des verwendeten Lösemittels.

Lokale Kettendynamik starrer Knäuel 59
4 Lokale Kettendynamik starrer Knäuel
Dieses Kapitel wird sich mit der Untersuchung der dynamischen Eigenschaften starrer
knäuelförmiger Polymere befassen. Im Vordergrund steht hierbei die lokale
Kettendynamik starrer Knäuel. Zu diesem Zweck werden Molekulardynamik-
Simulationen an Einzelketten im Vakuum und im kondensierten Zustand durchgeführt.
Das Ziel der Untersuchungen an ausgewählten Modellsystemen ist es, die
Temperaturabhängigkeit der lokalen Kettendynamik zu untersuchen und gegebenenfalls
Rückschlüsse auf makroskopische Eigenschaften zu ziehen. Weiterhin soll in diesem
Kapitel der Mechanismus der Glasumwandlung für starre Knäuel diskutiert werden.
4.1 Lokale Kettendynamik starrer Knäuel im Vakuum
Der kondensierte Zustand ist durch die Wechselwirkungen zwischen den einzelnen
Polymerketten charakterisiert. Die Molekulardynamik-Simulationen zur lokalen
Kettendynamik wurden im Vakuum durchgeführt, um eine schlechtes Lösemittel mit
einer niedrigen Viskosität zu simulieren [78]. Die Komplexität des Problems erfordert
die Auswahl eines geeigneten Modellsystems, daß einen sinnvollen Kompromiß
zwischen der Systemgröße und dem Rechenaufwand darstellt [95]. Eine wesentliche
Voraussetzung für Verwendbarkeit eines Modellsystems ist die Fähigkeit, die zu
untersuchende Eigenschaften exakt reproduzieren zu können. Das verwendete Modell
des Polynorbornens mit einem Polymerisationsgrad von 12 bietet eine Möglichkeit, die
lokale Kettendynamik unter adäquater Berücksichtigung der Nachbargruppeneffekte
hinreichend genau zu beschreiben.

Lokale Kettendynamik starrer Knäuel 60
Die Betrachtung der zentralen Torsion dieses Kettenfragments schließt, wie aus
Abbildung 4.1 hervorgeht, die Wechselwirkungen mit jeweils 6 benachbarten
Monomeren ein.
Abb. 4.1: Modellsystem für Molekulardynamik-Simulationen an 12er-Kette Polynorbornen
Durch die Beobachtung der Fluktuationen des mittleren Torsionswinkels mittels
Molekulardynamik-Simulationen ist es möglich, den Einfluß der komplexen n-
dimensionalen Potentialenergie-Hyperfläche des entsprechenden Modellsystems auf
eine bestimmte Torsion zu untersuchen. Aus der Halbwertsbreite dieser Torsionswinkel-
Fluktuationen läßt sich Breite der Potentialmulde abschätzen, die das dynamische
Verhalten der Torsion beeinflußt. Durch die Bestimmung der Halbwertsbreite der
Torsionswinkel-Fluktuationen als Funktion der Temperatur kann die
Aufweitungstendenz der Potentialmulden untersucht werden. Hierbei werden die
temperaturabhängigen Torsionswinkel-Fluktuationen gewissermaßen als Sonde für die
Exploration der Potentialenergie-Hyperfläche verwendet.
Die Simulationszeit der einzelnen Molekulardynamik-Simulationen für jede Temperatur
betrug 100 ps. Mögliche Partialladungen in den zu untersuchenden Modellsystemen
wurden mit Hilfe des Gasteiger-Marsili-Verfahrens [66] berücksichtigt.

Lokale Kettendynamik starrer Knäuel 61
4.1.1 Kettengestalt und Fluktuationen (12er-Kette)
Um die lokale Kettendynamik des Polynorbornens besser bewerten zu können, wurden
entsprechende Molekulardynamik-Simulationen an jeweils einem Modellsystem für
flexible Polymere, (Polyvinylchlorid, PVC) und für starre stäbchenförmige Polymere,
(Polyparaphenylen, PPP) durchgeführt.
4.1.1.1 Polyvinylchlorid (PVC)
Für die Molekulardynamik-Simulationen einer flexiblen Ketten wurde eine
Polyvinylchlorid-(PVC)-Einzelkette verwendet. Das untersuchte Modellsystem bestand
aus 12 isotaktisch verknüpften Vinylchlorid-Einheiten. Während der
Energieminimierung mittels des Conjugate Gradient-Algorithmus wurden die
Partialladungen alle 50 Rechenschritte neu berechnet. Um die dynamischen
Eigenschaften zwischen den einzelnen Monomeren zu untersuchen, wurde der
Torsionswinkel, der durch die beiden Chloratome einschließlich ihrer Kohlenstoffatome
aufgespannt wird, als Beobachtungsgröße gewählt.

Lokale Kettendynamik starrer Knäuel 62
-180 -150 -120 -90 -60 -30 0 30 60 90 120 150 1800
5
10
15
20
25
30
35
40
150K
50K
250K
300KHäu
figk
eit [
a.u.
]
Torsionswinkel φ [°]
Abb. 4.2: Torsionswinkelfluktuationen der 12er Einzelkette (PVC)
In Abbildung 4.2 ist die Häufigkeit eines Torsionswinkels bei einer bestimmten
Temperatur gegen den Torsionswinkel aufgetragen. Man erkennt, daß die virtuelle
Torsion zwischen zwei Chloratomen in benachbarten Monomeren bereits bei sehr tiefen
Temperaturen in einem Winkelbereich von 60° ungehindert drehbar ist. Ab 300 K ist
der gesamte Winkelbereich zu beobachten, die Polymerketten sind uneingeschränkt
beweglich. Die vollständige Rotation der beiden Chloratome um die virtuelle Torsion
sollte aus diesem Grund kaum behindert sein.
Zur Quantifizierung der Schwankungsbreite des Torsionswinkels wurden die
Torsionswinkel-Verteilungen um das Potentialminimum im Bereich von φ = −150°
durch Gauß-Funktionen angenähert. In Abbildung 4.3 wurde die Halbwertsbreite der
Gauß-Funktionen als Funktion der Temperatur aufgetragen.

Lokale Kettendynamik starrer Knäuel 63
50 100 150 200 2500
5
10
15
20
25
30
35
40
45
50
55
60
Hal
bwer
tsbr
eite
der
Gau
ß-Fu
nktio
n [°
]
Temperatur [K]
Abb. 4.3: Halbwertsbreiten der Gauß-Funktionen als Funktion der Temperatur (PVC)
Durch lineare Regression erhält man für die Region unterhalb von 300 K eine Steigung
von 0,0508°/K ± 0,0081°/K. Der Achsenabschnitt beträgt 13,02 ± 1,35. Auf die
Bedeutung der Steigung der Regressionsgerade und des Achsenabschnitts, der durch
lineare Regression erhalten wurde, wird an späterer Stelle eingegangen.
4.1.1.2 Polyparaphenylen (PPP)
Das verwendete Modellsystem aus 12 para-verknüpften Phenylen-Einheiten wurde
mittels des Conjugate Gradient-Algorithmus und unter Berücksichtigung von
Partialladungen mit Hilfe des Gasteiger-Schemas hinsichtlich der Energie minimiert.

Lokale Kettendynamik starrer Knäuel 64
-180 -150 -120 -90 -60 -30 0 30 60 90 120 150 1800
10
20
30
40
50
150K
100K
50KH
äufi
gkei
t [a.
u.]
Torsionswinkel φ [°]
Abb. 4.4: Torsionswinkelfluktuationen der 12er Einzelkette (PPP)
Die Ergebnisse der Molekulardynamik-Simulationen am Polyparaphenylen (PPP) in
Abbildung 4.4 zeigen, daß bei sehr niedrigen Temperaturen nur ein schmaler
Winkelbereich zu beobachten ist. Ab einer Temperatur von 150 K tritt ein zweites
diskretes Maximum auf. Die Rotationsbarriere unterhalb von 150 K wird durch eine
Kombination aus elektronischen Wechselwirkungen des π-Elektronensystems und den
sterischen Wechselwirkungen zwischen den ortho-ständigen Wasserstoffatomen der
Phenylringe verursacht.
Zur Beschreibung der dynamischen Eigenschaften wurde wiederum die
Schwankungsbreite der zentralen Torsion als Funktion der Temperatur aufgetragen. Die
Verteilung der Torsionswinkel in Abbildung 4.4 wurde durch eine Gauß-Funktionen
angenähert. Abbildung 4.5 zeigt die Auftragung der Halbwertsbreite als Funktion der
Temperatur.

Lokale Kettendynamik starrer Knäuel 65
50 100 150 2000
5
10
15
20
25
30
35
40
45
50
55
60
Hal
bwer
tsbr
eite
der
Gau
ß-Fu
nktio
n [°
]
Temperatur [K]
Abb. 4.5: Halbwertsbreiten der Gauß-Funktionen als Funktion der Temperatur (PPP)
Durch lineare Regression erhält man für Temperaturen unterhalb von 250 K eine
Steigung von 0,065°/K ± 0,01°/K. Der Achsenabschnitt beträgt 5,88 ± 1,52.
4.1.1.3 Polynorbornen (PN)
Zur Durchführung der Molekulardynamik-Simulation an Polynorbornen wurde ein
Modellsystem aus 12 cis-exo-verknüpften Norbornan-Einheiten verwendet.
In Abbildung 4.6 wurde die Häufigkeit eines Torsionswinkels bei verschiedenen
Temperaturen gegen den Torsionswinkel aufgetragen. Bei sehr tiefen Temperaturen, wie
etwa 50 K, ist nur ein sehr schmaler Winkelbereich zu beobachten. Bis 600 K tritt

Lokale Kettendynamik starrer Knäuel 66
lediglich eine Potentialverbreiterung auf. Erst oberhalb von 800 K wird ein zweiter
diskreter Torsionswinkel-Bereich besetzt. Aus diesem Diagramm erkennt man, daß für
dieses Modellsystem erst oberhalb 800 K Konformationsübergänge über
Potentialbarrieren hinweg möglich sind.
0 30 60 90 120 150 180 2100
50
100
150
200
250
300
350
800K
600K
300KHäu
figk
eit [
a.u.
]
Torsionswinkel φ [°]
Abb. 4.6: Torsionswinkelfluktuationen der 12er Einzelkette (PN)
Zur Quantifizierung der Schwankungsbreite bzw. Fluktuationsbreite wurden die
Torsionswinkel-Verteilungen durch Gauß-Funktionen genähert. In Abbildung 4.7 wurde
die Halbwertsbreite der Gauß-Funktionen als Funktion der Temperatur aufgetragen.

Lokale Kettendynamik starrer Knäuel 67
0 100 200 300 400 500 600 700 800 9000
5
10
15
20
25
30
35
40
45
50
55
60
Hal
bwer
tsbr
eite
der
Gau
ß-F
unkt
ion
(°)
Temperatur [K]
Abb. 4.7: Halbwertsbreiten als Funktion der Temperatur (PN, 12er Kette)
Lineare Regression liefert für das Polynorbornen eine Temperaturabhängigkeit der
Halbwertsbreite von 0,033/K ± 0,008°/K. Die Regressionsgerade besitzt einen
Achsenabschnitt von 2,43° ± 0,65°.
Das Auftreten des zweiten Maximums der Torsionswinkel-Population kann mit
Konformationsübergängen auf der n-dimensionalen Potentialenergie-Hyperfläche erklärt
werden. Interpretiert man diese Konformationsübergänge auf mikroskopischem Niveau
mit der makroskopischen Glasumwandlung, sollte für die Polynorbornen-Einzelkette
eine Glastemperatur oberhalb von 400°C bzw. 800 K zu erwarten sein. De Gennes hat
zur Beschreibung dieser Eigenschaft den Begriff der „Einzelketten-Glastemperatur“
geprägt [4]. Dieser Begriff basiert auf der Vorstellung, daß in einer rotationsbehinderten
Polymer-Einzelkette unterhalb dieser „Einzelketten-Glastemperatur“ keinerlei
Konformationsübergänge auftreten können. Unterhalb dieser Temperatur ist der

Lokale Kettendynamik starrer Knäuel 68
Rotationsfreiheitsgrad auf die Breite der Potentialsenke beschränkt. Die Konformation
der Polymerkette ist in den Potentialminima „ eingefroren“ . Die zunehmende
Beweglichkeit des einzelnen Kettenmoleküls bei steigender Temperatur ist in diesem
Temperaturbereich auf zunehmende Torsionswinkel-Fluktuationen um den Mittelwert
zurückzuführen.
Die lokale Kettendynamik des Polynorbornens wird durch das Auftreten hoher
Rotationsbarrieren charakterisiert, die die einzelnen Minima auf der n-dimensionalen
Energiehyperfläche voneinander trennen.
4.2 Kettendynamik starrer Knäuel im kondensierten Zustand
Der Vergleich der Ergebnisse von Molekulardynamik-Simulationen der Einzelkette im
Vakuum mit denen von Molekulardynamik-Simulationen im kondensierten Zustand,
sollte es ermöglichen, den Einfluß der Wechselwirkungen benachbarter Polymerketten
auf die lokale Kettendynamik abzuschätzen.
4.2.1 Lokale Kettendynamik der 12er-Kette im kondensierten Zustand
Umgibt man das in Abschnitt 4.1.1.3. verwendete Modellsystem aus 12 cis-exo-
verknüpften Norbornan-Einheiten in allen drei Raumrichtungen mit dessen identischen
Abbildern, kann die lokale Kettendynamik im kondensierten Zustand untersucht
werden. Die resultierende amorphe Zelle mit periodischen Randbedingungen und unter
Berücksichtigung der experimentellen Dichte wurde zunächst hinsichtlich der Energie
minimiert. Die Zellparameter waren variabel. Auf das Modellsystem wurde ein externer
Korrekturdruck angelegt. Die Startstruktur für die Molekulardynamik-Simulation wurde
durch wiederholtes Simulated Annealing und anschließendes Minimieren generiert. Die

Lokale Kettendynamik starrer Knäuel 69
Simulationszeit betrug für jede Temperatur 100 ps. Abbildung 4.8 zeigt, daß die
Temperaturabhängigkeit der Torsionswinkel-Fluktuationen im kondensierten Zustand
wesentlich geringer ist als dies im Fall der Einzelkette beobachtet wurde.
0 100 200 300 400 500 600 7000
5
10
15
20
25
30
35
40
45
50
55
60
Hal
bwer
tsbr
eite
der
Gau
ß-Fu
nktio
n [°
]
Temperatur [K]
Abb. 4.8: Halbwertsbreiten als Funktion der Temperatur (PN, kondensierte Phase)
Die Berechnung der Temperaturabhängigkeit der Halbwertsbreite durch lineare
Regression liefert einen Wert von 0,017°/K ± 0,0013°/K. Der Achsenabschnitt der
Regressionsgerade beträgt 5,1°± 0,03°. Der Vergleich mit dem entsprechenden Wert für
die Molekulardynamik-Simulation im Vakuum zeigt, daß die zusätzlichen
intermolekularen Wechselwirkungen im kondensierten Zustand zu einer weiteren
Behinderung der Kettendynamik führen und der Einfluß der Temperatur auf die
Potentialerweiterung um weitere 50 Prozent abnimmt. Die Steigung der
Regressionsgeraden für den kondensierten Zustand beträgt nur noch die Hälfte
derjenigen für die Einzelkette. Der Achsenabschnitt ist hingegen von 2,4° auf 5,1°
angewachsen.

Lokale Kettendynamik starrer Knäuel 70
4.2.2 Auswertung der Torsionswinkel-Fluktuationen
Bei den vorausgegangenen Molekulardynamik-Simulationen wurde die Verbreiterung
der Torsionswinkel um einen Mittelwert als Funktion der Temperatur untersucht. Zur
Quantifizierung der Torsionswinkel-Fluktuationen wurde die Halbwertsbreite σ einer
Gauß-förmigen Verteilung f(x) (Gleichung 4.1) verwendet, die in die
Häufigkeitsverteilung des Torsionswinkels gefittet wurde.
( )( )
f xA
e yx x
wc
= +−
σ π2
2
2
0 (4.1)
In Gleichung 4.1 bezeichnet A die Fläche unterhalb der Gauß-Funktion, σ die Weite der
Gauß-Funktion bei halber Höhe, x0 das Zentrum des Peaks und y0 den Offset von der
Basislinie. Die Beobachtungen waren, daß die Halbwertsbreite σ linear mit der
Temperatur ansteigt und daß σ bei einer Temperatur T = 0 K einen endlichen Wert
besitzt. Beide Beobachtungen sind überraschend.
Nach der statistischen Thermodynamik kann das Auftreten von Abweichungen x vom
Mittelwert durch Gleichung 4.2 beschrieben werden [96,97]:
( )p x ex
=
−1
2 2
1
22
2
2
πσσ (4.2)
Für die Mittelwerte von x gilt:
x = 0 (4.3)

Lokale Kettendynamik starrer Knäuel 71
x 2 2= σ (4.4)
Für das Schwankungsquadrat der Fluktuationen ergibt sich daraus:
x x2 2 2− = σ (4.5)
Nach der Fluktuationstheorie sollte das Schwankungsquadrat σ2 linear mit der
Temperatur T ansteigen. Die Temperaturabhängigkeit der Schwankungsbreite soll am
Beispiel der Freien Energie V untersucht werden. Der Potentialverlauf soll hierbei dem
eines harmonischen Oszillators entsprechen (s. Abbildung 3.8). Es gilt:
V V Dx= +021
2(4.6)
Für das Schwankungsquadrat folgt hieraus:
σ ∂∂
2 2 22
2
1
= − ∝
∝
−
x xV
xkT
kT
D(4.7)
Nach Gleichung 4.7 wäre zu erwarten, daß das Schwankungsquadrat σ2 linear mit der
Temperatur T ansteigt. Bei den Ergebnissen der Computersimulationen beobachtet man
aber, daß die Schwankungsbreite σ der Fluktuation linear mit der Temperatur T, das
Schwankungsquadrat σ2 mit T2 ansteigt.
Für den Potentialverlauf des harmonischen Oszillator bedeutet dies, daß die Krümmung
des Potentials mit zunehmender Temperatur (Abbildung 4.9) geringer wird.

Lokale Kettendynamik starrer Knäuel 72
-60 -40 -20 0 20 40 600
10
20
30
40
50
Ene
rgie
[a.
u.]
Potentialverlauf (niedrige Temperatur)
→
-60 -40 -20 0 20 40 600
5
10
15
20
25
30
35
40
45
50
Ene
rgie
[a.
u.]
Potentialverlauf (hohe Temperatur)
Abb. 4.9: Änderung der Potentialkrümmung mit zunehmender Temperatur
Das dynamische Verhalten wird nicht durch den Potentialverlauf des eindimensionalen
Rotationspotentials sondern durch die n-dimensionale Potentialenergie-Hyperfläche
charakterisiert. Diese komplexe Potentialenergie-Hyperfläche wird u.a. von allen im
Modellsystem vorhandenen Torsionsfreiheitsgraden aufgespannt. Die beobachtete
Temperaturabhängigkeit des Schwankungsquadrates ist demnach ein Maß für die
Aufweitungstendenz der Potentialmulde für den n-dimensionalen Fall, die für die
verschiedenen Polymere deutlich verschieden ist.
Der Vergleich der Steigungen der Regressionsgeraden von Polyvinylchlorid (PVC),
Polyparaphenylen (PPP) und Polynorbornen (PN) zeigt, daß die Aufweitungstendenz
der Potentialmulden beim Polynorbornen am geringsten ist.
Die Achsenabschnitte der Regressionsgeraden zeigen bei Temperaturen von T = 0 K
Schwankungsbreiten σ ungleich Null. Im quantenmechanischen Modell des
harmonischen Oszillators findet man eine sogenannte Nullpunkts-Energie. Man erwartet
für die Torsionswinkel-Fluktuationen endliche Werte bei einer Temperatur T von Null
Kelvin. Dies trifft für Systeme, die gemäß der klassischen Mechanik behandelt werden
nicht zu, es sei denn, das System befindet sich nicht im thermodynamischen
Gleichgewicht. Überträgt man diese Betrachtungen auf die Glasumwandlung amorpher
Polymere, so bedeutet dies, daß die Polymerkette in einem Nicht-Gleichgewichtszustand
eingefroren ist. Das System ist in einem kleinem Teilbereich des n-dimensionalen

Lokale Kettendynamik starrer Knäuel 73
Konfigurationsraums der unterkühlten Flüssigkeit hängengeblieben (Abbildung 4.10),
die Fluktuationen bleiben endlich [98-100].
0 100 200 300
200
400
600
800
1000
1200
1400
1600
1800
ideal(harm. Potential)
real
σ
Frei
e E
nerg
ie [
a. u
.]
n-dimensionale Konfigurationskoordinate
Abb. 4.10: Schematische Darstellung der Energiebarrieren im Nicht-Gleichgewichtszustand
Die gleichen Überlegungen gelten für Einzelketten bei tiefen Temperaturen. Die
unterschiedlichen Achsenabschnitte spiegeln die Breite der Potentiale wider.

Lokale Kettendynamik starrer Knäuel 74
4.2.3 Molekulardynamik-Simulationen zur Glasumwandlung
Die physikalischen Eigenschaften amorpher glasartiger Polymere lassen sich durch die
unterhalb der Glastemperatur eingefrorene Kettenkonformation charakterisieren. Die
Verglasung ist durch eine extrem lange Relaxationszeit relativ zum
Beobachtungszeitraum charakterisiert. Boyd, Rigby und Roe haben an einer Reihe
polymerer Modellsysteme gezeigt, daß sich die temperaturabhängigen volumetrischen
Eigenschaften amorpher Polymere durch Molekulardynamik-Simulationen (V-T-
Simulationen) erfolgreich untersuchen lassen [101-103]. Voraussetzung für
Molekulardynamik-Simulationen zur Glasumwandlung ist die Generierung eines NPT-
Ensembles, d.h. eines Modellsystems unter konstantem Druck, konstanter Teilchenzahl
und konstanter Temperatur. Kühlt man z.B. ein solches amorphes Modellsystem mit
periodischen Randbedingungen unterhalb dessen experimentellen Glastemperatur ab,
kann die spezifische Dichte des Modellsystems als Funktion der Temperatur beobachtet
werden. Die Glasumwandlung ist im resultierenden V-T-Diagramm an einem Knick zu
erkennen. Der Verlauf des thermischen Ausdehnungskoeffizienten α, der sich aus dem
V-T-Diagramm als Geradensteigung ergibt, zeigt im Bereich der Glasumwandlung
einen Sprung. Diese Simulationsmethode ist mit dem experimentellen Verfahren der
Dilatometrie, d.h. Bestimmung des Volumens als Funktion der Temperatur, zu
vergleichen.
Zur Temperierung der amorphen Zelle mit periodischen Randbedingungen wurde bei
der Simulationen der Berendsen-Thermostat verwendet. Das verwendete Modellsystem
für Polynorbornen besaß einen Polymerisationsgrad von 100. Entscheidend bei der
Durchführung dieser Molekulardynamk-Simulationen ist die Einhaltung der isobaren
Simulationsbedingungen, was durch die Anwendung eines externen Korrekturdrucks
realisiert wurde [59].

Lokale Kettendynamik starrer Knäuel 75
250 300 350 400 450 500 550 600 6500,85
0,90
0,95
1,00
1,05
1,10
1,15
spez
. Vol
umen
[cm
3 /g]
Temperatur [K]
Abb. 4.11: Temperaturabhängigkeit des spezifischen Volumens aus Molekulardynamik-Simulation
Die Simulationszeit für jede Temperatur in Abbildung 4.11 betrug 500 ps. Vor jeder
Simulation wurde das amorphe Modellsystem für 100 ps äquilibriert. Die Auswertung
der Molekulardynamik-Simulationen liefert im Bereich von rund 220°C eine
Diskontinuität der Temperaturabhängigkeit des spezifischen Volumens. Die
Auswertung durch lineare Regression oberhalb und unterhalb dieser Diskontinuität
liefert einen Schnittpunkt beider Regressionsgeraden bei 205°C. Der entsprechende
thermische Ausdehnungskoeffizient α, der aus der Steigung der Regressionsgeraden
ermittelt wurde, beträgt unterhalb der Glastemperatur rund 5,0⋅10-5 K-1, oberhalb rund
7,9⋅10-5K-1. Der experimentelle thermische Ausdehnungskoeffizient α von
Polyvinylchlorid (PVC) beträgt 7,0⋅10-5 K-1 unterhalb der Glastemperatur und 17,0⋅10-5
K-1 darüber [104], für Polystyrol (PS) findet man 17,0⋅10-5 K-1 unterhalb von TG und
51⋅10-5 K-1 darüber [105].

Lokale Kettendynamik starrer Knäuel 76
Der Vergleich des experimentellen thermischen Ausdehnungskoeffizienten α oberhalb
der Glastemperatur TG besitzt in der Regel den doppelten bis dreifachen Wert des
thermischen Ausdehnungskoeffizient α unterhalb von TG [106-109]. Derselbe Vergleich
der thermischen Ausdehnungskoeffizienten α für das Polynorbornen zeigt lediglich
einen Anstieg von 50%. Diese niedrige Änderung des thermischen
Ausdehnungskoeffizienten α könnte darauf zurückzuführen sein, daß die
Glasumwandlung nicht durch Konformationsübergänge hervorgerufen wird .
4.2.4 Experimentelle Bestimmung der Glasumwandlung
Die experimentelle Untersuchung der Glasumwandlung von Polynorbornen liefert mit
Hilfe differentialkalorimetrischer Methoden eine Glastemperatur von 210°C [82]. Die
Glasumwandlung ist durch eine sehr geringe Änderung der Wärmekapazität ∆Cp von
rund 0,1J/gK charakterisiert, die Erklärung ist analog zum Fall der
Ausdehnungskoeffizienten (s.o.).
80 100 120 140 160 180 200 220 24010
20
30
40
50
60
70
80
90
100
Str
euin
tens
ität
[a.
u.]
Temperatur [°C]
Abb. 4.12: Messung der Glastemperatur von Polynorbornen durch therm. Dichtefluktuationen (SAXS)

Lokale Kettendynamik starrer Knäuel 77
Die in Abbildung 4.12 dargestellte Bestimmung der Glastemperatur durch Messung der
thermischen Dichtefluktuationen im Röntgenkleinwinkel-Experiment (SAXS)
[100,110,111] liefert eine Glastemperatur von 205°C [82]. Die starke Zunahme der
Streuintensität im Bereich der Glastemperatur wird durch die steigende mikroskopische
Beweglichkeit der Polymerketten verursacht.
Die differentialkalorimetrische Bestimmung der Glastemperatur von Polynorbornen
durch Goddall et al. bei B.F. Goodrich lieferte hingegen einen Wert von 370°C [112].
Die Polymere bei B.F. Goodrich wurden durch Polymerisation mit Metallocen-
Katalysatoren dargestellt. Differentialkalorimetrische Untersuchungen von Kaminsky et
al. haben bei Polynorbornen, das mit Hilfe von Zirkonocenen dargestellt wurde,
ebenfalls sehr schwache Glasstufen bei rund 210°C ergeben [113].
4.3 Glasumwandlung starrer Knäuel
Die Glasumwandlung ist eine charakteristische Eigenschaft amorpher Materialien
[2,92]. Hierbei handelt es sich um einen kinetisch bedingten Übergang von der
Schmelze in einen unterkühlten, ungeordneten Zustand [98,114]. Die Glastemperatur ist
von der thermischen Vorgeschichte des Materials abhängig und somit kein
thermodynamischer Fixpunkt. Die eingeschränkte Beweglichkeit unterhalb der
Glastemperatur wird wesentlich durch die Rotationsfreiheitsgrade des Polymerrückgrats
beeinflußt [115]. Oberhalb der Glastemperatur sind Konformationsübergänge möglich.
Bei der Glasumwandlung tritt eine Veränderung in der Temperaturabhängigkeit der
Enthalpie auf, was sich z.B. mit Hilfe kalorimetrimetrischer Verfahren experimentell
beobachten läßt. Bezüglich der mechanischen Eigenschaften ist im Bereich der
Glasumwandlung eine starke Abnahme des Elastizitätsmoduls zu beobachten [116].

Lokale Kettendynamik starrer Knäuel 78
Die Ergebnisse der Molekulardynamik-Simulationen aus Kapitel 4.1.1.3 zeigen, daß im
Falle der Polynorbornen-Einzelkette im Vakuum ab einer Temperatur von 800 K ein
zweites diskretes Maximum des zentralen Torsionswinkels zu beobachten ist.
Interpretiert man dieses mikroskopische Phänomen mit der makroskopischen
Glasumwandlung, d.h. mit Konformationsübergängen über Potentialbarrieren hinweg,
so wäre für das Polynorbornen eine Glastemperatur oberhalb von 400°C zu erwarten.
Der Vergleich mit der experimentellen Untersuchung der Glasumwandlung des
Polynorbornens zeigt aber, daß die Glastemperatur im Bereich von 210°C beobachtet
wird. Der experimentelle Befund steht scheinbar im Widerspruch zu den Ergebnissen
aus den Molekulardynamik-Simulationen. Um den Mechanismus der Glasumwandlung
bei starren Knäuel zu verstehen, ist es notwendig, zunächst den Mechanismus der
Glasumwandlung für flexible Polymerketten zu diskutieren.
4.3.1 Mechanismus der Glasumwandlung bei flexiblen Polymeren
Der Glasübergang ist auf molekularer Ebene durch den Beginn der langreichweitigen
Bewegungen der Polymerketten bedingt durch Konformationsänderungen
gekennzeichnet [98,114]. Als Konsequenz der zunehmenden Beweglichkeit der
Polymerketten wächst das freie Volumen, d.h. der den Polymersegmenten zur
Verfügung stehende Raum, und damit letztendlich auch das spezifische Volumen am
Glasübergang charakteristisch an.
Die Glasumwandlung tritt bei flexiblen Polymeren durch Konformationsübergänge über
Potentialbarrieren hinweg auf. Abbildung 4.13 zeigt eine schematische Darstellung der
Glasumwandlung bei flexiblen Ketten.

Lokale Kettendynamik starrer Knäuel 79
0 30 60 90 120 150 180 210 240 270 300 330 3600
200
400
600
800
1000
1200
1400
rel.
Ene
rgie
[a.
u.]
rel. Torsionswinkel φ [°]
Abb. 4.13: Schematischer Glasübergang bei flexiblen Polymerketten
Aus Abbildung 4.13 geht hervor, daß die Glasumwandlung eines flexiblen Polymers auf
mikroskopischer Ebene im wesentlichen durch das Überwinden der Potentialbarrieren
hervorgerufen wird. Theoretische Untersuchungen zur Dynamik in Polymerketten [117-
119] haben zu dem Ergebnis geführt, daß Konformationsübergänge zu den wichtigsten
Torsionsbewegungen zu zählen sind. Experimentelle Untersuchungen zur Dynamik von
Polymeren mit Hilfe der Kernresonanzspektroskopie von Spiess et al. haben gezeigt,
daß Konformationsübergänge nicht isoliert, sondern stets, mehr oder weniger, gekoppelt
mit Librationsbewegungen auftreten [120,121].

Lokale Kettendynamik starrer Knäuel 80
4.3.2 Mechanismus der Glasumwandlung bei starren Knäueln
Vergleicht man die experimentell beobachtete Glastemperatur des Polynorbornens mit
den Ergebnissen der Molekulardynamik-Simulationen zur lokalen Kettendynamik, so
findet man erst bei Temperaturen im Bereich von etwa 800 K Konformationsübergänge.
Die Computersimulationen zur Untersuchung der Glasumwandlung zeigen eine
Glastemperatur im Bereich von 210°C. Als Konsequenz dieser Resultate scheiden
Konformationsübergänge als Ursache für die Glasumwandlung des Polynorbornens bei
dieser Temperatur offensichtlich aus.
Theoretische Arbeiten von Moro et al. haben gezeigt, daß die Glasumwandlung von
Polymeren nicht notwendigerweise mit dem Auftreten von Konformationsübergängen
verknüpft sein müssen [122-126]. Grundlage dieses Mechanismus ist nicht der streng
lokalisierte Konformationsübergang einer einzelnen Torsion sondern vielmehr das
Auftreten kooperativer Bewegungen von benachbarten Torsionen mit niedriger
Amplitude. Die Annahme basiert auf dem in Abbildung 4.14 dargestellten Modell der
starren Rotorkette, deren starre Monomere eine Behinderung für die Rotation um die
gemeinsame Bindung besitzen.
Abb. 4.14: Modell der starren Rotorkette

Lokale Kettendynamik starrer Knäuel 81
Für die Glasumwandlung starrer Knäuel bzw. des Polynorbornens ist aufgrund der
Barrierenhöhe demnach ein Mechanismus zu erwarten, der nicht auf
Konformationsübergängen basiert. Die Untersuchung der Potentialenergie-Hyperflächen
mit unterschiedlichen theoretischen Verfahren liefert sehr hohe Potentialwälle um die
Potentialminima. Es ist zu vermuten, daß die Glasumwandlung der starren Knäuel nach
dem Mechanismus der gekoppelten Librationen verläuft.
0 30 60 90 120 150 180 210 240 270 300 330 3600
200
400
600
800
1000
1200
1400
rel.
Ene
rgie
[a.
u.]
rel. Torsionswinkel φ [°]
Abb. 4.15: Schematische Glasumwandlung bei starren Knäuelmolekülen
Abbildung 4.15 zeigt eine schematische Darstellung der lokalen Kettendynamik starrer
Knäuel bei der Glasumwandlung. Der Pfeil in Abbildung 4.15 soll andeuten, daß die
Beweglichkeit der Polymerkette auf der mikroskopischen Skala auf einen relativ
schmalen Potentialbereich beschränkt ist.
Die Beweglichkeit der starren Knäuel auf der mesoskopischen Skala wird durch
gekoppelte Librationen innerhalb der Potentialmulden mehrerer benachbarter Torsionen

Lokale Kettendynamik starrer Knäuel 82
realisiert. Durch diesen Mechanismus sind auch die Beobachtungen aus
Molekulardynamik-Simulationen langer Polynorbornenketten zu erklären, bei denen
große Fluktuationen der Kettenendenabstände und Gyrationsradien auftreten, ohne daß
die Kette kollabiert oder diskrete Konformationsübergänge zu beobachten sind.
Logische Konsequenz ist, daß starre Knäuel, und damit auch das Polynorbornen, zwei
Glasumwandlungen zeigen könnten. Die erste, deutlich schwächere Glasumwandlung
würde bei niedrigen Temperaturen nach dem Mechanismus der gekoppelten Librationen
erfolgen. Eine zweite Glasumwandlung könnte nach dem „ konventionellen“
Mechanismus durch Konformationsübergänge über Potentialbarrieren hinweg bei
höheren Temperaturen erfolgen. Dieser Glasübergang sollte aufgrund des anderen
Mechanismus durch eine deutlich größere Änderung der Wärmekapazität und einer
größeren Änderung des thermischen Ausdehnungskoeffizienten α charakterisiert sein.
4.4 Makroskopische Beweglichkeit
Die Konformationsänderungen auf mikroskopischem Niveau im Bereich der
makroskopischen Glasumwandlung führen gleichzeitig zu einer Erweichung des
kondensierten Polymers. Verfolgt man diese makroskopische Erweichung mit Hilfe der
Thermisch-Mechanischen Analyse (TMA), d.h. dem Eindringen einer Metallspitze in
eine Polymerprobe, so zeigt sich in der Regel im Bereich der Glasumwandlung ein
deutliches Eindringen der Metallspitze.

Lokale Kettendynamik starrer Knäuel 83
40 60 80 100 120 140 160 1800,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
Polycarbonat (PC)
Polystyrol (PS)
Pen
etra
tion
[mm
]
Temperatur [°C]
Abb. 4.16: Thermisch-Mechanische Analyse von Polystyrol und Polycarbonat (TMA)
Die Polycarbonat-Probe wie auch die Polystyrol-Probe zeigen im Temperaturbereich
unterhalb deren Glasumwandlungs-Temperaturen von 150°C bzw. 105°C eine deutliche
Erweichung. In Abbildung 4.16 ist dies an der deutlichen Penetration zu erkennen. Die
beobachtete Erweichung wird durch Konformationsübergänge verursacht und entspricht
somit dem bekannten Mechanismus der Glasumwandlung.
Vergleicht man das thermisch-mechanische Verhalten des Polynorbornens mit dem von
Polycarbonat (PC) und Polystyrol (PS) aus Abbildung 4.16, so zeigen sich deutliche
Unterschiede:

Lokale Kettendynamik starrer Knäuel 84
140 160 180 200 220 240 260 280 300 320 340 360 380 4000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
1,1
1,2
1,3P
enet
rati
on [
mm
]
Temperatur [°C]
Abb. 4.17: Thermisch-Mechanische Analyse von Polynorbornen (TMA)
Abbildung 4.17 zeigt das Ergebnis der Thermisch-Mechanischen Analyse an einer
Polynorbornen-Filmprobe. Man erkennt, daß im Bereich der kalorimetrischen Glasstufe
bei 210°C lediglich ein sehr geringes Eindringen der Wolfram-Nadel in die Probe zu
beobachten ist. Ein deutliche makroskopische Erweichung ist hingegen erst bei einer
Temperatur oberhalb von 330°C zu beobachten, was vermutlich durch den Abbau der
Probe verursacht wird.
Dieses experimentelle Ergebnis bietet einen weiteren Hinweis darauf, daß die
Glasumwandlung starrer Knäuel nach einem anderem Mechanismus verlaufen könnte.
Es ist zu erwarten, daß das Polynorbornen, wie bereits erwähnt, bei höheren
Temperaturen eine weitere Glasumwandlung bzw. Relaxation zeigt, die durch
Konformationsübergänge verursacht wird.

Lokale Kettendynamik starrer Knäuel 85
4.5 Zusammenfassung Kapitel 4
Die Molekulardynamik-Simulationen zur lokalen Kettendynamik zeigen bei allen
untersuchten Modellsystemen, daß die Fluktuationen der Torsionswinkel bei tiefen
Temperaturen auf einen relativ schmalen Bereich beschränkt sind. Die einzelnen
Polymerketten unterscheiden sich deutlich hinsichtlich der Schwankungen der
Torsionswinkelfluktuationen und in der Temperatur, bei der das Auftreten eines zweiten
diskreten Maximums der Torsionswinkelpopulation beobachtet wird. Das
Schwankungsquadrat der Torsionswinkelfluktuationen ist ein Maß für die Breite des
Potentials auf der n-dimensionalen Energie-Hyperfläche, in der sich die betrachtete
Bindung befindet. Das Auftreten eines zweiten besetzten, diskreten Maximums erlaubt
unter Berücksichtigung der Temperatur, bei der der Übergang auftritt, eine Abschätzung
der Energie, die notwendig ist, um von einem Minimum auf der n-dimensionalen
Energie-Hyperfläche in ein anderes Minimum zu gelangen. Polynorbornen besitzt im
Vergleich zum Polyparaphenylen und zum Polyvinylchlorid die niedrigste
Temperaturabhängigkeit der Halbwertsbreite und die niedrigste Aufweitungstendenz der
Potentialmulde.
Der in diesem Kapitel vorgestellte Mechanismus der gekoppelten Librationen für die
Glasumwandlung bei starren Knäuel bietet eine Möglichkeit, die, auf den ersten Blick,
widersprüchlichen Befunde bezüglich der experimentellen Glastemperaturen in
Einklang zu bringen. Die Ergebnisse der experimentellen Untersuchungen zur
Glasumwandlung der Polynorbornene und der Molekulardynamik-Simulationen zur
lokalen Kettendynamik läßt die Spekulation über eine zweite Relaxation bzw. eine
zweite Glasumwandlung zu.
Sollten starre Knäuel wie z.B. das Polynorbornen tatsächlich zwei Glasumwandlungen
zeigen, ist zu vermuten, daß sich die sehr schwache, erste Glasstufe aufgrund ihrer
geringen Änderung der Wärmekapazität ∆Cp nur äußerst schwer identifizieren läßt. Die
zweite Glasstufe sollte aufgrund ihres Mechanismus und der damit verbundenen,
besseren Dissipation der thermischen Energie mit einer wesentlich höheren Änderung
der Wärmekapazität einhergehen.

Der kondensierte Zustand starrer Knäuel 86
5 Der kondensierte Zustand starrer Knäuel
Das folgende Kapitel wird sich mit der Aufklärung von Struktur-Eigenschafts-
Beziehungen mit Hilfe von Experimenten und Computersimulationen beschäftigen.
Ausgewählte makroskopische Eigenschaften des Polynorbornens, die mit der
Kettenkonformation, den dynamischen Eigenschaften und den verschiedenen
Wechselwirkungen verknüpft sind, sollen untersucht werden. Hierzu gehören die
statischen strukturellen Eigenschaften des kondensierten Zustandes, die Untersuchung
des Hildebrandschen Löslichkeitsparameters, die Oberflächenenergie und die
Permeationseigenschaften des Polynorbornens sowie eine Abschätzung der
mechanischen Eigenschaften. Ein Ziel ist der Vergleich der Eigenschaften des
Polynorbornens mit anderen flexiblen Polymeren.
5.1 Statische Eigenschaften im kondensierten Zustand
Die starre Kettenkonformation des Polynorbornens führt in Kombination mit der hohen
Dichte zu einer besonderen Anordnung der Ketten im kondensierten Zustand. Frühere
Untersuchungen zur Korrelation von molarem Volumen und Van-der-Waals-Volumen
haben gezeigt, daß das Polynorbornen für dieses Verhältnis einen Wert von 1,43 besitzt
[89]. Dieser Wert entspricht typischerweise kristallinen Polymeren [70]. Polynorbornen
weist demnach also eine ungewöhnlich dichte Packung der Polymerketten im
kondensierten Zustand auf. Zur Untersuchungen der strukturellen Eigenschaften des
unsubstituierten Polynorbornens wurden Röntgenstreu-Experimente durchgeführt, um
zu überprüfen, ob diese dichte Packung zu molekularen Überstrukturen führt.

Der kondensierte Zustand starrer Knäuel 87
Mit Hilfe von Röntgenweitwinkel-(WAXS) und Röntgenkleinwinkel-Streuung (SAXS)
an ungetemperten und getemperten Pulverproben bzw. an Filmen wurden die statischen,
strukturellen Eigenschaften des Polynorbornens charakterisiert.
5.1.1 Strukturelle Eigenschaften im kondensierten Zustand
5.1.1.1 Röntgenweitwinkelstreuung (WAXS)
Die Strukturanalyse durch Röntgenweitwinkelstreuung ergab für das pulverförmige
Polynorbornen vollständig amorphe Beugungsdiagramme. Auffällig für die erhaltenen
Diffraktogramme ist das Auftreten zweier amorpher Halos. Abbildung 5.1 zeigt eine
Auswahl von Streukurven für Polynorbornene, die typischerweise zwei breite amorphe
Halos mit Intensitätsmaxima im Bereich von 2Θ = 10° und 2Θ = 18° besitzen. Diese
Maxima lassen sich mit Hilfe der, für die Streuung an amorphen Festkörpern,
modifizierten Bragg-Beziehung in Gleichung 5.1. intramolekularen Abständen von 6 Å
bzw. 10 Å zuordnen.
r ≈ ⋅5
4 2
λsin Θ
(5.1)
In Gleichung 5.1 bezeichnet λ die Wellenlänge der verwendeten Röntgenstrahlung und
Θ den Streuwinkel.

Der kondensierte Zustand starrer Knäuel 88
5 10 15 20 25 300
10
20
30
40
50
60
70
80
Polynorbornen (Mw: 10500g/mol)
Polynorbornen (Mw: 4500g/mol)
Polynorbornen (Mw: 37000g/mol)
Stre
uint
ensi
tät [
a.u.
]
Streuwinkel 2Θ [°]
Abb. 5.1: Pulver-Diffraktogramme von Polynorbornenen (WAXS)
Durch längeres Tempern im Bereich unterhalb der Glastemperatur konnten bei
pulverförmigen Polynorbornenproben keine Veränderungen des Ordnungszustands
erzielt werden. Das Auftreten zweier amorpher Halos wird auch bei anderen Polymeren
wie z.B. Poly-α-methylstyrol [127,128] beobachtet. Die beiden amorphen Halos im
Röntgenweitwinkel-Streudiagramm in Abbildung 5.1 lassen sich durch den Vergleich
mit Ergebnissen aus Computersimulationen intramolekularem Ursprung zuordnen
[129,130].
Die Struktur eines Festkörpers läßt sich auf mikroskopischer Ebene mit Hilfe einer
radialen Verteilungsfunktionen g r( )r
beschreiben [2,7,11]. Diese Verteilungsfunktion
beschreibt die Wahrscheinlichkeit ein Teilchen bzw. ein Atom an einer Position r
r zu
finden. Die Verteilungsfunktion liefert die Summe alle interatomaren Abstände einer
Struktur (Gleichung 5.2)

Der kondensierte Zustand starrer Knäuel 89
( ) ( )g r r r i
i
Nr r r= −
=∑δ 1
1
( ) (5.2)
In Gleichung 5.2 bezeichnet N die Anzahl, r
r i( ) die Positionen der Teilchen und δ die
atomaren Abstände. Dementsprechend liefert die atomare radiale Verteilungsfunktion
Informationen über alle im System auftretenden interatomaren Abstände. Bei der
Berechnung der elektronischen radialen Verteilungsfunktion erfolgt noch eine
zusätzliche Gewichtung der Abstände mit der Elektronenzahldichte der jeweiligen
Atome. Radiale Verteilungsfunktionen sind experimentell sowohl über Röntgenstreu-
Experimente und Neutronenstreuung zugänglich [131,132].
Zur Untersuchung des Ursprungs der Streudiagramms des Röntgendiffraktogramms
wurde die elektronische radiale Verteilungsfunktion (RDF) für ein periodisches
Modellsystem des Polynorbornens berechnet und mit der experimentellen Streukurve
verglichen.
4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 1930
40
50
60
70
80
90
100
Häu
figk
eit [
a.u]
Abstand [Å]
Abb. 5.2: Elektronische radiale Verteilungsfunktion (RDF) für Polynorbornen aus Simulation

Der kondensierte Zustand starrer Knäuel 90
Abbildung 5.2 zeigt die aus der Molekularmechanik-Simulation erhaltene elektronische
radiale Verteilungsfunktion für das Polynorbornen. Diese radiale Verteilungsfunktion
liefert die Verteilung der inter- und intramolekuaren Abstände im System. Die beiden
stärksten lokalen Maxima der radialen Verteilungsfunktion liefern Abstände im Bereich
von rund 6 Å und 10 Å. Durch den Vergleich der elektronischen radialen
Verteilungsfunktionen für eine Einzelkette und für den kondensierten Zustand konnten
die Maxima intramolekularen Ursprungs zugewiesen werden. Im Bereich unterhalb von
2 Å findet man in der Verteilungsfunktion die atomaren Bindungslängen im
Modellsystem. Die enge Packung der Polynorbornene führt also nicht zu einer
besonderen Überstruktur, die sich in der Paarkorrelationsfunktion zeigt. Überstrukturen
auf größeren Skalen sollten sich in der Röntgenkleinwinkel-Streuung (Partikelstreuung)
zeigen.
5.1.1.2 Röntgenkleinwinkelstreuung (SAXS)
Mit Hilfe der Röntgenkleinwinkelstreuung wurde ein aus Lösung präparierter
Polynorbornenfilm auf das Vorliegen von molekularen Überstrukturen untersucht. Die
Dicke des verwendeten Polynorbornenfilmes, der aus 1,2-Dichlorbenzol präpariert
wurde, betrug 1 mm.
Das Streudiagramm in Abbildung 5.3 liefert keine Hinweise auf eine supermolekulare
Ordnung im Polynorbornenfilm.

Der kondensierte Zustand starrer Knäuel 91
0 1 2 3 4 5 60
1000
2000
3000
4000
5000
6000
7000
8000
9000
10000
Stre
uint
ensi
tät [
a.u.
]
Streuvektor S [Å-1]
Abb. 5.3: Röntgenkleinwinkel-Diffraktogramm eines Polynorbornenfilms (SAXS)
Der Anstieg der Streuintensität bei sehr kleinem Streuwinkel ist auf den Abfall der
Intensität des Primärstrahls zurückzuführen und der Anstieg der Streuintensität bei
größerem Streuwinkel auf den ersten amorphen Halo im Diffraktogramm des
Röntgenweitwinkel-Experiments (Abbildung 5.1).
5.2 Thermodynamische Eigenschaften im kondensierten Zustand
Zur Charakterisierung und Einordnung der thermodynamischen Eigenschaften im
kondensierten Zustand soll das Polynorbornen mit einem rein aliphatischen Polymer,
wie dem Polyethylen, und einem aromatischen Polymer, wie dem Polystyrol, verglichen
werden. Zu diesem Zweck werden thermodynamische Kenngrößen wie der
Hildebrandsche Löslichkeitsparameter δ und die Oberflächenenergie σ charakterisiert.
Der Hildebrandsche Löslichkeitsparameterδ wird mit Hilfe von Computersimulationen

Der kondensierte Zustand starrer Knäuel 92
bestimmt werden während die Charakterisierung der Oberflächenenergie σ durch
experimentelle Untersuchung erfolgt.
5.2.1 Hildebrandscher Löslichkeitsparameter
Der Lösungsprozeß eines amorphen Polymers in einem Lösemittel wird durch eine
entsprechende Änderung der Freien Mischungsenergie ∆Gmix charakterisiert
[92,96,133]:
∆ ∆ ∆G H T Smix mix mix= − (5.3)
In Gleichung 5.3 ist ∆Gmix die Änderung der Freien Mischungsenergie, ∆Hmix die
Änderung der Mischungsenthalpie und ∆Smix die Änderung der Mischungsentropie bei
der Mischung der Komponenten bei der absoluten Temperatur T. Tritt eine spontane
Lösung der beiden Komponenten auf, besitzt die Freie Mischungsenergie ∆Gmix einen
negativen Wert. Da das Lösen von hochmolekularen Polymerketten häufig nur mit
einem geringen Anstieg der Entropie verbunden oder ∆Smix gar negativ ist, spielt der
enthalpische Anteil den entscheidenden Faktor für das Vorzeichen der Freien
Mischungsenergie ∆Gmix. Zur Beschreibung der Mischungsenthalpie ∆Hmix werden u.a.
Löslichkeitsparameter verwendet. Die bei der Mischung eines binären Systems
auftretende Mischungsenthalpie ∆Hmix kann im Rahmen der Hildebrand-Scott-Theorie
durch die folgende Beziehung behandelt werden [134]:
( )∆H Vmix mix= −δ δ φ φ1 2
2
1 2 (5.4)

Der kondensierte Zustand starrer Knäuel 93
In Gleichung 5.4 steht Vmix für das Volumen der Mischung und φi ist der Molenbruch
der i-ten Komponente. Der Hildebrandsche Löslichkeitsparameter δ einer Komponente
bzw. eines Polymers gibt darüber Auskunft, in welchen Lösemittel sich diese
Komponente löst.
Zur Bestimmung des Hildebrandschen Löslichkeitsparameters δ ist es erforderlich, die
Kohäsivenergiedichte ECoh zu berechnen. Anschaulich repräsentiert die
Kohäsivenergiedichte diejenige Energie, die aufzuwenden ist, um eine Polymerkette von
dessen nächsten Nachbarn zu entfernen (Gleichung 5.5).
E U UCoh ra gesamt= −int (5.5)
Mit experimentellen Verfahren kann der Löslichkeitsparameter δ über die
Verdampfungsenthalpie erhalten werden. Polymere besitzen jedoch nur eine geringe
Flüchtigkeit und bauen bereits vor Erreichen der Verdampfungstemperatur ab.
Die Löslichkeitsparameter von Polymeren lassen sich näherungsweise entweder über
Inkrement-Verfahren [70] oder aus geeigneten Molekulardynamik-Simulationen
[129,130] durch Anwendung der Gleichung 5.5 und 5.6 erhalten.
δ =E
VCoh
mol
(5.6)
Aus Molekulardynamik-Simulationen kann mit Hilfe von Gleichung 5.5 die
Kohäsivenergiedichte als Differenz der Gesamtenergie von der amorphen Struktur und
der freien Kette berechnet werden. Für die Berechnung der Hildebrandschen
Löslichkeitsparameter δ der einzelnen Polymere wurden ausreichend energieminimierte
Modellsysteme mit periodischen Randbedingungen unter Verwendung der
experimentellen Dichte verwendet. Die isolierte Einzelkette konnte durch das
Weglassen der periodischen Randbedingungen direkt aus der Elternkette der amorphen
Zelle generiert werden. Durch dieses Verfahren wird gewährleistet, daß sich die

Der kondensierte Zustand starrer Knäuel 94
Kohäsivenergiedichte ECoh ausschließlich aus den nichtbindenden Wechselwirkungen
zusammensetzt.
Die Ergebnisse der Molekulardynamik-Simulationen, für die Polymerketten bestehend
aus jeweils 50 cis-exo-verknüpften Norbornan-Einheiten als Parent-Chain verwendet
wurden, sind in Tabelle 5.1 zusammengestellt. Die angegebenen Hildebrandschen
Löslichkeitsparameter δ wurden über die Kohäsivenergiedichte ECoh berechnet.
Tabelle 5.1:
Kohäsionsenergiedichten ECoh und Hildebrandsche Löslichkeitsparameter δ für
Polynorbornene aus Molekulardynamik-Simulationen
Meßgröße Zelle 1 Zelle 2 Zelle 3
Dichte [g/cm3] 1,092 1,091 1,091
Vmol [cm3] 4313,07 4317,02 4305,18
ECoh [kcal/mol] 114,36 127,67 119,79
δ [cal/cm3]1/2 5,15 5,43 5,28
Aus den Molekulardynamik-Simulationen wurde für drei amorphen Zellen des
Polynorbornens ein relativ niedriger, mittlerer Löslichkeitsparameter δ von rund 5,3
(cal/cm3)1/2 erhalten. Die statistischen Schwankungen betragen rund drei Prozent.
Der Vergleich mit den Löslichkeitsparametern der verwendeten Lösemittel für das
Polynorbornen in Tabelle 3.8 zeigt jedoch große Unterschiede. Die Differenz der
Hildebrandschen Löslichkeitsparameter δ zwischen Polynorbornen und dem
Chlorbenzol beträgt 4,2 (cal/cm3)1/2 sowie 4,0 (cal/cm3)1/2 für den Vergleich mit
Cyclohexan [94]. Die Berechnung des Hildebrandschen Löslichkeitsparameters δ mit
Hilfe von Computersimulationen versagt offenbar beim Polynorbornen. Die Werte sind
zu klein. Im Vergleich dazu besitzt Polyethylen (PE) (berechnet mit

Der kondensierte Zustand starrer Knäuel 95
Inkrementverfahren) einen Hildebrandschen Löslichkeitsparameter von 8,4 (cal/cm3)1/2
und Polystyrol 9,4 (cal/cm3)1/2 [70].
In der Literatur findet man zwischen den berechneten Hildebrandschen
Löslichkeitsparametern und den experimentell beobachteten Löslichkeitsparametern
Abweichungen in der Größenordnung von rund 20% [84].
Für das Polynorbornen scheint jedes Lösemittel ein Theta-Lösemittel zu sein, da das
Lösemittel keinen Einfluß auf die starre knäuelförmige Kettenkonformation des
Polynorbornens ausüben kann.
5.2.1.1 Oberflächenspannung
Die Freie Oberflächenenthalpie ∆G ist diejenige reversible Arbeit, die bei konstantem
Volumen und Temperatur aufgewandt werden muß, um die Oberfläche einer Flüssigkeit
zu vergrößern. Die Änderung der Freien Oberflächenenthalpie ∆G ist demnach eine
Funktion der Temperatur T und des äußeren Druckes P [91,96]:
Freie Enthalpie ( )G f T P= , (5.5)
Experimentell ist die Freie Oberflächenenthalpie ∆G durch die Messung eines
Kontaktwinkels Θ zugänglich, den eine Flüssigkeit mit einer Oberfläche bildet. Im Falle
eines Kontaktwinkels von Θ = 0° spricht man von vollständiger Benetzung, bei einem
Kontaktwinkel Θ ≥ 90° von Nicht-Benetzung.
Die Grundlage der folgenden Bestimmung der Freie Oberflächenenthalpie mit Hilfe des
Kontaktwinkels stellt die Gleichung von Young [136] dar:
σ σ σ θSV SL LV= + ⋅ cos (5.6)

Der kondensierte Zustand starrer Knäuel 96
In Gleichung 5.6 steht σSV für die Grenzflächenspannung zwischen dem Festkörper und
der Gasphase, σLV für die Grenzflächenspannung zwischen der flüssigen Phase und der
Gasphase und σSL für die Grenzflächenspannung zwischen der flüssigen Phase und dem
Festkörper.
Nach Owens, Wendt, Rabel und Kaelble kann die Grenzflächenspannung als Summe aus
dispersen und polaren Anteilen formuliert werden (Gleichung 5.7):
σ σ σ= +dispers polar (5.7)
Die Kombination beider Ausdrücke liefert die Owens-Wendt/Young Gleichung (5.8)
[137]:
( )σ θ
σσ σ σ
σL
ld s
dsp l
p
ld
1
2
+= +
cos(5.8)
Die Messung der Kontaktwinkel wurde an mehreren Polymerfilmen durchgeführt, die
mittels Spincoating präpariert wurden. Als Flüssigkeiten wurden Wasser und
Methyleniodid verwendet [138]. Die Ergebnisse dieser Messungen befinden sich in
Tabelle 5.3.
Tabelle 5.3:
Experimentelle Bestimmung des Kontaktwinkels [139]
Substanz Kontaktwinkel Θ
Wasser Methyleniodid
Polynorbornen 94 ± 2° 37 ± 2°

Der kondensierte Zustand starrer Knäuel 97
Mit Hilfe der Literaturwerte für die verwendeten Testflüssigkeiten läßt sich nach der
Methode von Owens und Wendt die Oberflächenspannung von Polynorbornen
berechnen. Zum Vergleich wurden die Oberflächenspannungen für Polystyrol und
Polyethylen (LDPE) [137] in Tabelle 5.4 angegeben.
Tabelle 5.4:
Oberflächenspannungen für Polynorbornen, Polystyrol und Polyethylen (LDPE)
Probe σ [mN/m] σdispers [mN/m] σpolar [mN/m]
Polynorbornen 40,2 39,8 0,4
Polystyrol 42,0 41,4 0,6
LDPE 33,1 32,0 1,1
In Tabelle 5.4 ist zu erkennen, daß das Polynorbornen eine Oberflächenspannung von
40,2 Nm/m besitzt. Dieser Wert ist näherungsweise mit dem des Polystyrols mit 42,0
Nm/m vergleichbar. Überraschend ist jedoch der Vergleich mit der
Oberflächenspannung von Polyethylen (LDPE) mit 33,1 Nm/m, da es sich sowohl beim
Polynorbornen als auch beim Polyethylen um rein aliphatische Systeme handelt.

Der kondensierte Zustand starrer Knäuel 98
5.3 Mechanische Eigenschaften
5.3.1 Computersimulationen
Die mechanische Eigenschaften von Polymeren sind für technische Anwendungen von
großem Interesse [116,135]. Die makroskopischen Eigenschaften von
Polynorbornenfilmen sind aufgrund deren Sprödheit experimentell schwer zugänglich.
Die Berechnung der elastischen Konstanten mit Hilfe von Computersimulationen ist
hingegen recht einfach zu realisieren [140].
Die elastischen Konstanten eines Festkörpers sind bei konstanter Temperatur durch die
Bildung der zweiten Ableitung der Freien Energie A nach den Komponenten eines
Verzerrungstensors ελ zu erhalten.
cV
AT
T
γλγ λ
∂∂ε ∂ε
=
1 2
(5.9)
Dieser Tensor vierter Ordnung kann aufgrund der Voigt-Symmetrie-Beziehung [141] in
der Form einer 6×6-Matrix geschrieben werden. Diese 6×6-Matrix cT der elastischen
Konstanten, die als Steifigkeitsmatrix bezeichnet wird, erlaubt die Berechnung aller
bekannten Materialkonstanten. Für ein isotropes Material, wie z.B. amorphe glasartige
Polymere, besitzt der Tensor der isothermen Elastizitätskoeffizienten die folgende
Form:
C =
++
+
2 0 0 0
2 0 0 0
2 0 0 0
0 0 0 0 0
0 0 0 0 0
0 0 0 0 0
µ λ λ λλ µ λ λλ λ µ λ
µµ
µ
(5.10)

Der kondensierte Zustand starrer Knäuel 99
Im Tensor der isothermen elastischen Konstanten (Gleichung 5.10) sind λ und µ die
Lamé-Konstanten. Der Zug-Modul (Youngs Modul) E (Gleichung 5.11), der
Schermodul G (Gleichung 5.12), der Bulk-Modul B (Gleichung 5.13) und das Poisson-
Verhältnis ν (Gleichung 5.14) stehen mit λ und µ in dem folgenden Verhältnis:
E = ++
µ λ µλ µ
3 2(5.11)
G = µ (5.12)
B = +λ µ2
3(5.13) ( )υ λ
λ µ=
+2(5.14)
Im einfachsten Fall kann die Berechnung direkt nach dem Hookschen Gesetz erfolgen.
Dies setzt allerdings voraus, daß sich das entsprechende Modellsystem mit periodischen
Randbedingungen im thermodynamischen Gleichgewicht befindet. Dieser Zustand
wurde mit Hilfe des Simulated Annealing-Verfahrens und anschließender Minimierung
hinischtlich der Konformationsenergie erzeugt. Mit Hilfe dieses Vorgehens wurde ein
sehr großer Konfigurationsraum durchmustert und eine realistische Startstruktur
generiert. Durch dieses Molekularmechanik-Verfahren erhält man die entsprechenden
Eigenschaften bei einer Temperatur von 0 K.
Zur Berechnung der mechanischen Eigenschaften wurde die energieminimierte amorphe
Zelle mit periodischen Randbedingungen durch kleine Auslenkungen deformiert. Für
die verwendete amorphe Zelle wurde eine kubische Elementarzelle verwendet. Die
Parent-Chain des Modellsystems besaß einen Polymerisationsgrad von 100. Die
Struktur wurde unter konstanten Gitterparametern erneut minimiert. Die resultierende
Spannung dieser minimierten Struktur wird zur Berechnung der Steifigkeitsmatrix
herangezogen [140].

Der kondensierte Zustand starrer Knäuel 100
Die Berechnung der Steifigkeitsmatrix des Polynorbornens erfolgte durch Mittelung der
Ergebnisse von vier Simulationen an derselben amorphen Zelle. Gleichung (5.15) zeigt
die gemittelte Steifigkeitsmatrix des Polynorbornens aus den Simulationen:
[ ]C GPa=
−−
−− −
−
8 30 3 12 3 68 0 40 0 14 0 37
312 10 02 3 41 0 42 0 22 0 342
3 68 3 41 9 58 0 19 0 34 0 06
0 40 0 42 019 2 98 0 09 0 13
0 14 0 22 0 34 0 09 3 73 0 17
0 37 0 342 0 06 0 13 0 17 3 38
, , , . , ,
, , , , , ,
, , , , , ,
, , . , , ,
, , , , , ,
, , , , , ,
(5.15)
Im Idealfall, d.h. bei völlig isotropen Strukturen, sollten die Nebenelemente Null
ergeben, wie dies in Gleichung 5.10 zu erkennen ist. Die in Gleichung 5.15 auftretenden
Abweichungen sind darauf zurückzuführen, daß die verwendete amorphe Zelle nicht
isotrop ist. Die Nebenelemente können aber im Vergleich mit den Diagonalelementen
und den Matrixelementen C12, C13, C21, C23, C31 und C32 vernachlässigt werden.
Die Schwankungen der Lamé-Konstanten λ und µ betragen rund 20%. Aus den Lamé-
Konstanten λ und µ lassen sich für das Polynorbornen die folgenden
Materialeigenschaften berechnen, die in Tabelle 5.5 zusammengefaßt sind:
Tabelle 5.5:
Berechnete Materialeigenschaften des Polynorbornens
Materialeigenschaft berechneter Wert
Zug-Modul (Youngs Modul) E 8,41 GPa
Schermodul G 3,36 GPa
Bulk-Modul B 5,643 GPa
Poisson-Verhältnis ν 0,252

Der kondensierte Zustand starrer Knäuel 101
5.3.2 Experimentelle Untersuchung der mechanischen Eigenschaften
Aufgrund der schwierigen Präparation und der hohen Sprödheit des unsubstituierten
Polynorbornens sind dessen mechanischen Eigenschaften durch Spannungs-Dehnungs-
Messungen oder Dynamisch-Mechanisch-Thermische Analyse (DMTA) experimentell
nur sehr schwer zugänglich. Infolge der Sprödheit der Polynorbornenfilme tritt bei
Filmproben mit größerer Dicke bei der Messung in der Regel Probenbruch auf.
Die Untersuchung der mechanischen Eigenschaften des Polynorbornens wurde mit
einem Dynamisch-Mechanisch-Thermischen Analysegerät der Firma Polymer
Laboratories durchgeführt. Abbildung 5.3. zeigt als Ergebnisse dieser Dynamisch-
Mechanischen-Thermischen Analyse den Realteil des Elastizitätsmoduls bei zwei
verschiedenen Frequenzen als Funktion der Temperatur.
150 160 170 180 190 200 210 220 230
7,0
7,2
7,4
7,6
7,8
8,0
8,2
8,4
8,6
8,8
9,0
0.1 Hz 1.0 Hz
log
(E’)
[Pa]
Temperatur [°C]
Abb. 5.4: Dynamischer Elastizitätsmodul von Polynorbornen (DMTA)

Der kondensierte Zustand starrer Knäuel 102
Aus dem Verlauf des Realteils des Elastizitätsmoduls in Abbildung 5.4 erkennt man,
daß das Polynorbornen einen Elastizitätsmodul von etwa 109 Pa bzw. 1 GPa besitzt.
Weiterhin ist in dieser Abbildung die Abnahme des Elastizitätsmoduls als Folge der
Glasumwandlung im Bereich von 210°C zu beobachten. Der Elastizitäsmodul von
Polymethylmethacrylat (PMMA) beträgt im Vergleich 3,3 GPa [142].
Die Untersuchung des Schallgeschwindigkeit von Polynorbornen liefert einen Wert von
2400m/s [143]. Dieser Wert entspricht typischerweise dem von amorphen Polymeren.
Andere amorphe Polymere, wie z.B. Polystyrol (PS) oder Polymethylmethacrylat
(PMMA) besitzen bei Raumtemperatur Schallgeschwindigkeiten von 2100 m/s [144]
bzw. 2700 m/s [145]. Den Zusammenhang zwischen der experimentellen
Schallgeschwindigkeit vs und dem Elastizitätsmoduls E liefert Gleichung 5.16.
[146,147]:
vE
s =ρ
(5.16)
In Gleichung 5.16 bezeichnet ρ die Dichte des Polymers. Setzt man die gemessene
Schallgeschwindigkeit und die Dichte des Polynorbornens in Gleichung 5.16 ein, erhält
man einen E-Modul von 0,63 GPa.
Das verwendete statische Verfahren zur Berechnung der elastischen Konstanten, das auf
der Anwendung des Hookschen Gesetz basiert, ist prinzipiell nur für kleine
Auslenkungen gültig. Jede Rauhigkeit der Potentialenergie-Hyperfläche wird sich
deshalb direkt als Fehler auf die Berechnung der elastischen Konstanten auswirken. Die
auf diese Weise berechneten Werte werden zu hoch liegen [148]. Die Schlußfolgerung
der experimentellen Untersuchungen ist, daß sich die mechanischen Eigenschaften des
Polynorbornens mit denen von flexiblen Polymeren vergleichen lassen.

Der kondensierte Zustand starrer Knäuel 103
5.4 Permeationseigenschaften
5.4.1 Molekulardynamik-Simulationen zum Permeationsverhalten
Polynorbornene besitzen, wie aus den bisherigen Untersuchungen hervorgegangen ist,
eine Reihe von Eigenschaften, die in dieser Kombination einzigartig sind. Hierzu zählen
z.B. eine hohe Dichte, ein sehr enge Packung der Polymerketten, die starre
knäuelförmige Gestalt und eine eingeschränkte Beweglichkeit. Diese Kombination
mikroskopischer Eigenschaften sollte zu guten Barriereeigenschaften gegenüber kleinen
Molekülen führen. Die Diffusionseigenschaften bzw. Barriereeigenschaften sind für
potentielle industrielle Anwendungen des Polynorbornen als inertes Matrixmaterial von
großer Bedeutung.
Im einfachsten Fall kann die Diffusionskonstante D experimentell über die Stokes-
Einstein-Beziehung [83,96] bestimmt werden:
Dk T
RB
H
=6πη
(5.17)
In Gleichung 5.17 bedeutet η die Viskosität des Lösemittels und RH den
hydrodynamischen Radius des Moleküls. Mit Hilfe von Molekulardynamik-
Simulationen kann die Berechnung der Diffusionskonstante D über die mittlere
quadratische Verschiebung durchgeführt werden [149-156]. Für die Diffusion einer
Komponente a in einer Matrix ist deren Diffusionskonstante Da durch die Einstein-
Smoluchowski-Beziehung gegeben [83,96]:
( ) ( )[ ]DN
d
dtr t r ta
at
i ii
Na
=→∞
=∑1
6 0
2
1
lim (5.18)

Der kondensierte Zustand starrer Knäuel 104
In Gleichung 5.18 bedeuten ri die Ortsvektoren der diffundierenden Partikel, Na ist die
Anzahl der diffundierenden Partikel a und der Klammerausdruck ⟨...⟩ die zeitlichen
Mittelwerte. Die Diffusionskonstante Da ergibt sich durch eine entsprechende
Auftragung der o.g. Gleichung.
Die Berechnung der Diffusionskonstante für Sauerstoff in Polynorbornen wurde durch
eine Molekulardynamik-Simulation mit periodischen Randbedingungen durchgeführt.
Das Modellsystem wurde, wie in Abbildung 5.5 dargestellt, aus einer
energieminimierten amorphen Zelle durch Einfügen eines Sauerstoffmoleküls generiert.
Abb. 5.5: Molekulardynamik-Simulation zur Diffusion von Sauerstoff in Polynorbornen
Der Übersichtlichkeit halber wurde in Abbildung 5.5 nur eine Polynorbornenkette und
ein Sauerstoff-Molekül abgebildet. Dieses Modellsystem wurde hinsichtlich der Energie
minimiert. Die eigentliche Molekulardynamik-Simulation wurde anschließend unter
Berücksichtigung bestimmter Randbedingungen, d.h. konstanter Temperatur und
konstantem Volumen (NVT) durchgeführt. Die Simulationszeit betrug 1 ns.
In Abbildung 5.6 wurde das mittlere Verschiebungsquadrat ⟨r2⟩ als Funktion der
Simulationszeit aufgetragen.

Der kondensierte Zustand starrer Knäuel 105
0 100 200 300 400 500 600 700 800 900 10000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
D [
Å2 ]
Simulationszeit [ps]
Abb. 5.6: Molekulardynamik-Simulation zum Diffusionsverhalten von Sauerstoff in Polynorbornen
Die Berechnung des mittleren Verschiebungsquadrates für die Diffusion von Sauerstoff
durch Polynorbornen liefert bei einer Temperatur von 300 K eine Diffusionskonstante D
von 0,33⋅10-7 cm2⋅s-1. In der Tabelle 5.6 sind experimentelle Werte für die
Diffusionskonstanten einiger Standard-Polymere für die Diffusion von Sauerstoff
aufgeführt.
Van Gunsteren et al. konnten zeigen, daß die Diffusionskoeffizienten aus
Molekulardynamik-Simulationen mit einem Fehler von 5 bis 10 Prozent von den
experimentell bestimmten Werten abweichen [155].

Der kondensierte Zustand starrer Knäuel 106
Tabelle 5.6:
Experimentelle Diffusionskonstanten einiger Standard-Polymere
Polymer Diffusionskonstante D für Sauerstoff
Polyethylen (LDPE) [142] 4,6⋅10-7 cm2⋅s-1
Polyethylen (HDPE) [142] 1,7⋅10-7 cm2⋅s-1
Teflon [143] 1,5⋅10-7 cm2⋅s-1
Der Vergleich mit den Diffusionskonstanten einiger Standardpolymere in Tabelle 5.6
zeigt, daß das Polynorbornen ausgezeichnete Barriereeigenschaften besitzen sollte. Es
ist zu erwarten, daß Polynorbornene Barriereeigenschaften besitzen, die um rund einen
Faktor fünf besser sind als die der oben genannten Standardpolymere, was durch
experimentelle Untersuchungen noch zu verifizieren ist.
5.5 Zusammenfassung Kapitel 5
Das Ergebnis der Untersuchung der strukturellen Eigenschaften des Polynorbornens mit
Hilfe von Röntgen-Streuexperimenten zeigt den amorphen Charakter des
Polynorbornens im kondensierten Zustand. Die Berechnung des Hildebrandschen
Löslichkeitsparameters δ liefert einen relativ niedrigen Wert von 5,28 (cal/cm3)1/2, der
sich deutlich von denen der verwendbaren organischen Lösemittel unterscheidet. Die
experimentell durch Kontaktwinkelmessung bestimmte Oberflächenspannung ist mit der
des Polystyrols (PS) vergleichbar, obwohl es sich beim Polynorbornen um ein rein
aliphatisches System handelt. Sowohl die experimentelle als auch die theoretische
Bestimmung des Elastizitätsmodul von Polynorbornen zeigen übereinstimmend einen
niedrigen Wert. Der experimentell beobachtete Elastizitätsmodul liegt in einem Bereich

Der kondensierte Zustand starrer Knäuel 107
von rund 1 GPa, der berechnete Elastizitätsmodul besitzt einen Wert von rund 8 GPa.
Die große Abweichung des berechneten Wertes resultiert aus der statischen Methode zur
Berechnung des Elastizitätsmoduls. Das mechanische Verhalten des Polynorbornens
wird durch die Abnahme des Elastizitätsmoduls im Bereich der Glasumwandlung bei
210°C charakteristisch beeinflußt. Die Simulationen zum Permeationsverhalten des
Polynorbornens zeigen eine niedrige Diffusionskonstante für die Diffusion von
Sauerstoff. Diese eingeschränkte Diffusivität war aufgrund der hohen Dichte, der
geringen Beweglichkeit und der dichten Packung der Polymerketten zu erwarten.

Modifikation starrer Knäuel durch Katalysatorwahl 108
6 Modifikation starrer Knäuel durch Katalysatorwahl
Das zweite Kapitel hat gezeigt, daß die Drehung um die Bindungen zwischen den
Monomeren im Polynorbornen durch hohe Rotationsbarrieren erheblich erschwert bzw.
verhindert wird. Als logische Konsequenz aus diesen Ergebnissen ist zu vermuten, daß
die Kettenkonformation der starren Knäuel, bzw. vom Polynorbornen als Prototyp,
bereits während der Polymerisation festgelegt wird. Eine Untersuchung der
Polymerisation am aktiven Katalysatorsystem und des anschließenden Kettenwachstums
ist mit experimentellen Methoden in situ sehr schwierig. Um diesen, für die
Kettenkonformation starrer Knäuel wahrscheinlich charakteristischen Vorgang zu
untersuchen, wurden eine Reihe von Computersimulationen durchgeführt, um den
Einfluß der Katalysatorstruktur auf die Kettenkonformation des resultierenden Polymers
zu überprüfen.
Durch die Starrheit der Polymerketten sollte das Packungsverhalten und damit auch die
Ordnung der Polynorbornene im kondensierten Zustand beeinflußt werden. Neben
Computersimulationen werden experimentelle Untersuchungen mit Hilfe von Streu-
Methoden Aufschluß über den Ordnungszustand der Polynorbornene im festen Zustand
geben.
Die Durchführung dieser theoretischen Untersuchungen zur Polymerisation von
Norbornen erfolgt durch eine Kombination aus quantenmechanischen Verfahren und
Molekularmechanik-Verfahren unter Verwendung eines empirischen Kraftfeldes. Dieses
kombinierte Verfahren wurde von Rappé und Castonguay zur theoretischen
Untersuchung der isotaktischen Polymerisation von Propylen erfolgreich eingesetzt
[159].
Zur Berechnung der geometrischen Eigenschaften der verwendeten Übergangsmetall-
Katalysatoren wurden zunächst quantenmechanische Programme verwendet. Das für

Modifikation starrer Knäuel durch Katalysatorwahl 109
diese Computersimulationen in Frage kommende Universal Force Field (UFF) von
Rappé et al. [50] wurde unter Berücksichtigung der Ergebnisse der
quantenmechanischen Rechnungen für ausgewählte Systeme angepaßt und in dieser
Form für die folgenden Molekularmechanik-Simulationen eingesetzt.
6.1 Polymerisation von Olefinen mit Metallocenen
Die Untersuchung der Korrelation zwischen der Mikrostruktur der Polyolefinketten und
der Struktur des verwendeten Metallocen-Katalysators zeigt, daß die Ligandensphäre
des Katalysators in charakteristischer Weise die Struktur des resultierenden Polymers
beeinflußt. Eine Vielzahl experimenteller Untersuchungen zur Ziegler-Natta-
Polymerisation von Olefinen hat gezeigt, daß die räumliche Umgebung um das zentrale
Metallatom einen charakteristischen Einfluß auf die Orientierung des eintretenden
Monomers und damit direkt auf die Taktizität besitzt. [160-165]
Metallocen-Katalysatoren lassen sich aufgrund ihrer Ligandensphäre unterschiedlichen
Symmetrien zuordnen, die sich auf die Mikrostruktur der entstehenden Polymerketten
auswirkt. Tabelle 6.1 zeigt den Vergleich zwischen den generellen Formen der
Metallocene, deren Notation und die zu erwartende Taktizität der Polymerketten.
Die in Tabelle 6.1 aufgezeigten Korrelationen zwischen der Katalsysatorstruktur und der
Polymerstruktur wurden am Beispiel von Polynorbornen, die mit Zirkonocenen
polymerisiert wurden, verifiziert [159,166].

Modifikation starrer Knäuel durch Katalysatorwahl 110
Tabelle 6.1:
Katalysator-Symmetrien von Metallocenen und zu erwartende Taktizitäten der Polymere
Allgemeine Struktur Symmetrie und Notation vermutl. Taktizität
MC2-Katalysator isotaktisch
Mmeso-Katalysator ataktisch
M
σs-Katalysator syndiotaktisch
Bei den Metallocenen werden generell verschiedene Möglichkeiten zur Stereokontrolle
der Polymerisation diskutiert:
• Stereochemische Kontrolle durch die Chiralität des Katalysators (enantiomorphe
Kontrolle)
• Stereochemische Kontrolle durch die Konfiguration des letzten insertierten
Monomers (Kettenenden-Kontrolle)
6.2 Modifikation des Kraftfeldes
Zur Untersuchung der sterischen Verhältnisse bei der Polymerisation von Norbornen
mit dem Tetrakis(acetonitril)-Palladium(II)-Komplex in Abbildung 6.1 und den
Chrom(III)-Komplexen in Abbildung 6.2 ist es erforderlich, daß das verwendete
Kraftfeld die molekularen Geometrien hinreichend genau reproduzieren kann.

Modifikation starrer Knäuel durch Katalysatorwahl 111
Abb. 6.1: Tetrakis[acetonitril]Palladium (II)-Komplex
Bei der Verwendung des Palladium-Katalysators in Abbildung 6.1 führen die relativ
kleinen Acetonitril-Liganden in der Ligandensphäre nur zu einem geringen sterischen
Anspruch.
Bei den verwendeten Chrom(III)-Verbindungen wird der sterische Anspruch des
Katalysators durch die unterschiedliche Ligandensphäre bestimmt. Die untersuchten
Chrom(III)-Katalysatoren sind in Abbildung 6.2 abgebildet.
η5-Cyclopentadienyl η5-Indenyl- η5-Fluorenyl- η5-Cp*-
Abb. 6.2: Liganden der Chrom(III)-Katalysatoren
Der Aufbau entsprechender Modellsysteme für die verwendeten Katalysatoren zeigte,
daß die experimentell bestimmten Geometrien der Chrom(III)-Verbindungen nur
ungenügend, d.h. mit Abweichungen von bis zu zehn Prozent, wiedergegeben werden.
Zur Modellierung der Katalysator-Komplexe wurde die Ladung durch das Charge
Equilibration-Schema von Rappé et al. verwendet. Die entsprechenden Strukturen

Modifikation starrer Knäuel durch Katalysatorwahl 112
wurden zur Lokalisierung des Energieminimums unter Verwendung des Universal
Force Field 1.01 einem mehrfachen Simulated Annealing unterworfen. Die minimierten
Strukturen aus der entsprechenden Annealing-Trajektorie wurden mittels des Conjugate
Gradient-Algorithmus hinsichtlich der Energie weiter minimiert. Dieses kombinierte
Verfahren mußte eingesetzt werden, da die minimierten Strukturen, die aus der
Annealing-Trajektorie extrahiert wurden, hinsichtlich der Energie noch nicht
konvergiert waren.
Die Anpassung des Universal Force Field 1.01 (UFF) erfolgte durch Verwendung der
Bindungslängen bzw. -winkel, die aus den quantenmechanischen Rechnungen erhalten
wurden. Aufgrund der eingeschränkten Rechenkapazität wurde auf eine weitergehende
Parametrisierung über diese geometrischen Eigenschaften hinaus verzichtet. Zur
Durchführung der folgenden Molekularmechanik-Simulationen an den Katalysatoren
wurde das Universal Force Field 1.01 durch die Verwendung von optimierten
Molekülgeometrien, d.h. durch gemittelte Werte für die Bindungslängen der Chrom(III)-
Katalysatoren und des Palladium(II)-Komplexes, modifiziert.
Zur Modifikation des Universal Force Field 1.01 wurden die in Tabelle 6.2 dargestellten
Bindungslängen für die Palladium(II)-Verbindung verwendet.
Tabelle 6.2:
Bindungslängen für den Palladium(II)-Komplex aus B3LYP/6-31G(d)//HF/3-21G(d)-
Rechnungen [139] sowie aus der Röntgenstrukturbestimmung [167]
Bindung berechnete Bindungslänge
B3LYP/6-31G(d)//HF/3-21G(d)
Bindungslänge (X-RAY)
Pd - N 2,009 Å 1,956 Å
N - C 1,155 Å 1,121 Å
C - C 1,453 Å 1,485 Å

Modifikation starrer Knäuel durch Katalysatorwahl 113
Der Vergleich der berechneten Bindungslängen mit denen aus der Kristallstruktur-
Bestimmung in Tabelle 6.2 zeigten eine gute Übereinstimmung. Der verwendete
Basissatz stellt einen guten Kompromiß zwischen der Genauigkeit der Ergebnisse und
der Rechenzeit dar.
Zur Berechnung der Molekülgeometrien der Chromkomplexe wurde der in GAMESS
implementierte Triple Zeta Basissatz von Wachters in der Kontraktion
[14s11p6d/10s8p3d] verwendet [31,32]. Diese Kontraktion wurde von Jensen et al.
[168] zur Behandlung von Chrom(III)-Katalysatoren eingesetzt. Die Atome der
Liganden wurden durch die Double-Zeta-Basissätze von Huzinaga et al. in Kombination
mit den Kontraktionsschemata von Dunning et al. verwendet [29]. Der Vergleich der
berechneten mittleren Bindungslängen in Tabelle 6.3 erfolgte mit Daten aus
Röntgenstrukturuntersuchungen an den entsprechenden Chromverbindungen [169].
Für die Molekularmechanik-Simulationen an den Chrom(III)-Verbindungen wurden die
in Tabelle 6.3 aufgeführten Bindungslängen verwendet.
Tabelle 6.3:
Gemittelte Bindungslängen für die Chrom(III)-Komplexe und das monomere Norbornen
aus quantenmechanischen Rechnungen sowie Röntgenstrukturbestimmungen
Bindung berechnete Bindungslänge Bindungslänge (X-RAY)
Cr - CH3 1,975 Å 2,056 Å
Cr - C (Ligand) 2,200 Å 2,211 Å
C = C (Norbornen) 1,323 Å 1,366 Å [89]
Die berechneten mittleren Bindungslängen in Tabelle 6.3 zeigen sehr gute
Übereinstimmung mit experimentellen Ergebnissen.

Modifikation starrer Knäuel durch Katalysatorwahl 114
6.3 Computersimulationen zur Polymerisation von Bicylo[2.2.1]hept-
2-en (Norbornen)
6.3.1 Mechanismus der Polymerisation von Polynorbornen
Polynorbornene lassen sich wie viele andere Olefine, durch eine Reihe von
Übergangsmetallkatalysatoren in homogener Phase darstellen. Die Untersuchungen
beschränken sich auf die Behandlung von Pd(II)- und Cr(III)-Katalysatoren, da im
Rahmen dieser Arbeit im wesentlichen Polynorbornene untersucht wurden, die mit
diesen Katalysator-Systemen hergestellt wurden. Die Polymerisation erfolgt in beiden
Fällen nach dem Cossee-Mechanismus [170] für die Ziegler-Natta-Polymerisation von
Olefinen.
M
P CH3P
M
CH3
M
CH3
P
M
P
+CH3
= vakante Bindungsstelle P = Polymerkette
Abb. 6.3: Cossee-Mechanismus für die Ziegler-Natta-Polymerisation von Olefinen
Bei dem in Abbildung 6.3 gezeigten Cossee-Mechanismus wird zu Beginn der
Polymerisation die Bildung einer vakanten Bindungsstelle initiiert. Im nächsten Schritt
wird das Olefin über eine π-Bindung an den Übergangsmetall-Komplex bzw. das
zentrale Metallatom gebunden. Die Migration der Endgruppe P und die Ausbildung der
Metall-Kohlenstoff-Bindung erfolgen konzertiert über einen sogenannten Vier-Zentren-
Übergangszustand. Durch diesen Vorgang wird erneut eine vakante Bindungsstelle am
Metallatom erzeugt und die Insertion des nächsten Monomers kann beginnen. Die
aktuelle vakante Bindungsstelle befindet sich an der Stelle, die vorher von der

Modifikation starrer Knäuel durch Katalysatorwahl 115
Endgruppe bzw. der bereits existierenden Polymerkette eingenommen wurde. Das Ende
der wachsenden Polymerkette springt also zwischen diesen beiden Positionen hin und
her.
6.3.2 Polymerisation mit Pd(II)-Komplexen
Die vinylische Polymerisation von Norbornen mit Palladium(II)-Verbindungen in
homogener Phase verläuft mit einer Reihe von Liganden, die einen wesentlichen Einfluß
auf Ausbeute und Polymerisationszeit besitzen. Als Gegenion wird das schwach
koordinierende Tetrafluoroborat-Anion eingesetzt, so daß die Polymerisation mit einem
nahezu „nackten“ Palladium-Kation erfolgt [82,93,171-173].
L= -CH3, -Ch2-CH3, C(CH3)3, -Phenyl
* *n
[(L-CN)4Pd][BF4]2
Abb.6.4: Reaktionsschema für vinylische Polymerisation mit Pd(II)-Komplexen
Abbildung 6.4 zeigt das Reaktionsschema für die vinylische Polymerisation von
Polynorbornen mit Palladium(II)-Komplexen. Die auf diesem Weg erhaltenen
Polynorbornene lösen sich in Chlorbenzol, 1,2-Dichlorbenzol, 1,2,4-Trichlorbenzol, p-
Xylol und Cyclohexan [82].

Modifikation starrer Knäuel durch Katalysatorwahl 116
6.3.3 Polymerisation mit Cr(III)-Komplexen
Die vinylische Polymerisation von Norbornen mit Chrom(III)-Verbindungen verläuft
mit einer Reihe von sterisch anspruchsvollen Liganden, die einen wesentlichen Einfluß
auf Ausbeute, Polymerisationszeit und Struktur des entstehenden Polymers besitzen. Bei
der Polymerisation wird Methylaluminoxan (MAO) als Co-Katalysator eingesetzt, der
die ursprünglich zweikernige Chromverbindung unter Abspaltung von Chlorid-Ionen
spaltet. Als Liganden lassen sich η5-Cyclopentadienyl (Cp)-, η5-Indenyl (In)-, η5-
Fluorenyl (Flu)- und η5-Pentamethyl-cyclopentadienyl (Cp*)-Gruppen einsetzen
[174,175].
R
Cr
CH3
Cl
ClCr
CH3
R
+MAO
R
CrCH3
R
Cr
CH3
+
Abb. 6.5: Reaktionsschema für die vinylische Polymerisation mit Chrom(III)-Komplexen
Abbildung 6.5 zeigt das Reaktionsschema für die vinylische Polymerisation von
Polynorbornen mit Chrom(III)-Verbindungen.
6.3.4 Sterischer Anspruch des Katalysators
Vergleicht man die Ligandensphären der in den Abbildungen 6.1 und 6.2 abgebildeten
Übergangsmetallkatalysatoren, zeigt sich der unterschiedliche sterische Anspruch für
das neu eintretende Norbornen-Monomer.
Die Ligandensphäre des Tetrakis(acetonitril)-Palladium(II)-Komplexes besitzt aufgrund
der relativ kleinen Acetonitril-Liganden nur einen relativ geringen sterischen Anspruch
an das erste eintretende Monomer. Mit fortschreitender Polymerisation und der

Modifikation starrer Knäuel durch Katalysatorwahl 117
wachsenden voluminösen Polynorbornenkette ist zu erwarten, daß die Orientierung der
jeweils neu eintretenden Monomeren im wesentlichen durch die bereits vorhandene
Polymerkette festgelegt wird.
Bei den Chrom(III)-Katalysatoren wird der sterische Anspruch durch die variable
Ligandensphäre mehr oder weniger stark beeinflußt. Es ist zu erwarten, daß der
voluminöse Cp*-Ligand den größten sterischen Anspruch an das Monomer stellt.
Weiterhin ist zu vermuten, daß die voluminöse, starre Polymerkette, die an dem
Katalysatorzentrum durch Insertion heranwächst, diesen sterischen Anspruch an die neu
eintretenden Monomeren noch weiter erhöht. Als Folge dieser räumlichen Ausfüllung
des aktiven Zentrums kann ein direkter Einfluß auf die Taktizität des Polymers erwartet
werden.
6.4 Molekularmechanik-Simulationen zur Polymerisation von
Norbornen
Das Ziel der folgenden Molekularmechanik-Simulationen war die Untersuchung des
Polymerisationsvorgangs auf mikroskopischer Ebene. Durch die Verwendung von
Molekularmechanik-Verfahren lassen sich Informationen über den Zusammenhang
zwischen dem sterischen Anspruch des Katalysators und der resultierenden
Mikrostruktur der Polymerkette erhalten. Im Vordergrund dieser Kraftfeldrechnungen
steht also weniger die Beschreibung der elektronischen Eigenschaften als vielmehr die
Untersuchung der sterischen Verhältnisse, die durch die Ligandensphäre entscheidend
beeinflußt wird. Für die genaue Untersuchung der elektronischen Eigenschaften sind
wesentlich aufwendigere Simulationsverfahren bzw. Hybrid-Methoden wie z.B.
gekoppelte Quantenmechanik/Molekulardynamik-Verfahren [176] oder das ab initio
Molekulardynamik-Verfahren von Car und Parinello [177] erforderlich.

Modifikation starrer Knäuel durch Katalysatorwahl 118
6.4.1 Allgemeines zu den Molekularmechanik-Simulationen
Für die Computersimulationen zum Verlauf der vinylischen Polymerisation von
Norbornen mit Hilfe von Pd(II)- und Cr(III)-Katalysatoren wurden die in Tabelle 6.4
aufgeführten Übergangsmetall-Verbindungen ausgewählt.
Tabelle 6.4:
Übersicht über die ausgewählten Katalysatorsysteme für die Molekularmechanik-
Simulationen
Struktur Bezeichnung
Tetrakis(acetonitril)palladium(II)-dikation
η5-Cyclopentadienyl-methyl-Chrom(III)-
kation
(Cp-Me-Cr)
η5-Pentamethylcyclopentadienyl-methyl-
Chrom (III)-kation
(Cp*-Me-Cr)

Modifikation starrer Knäuel durch Katalysatorwahl 119
Aufgrund der Tatsache, daß Molekularmechanik-Verfahren auf der expliziten
Verwendung von definierten Bindungen basieren, sind keinerlei Aussagen über
Vorgänge möglich, die mit der Veränderung elektronischer Eigenschaften einhergehen.
Aus diesem Grund können keine Prozesse modelliert werden, bei denen Bindungen
entstehen oder verloren gehen [178]. Der hohe sterisch Anspruch des monomeren
Norbornens in Kombination mit den hohen Rotationsbarrieren erlaubt genaue Aussagen
bezüglich der relativen Energieunterschiede der resultierenden Konformationen. Die
folgenden Computersimulationen wurden im Vakuum durchgeführt. Bei der
Polymerisation sind demnach keinerlei Lösemittel-Einflüsse berücksichtigt. Bei den
Molekularmechanik-Simulationen wurden die Ladungen der Komplexe über das
Charge-Equilibration-Verfahren berücksichtigt [67].
Für die Molekularmechanik-Simulationen an den Chrom(III)-Katalysatoren wurden
ausschließlich die aktiven Katalysatorspezies verwendet. Das Methylaluminoxan
(MAO), das bei der Polymerisation als Co-Katalysator verwendet wird, bleibt bei diesen
Molekularmechanik-Simulationen unberücksichtigt.
Um den Konfigurationsraum der verwendeten Modellstrukturen trotz der hohen
Komplexität der Potentialenergie-Hyperfläche hinreichend genau zu untersuchen, wurde
das Simulated Annealing in Kombination mit einem Minimierungs-Algorithmus
eingesetzt. Bei der Durchführung der Simulated Annealing-Simulationen wurden aus
den 50 erzeugten Strukturen die jeweils zehn energetisch niedrigstliegenden ausgewählt
und diese Modellsysteme hinsichtlich deren Potentialenergie weiter minimiert. Für die
Annealing-Zyklen wurde ein Temperaturintervall von 300 K bis 1200 K verwendet. Die
Partialladungen wurden durch das Charge-Equilibration-Schema alle 50 Rechenschritte
berechnet. Zur Untersuchung der Konformationen wurden die mit der Modellgeometrie
verbundene Energie und die Connolly-Oberfläche [71] am zentralen Metallatom
berechnet, um die sterischen Verhältnisse am aktiven Zentrum zu dokumentieren. Die
Connolly-Oberfläche ist die Oberfläche, die man in der Näherung interpenetrierender
Kugeloberflächen durch Abtastung mit einer Sonde mit infinitesimalen Radius erhält.

Modifikation starrer Knäuel durch Katalysatorwahl 120
Aus Hardwaregründen und wegen der beschriebenen Schwierigkeiten bei der
Lokalisierung der Potentialminima konnten lediglich Modellsysteme betrachtet werden,
die bis zu sieben Monomere enthielten. In den Vorarbeiten zu Kapitel 3 konnte gezeigt
werden, daß die Berücksichtigung von jeweils 6 Monomeren eine adäquate
Beschreibung der Nachbargruppen-Effekte des Systems liefert. Die Simulationen sollten
auf diese Weise den sterischen Einfluß der Ligandensphäre und der an dem Katalysator
herauswachsenden Polymerkette berücksichtigen. Bei den Computersimulationen zur
Polymerisation ist stets zu bedenken, daß es sich um das Modell einer Kette handelt.
Die Ergebnisse, die durch die Modellierung mit dem Molekularmechanik-Verfahren
einer Polymerkette erhalten wurden, geben dementsprechend einen Trend wieder.
Die Auswahl der drei Katalysatorsysteme für die Molekularmechanik-Simulationen
(Tabelle 6.4) erfolgte unter dem Gesichtspunkt des unterschiedlichen sterischen
Anspruchs. Der Palladium(II)-Komplex stellt aufgrund seiner quadratisch-planaren
Struktur nur einen sehr niedrigen sterischen Anspruch an das Monomer. Um den
Einfluß des Cp- und des Cp*-Liganden auf die Polymerisation zu untersuchen, sollen
für die Chrom(III)-Katalysatoren diese beiden Extremfälle betrachtet werden.
6.4.2 Polymerisation mit Pd(II)tetrakisacetonitril-bis-tetrafluoroborat
Bei der Polymerisation mit Tetrakis(acetonitril)-Palladium(II)-bis-tetrafluoroborat sind
die ersten Schritte der Polymerisation weitestgehend unbeeinflußt von der
Ligandensphäre. Abbildung 6.6 zeigt das System nach der Insertion des ersten
Norbornens und die Anlagerung des zweiten Norbornens. Die frei zugängliche
Connolly-Oberfläche im ersten σ-Komplex hat eine Fläche von 12,0 Å2 (linke Seite in
Abb. 6.6).

Modifikation starrer Knäuel durch Katalysatorwahl 121
Abb. 6.6: Anlagerung des zweiten Norbornens am Palladium(II)-Komplex
In Abbildung 6.6 ist zu erkennen, daß sich das zweite Monomer ungestört am
Katalysator anlagern kann. Das bereits kovalent gebundene Norbornen führt zu einer
Orientierung des neueintretenden Monomers, die im zweiten Insertionsschritt zu einer
meso-Verknüpfung führt. Die Anlagerung des dritten Norbornens kann ebenfalls fast
ohne Einschränkung erfolgen, die frei zugängliche Connolly-Oberfläche am Palladium
beträgt 10,5 Å2. Die folgenden Anlagerungen der Norbornene unter Ausbildung
entsprechender π-Komplexe und Insertionen führen zu einer racemischen Form des cis-
exo-verknüpften Polynorbornens. Diese Form der Verknüpfung zeigt aufgrund
sterischer Effekte gegenüber der meso-Verknüpfung eine günstigere Energie im Bereich
von 20 bis 30 kJ/mol. Abbildung 6.7 zeigt die Anlagerung und Insertion des fünften
Monomers.
Abb. 6.7: Anlagerung und Insertion des 5. Norbornens

Modifikation starrer Knäuel durch Katalysatorwahl 122
In Abbildung 6.7 wurde das zuletzt insertierte Monomer eingefärbt, um die
Übersichtlichkeit der Darstellung zu erhöhen. Die Verknüpfung der Monomeren erfolgt
nahezu ausschließlich in racemischer cis-exo-Verknüpfung, die energetisch günstiger als
die meso-Verknüpfung ist. Die Molekularmechanik-Simulationen zur Palladium-
katalysierten Polymerisation von Norbornen haben zu dem Ergebnis geführt, daß die
Insertion der eintretenden Monomere keinen sterischen Restriktionen unterliegt.
6.4.3 Polymerisation mit Cylopentadienyl-methyl-Cr(III)
Dieser Katalysator besitzt unter den in Abbildung 6.2 abgebildeten Chrom(III)-
Katalysatoren den geringsten sterischen Anspruch der Ligandensphäre. Die Anlagerung
des ersten Monomers und dessen Insertion erfolgt ungehindert. Die Anlagerung des
ersten Norbornens an das zentrale Metallatom ist in zwei Orientierungen möglich, die
sich um rund 25 kJ/mol unterscheiden.
anti-parallelparallel
Abb. 6.8: Anlagerung des ersten Norbornens an Cp-Cr-Katalysator
Die beiden in Abbildung 6.8 abgebildeten Strukturen unterscheiden sich durch die
Orientierung der Methyl-Gruppe relativ zur C7-Brücke im Norbornen. Die anti-parallele
Ausrichtung besitzt aus sterischen Gründen die niedrigere Energie beider Strukturen.

Modifikation starrer Knäuel durch Katalysatorwahl 123
Das Kettenwachstum, durch wiederholtes Anlagern und Insertieren der Monomere, wird
im wesentlichen durch die Zugänglichkeit des zentralen Metallatoms geprägt. Die freie
Connolly-Oberfläche der anti-parallelen Struktur in Abbildung 6.8 resultiert mit 5,2 Å2
aus einem engeren Kontakt des Monomers zum Katalysatorkomplex als die parallele
Anordnung mit einer Connolly-Oberfläche von 6,0 Å2. Das Wachstum der
Polynorbornenkette erfolgt bis zum Einbau des dritten Monomers aus energetischen
Gründen ausschließlich in der racemischen cis-exo-Verknüpfung. Der Einbau des dritten
Monomers wird nur durch geringe energetische Unterschiede zwischen der meso- und
rac-Verknüpfung der Norbornan-Einheiten charakterisiert. Die meso-Form
unterscheidet sich nur um einen geringen Energiebetrag von 5 kJ/mol von der rac-Form.
Die Anlagerung des folgenden Monomers ist in der rac-Verknüpfung wieder um 30
kJ/mol energetisch begünstigt.
Abb. 6.9: Anlagerung des 4. Monomers an den Cp-Cr-Katalysator
Die folgenden Monomere werden nach dem beschriebenen Schema eingebaut. Die
resultierende Polymerkette wird einen ausgeprägten ataktischen Charakter besitzen, da
etwa alle zwei Monomer-Einheiten sowohl die meso-Verknüpfung als auch die rac-
Verknüpfung mit der gleichen Wahrscheinlichkeit auftreten.

Modifikation starrer Knäuel durch Katalysatorwahl 124
6.4.4 Polymerisation mit Cp*-methyl-Cr(III)
Bei der Polymerisation mit dem voluminösen Cp*-Me-Cr(III)-Katalysator ist der Schritt
zur Anlagerung des zweiten Monomers, die zur Ausbildung eines π-Komplexes führt,
von hoher Selektivität. Die wachsende Polymerkette beschränkt die Orientierung des
neu eintretenden Monomers auf eine einzige Anordnung. Abbildung 6.10 zeigt die
beiden Minimumstrukturen, die sich als parallele bzw. anti-parallele Orientierung der
C7-Brücke des Norbornens relativ zu den Methylgruppen des Cp*-Liganden
beschreiben lassen.
anti-parallel parallel
Abb. 6.10: Anlagerung des zweiten Norbornens an Cp*-Cr-Katalysator
Die Energie der parallelen Struktur ist um rund 80 kJ/mol niedriger als die der anti-
parallelen Anordnung des Norbornen-Monomers. Diese drastischen energetischen
Unterschied von 60 bis 80 kJ/mol treten stets bei der Anlagerung eines Norbornens
unter Ausbildung des π-Systems auf. Die Verknüpfung der Monomeren erfolgt durch
eine racemische cis-exo-Verknüpfung. Die resultierende Polymerkette wird eine
isotaktische Konfiguration zeigen, die sich auf die Kristallinität der Polymere auswirken
wird [92,160].

Modifikation starrer Knäuel durch Katalysatorwahl 125
6.5 Experimentelle Charakterisierung der Polynorbornene
Die Röntgendiffraktogramme der mit Palladium(II) dargestellten Polynorbornene
wurden bereits im Abschnitt 5.2 eingehend vorgestellt.
Abbildung 6.11 zeigt die Röntgenweitwinkel-Diffraktogramme von Polynorbornenen,
die mit den vorgestellten Chrom-Katalysatoren dargestellt wurden.
5 10 15 20 25 300
20
40
60
80
100
120
140
160
180
200
[IndCrMeCl]2
[CpCrMeCl]2
[FluCrMeCl]2
[Cp*CrMeCl]2
Str
euin
tens
ität [
a.u.
]
Streuwinkel 2Θ [°]
Abb. 6.11: Röntgenweitwinkel-Diffraktogramme der Polynorbornene (Cr-Katalysatoren)
Die Diffraktogramme der verschiedenen Polynorbornene unterscheiden sich in der Lage
der Intensitätsmaxima und vor allem durch die Form. Das Streubild des Polynorbornens,
das mit Hilfe des Cyclopentadienyl-Katalysators hergestellt wurde, zeigt den für das
Norbornan typischen amorphen Halo bei einem Streuwinkel von rund 2Θ = 18°. Der
zweite amorphe Halo bei kleinerem Streuwinkel ist deutlich breiter als bei dem mit

Modifikation starrer Knäuel durch Katalysatorwahl 126
Palladium oder mit den anderen Chrom-Katalysatoren hergestellten Polynorbornenen.
Unter den Chrom-Katalysatoren besitzt der Cp*-Katalysator offensichtlich den größten
sterischen Anspruch. Das resultierende Polymer zeigt im Vergleich zu den anderen
Polynorbornenen in Abbildung 6.11. die höchste Ordnung bezüglich der Mikrostruktur,
was sich an den schärferen Peaks im Diffraktogramm äußert.
Die Streudiagramme spiegeln offensichtlich die unterschiedliche Kettenkonformation
wider. Genaue Rechnungen stehen noch aus.
6.6 Zusammenfassung Kapitel 6
Die Untersuchungen zur Modifikation der Polynorbornene durch die Wahl des
Katalysators zeigt, daß die Kombination aus dem voluminösen monomeren Norbornen
und dem sterischen Anspruch des Katalysators einen charakteristischen Einfluß auf die
Struktur des Polymers besitzt.
Ein wesentliches Ergebnis der Molekularmechanik-Simulationen zur Übergangsmetall-
katalysierten Polymerisation von Norbornen ist, daß die Konformation des
Polynorbornens bereits bei der Polymerisation festgelegt wird. Die Simulationen haben
gezeigt, daß allein die Orientierung des Monomers einen sehr großen Einfluß auf die
Energie des π-Systems besitzt. Der Einbau der Monomeren in die Kette erhält durch
eine sterisch sehr anspruchsvolle Katalysatorstruktur, wie am Beispiel des Cp*-
Katalysators exemplarisch dargestellt, einen sehr selektiven Charakter. Polynorbornen,
das mit dem sterisch sehr anspruchsvollen Cp*-Katalysator hergestellt wurde, zeigt im
Röntgenweitwinkel-Diffraktogramm die schärfsten Reflexe.

Modifikation starrer Knäuel durch Katalysatorwahl 127
Die Konformation der Polymerkette wird durch einen einmaligen und unveränderbaren
Vorgang festgelegt. Die Mikrostruktur der Polymerkette ist somit ein Fingerabdruck des
verwendeten Katalysators.

Modifikation der Rotationsbehinderung starrer Knäuel 128
7 Modifikation der Rotationsbehinderung starrer Knäuel
Die bisherigen Abschnitte haben gezeigt, daß das Polynorbornen als starres,
knäuelförmiges Polymer Eigenschaften besitzt, die zwischen denen der flexiblen Knäuel
und denen der starren Stäbchen anzusiedeln sind. Durch eine geeignete Modifikation der
chemischen Struktur sollte es möglich sein, die Steifigkeit des Polynorbornens zu
variieren. Mit dem Norbornen, als leicht zugänglicher Grundstruktur, sollte eine große
Variationsbreite unterschiedlicher Monomere zugänglich sein. Hierdurch sollten sich
die strukturellen Eigenschaften des bicyclischen Grundgerüsts sowohl verstärken als
auch abschwächen lassen.
Der folgende Abschnitt wird den Einfluß unterschiedlicher Modifikationen des
Polynorbornens beschreiben und die resultierenden Eigenschaften durch die
Untersuchung der entsprechenden Struktur-Eigenschaftsbeziehungen aufklären. Als
Modifikation der Eigenschaften starrer Knäuel bzw. des Polynorbornens kommen die
folgenden Möglichkeiten in Frage:
• Modifikation des Monomers durch Einführung lateraler Substituenten, z.B. Ester der
Norbornencarbonsäure und der Polynorbornenimide
• Modifikation des Polymerbackbones durch Einführung von Heteroatomen, z.B.
durch die Polymerisation von Polynorbornenamiden und -epoxiden
• Copolymerisation mit anderen Comonomeren, wie z.B. Ethylen, zur Darstellung von
statistischen oder Block-Copolymeren
Weiterhin ist die chemische Modifikation des Norbornangerüsts durch die Einführung
von Heteroatomen denkbar, wobei diese Variationsmöglichkeit zur Zeit experimentell

Modifikation der Rotationsbehinderung starrer Knäuel 129
noch nicht realisiert wurde. Dieses Kapitel wird Modifikationsmöglichkeiten aufzeigen,
die bereits synthetisch-präparativ realisiert worden sind. Die Beschreibung der
Modellverbindungen wird sich auf einige ausgewählte Eigenschaften beschränken. Mit
Hilfe dieser Modellsysteme soll die enge Beziehung zwischen mikroskopischer
Strukturvariation und deren makroskopischen Auswirkungen beschrieben werden.
7.1 Modifikation durch Einführung lateraler Substituenten
Die in den Kapiteln 2 und 3 beschriebenen Ergebnisse belegen den Einfluß der starren
dreidimensionalen Struktur des Monomeren auf die dynamischen Eigenschaften starrer
Knäuel. Die Starrheit des Polynorbornen-Grundgerüstes kann auf zwei verschiedene
Arten modifiziert werden:
• Einführung eines lateralen Substituenten mit einer flexiblen Seitengruppen
• Einführung eines lateralen Substituenten mit einer starren, voluminösen und
zusätzlich elektronenreichen Seitengruppe
Für die Gruppe der lateral substituierte Polynorbornene soll der Polynorbornen-
carbonsäuremethylester (PNme) als Beispiel für einen flexiblen lateralen Substituenten
und das Poly-N-para-tolyl-bicyclo[2.2.1]hept-2-en-5,6-dicarbonsäureimid (PNim) als
Beispiel für einen starren lateralen Substituenten betrachtet werden.

Modifikation der Rotationsbehinderung starrer Knäuel 130
O
O N CH3
O
O
Norbornenmethylester Norbornenimid
Abb. 7.1: Lateral substituierte Norbornene
7.1.1 Polynorbornencarbonsäuremethylester (PNme)
Polynorbornencarbonsäureester lassen sich durch vinylische Polymerisation der
entsprechenden 5-Norbornen-2-carbonsäureester mit einer Reihe unterschiedlicher
Palladium-Katalysatoren darstellen. Die hochmolekularen Polymere lösen sich z.B. in
Dichlormethan, Tetrahydrofuran, Chlorbenzol und Toluol [179,180]. Das Verhältnis
zwischen der exo- und der endo-Form des 5-Norbornen-2-carbonsäuremethylesters
beträgt exo : endo = 70 : 30. In den nachfolgenden Computersimulationen wurden
Modellsysteme mit ausschließlich exo-ständigen Carbonsäuremethylester-Gruppen
verwendet.
7.1.1.1 1D-Rotationspotential
Die Berechnung des eindimensionalen Rotationspotentials des Polynorbornen-
carbonsäuremethylesters (PNme) erfolgt durch Molekularmechanik-Rechnungen. Für
die Molekularmechanik-Simulationen wurde das Dreiding 2.21-Kraftfeld [51]
verwendet. Intramolekulare Partialladungen wurden durch das Charge Equilibration-
Verfahren jeweils alle 50 Rechenschritte neu berechnet. In Abbildung 7.2 ist der
Torsionswinkel zwischen den beiden Monomeren bezeichnet.

Modifikation der Rotationsbehinderung starrer Knäuel 131
O
O
O
O
1
2
3
4
Abb. 7.2: Definition des Torsionswinkels für den dimeren Norbornen-2-carbonsäuremethylester
Der Verlauf der Potentialenergie entspricht im wesentlichen dem Potentialverlauf des
unsubstituierten dimeren Norbornans. Wie man aus Abbildung 7.3 erkennen kann, ist
das globale Minimum bei rund 60° lokalisiert.
0 30 60 90 120 150 180 210 240 270 300 330 3600
10
20
30
40
50
60
70
80
90
100
110
IIIII
I
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 7.3: Eindimensionales Rotationspotential für dimeren Norbornen-2-carbonsäuremethylester

Modifikation der Rotationsbehinderung starrer Knäuel 132
Die flexible Seitengruppe des Norbornen-2-carbonsäuremethylesters zeigt im Vergleich
zum Rotationspotential des unsubstituierten dimeren Norbornens offensichtlich keinen
Einfluß auf den Potentialverlauf. Die Lage der Potentialminima (Abbildung 7.3) gleicht
der des Rotationspotentials des unsubstituierten Norbornens. Die relativen energetischen
Unterschiede zwischen den Minima I und II bzw. III betragen 40 bzw. 50 kJ/mol und
sind damit deutlich höher als im Fall des unsubstituierten Norbornens mit 17 bzw. 18
kJ/mol. Im Potentialverlauf fällt die hohe Rotationsbarriere zwischen den Minima I und
II mit rund 100 kJ/mol, deutlich höher aus beim unsubstituierten Norbornen mit nur 55
kJ/mol.
7.1.1.2 2D-Rotationspotential trimerer Norbornen-2-carbonsäuremethylester
Das Resultat der Berechnung des zweidimensionalen Rotationspotentials für den
trimeren Norbornen-2-carbonsäuremethylester in Abbildung 7.4 zeigt, daß die diskreten
Minima, die beim trimeren Norbornan zu beobachten sind, durch die flexiblen
Seitengruppen „delokalisiert“ werden. Die intramolekularen Partialladungen im
trimeren Modellsystem, die durch die Anwesenheit der Heteroatome zu erwarten sind,
wurden bei den Molekularmechanik-Rechnungen durch den Gasteiger-Marsili-Ansatz
berücksichtigt.
Die Potentialenergie-Hyperfläche des trimeren Norbornen-2-carbonsäuremethylesters
(Abbildung 7.4) zeichnet sich im Vergleich mit der des unsubstituierten Norbornens
durch höhere Potentialbarrieren aus.

Modifikation der Rotationsbehinderung starrer Knäuel 133
0 30 60 90 120 150 180 210 240 270 300 330 3600
30
60
90
120
150
180
210
240
270
300
330
360
Tor
sion
swin
kel 2
[°]
Torsionswinkel 1 [°]
120.0 -- 125.0 115.0 -- 120.0 110.0 -- 115.0 105.0 -- 110.0 100.0 -- 105.0 95.00 -- 100.0 90.00 -- 95.00 85.00 -- 90.00 80.00 -- 85.00 75.00 -- 80.00 70.00 -- 75.00 65.00 -- 70.00 60.00 -- 65.00 55.00 -- 60.00 50.00 -- 55.00 45.00 -- 50.00 40.00 -- 45.00 35.00 -- 40.00 30.00 -- 35.00 25.00 -- 30.00 20.00 -- 25.00 15.00 -- 20.00 10.00 -- 15.00 5.000 -- 10.00 0 -- 5.000
Abb. 7.4: 2D-Rotationspotential für trimeren Norbornen-2-carbonsäuremethylester (PNme)
Die Energie, die für eine Konformationsänderung aufzuwenden ist, liegt bei 120 kJ/mol.
Der bei Raumtemperatur zugängliche Konfigurationsraum ist, vergleichbar mit dem des
unsubstituierten Norbornens, ebenfalls sehr stark eingeschränkt. Die Rauhigkeit der n-
dimensionalen Potentialenergie-Hyperfläche ist größer als die des unsubstituierten
Norbornens.
7.1.1.3 Lokale Kettendynamik Polynorbornencarbonsäuremethylester (12mer)
Die Untersuchung der lokalen Kettendynamik an einem Modellsystem bestehend aus 12
cis-exo-verknüpften Norbornen-2-carbonsäuremethylester-Einheiten liefert die in
Abbildung 7.5 dargestellte Verteilung der Torsionswinkel bei verschiedenen
Temperaturen. Aus Abbildung 7.5 ist zu erkennen, daß die erhöhte Beweglichkeit des

Modifikation der Rotationsbehinderung starrer Knäuel 134
Polymer-Backbones bei zunehmender Temperatur auf einer Potentialaufweitung beruht.
Bis zu einer Temperatur von 1100 K sind keinerlei Konformationsübergänge zu
beobachten.
-180 -150 -120 -90 -60 -30 00
20
40
60
80
100
120
600 K
300 K
Häu
figk
eit
Torsionswinkel φ [°]
Abb. 7.5: Häufigkeit der Torsionswinkel bei verschiedenen Temperaturen (PNme)
Nähert man die in Abbildung 7.5 dargestellten Torsionswinkel-Verteilungen durch
Gauß-Funktionen an, ergibt sich für die Halbwertsbreiten der eingefitteten Gauß-
Funktionen eine Temperaturabhängigkeit, die aus Abbildung 7.6 durch lineare
Regression erhalten wurde.

Modifikation der Rotationsbehinderung starrer Knäuel 135
0 100 200 300 400 500 600 700 800 900 1000 11000
5
10
15
20
25
30
35
40
45
50
55
60
Hal
bwer
tsbr
eite
der
Gau
ß-F
unkt
ion
[°]
Temperatur [K]
Abb. 7.6: Halbwertsbreite der Gauß-Funktionen (Pnme)
Die Steigung der Regressionsgerade in Abbildung 7.6. beträgt 0,012°/K ± 0,002°/K. Der
Vergleich mit dem unmodifizierten Polynorbornen in Kapitel 4.1.1.1 liefert eine
niedrigere Temperaturabhängigkeit der Torsionswinkelfluktuationen. Der
Achsenabschnitt der Regressionsgerade besitzt mit 2,52° ± 0,72° einen Wert, der
geringfügig größer als der des Polynorbornens ist. Die Aufweitungstendenz des
Potentials des Polynorbornencarbonsäuremethylesters ist also für den n-dimensionalen
Fall mit dem des unsubstituierten Polynorbornens näherungsweise vergleichbar.

Modifikation der Rotationsbehinderung starrer Knäuel 136
7.1.1.4 Monte Carlo-Simulation Polynorbornencarbonsäuremethylester
Da die Beschreibung der Konformationseigenschaften einer Polymerkette aus 100
Norbornen-2-carbonsäuremethylester-Einheiten mit Hilfe von Molekulardynamik-
Simulationen zu fehlerhaften Ergebnissen führt, wurden Monte Carlo-Simulationen
eingesetzt [181].
Hierzu wurde ein Ensemble aus 1000 Polynorbornencarbonsäuremethylester-
Einzelketten, bestehend aus jeweils 100 Monomeren, generiert. Die Polymerketten
wurden nach dem Rotational Isomeric State-Modell aufgebaut. Die Verteilung der
Kettenendenabstände besitzt ein Maximum bei einem Abstand von rund 72 Å. Das
charakteristische Verhältnis C100 berechnet für den Polynorbornen-
carbonsäuremethylester sich zu 7,1. Der geringere Wert für C100 resultiert aus der
größeren Anzahl von Freiheitsgraden verglichen mit dem unsubstituierten
Polynorbornen, die zu einer Aufweitung der Minima führt.
7.1.1.5 Experimentelle Charakterisierung
Bei der Untersuchung der strukturellen Eigenschaften der Polynorbornencarbonsäure-
methylester durch Röntgenweitwinkelstreuung erhält man das in Abbildung 7.8
dargestellte Diffraktogramm. Die untersuchten Proben zeigten die für das
unsubstituierte Polynorbornen charakteristischen zwei amorphen Halos. Diesen Halos
konnten Abstände von 4,5 Å bzw. 11 Å zugeordnet werden.

Modifikation der Rotationsbehinderung starrer Knäuel 137
5 10 15 20 25 300
20
40
60
80
100
120
140
160
180
200
220
240
rel.
Inte
nsit
ät [
a.u.
]
Streuwinkel 2Θ [°]
Abb. 7.8: Röntgenweitwinkel-Diffraktogramm von Polynorbornencarbonsäuremethylester (Pnme)
Die experimentelle Bestimmung der Dichte mit Hilfe der Schwebemethode liefert einen
Wert von 1,19 g/cm3. Dieser Wert liegt mit rund 0,1 g/cm3 über der Dichte des
unsubstituierten Polynorbornens. Das Verhältnis aus molarem Volumen Vm und Van-
der-Waals-Volumen Vw liefert einen Wert von 1,41 [181]. Dieser Wert liegt noch
unterhalb dem Wert von unsubstituierten Polynorbornen mit 1,43 und entspricht einem,
für kristalline Polymere typischen, sehr dichten Packungsverhalten der Polymerketten.
Thermogravimetrische und differentialkalorimetrische Untersuchungen haben gezeigt,
daß die Glastemperatur des Polynorbornencarbonsäuremethylesters vermutlich oberhalb
der Zersetzungstemperatur von 250°C liegt. Dieses Verhalten steht im Einklang mit der
dichten Packung, die die Beweglichkeit der Polymerketten stark einschränkt.
Bei der experimentellen Untersuchung des Relaxationsverhaltens des Polynorbornen-
carbonsäuremethylesters durch dielektrische Relaxationsspektroskopie konnte eine
Relaxation beobachtet werden. Hierbei handelt es sich um eine Sekundärrelaxation der

Modifikation der Rotationsbehinderung starrer Knäuel 138
lateralen Methylestergruppe. Die Aktivierungsenergie dieser thermisch aktivierten
Relaxation beträgt 47 kJ/mol. Die Beweglichkeit der lateralen Estergruppe wird durch
die Starrheit des Polymerbackbones und die dichte Packung des Polymers nicht
eingeschränkt. Die gemessene Aktivierungsenergie liegt innerhalb des Energiebereichs,
die bei glasig erstarrten Polymeren mit Estergruppen beobachtet werden [182,183].
7.1.2 Polynorbornenimid (PNim)
Polynorbornenimide (PNim) lassen sich durch vinylische Polymerisation von N-para-
tolyl-bicyclo[2.2.1]hept-2-en-5,6-dicarbonsäureimid unter Verwendung von Tetrakis-
acetonitril-Palladium(II)-Komplexen darstellen. Als Gegenionen der Übergangsmetall-
Komplexe kommen für die Polymerisation in Eisessig BF4-, SbF6-, AsF6
-, PF6- und
CF3SO3- in Frage [184]. Bei den nachfolgenden Computersimulationen wurden die
Modellsysteme ausschließlich aus Monomeren mit exo-ständigen Imid-Gruppen
generiert.
7.1.2.1 1D-Rotationspotential
Zur Berechnung des eindimensionalen Rotationspotentials des Polynorbornenimids
wurde, analog zum Polynorbornen, davon ausgegangen, daß die Monomeren
ausschließlich durch eine cis-exo-Verknüpfung miteinander verbunden sind.

Modifikation der Rotationsbehinderung starrer Knäuel 139
N
N
O
O
O
O
CH3
CH3
1
2
3
4
Abb. 7.10: Definition des Torsionswinkels für dimeres Norbornenimid (PNim)
Bereits aus der Strukturformel des dimeren Norbornenimids in Abbildung 7.10 läßt sich
der sterische Anspruch des Monomers erkennen.
0 30 60 90 120 150 180 210 240 270 300 330 3600
5
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
IIIII
I
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 7.11: Potentialverlauf 1D-Rotationspotential PNim
Der Vergleich des eindimensionalen Rotationspotentials in Abbildung 7.11 mit dem des
unsubstituierten Polynorbornens zeigt qualitativ denselben Potentialverlauf. Der
Unterschied zum unsubstituierten Norbornen besteht darin, daß die Rotationsbarriere für

Modifikation der Rotationsbehinderung starrer Knäuel 140
die vollständige Rotation um die zentrale Bindung im dimeren Norbornenimid mit rund
80 kJ/mol deutlich höher liegt als im eindimensionalen Rotationspotential des
unsubstituierten dimeren Norbornans. Im Potentialverlauf fällt die, im Vergleich zum
unsubstituierten Norbornen, um 15 kJ/mol höhere Potentialbarriere zwischen den
Minima I und II auf.
7.1.2.2 2D-Rotationspotential des trimeren Norbornenimids
Das Modellsystem des trimeren Norbornenimids unterscheidet sich von den bisherigen
durch das Vorliegen eines voluminösen, starren lateralen Substituenten mit hohem
sterischen Anspruch in Kombinationen mit elektronenreichen Heteroatomen bzw. einem
aromatischen Ringsystem. Das in Abbildung 7.12 dargestellte Modellsystem zur
Berechnung des zweidimensionalen Rotationspotentials läßt erwarten, daß sich die
elektronischen Wechselwirkungen zwischen den Sauerstoffatomen und den
aromatischen Systemen der lateralen Substituenten charakteristisch auf das
zweidimensionale Rotationspotential auswirken.
N
N
O
O
O
O
CH3
CH3
N
O
O
CH3
1
2
3
45
6
Abb. 7.12: Definition der Torsionswinkel für das zweidimensionale Rotationspotential für PNim

Modifikation der Rotationsbehinderung starrer Knäuel 141
Die Potentialenergie-Hyperfläche des trimeren Norbornenimids in Abbildung 7.13 zeigt
nur einige wenige energetisch bevorzugte Konformationen, die an den dunkelblauen
Farbtönen zu erkennen sind. Bei höheren Energien von rund 25 kJ/mol bilden diese
Minima einen einzigen großen, energetisch bevorzugten Konfigurationsraum.
0 30 60 90 120 150 180 210 240 270 300 330 3600
30
60
90
120
150
180
210
240
270
300
330
360
Torsionswinkel 1 [°]
Tor
sion
swin
kel 2
[°]
76.00 -- 80.00 72.00 -- 76.00 68.00 -- 72.00 64.00 -- 68.00 60.00 -- 64.00 56.00 -- 60.00 52.00 -- 56.00 48.00 -- 52.00 44.00 -- 48.00 40.00 -- 44.00 36.00 -- 40.00 32.00 -- 36.00 28.00 -- 32.00 24.00 -- 28.00 20.00 -- 24.00 16.00 -- 20.00 12.00 -- 16.00 8.000 -- 12.00 4.000 -- 8.000 0 -- 4.000
Abb. 7.13: 2D-Rotationspotential für trimeres PNim
(Energien in kJ/mol)
Die niedrigliegenden Minima auf dieser Potentialenergie-Hyperfläche entstehen
typischerweise durch Kombinationen der Potentialminima des eindimensionalen
Rotationspotentials, wie z.B. 60° und 200°, 60° und 320°, 320° und 220° oder 320° und
320°. Konformationsübergänge sind in diesem Rotationspotential erst mit Energien von
mindestens 80 kJ/mol möglich. Der Übergang erfolgt dann stets in dasselbe Minimum,
da keine weiteren Minima auf der Potentialenergie-Hyperfläche vorhanden sind.

Modifikation der Rotationsbehinderung starrer Knäuel 142
7.1.2.3 Lokale Kettendynamik 12er-Kette Polynorbornenimid
Untersucht man die lokale Kettendynamik der Polynorbornenimide mit einem
Modellsystem aus 12 Monomeren, so zeigt sich die in Abbildung 7.14 abgebildete
Torsionswinkel-Verteilung als Funktion der Temperatur.
Man erkennt, daß ähnlich wie beim unsubstituierten Polynorbornen, unterhalb 750 K
lediglich eine Potentialverbreiterung zu beobachten ist. Oberhalb dieser Temperatur
wird ein zweites diskretes Maximum beobachtet.
-180 -150 -120 -90 -60 -30 0 30 60 90 120 150 1800
20
40
60
80
100
120
140
50K 150K 250K 350K 450K 550K 650K 750K
Häu
figk
eit [
a.u.
]
Torsionswinkel φ [°]
Abb. 7.14: Temperaturabhängigkeit der Torsionswinkel-Verteilung für PNim
Nähert man die Häufigkeitsverteilungen durch Gauß-Funktionen an, so erhält man eine
Gerade, die in Abbildung 7.15 abgebildet ist.

Modifikation der Rotationsbehinderung starrer Knäuel 143
0 100 200 300 400 500 600 7000
5
10
15
20
25
30
35
40
Hal
bwer
tsbr
eite
der
Gau
ßfun
ktio
n [°
]
Temperatur [K]
Abb. 7.15: Temperaturabhängigkeit der Halbwertsbreite der Fluktuationen
Die Temperaturabhängigkeit der Halbwertsbreite beträgt im Falle des
Polynorbornenimids 0,018°/K ± 0,001°/K. Der Achsenabschnitt beträgt 4,1° ± 0,5°. Die
Steigung der Regressionsgerade liegt unterhalb der des Polynorbornens. Auf die
Bedeutung dieser beiden Parameter wurde weiter oben eingegangen.
7.1.2.4 Molekulardynamik-Simulation Polynorbornenimid 50er-Kette
Aus Hardwaregründen konnte bei den Molekulardynamik-Simulationen des
Polynorbornenimids lediglich eine Polymerkette bestehend aus 50 Monomeren (1700
Atome) unter Berücksichtigung der Partialladungen durch den Gasteiger-Marsili-Ansatz
[66] behandelt werden. Die Beschränkung war erforderlich, um die Polymerkette in
einer hinreichend kurzen Zeit mit Ladungen zu simulieren. Zur Untersuchung der

Modifikation der Rotationsbehinderung starrer Knäuel 144
zeitlichen Entwicklung der Kettendimensionen wurde ein repräsentatives Intervall von
100 ps Simulationszeit aus der Molekulardynamik-Trajektorie ausgewählt.
100 110 120 130 140 150 160 170 180 190 20015
20
25
30
35
40
45
50
55
60
65
Gyrationsradius
Kettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 7.16: Molekulardynamik-Simulation PNim bei 300 K
Abbildung 7.16 zeigt, daß der Abstand der Kettenenden und der Gyrationsradius der
Polynorbornenimid-Kette nur sehr wenig um deren Mittelwerte von rund 47,5 Å bzw.
18,1 Å fluktuieren. Die thermischen Fluktuationen belaufen sich auf 1,3 Å bzw. auf 0,1
Å. Das Verhältnis aus ⟨RE2⟩ / ⟨RG
2⟩ liefert einen Wert von 7,1, was auf eine Kettengestalt
zwischen einem Gauß-Knäuel und einem starren Stäbchen hinweist. Die Berechnung
des charakteristischen Verhältnisses C50 liefert bei 300 K einen Wert von 6,1.
Führt man dieselbe Molekulardynamik-Simulation bei 600 K durch (Abb. 7.17), zeigen
sich interessante Effekte. Die Kombination aus sterischem Anspruch der Monomere und
den elektronischen Wechselwirkungen der lateralen Substituenten führt zu einer
Expansion des Kettenendenabstandes und des Gyrationsradius.

Modifikation der Rotationsbehinderung starrer Knäuel 145
100 110 120 130 140 150 160 170 180 190 20010
15
20
25
30
35
40
45
50
55
60
65
Gyrationsradius
Kettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 7.17: Molekulardynamik-Simulation PNim bei 300 K
Bei erhöhter Temperatur, d.h. 600 K, zeigt die Polynorbornenimid-Kette im Mittel einen
Kettenendenabstand von rund 59,5 Å, während dessen Fluktuationen etwa 3,1 Å
betragen. Für den Gyrationsradius findet man einen Mittelwert von 21,5 Å bzw.
Fluktuationen von 0,5 Å. Das Verhältnis aus ⟨RE2⟩ / ⟨RG
2⟩ liefert bei einer Temperatur
von 600 K einen Wert von 7,5, was auf einen Übergang zu einer starren, stärker
stäbchenförmigeren Kettengestalt hinweist. Die Berechnung des charakteristischen
Verhältnisses C50 liefert bei 300 K einen Wert von 9,5.
Der Vergleich der Kettendimensionen bei unterschiedlichen Temperaturen im Vakuum
zeigt, daß sowohl der Kettenendenabstand als auch der Gyrationsradius der
Polynorbornenimidkette mit steigender Temperatur zunehmen. Durch die thermisch
bedingte, erhöhte Beweglichkeit des Polymerbackbones führt die elektrostatische
Abstoßung der lateralen Substituenten zu einer Aufweitung der Kettendimensionen. Der

Modifikation der Rotationsbehinderung starrer Knäuel 146
Temperaturkoeffizient τ der ungestörten Kettendimension berechnet sich nach der
folgenden Gleichung [1]:
τ =d r
dT
ln 02
(7.1)
Aus den Molekulardynamik-Simulationen für die Polynorbornenimide bei 300 K und
600 K berechnet sich dieser Koeffizient zu 1,502�103dln⟨r02⟩/dT. Ähnliche positive
thermische Ausdehnungskoeffizienten der Kettendimension findet man bei ataktischem
Polystyrol (aPS) mit 0,3�103dln⟨r02⟩/dT [185] oder bei Polydimethylsiloxan (PDMS) mit
0,78�103dln⟨r02⟩/dT [186].
7.1.2.5 Experimentelle Charakterisierung
Polynorbornenimide zeichnen sich durch eine Reihe von Besonderheiten gegenüber den
unsubstituierten Polynorbornenen aus.
Das Röntgenweitwinkel-Diffraktogramm (WAXS) in Abbildung 7.18 dokumentiert
grundsätzlich den amorphen Charakter des Polynorbornenimids. Überraschend ist
jedoch die Tatsache, daß das Polynorbornenimid im Gegensatz zu den beiden Halos des
unsubstituierten Polynorbornens drei amorphe Halos aufweist. Im Diffraktogramm des
Polynorbornenimids sind die beiden Halos bei großen Winkel sehr breit. Die
entsprechenden Abstände auf mikroskopischer Ebene lassen sich 16,2 Å, 6,7 Å sowie
5,2 Å zuordnen.

Modifikation der Rotationsbehinderung starrer Knäuel 147
2 4 6 8 10 12 14 16 18 20 22 240
20
40
60
80
100
120
140
160
180
200
rel.
Inte
nsit
ät [
a.u.
]
Streuwinkel 2Θ [°]
Abb. 7.18: Röntgen-Weitwinkel-Diffraktogramm (WAXS) von Polynorbornenimid (PNim)
Der größte Abstand läßt sich mit Hilfe von quantenmechanischen Rechnungen und
Molekulardynamik-Simulationen an der Einzelkette als Durchmesser einer starren,
röhrenartigen Kettenkonformation zuordnen (Abbildung 7.19).
N
O
O
CH3
8,1 Å
Abb. 7.19. Länge des monomeren Norbornenimids aus quantenmechanischen Rechnungen

Modifikation der Rotationsbehinderung starrer Knäuel 148
Die differentialkalorimetrische Untersuchung (DSC) der Glasumwandlung liefert für
das Polynorbornenimid eine hohe Glasumwandlungstemperatur von 370°C (Abb. 7.20).
Die Glasumwandlung des unsubstituierten Polynorbornens konnte bei einer Temperatur
von 210°C beobachtet werden [82].
230 240 250 260 270 280 290 300 310 320 330 340 350 360 370 3806,0
6,5
7,0
7,5
8,0
8,5
9,0
9,5
10,0
10,5
11,0
11,5
12,0
mW
Temperatur [°C]
Abb. 7.20: Differentialkalorimetrische Bestimmung der Glastemperatur von Polynorbornenimid
Die Bestimmung der Dichte mit Hilfe der Schwebemethode liefert einen sehr hohen
Wert von 1,247 g/cm3. Das Verhältnis aus molarem Volumen Vm und Van-der-Waals-
Volumen liefert Vw einen Wert von 1,39, was auf ein sehr dichtes Packungsverhalten der
Polymerketten schließen läßt.

Modifikation der Rotationsbehinderung starrer Knäuel 149
7.2 Modifikation des Polymerbackbones durch Heteroatome
Eine weitere Möglichkeit zur Modifikation der Flexibilität der Polynorbornene bietet
der Einbau von Heteroatomen in das Polymerbackbone. Durch diese Möglichkeit zur
Strukturvariation lassen sich verschiedene funktionelle Gruppen in das Polymer
einfügen, die weitergehende Modifikationen erlauben. Am Beispiel von zwei
ausgewählten Modellsystemen, dem Polynorbornenoxid (PNox) und
Polynorbornenamid (PNam), sollen die in Abbildung 7.21 gezeigten Möglichkeiten zur
Modifikation des Polymerbackbones vorgestellt werden.
O ** n
NH
O
*
On
NHm
Abb. 7.21: Backbone-modifizierte Polynorbornene (PNox, PNam)
7.2.1 Polynorbornenamide (PNam)
Polynorbornenamide lassen sich in homogener Phase durch Polykondensation von
Norbornandicarbonsäurechlorid zusammen mit Ethandiamin-1,2 darstellen [187]. Diese
Reaktion erlaubt die einfache Variation der Eigenschaften über den Spacer durch eine
geeignete Wahl der Amin-Komponente. Die Polynorbornenamide sind in wasserfreier
Ameisensäure löslich. Die Präparation der Polyamide aus Lösung ist aufgrund des
Lösemittels nur eingeschränkt möglich.

Modifikation der Rotationsbehinderung starrer Knäuel 150
7.2.1.1 1D-Rotationspotential
Zur Betrachtung des eindimensionalen Rotationspotentials wurde vom einfachsten Fall
ausgegangen, d.h. von der Annahme, daß die verwendete Amin-Komponente keine
Methylen-Gruppen enthält. Betrachtet man die Amid-Gruppe zwischen zwei
Norbornan-Einheiten als planar und starr, finden sich im Polynorbornenamid zwei
rotierbare Bindungen.
NH
O
1 2
Abb. 7.22: Rotierbare Bindungen im dimeren Norbornenamid
Bei der Berechnung der einzelnen Rotationspotentiale mit Hilfe von
Molekularmechanik-Rechnungen wurde der Torsionswinkel schrittweise um 1° gedreht
und das resultierende molekulare System hinsichtlich der Energie minimiert. Die
Partialladungen im Modellsystem wurden mit Hilfe des Gasteiger-Marsili-Ansatzes
berücksichtigt.

Modifikation der Rotationsbehinderung starrer Knäuel 151
0 30 60 90 120 150 180 210 240 270 300 330 3600
5
10
15
20
25
30
35
40
45
50
Norbornen-NH (1) Norbornen-CO (2)
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 7.23: Eindimensionale Rotationspotentiale im dimeren Norbornenamid
Aus Abbildung 7.23 ist erkennbar, daß die planare Anordnung der an der Definition der
Torsion 1 beteiligten Atome eine energetisch bevorzugte Konformation bildet. Die
Planarität wird durch das Potentialminimum bei einem Torsionswinkel von 180°
gewährleistet. Für Torsion 2, d.h. Drehung der Bindung zwischen der Carboxyl-Gruppe
und dem Norbornangerüst, ergibt sich ein globales Minimum bei 310°. Da die
Potentialminima bei 305° und 245° näherungsweise dieselbe Energie besitzen, sollten
beide Minima gleich stark besetzt sein.
Die dynamischen Eigenschaften der Polynorbornenamide werden durch zwei Torsionen
1 und 2 (Abb. 7.22) bestimmt. Es ist zu erwarten, daß die Rotationspotentiale der
Bindungen zwischen der starren Amid-Gruppe und den Norbornan-Einheiten
unabhängig voneinander sind. Diese Vermutung kann durch die Berechnung des
zweidimensionalen Rotationspotentials unter Berücksichtigung dieser beiden Bindungen
überprüft werden.

Modifikation der Rotationsbehinderung starrer Knäuel 152
7.2.1.2 2D-Rotationspotential Polynorbornenamid(PNam)
Bei der Berechnung des zweidimensionalen Rotationspotentials wurde dasselbe
Modellsystem wie in Abschnitt 7.2.1.1 verwendet. Beide Torsionswinkel wurden
schrittweise um jeweils 5° geändert und anschließend die entsprechende
Potentialenergie unter Berücksichtigung der Partialladungen berechnet. Die
Potentialenergie-Hyperfläche in Abbildung 7.24 wurde durch 1369 Rechnungen an
einzelnen Strukturen generiert.
0 30 60 90 120 150 180 210 240 270 300 330 3600
30
60
90
120
150
180
210
240
270
300
330
360
Tor
sion
swin
kel 2
PN
-CO
[°]
Torsionswinkel1 PN-NH [°]
38.00 -- 40.00 36.00 -- 38.00 34.00 -- 36.00 32.00 -- 34.00 30.00 -- 32.00 28.00 -- 30.00 26.00 -- 28.00 24.00 -- 26.00 22.00 -- 24.00 20.00 -- 22.00 18.00 -- 20.00 16.00 -- 18.00 14.00 -- 16.00 12.00 -- 14.00 10.00 -- 12.00 8.000 -- 10.00 6.000 -- 8.000 4.000 -- 6.000 2.000 -- 4.000 0 -- 2.000
Abb. 7.24: 2D-Rotationspotential von Norbornenamid
(Energien in kJ/mol)
Aus der in Abbildung 7.24 dargestellten Potentialenergie-Hyperfläche erkennt man, daß
die Drehung um die PN-CO-Bindung (Abb. 7.22 (1)) eine deutlich niedrigere
Potentialbarriere besitzt als die Drehung um die PN-NH-Bindung (Abb. 7.22 (2)). Die
zweidimensionale Potentialenergie-Hyperfläche zeigt weiterhin, daß die beiden
betrachteten Torsionen weitgehend unabhängig voneinander sind. Der Verlauf des

Modifikation der Rotationsbehinderung starrer Knäuel 153
eindimensionalen Rotationspotentials von Torsion 1 bzw. Torsion 2 läßt sich in
Abbildung 7.24 wiederfinden. Über einen Schnitt der Potentialenergie-Hyperfläche bei
konstantem Torsionswinkel φ erhält man wieder den Potentialverlauf des
eindimensionalen Rotationspotentials. Die Drehung um die PN-CO-Bindung hat eine
um rund 10 kJ/mol niedrigere Potentialbarriere als die Drehung um die PN-NH-
Bindung. Die Höhe der Rotationsbarriere für die vollständige Drehung um die PN-CO-
Bindung besitzt eine Potentialbarriere von 10 kJ/mol. Es ist daher zu erwarten, daß
längere Ketten kollabieren.
7.2.1.3 Lokale Kettendynamik 12er-Kette Polynorbornenamid (PNam)
Zur Untersuchung der lokalen Kettendynamik wurde ein Modellsystem aus 12 cis-exo-
verknüpften Monomeren verwendet. Die beiden unterschiedlichen Bindungen, mit
denen die Norbornan-Einheiten mit dem jeweiligen „Comonomer“ verbunden sind,
wurden einzeln untersucht. Abbildung 7.25 zeigt die lokale Kettendynamik für die
„Amid-Torsion“.
-180 -150 -120 -90 -60 -30 0 30 60 90 120 150 1800
20
40
60
80
100
120
600K
300K
Häu
figk
eit
Torsionswinkel φ Norbornan-NH [°]
Abb. 7.25: Temperaturabhängigkeit der Torsionswinkelfluktuationen (PNam, NH-Torsion)

Modifikation der Rotationsbehinderung starrer Knäuel 154
Abbildung 7.26 zeigt die Zunahme der Torsionswinkel-Fluktuationen der „ Carboxyl-
Torsion“ mit zunehmender Temperatur.
-180 -150 -120 -90 -60 -30 0 30 60 90 120 150 1800
20
40
60
80
100
120
140
50K
600K
300K
Häu
figk
eit
Torsionswinkel φ Norbornen-CO [°]
Abb. 7.26: Temperaturabhängigkeit der Torsionswinkelfluktuationen (PNam, CO-Torsion)
Zur Quantifizierung der Torsionswinkel-Fluktuationen wurden in Abbildung 7.27 die
entsprechenden Halbwertsbreiten gegen die Temperatur aufgetragen.

Modifikation der Rotationsbehinderung starrer Knäuel 155
0 100 200 300 400 500 600 700 800 9000
5
10
15
20
25
30
35
40
45
50
55
60
Torsion PN-CO Torsion PN-NH
Hal
bwer
tsbr
eite
der
Gau
ß-Fu
nktio
n [°
]
Temperatur [K]
Abb. 7.27: Halbwertsbreiten der Torsionswinkel-Fluktuationen als Funktion der Temperatur
Die Steigung der Regressionsgeraden für die „ Amid-Torsion“ beträgt 0,066°/K ±
0,053°/K , die der „ Carboxyl-Torsion“ 0,035°/K ± 0,034. Die Aufweitungstendenz des
Konfigurationsraums der PN-NH-Bindung ist gegenüber der PN-CO-Bindung stärker
ausgeprägt, der extrapolierte Bereich bei T = 0 K ist allerdings ungewöhnlich breit.
7.2.1.4 Molekulardynamik-Simulation von Polynorbornenamid (PNam)
Die Molekulardynamik-Simulation eine Polynorbornenamid-Kette aus 100 cis-exo-
verknüpften Monomeren wurde bei einer Temperatur von 300 K durchgeführt. In
Abbildung 7.28 ist die zeitliche Entwicklung des Kettenendenabstandes und des
Gyrationsradius dargestellt.

Modifikation der Rotationsbehinderung starrer Knäuel 156
0 10 20 30 40 50 60 70 80 90 1000
20
40
60
80
100
120
140
160
180
200
220
240
Gyrationsradius
Kettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 7.28: Molekulardynamik-Simulation von Polynorbornenamid (PNam)
Ein wesentliches Ergebnis dieser Molekulardynamik-Simulation im Vakuum ist, daß die
Polymerkette kollabiert. Abbildung 7.28 zeigt, daß der Abstand der Kettenenden und
der Gyrationsradius der Polynorbornenamid-Kette nach rund 80 ps um deren
Mittelwerte von rund 26,8 Å bzw. 15,2 Å fluktuieren. Die thermischen Fluktuationen
belaufen sich auf 1,1 Å bzw. auf 0,2 Å. Das Verhältnis aus ⟨RE2⟩ / ⟨RG
2⟩ liefert einen
Wert von 3,1, welcher charakteristisch für eine vollständig kollabierte Kette ist. Die
Berechnung des charakteristischen Verhältnisses C50 liefert bei 300 K einen Wert von
1,0. Das Kollabieren der Polymerkette ist auf das Vorliegen von zwei näherungsweise
unabhängigen Torsionen mit niedrigen Potentialbarrieren zurückzuführen.

Modifikation der Rotationsbehinderung starrer Knäuel 157
7.2.1.5 Experimentelle Charakterisierung
Die strukturellen Eigenschaften der Polynorbornenamide variieren mit der Spacerlänge
der verwendeten Amin-Komponente. Aus den Ergebnissen der Röntgenweitwinkel-
Experimente erkennt man eine deutliche Korrelation zwischen der Lage der amorphen
Halos und der Spacerlänge der Amin-Komponente. Abbildung 7.29 zeigt zwei
Streudiagramme von Polynorbornenamiden mit unterschiedlichen aliphatischen
Spacern.
5 10 15 20 25 300
50
100
150
200
250
300
350
400
450
500
10 -CH2-Spacer
4 -CH2-Spacer
Inte
nsit
ät [
a.u.
]
Streuwinkel 2Θ [°]
Abb. 7.29: Röntgenweitwinkel-Diffraktogramme verschiedener Polynorbornenamide (PNam)
Die relativ schmalen amorphen Halos in Abbildung 7.29 lassen die Spekulation über
eine schwache hexagonale Ordnung der Polynorbornenamide zu [188]. Durch Tempern
der Proben konnte keine Verbesserung der Ordnung herbeigeführt werden.

Modifikation der Rotationsbehinderung starrer Knäuel 158
Die differentialkalorimetrische Untersuchung der Polynorbornenamide liefert ebenfalls
eine deutliche Korrelation der gemessenen Glastemperatur mit der Länge des
verwendeten aliphatischen Spacers in der Polymerkette. Tabelle 7.1 zeigt eine Übersicht
über die gemessenen Glastemperaturen der Polynorbornenamide [187].
Tabelle 7.1:
Abhängigkeit der Glastemperatur als Funktion der Länge der aliphatischen Spacer
Anzahl der (-CH2-)-Gruppen Glastemperatur [°]
10 119
8 132
6 134
4 155
3 250
Aus Tabelle 7.1 geht hervor, daß die Glastemperatur der Polynorbornenamide mit
wachsender Länge des aliphatischen Spacers abnimmt.
Die Dichte der Polynorbornenamide konnte mit Hilfe der Schwebemethode zu 1,13
g/cm3 bestimmt werden. Die Untersuchung des Packungsverhaltens durch das
Verhältnis aus molarem Volumen Vm und Van-der-Waals-Volumen Vw liefert einen
Wert von 1,46. Dieser Wert entspricht einem Packungsverhalten, der typischerweise bei
kristallinen Polymeren gefunden wird.

Modifikation der Rotationsbehinderung starrer Knäuel 159
7.2.2 Polynorbornenoxide (PNox)
Polynorbornenoxide lassen sich durch kationische Polymerisation von exo-2,3-
Epoxybicylo[2.2.1]heptan darstellen [188]. Die Betrachtung der Polynorbornenoxide
wird sich auf den Grenzfall des cis-exo-verknüpften Polymers beschränken. Aufgrund
des kationischen Mechanismus sind bei der Polymerisation ist neben der 2,3- auch die
2,7-Verknüpfung zu beobachten [189].
Polynorbornenoxide besitzen als charakteristisches Merkmal den sp3-hybridisierten
Sauerstoff als flexiblen Spacer zwischen den einzelnen Norbornan-Einheiten.
Abbildung 7.30 zeigt einen entsprechenden Kettenausschnitt.
O
Abb. 7.30: Kettenausschnitt aus Polynorbornenoxid (PNox)
Es ist zu erwarten, daß die dynamischen Eigenschaften des Polynorbornenoxids durch
die zusätzlichen Rotationsfreiheitsgrade des Sauerstoffs bzw. durch das sehr breite
Potentialminimum des eindimensionalen Rotationspotentials beeinflußt werden.
Bei den folgenden Computersimulationen wurden die Partialladungen durch
Anwendung des Charge Equilibration-Verfahrens alle 50 Rechenschritte neu berechnet.

Modifikation der Rotationsbehinderung starrer Knäuel 160
7.2.2.1 1D-Rotationspotential Polynorbornenoxid
Aufgrund der Symmetrieebene in Abbildung 7.30 wurde zur Berechnung des
eindimensionalen Rotationspotentials der Torsionswinkel durch das Sauerstoffatom
gelegt.
0 30 60 90 120 150 180 210 240 270 300 330 3600
5
10
15
20
25
30
35
40
III
III
rel.
Ene
rgie
[kJ
/mol
]
Torsionswinkel φ [°]
Abb. 7.31: 1D-Rotationspotential für Polynorbornenoxid (PNox)
Bei der Berechnung des Rotationspotentials für das dimere Norbornenoxid führt die
Flexibilität des sp3-hybridisierten Sauerstoffspacers zu einem Potentialverlauf, der
deutlich von dem des dimeren Norbornans abweicht (Abbildung 7.31). Die Minima I
und II bei 80° bzw. 170° sollten aufgrund ihrer geringen energetischen Unterschiede bei
Raumtemperatur gleichmäßig besetzt sein. Die niedrige Potentialbarriere zwischen
diesen Minima wird mit einer Höhe von rund 5 kJ/mol bereits bei Raumtemperatur
überwunden werden [1]. Hierdurch ist ein breiter Torsionswinkel von rund 90° besetzt.
Die Potentialbarriere für die vollständige Rotation der Bindung besitzt eine Höhe von
rund 33 kJ/mol.

Modifikation der Rotationsbehinderung starrer Knäuel 161
7.2.2.2 2D-Rotationspotential Polynorbornenoxid
Die Berechnung des zweidimensionalen Rotationspotentials mit Hilfe von
Molekularmechanik-Rechnungen liefert die in Abbildung 7.32 dargestellte
Potentialenergie-Hyperfläche.
0 30 60 90 120 150 180 210 240 270 300 330 3600
30
60
90
120
150
180
210
240
270
300
330
360
Tor
sion
swin
kel 2
[°]
Torsionswinkel 1 [°]
84.00 -- 87.00 81.00 -- 84.00 78.00 -- 81.00 75.00 -- 78.00 72.00 -- 75.00 69.00 -- 72.00 66.00 -- 69.00 63.00 -- 66.00 60.00 -- 63.00 57.00 -- 60.00 54.00 -- 57.00 51.00 -- 54.00 48.00 -- 51.00 45.00 -- 48.00 42.00 -- 45.00 39.00 -- 42.00 36.00 -- 39.00 33.00 -- 36.00 30.00 -- 33.00 27.00 -- 30.00 24.00 -- 27.00 21.00 -- 24.00 18.00 -- 21.00 15.00 -- 18.00 12.00 -- 15.00 9.000 -- 12.00 6.000 -- 9.000 3.000 -- 6.000 0 -- 3.000
Abb. 7.32: 2D-Rotationspotential von Polynorbornenoxid (PNox)
(Energien in kJ/mol)
Das zweidimensionale Rotationspotential in Abbildung 7.32 zeigt, daß die beiden
Bindungen nur schwach gekoppelt sind. Der Potentialverlauf des eindimensionalen
Rotationspotentials in Abbildung 7.31 kann mit Hilfe eines geeigneten Schnittes durch
die Potentialenergie-Hyperfläche wiedergefunden werden. Durch das zweidimensionale
Rotationspotential in Abbildung 7.32 kann ein Minimumspfad so gelegt werden, daß ein
Konformationsübergang, der in diesem Fall mit einer vollständigen Rotation verbunden
wäre, eine Energie von maximal 15 kJ/mol erfordert.

Modifikation der Rotationsbehinderung starrer Knäuel 162
7.2.2.3 Lokale Kettendynamik 12er-Kette Polynorbornenoxid (PNox)
Bei den Molekulardynamik-Simulationen mit dem Modellsystem aus 12
Norbornenoxid-Monomeren wurde der Torsionswinkel über das Sauerstoffatom hinweg
ausgewertet. Die Torsion ist analog zum Norbornensystem über dieselben
Kohlenstoffatome definiert, um einen direkten Vergleich mit dem unsubstituierten
Polynorbornen zu ermöglichen.
0 100 200 300 400 500 600 700 800 900 1000 11000
5
10
15
20
25
30
35
40
45
50
55
60
Hal
bwer
tsbr
eite
der
Gau
ß-Fu
nkti
on [
°]
Temperatur [K]
Abb. 7.33: Temperaturabhängigkeit der Halbwertsbreite von PNox
Die in Abbildung 7.33 aufgetragene Regressionsgerade besitzt eine Steigung von
0,033°/K ± 0,002°/K und einen Achsenabschnitt von 2,6°± 0,73°. Bei diesen
Untersuchungen zur Temperaturabhängigkeit der Halbwertsbreiten wurde nur eine
Torsion des symmetrischen Modellsystems berücksichtigt.

Modifikation der Rotationsbehinderung starrer Knäuel 163
7.2.2.4 Molekulardynamik-Simulationen an Polynorbornenoxid (PNox)
Zur Durchführung der Molekulardynamik-Simulationen wurde eine Polymerkette aus
100 2,3-cis-exo-verknüpften Norbornenoxid-Monomeren verwendet. Die
Partialladungen wurden durch den Gasteiger-Marsili-Ansatz berücksichtigt.
0 50 100 150 200 250 300 350 4000
20
40
60
80
100
120
140
160
180
200
Gyrationsradius
Kettenendenabstand
Abs
tand
[Å
]
Simulationszeit [ps]
Abb. 7.34: Molekulardynamik-Simulation von Polynorbornenoxid (PNox)
Aus Abbildung 7.34 kann man erkennen, daß die Polymerkette bereits nach 50 ps
vollständig kollabiert. Dieses Verhalten wird durch die erhöhte Beweglichkeit des
Polymerbackbones hervorgerufen. Ursache für diese Beweglichkeit sind die niedrigen
Potentialbarrieren für die Drehung um die beiden Bindungen zwischen dem
Sauerstoffatom und den beiden Bicyclen. Die Abnahme des Kettenendenabstandes
verläuft während der ersten 70 ps nahezu linear. Danach fluktuiert der
Kettenendenabstand sehr schnell um den Mittelwert von 17,7 Å. Nach dem Kollabieren
der Polymerkette beträgt der Gyrationsradius 14,7 Å. Das Verhältnis von ⟨RE2⟩ / ⟨RG
2⟩
liefert einen Wert von 1,5.

Modifikation der Rotationsbehinderung starrer Knäuel 164
7.2.2.5 Experimentelle Charakterisierung
Pulverförmige Polynorbornenoxide zeigen bei Röntgenweitwinkel-Untersuchungen als
Streudiagramm einen amorphen Halo bei 2Θ = 15°. Der in Abbildung 7.35 abgebildete
amorphe Halo ist im Gegensatz zu dem Streudiagramm des unsubstituierten
Polynorbornens, das zwei amorphe Halos zeigt, deutlich breiter und scheinbar durch
einen zweiten Halo überlagert. Durch Tempern des Polynorbornenoxids konnte keine
Veränderung des Diffraktogramms erhalten werden.
2 4 6 8 10 12 14 16 18 20 22 240
100
200
300
400
500
600
700
800
900
1000
Inte
nsitä
t [a.
u.]
Streuwinkel 2Θ [°]
Abb. 7.35: Röntgenweitwinkel-Diffraktogramm von Polynorbornenoxid (PNox)
Bei der Untersuchung des Polynorbornenoxids mit Hilfe der Thermisch-Mechanischen
Analyse (TMA) wurde ein Erweichungspunkt von 83°C beobachtet [139]. Die
Untersuchung der Glasumwandlung mittels differentialkalorimetrischer Untersuchungen
liefert eine Glastemperatur von 81°C. Der Glasübergang des Polynorbornenoxids ist

Modifikation der Rotationsbehinderung starrer Knäuel 165
ähnlich wie beim unsubstituierten Polynorbornen ebenfalls mit einer nur geringen
Änderung der Wärmekapazität ∆Cp von 0,23 J/gK verbunden.
Das Verhältnis aus molarem Volumen Vm und Van-der-Waals-Volumen Vw liefert einen
Wert von 1,47, was auf eine weniger dichte Packung im Vergleich mit dem
Polynorbornen hindeutet.
7.3 Zusammenfassung Kapitel 7
In diesem Kapitel konnte gezeigt werden, daß das Polynorbornen als Prototyp eines
starren Knäuels durch die Einführung lateraler Substituenten bezüglich der
Kettensteifigkeit über einen weiten Bereich variiert werden kann.
Die Modifikation durch laterale Substituenten führt zu höheren Potentialbarrieren und
zu sehr rauhen Potentialenergie-Hyperflächen. Die Polymerketten zeichnen sich durch
eine stark eingeschränkte Beweglichkeit aus. Die Durchmusterung der dazugehörigen
Konfigurationsräume wird zunehmend erschwert. Im Fall des Polynorbornen-
carbonsäuremethylesters ist die Beschreibung der Konformationseigenschaften nur mit
Hilfe von Monte Carlo-Methoden möglich, da die Verwendung von Molekulardynamik-
Verfahren zu fehlerhaften Ergebnissen führt.
Die Modifikation des Polymerbackbones führt zum Verlust der Kettensteifigkeit, da die
zusätzlichen Torsionsfreiheitsgrade zu einer mehr oder weniger starken Aufweitung der
Potentialenergie-Hyperflächen führt. Die Einführung eines einzelnen Atoms im
Polymerrückgrat, wie am Beispiel des Polynorbornenoxids gezeigt, liefert zwei
zusätzliche Torsionsfreiheitsgrade. Die entsprechenden Rotationspotentiale sind
weitgehend unabhängig voneinander. Eine analoge Situation zeigt sich im Fall der

Modifikation der Rotationsbehinderung starrer Knäuel 166
Polynorbornenamide, deren Bindungen zwischen dem Bicyclus und den Amidgruppen
weitgehend unabhängig voneinander rotieren können. Die Kettensteifigkeit des
Polymerbackbones kann also nur durch sterisch anspruchsvolle Gruppierungen erhalten
werden, die einen sterischen Anspruch besitzen, der mit dem des Norbornens oder
dessen lateral substituierten Derivaten vergleichbar ist.

Zusammenfassung 167
8 Zusammenfassung
Die vorliegende Arbeit befaßt sich mit dem Konzept starrer und gleichzeitig
knäuelförmiger Polymerketten. Die experimentellen und theoretischen Untersuchungen
dienten dem Zweck, die besonderen strukturellen und dynamischen Eigenschaften
solcher neuartigen Polymersysteme herauszuarbeiten und die Auswirkung dieser
Faktoren auf die makroskopischen Eigenschaften zu überprüfen.
Das Konzept der starren Knäuel basiert auf der Anforderung, daß die Bindungswinkel
zwischen den Monomeren ungleich 180° sind und daß die Potentialbarrieren für die
Drehung um diese Bindungen sehr hoch sind. Das Auftreten hoher Potentialbarrieren im
Rotationspotential von Polymerketten und der damit verbundenen stark eingeschränkten
Beweglichkeit wurde von de Gennes mit dem Begriff des „Einzelkettenglases“
charakterisiert.
Zur Durchführung der theoretischen Untersuchungen wurden Computersimulationen
eingesetzt. Mit Hilfe von quantenmechanischen Verfahren erfolgte die Berechnung
geometrischer Eigenschaften einzelner Monomere und die Berechnung der
eindimensionalen Rotationspotentiale. Durch die Verwendung von Molekularmechanik-
Simulationen konnte der Konfigurationsraum größerer Modellsysteme auf das
Vorliegen von Nachbargruppeneffekten und auf die Kopplung benachbarter
Rotationspotentiale untersucht werden. Die Durchführung von Molekulardynamik-
Simulationen an Modellsystemen unterschiedlicher Größe hat die Beobachtung der
zeitlichen Entwicklung bestimmter mikroskopischer Größen wie z.B.
Kettenendenabstand und Gyrationsradius, Torsionswinkel, usw. ermöglicht. Zur
Untersuchung der Glasumwandlung wurde ein Modellsystem mit periodischen
Randbedingungen eingesetzt. Schließlich wurden Untersuchungen zu der
Übergangsmetall-katalysierten Polymerisation von Norbornen mit Hilfe von Kraftfeld-
Methoden durchgeführt.

Zusammenfassung 168
Die Eigenschaften der starren knäuelförmigen Polymerketten werden charakteristisch
durch die hohen Rotationsbarrieren für die Drehung um die Einfachbindung zwischen
den Monomeren beeinflußt. Die Untersuchungen zeigten, daß das Rotationspotential des
dimeren Norbornans ein globales Minimum besitzt, dessen energetische Lage sich
deutlich von den beiden anderen lokalen Minima unterscheidet. Der hohe sterische
Anspruch des Norbornans führt zu charakteristischen Nachbargruppeneffekten, die eine
sehr rauhe n-dimensionale Potentialenergie-Hyperfläche verursachen. Die Ausbildung
diskreter, stark lokalisierter Potentialminima führt zu einigen wenigen, energetisch
bevorzugten Konformationen. Konformationsübergänge sind bei starren knäuelförmigen
Polymerketten bzw. beim Polynorbornen aufgrund der hohen Rotationsbarrieren auf der
n-dimensionalen Potentialenergie-Hyperfläche nur selten oder gar nicht zu beobachten.
Die Molekulardynamik-Simulationen an Polynorbornen-Einzelketten im Vakuum haben
gezeigt, daß die knäuelförmigen Polymerketten nicht kollabieren und die Gestalt der
Kettenmoleküle einem engen Gauß-Knäuel entspricht. Die experimentellen
Untersuchungen zur Kettenkonformation mit Hilfe von Lichtstreu-Experimenten und
der Viskosimetrie zeigten, daß die Kettengestalt weitgehend unabhängig von der Art des
verwendeten Lösemittels ist. Hierbei zeigten die Ergebnisse der Molekulardynamik-
Simulationen eine sehr gute Übereinstimmung mit den experimentellen Befunden.
Die dynamischen Eigenschaften des Polynorbornens werden durch die stark
eingeschränkte Beweglichkeit der Polymerketten geprägt. Die Molekulardynamik-
Simulationen an Polynorbornen-Modellsystemen zeigten, daß erst oberhalb von 800 K
Konformationsübergänge zu erwarten sind.
Die experimentelle Bestimmung der Glastemperatur mit Hilfe von Röntgenkleinwinkel-
Streuung, Differentialkalorimetrie und Dynamisch-Mechanisch-Thermischer Analyse
lieferten in guter Übereinstimmung mit dem Ergebnis der Molekulardynamik-
Simulationen eine Glasumwandlung im Bereich von 210°C. Diese Glasumwandlung
wird nach den Ergebnissen der Molekulardynamik-Simulationen jedoch nicht durch
Konformationsübergänge hervorgerufen, da die kinetische Energie noch nicht

Zusammenfassung 169
ausreichend ist, um die hohen Potentialbarrieren auf der n-dimensionalen
Potentialenergie-Hyperfläche zu überwinden. Diese Relaxation, die mit einer sehr
niedrigen Änderung der Wärmekapazität und einem geringen Anstieg des thermischen
Ausdehnungskoeffizienten einhergeht, verläuft vermutlich nach dem Mechanismus der
gekoppelten Librationen. Moro et al. haben diesen Mechanismus der gekoppelten
Librationen für Relaxationen einer starren Rotorkette postuliert. Die Untersuchung der
thermisch-mechanischen Eigenschaften des Polynorbornens zeigten, daß mit dieser
Relaxation im Bereich von 210°C keine erhöhte makroskopische Beweglichkeit
verbunden ist. Die Vermutung liegt nahe, daß starre Knäuel zwei Glasumwandlungen
zeigen, wobei die erste durch gekoppelte Librationen erfolgt und die zweite nach dem
herkömmlichen Mechanismus durch Konformationsübergänge. Durch die Annahme
zweier Glasumwandlungen starrer Knäuel lassen sich die experimentellen Befunde
dieser Arbeit mit den experimentellen Ergebnissen zur Glasumwandlung des
Polynorbornens von W. Kaminsky et al. und von B. L. Goddall (B.F. Goodrich) in
Verbindung bringen.
Starre Knäuel zeigen bei der Untersuchung des kondensierten Zustands die typischen
Eigenschaften eines amorphen Festkörpers. Die strukturelle Charakterisierung mittels
Streuexperimenten liefert zwei amorphe Halos und kein Anzeichen für supermolekulare
Strukturen im Röntgenkleinwinkel-Diffraktogramm. Die experimentellen und
theoretischen Untersuchungen zu den mechanischen Eigenschaften führen
übereinstimmend zu dem Ergebnis, daß das Polynorbornen durch einen niedrigen
Elastizitätsmodul im Bereich von einem GPa und durch eine hohe Sprödheit geprägt
wird. Die mechanischen Eigenschaften des Polynorbornens sind mit denen von flexiblen
Polymeren, wie z.B. Polystyrol zu vergleichen.
Die Oberflächenenergie des Polynorbornens ist mit der anderer amorpher Polymere, wie
z.B. Polystyrol, vergleichbar. Aufgrund der hohen Packungsdichte und der sehr
niedrigen Beweglichkeit der Polymerketten sollten starre knäuelförmige Polymere
besondere Barriereeigenschaften besitzen. Theoretische Untersuchungen zur Diffusion
von Sauerstoff im Polynorbornen sagen makroskopische Diffusionskonstanten vorher,

Zusammenfassung 170
die deutlich unterhalb denen von Teflon oder Polyethylen niedriger Dichte (LDPE)
liegen.
Ein weiteres, wesentliches Merkmal der starren Knäuel ist die Tatsache, daß die
Kettenkonformation des Polynorbornens bereits während der Polymerisation festgelegt
wird. Mit Hilfe von Molekularmechanik-Simulationen konnte gezeigt werden, daß die
Anlagerung des neu eintretenden Monomers sowohl durch die Ligandensphäre des
Katalysators als auch durch die bereits vorhandene Polymerkette charakteristisch
beeinflußt wird. Die Insertion des Norbornens in die Kette kann im Fall des Cp*-
Chrom-Katalysators nur in einer bestimmten, aus vermutlich sterischen Gründen,
energetisch bevorzugten Orientierung erfolgen. Die auf diese Weise erhaltenen
Polynorbornene zeigen in Abhängigkeit des verwendeten Katalysators unterschiedliche
Streudigramme im Röntgenweitwinkel-Experiment, was unterschiedliche lokale
Kettenkonformationen hinweist.
Eine Modifikation der Kettensteifigkeit des Polynorbornens wurde mit verschiedenen
Norbornenderivaten realisiert. Bei den Variationsmöglichkeiten lassen sich prinzipiell
die Einführung lateraler Substituenten am Norbornen und die Modifikation des
Polymerbackbones unterscheiden. Hierbei ist jeweils die Einführung starrer oder
flexibler Gruppen möglich, die es erlaubt die Eigenschaften der starren Knäuel über
einen weiten Bereich zu variieren. Am Beispiel des
Polynorbornencarbonsäuremethylesters und des Polynorbornenimids konnte gezeigt
werden, daß laterale Substituenten im Vergleich zum unsubstituierten Polynorbornen zu
höheren Potentialbarrieren führen. Gleichzeitig war es möglich, wie z.B. im Fall der
Polynorbornenimide die Temperaturabhängigkeit der Kettengestalt zu steuern. Durch
Copolymerisation bzw. Modifikation des Polymerbackbones konnten dynamische und
strukturelle Eigenschaften über einen großen Bereich eingestellt werden. Hierzu zählten
die Modifikation des Polymerbackbones z.B. durch Amidgruppen in den
Polynorbornenamiden oder durch Sauerstoffatome im Fall des Polynorbornenoxids. Der
Vergleich der beiden Modifikationsmöglichkeiten zeigte, daß die Einführung lateraler
Substituenten zum Erhalt der Kettensteifigkeit bzw. zu einer weiteren Erhöhung führt.

Zusammenfassung 171
Im Gegensatz dazu verursacht die Einführung von Atomen in das Polymerbackbone
durch die zusätzlichen Torsionsfreiheitsgrade den Verlust der Kettensteifigkeit.
Die vorliegende Arbeit hat gezeigt, daß starre knäuelförmige Kettenmoleküle
realisierbar sind und eine Reihe neuartiger Eigenschaften besitzen.

Anhang 172
Literaturhinweise
[1] P. J. Flory, "Statistical Mechanics of Chain Molecules"; HanserVerlag/Gardner Publications Inc. (1988)
[2] G. Strobl, "The Physics of Polymers", Springer Verlag, Berlin, Heidelberg(1996)
[3] P.-G. de Gennes, J. Prost "The Physics of Liquid Crystals", Clarendon Press,Oxford (1993)
[4] P.-G. de Gennes, "Scaling Concepts in Polymer Physics", Cornell UniversityPress, Ithaca (1979)
[5] W.L. Mattice, U.W. Suter; „Conformational properties of large molecules“, J.Wiley & Sons, New York (1994)
[6] N.A. Neuburger, W.L. Mattice; in „Computersimulations of Polymers“; R.J.Roe (Ed.) Prentice-Hall, Englewood Cliffs, NJ 1991
[7] M. P. Allen, D. J. Tildesley, „Computer Simulations of Liquids“, OxfordUniversity Press (1987)
[8] D. Frenkel, B. Smit; "Understanding Molecular Simulation: From Algorithmsto Applications"; Academic Press, San Diego (1996)
[9] J.M. Haile, "Molecular Dynamics Simulation", J. Wiley & Sons, New York(1997)
[10] D.C. Rapaport, "The Art of Molecular Dynamics Simulation", CambridgeUniversity Press (1997)
[11] A. R. Leach, "Molecular Modelling"; Addison Wesley Longman Ltd., London(1996)
[12] E. Schrödinger; Ann. Physik. 79 361 (1926)
[13] H. Born, J.R. Oppenheimer; Ann. Physik. 84 457 (1927)
[14] A. Szabo, N. S. Ostlund, "Modern Quantum Chemistry"; Mc Graw-Hill,(1989)
[15] I.N. Levine; "Quantum Chemistry" Prentice-Hall, Englewood Cliffs NJ (1991)
[16] W. J. Hehre, Radom, von Rague-Schleyer, J. A. Pople; "Ab Initio MolecularOrbital Theory"; J. Wiley & Sons, New York (1986)

Anhang 173
[17] Gaussian 94 Revision C.2, M. J. Frisch, G.W. Trucks, H. B. Schlegel, M.Head-Gordon, P. M. W. Gill, M. W. Wong, J. B. Foresman, B. G. Johnson, M.A. Robb, J. R. Cheeseman, T. Keith, G. A. Petersson, J. A. Montgomery, M.A. Al-Laham, V. G. Zakrzewski, J. V. Ortiz, J. Cioslowski, B.B. Stefanov, A.Nanayakkara, M. Challacombe, C. Y Peng, P. Y. Ayala, W. Chen, E. S.Replogle, R. Gomperts, L. L. Andres, K. Raghavachari, J. S. Binkley, C.Gonzales, R. L. Martin, D. J. Fox, D. J. Defrees, J. Baker, J. P. Stewart, J. A.Pople; Gaussian Inc., Pittsburgh PA (1995)
[18] M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H.Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M.Dupuis, J. A. Montgomery; J. Comput. Chem., 14, 1347 (1993)
[19] J. B. Foresman, A. Frisch, "Exploring Chemistry with Electronic StructureMethods" Gaussian Inc., Pittsburgh PA (1993)
[20] W. J. Hehre, R.F. Stewart, J. A. Pople; J. Chem. Phys., 51, 2657 (1969)
[21] W. J. Hehre, R. Ditchfield, R. F. Stewart; J. Chem. Phys., 52, 2769 (1970)
[22] J. S. Binkley, J. A. Pople, J. Hehre; J. Chem. Phys., 51, 2657 (1969)
[23] R. Ditchfield, W. J. Hehre, J. A. Pople; J. Chem. Phys., 54, 724 (1971)
[24] W. J. Hehre, R. Ditchfield, J. A. Pople; J. Chem. Phys., 56, 2257 (1971)
[25] P. J. Hay, W. R. Wadt; J. Chem. Phys., 82, 270 (1985)
[26] W. R. Wadt, P. J. Hay; J. Chem. Phys., 82, 284 (1985)
[27] P. J. Hay, W. R. Wadt; J. Chem. Phys., 82, 299 (1985)
[28] S. Huzinaga, J. Andzelm, M. Klobukowski, E. Radzio-Andselm, Y. Sakai, H.Tatewaki, „ Gaussian Basis Sets for Molecular Calculations“ ; Elsevier,Amsterdam (1984)
[29] T. H. Dunning jr., P. J. Hay; Chapter 1 in „ Methods of Electronic StructureTheory“ ; H.F. Shaefer III (Ed.), Plenum Press, New York (1977)
[30] R. C. Binning jr., L. A. Curtiss; J. Comput. Chem., 11, 1206 (1990)
[31] A. J. H. Watchers; J. Chem. Phys., 75, 1033 (1970)
[32] A. K. Rappé, T. A. Smedley, W. A. Goddard III; J. Phys. Chem., 85, 2607(1981)
[33] R. G. Parr; Ann. Rev. Phys. Chem., 34, 631 (1983)

Anhang 174
[34] E. Wimmer, in „ Density Functional Methods in Chemistry“ ; (Eds. J. R.Labanowski, J. W. Andzelm); Springer Verlag, Berlin (1991)
[35] P. Hohenberg, W. Kohn; Phys. Rev. B., 136, 864 (1964)
[36] W. Kohn, L. J. Sham; Phys. Rev., 140, A1133 (1965)
[37] A. D. Becke; Phys. Rev. A., 38, 3098 (1988)
[38] J. P. Perdew, Y. Wang; Phys. Rev. B., 45, 1324 (1992)
[39] T. Ziegler; Chem. Rev., 91, 651 (1991)
[40] P. Pulay; Mol. Phys., 17, 197 (1969)
[41] J. A. Pople, R. Krishnan, H. B. Schlegel, D. DeFrees, J. S. Binkley, M. J.Frisch, R. F. Whiteside, R. F. Hout, W. J. Hehre; Int. Quantum Chem., Symp.,15, 269 (1981)
[42] H.B. Schlegel; Adv. Chem. Phys. 67, 249 (1987)
[43] W. F. van Gunsteren, H. J. C. Berendsen; Angew. Chem., 102, 1020 (1990)
[44] D. Chandler, „ Introduction to Modern Statistical Mechanics“ , OxfordUniversity Press (1987)
[45] L. D. Landau, E. M. Lifschitz; „ Lehrbuch der Theoretischen Physik“ Band 5,Akademie Verlag Berlin (1987)
[46] U. Burkert, N. L. Allinger, „ Molecular Mechanics“ , ACS Monografie 177(1982)
[47] W. H. Press, B. P. Flannery, S. A. Teukolsky, W. T. Vetterling, „ NumericalRecipes in C“ ; Cambridge University Press (1988)
[48] S. Kirkpatrick, C. D. Gelatt, M. P. Vecchi; Science, 220 , 671 (1983)
[49] Cerius2, Version 2.0; Molecular Simulations Inc. (1995)
[50] A. K. Rappé, C. J. Casewit, K. S. Colwell, W. A. Goddard III, W. M. Skiff; J.Am. Chem. Soc., 114, 10024 (1992)
[51] S. L. Mayo, B. D. Olafson, W. A. Goddard III; J. Phys. Chem., 94, 8897(1990)
[52] J. R. Maple, M.-J. Hwang, T. P. Stockfisch, U. Dinur, M. Waldman, C. S.Ewig, A. T. Hagler; J. Comput. Chem., 15, 162 (1994)
[53] L. V. Woodcock; Chem. Phys. Lett., 10, 257 (1971)

Anhang 175
[54] H. C. Anderson; J. Chem. Phys., 72, 2384 (1980)
[55] W. G. Hoover; Phys. Rev., A31, 1695 (1985)
[56] S. Nosé; Mol. Phys., 53, 255 (1984)
[57] H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Di Nola, J. R.Haak; J. Chem. Phys., 81, 3684 (1984)
[58] D. J. Adams; Quarterly, 10, 30 (1983)
[59] P. V. K. Pant, R. M. Boyd; Macromolecules, 25, 494 (1992)
[60] L. Greengard, V.I. Roklin; J. Comput. Phys. 73, 325 (1987)
[61] H.-Q. Ding, N. Karasawa, W. A. Goddard III; J. Phys. Chem., 97, 4309 (1992)
[62] H.-Q. Ding, N. Karasawa, W. A. Goddard III; Chem. Phys. Lett. 196 6 (1992)
[63] H.L. Friedman; Mol. Phys. 29 1533 (1975)
[64] H. Alper, R. M. Levy; J. Chem. Phys., 99, 9847 (1993)
[65] B. Honig, A. Nicholls; Science, 268, 1144 (1995)
[66] J. Gasteiger, M. Marsili; Tetrahedron, 36, 3219 (1980)
[67] A. K. Rappé, W. A. Goddard III; J. Phys. Chem., 95, 3358 (1991)
[68] J.D. Roberts, J. B. Grutzner, M. Jantelat, J. B. Deuce, R. A. Smith; J. Am.Chem. Soc., 92, 7107 (1970)
[69] T. F. A. Haselwander; Dissertation Marburg (1996)
[70] D. W. van Krevelen, „ Properties of Polymers“ , Elsevier, Amsterdam (1990)
[71] M. L. Connolly; Science, 221, 709 (1983)
[72] K. Sturm; in „ Physik der Polymere“ , Vorlesungsmanuskript, 22. IFFFerienkurs, Forschungszentrum Jülich (1991)
[73] K. B. Wiberg, M. A. Murcko; J. Am. Chem. Soc., 110, 8029 (1988)
[74] D.A.C. Compton, S. Montero, W.F. Murphy; J. Phys. Chem. 84, 3587 (1980)
[75] G. D. Smith, R. C. Jaffe; J. Phys. Chem., 100, 18718 (1996)
[76] SYBYL, Tripos Associates, St. Louis, MO, 1991

Anhang 176
[77] K. N. Baker, A. V. Fratini, T. Resch, H. C. Knachel, W. W. Adams, E. P.Socci, B. L. Farmer; Polymer, 34, 1571 (1993)
[78] G. Tanaka, W. L. Mattice; Macromolecules, 28, 1049 (1995)
[79] T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff;Macromolecules, 30, 5345 (1997)
[80] K. Binder; „ Applications of The Monte Carlo Method in Statistical Physics“ ,Springer Verlag, Heidelberg (1987)
[81] D. W: Heermann, „ Computer Simulation Methods in Theoretical Physics“Springer Verlag, Heidelberg (1990)
[82] T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff; Macromol.Chem. Phys., 197, 3435 (1996)
[83] M. Doi, S. F. Edwards, „ The Theory of Polymer Dynamics“ , Clarendon Press,Oxford (1988)
[84] D. N. Theodorou, U. W. Suter; Macromolecules 18, 1467 (1085)
[85] G. D. Smith, D. Y. Yoon, W. Zhu, M. D. Ediger; Macromolecules, 27, 5563(1994)
[86] R. J. Roe, M. Mondello, H. Furuya, H. J. Yang; Macromolecules, 28, 2807(1995)
[87] H. Takeuchi, R. J. Roe; J. Chem. Phys., 94, 7446 (1991)
[88] R. H. Boyd, R. H. Gee, J. Han, Y. Jin; J. Chem. Phys., 101, 788 (1994)
[89] S. A. Krügel, Diplomarbeit Marburg (1995)
[90] H. Maletta, in „ Physik der Polymere“ , Vorlesungsmanuskripte, 22. IFFFerienkurs, Forschungszentrum Jülich (1991)
[91] H. D. Dörfler, „ Grenzflächen- und Kolloidchemie“ , Verlag Chemie,Weinheim (1994)
[92] M. D. Lechner, K. Gehrke, E. H. Nordmeier, "Makromolekulare Chemie";Birkhäuser Verlag, Basel (1993)
[93] T. F. A. Haselwander, W. Heitz, M. Maskos; Macromol. Rapid Commun., 18,689 (1997)
[94] I. Brandrup, E. H. Immergut; „ Polymer Handbook“ 3rd Edition, J. WileyInterscience (1989)

Anhang 177
[95] C. F. Fan, T. Cagin, W. Shi; Macromol. Theory Simul., 6, 83 (1997)
[96] P. W. Atkins, „ Physikalische Chemie“ , Verlag Chemie, Weinheim (1988)
[97] L. D. Landau, E. M. Lifschitz, „ Lehrbuch der Theoretischen Physik“ Band 5,Akademie Verlag Berlin (1979)
[98] U. Buchenau; in „ Physik der Polymere“ , Vorlesungsmanuskripte, 22. IFFFerienkurs, Forschungszentrum Jülich (1991)
[99] J. J. Jäckle; Rep. Prog. Phys., 49, 171 (1986)
[100] J. H. Wendorff, E. W. Fischer; Kolloid-Z. Z. Polym, 251, 876, (1973)
[101] D. Rigby, R. J. Roe; in „ Computer Simulation of Polymers“ , Prentice-Hall,Englewood Cliffs, New Jersey (1991)
[102] D. Rigby, R. J. Roe; J. Chem. Phys., 87, 7285 (1987)
[103] J. Han, R. H. Gee, R. H. Boyd; Macromolecules, 27, 7781 (1994)
[104] M. L. Dannis; Mod. Plastics, 31, 120 (1954)
[105] W. Patnode, W. J. Scheiber, J. Appl. Phys., 20, 502 (1949)
[106] R. Simha, R.F. Boyer; J. Chem. Phys. 37, 1003 (1962)
[107] T. G. Fox, P. J. Flory; J. Polym. Sci. 14, 314 (1954)
[108] T. G. Fox, P. J. Flory; J. Phys. Chem. 55, 221 (1955)
[109] R. N. Haward; „ The Physics of Glassy Polymers“ , Applied Science PublishersLtd, London 1973
[110] J. Rathje, W. Ruhland; Colloid Polym. Sci., 254, 358 (1976)
[111] W. Wiegand, Dissertation Marburg (1977)
[112] Persönliche Mitteilung B. Goddall, B. F. Goodrich
[113] Persönliche Mitteilung M. Arndt, Universität Hamburg
[114] M. P. Stevens, „ Polymer Chemistry“ , Oxford University Press, New York(1990)
[115] W. E. Moe, M. D. Ediger; Macromolekules, 29, 5484 (1996)
[116] I. M. Ward, D. W. Hadley, „ Mechanical Properties of solid Polymers“ ; J.Wiley & Sons, Chichester (1993)

Anhang 178
[117] A. A. Jones, W. H. Stockmayer; J. Polym. Sci., Polym. Phys. Ed., 15, 847(1977)
[118] B. Valeur, J.-P. Jarry, F. Geny, L. Monnerie; J. Polym. Sci., Polym. Phys. Ed.,13, 667 (1975)
[119] E. Helfand; J. Polym. Sci., Polym. Symp., 73,39 (1985)
[120] K. Zemke, B. F. Chmelka, K. Schmidt-Rohr, H. W. Spiess; Macromolecules,24, 6874 (1991)
[121] D. Schaefer, H. W. Spiess; J. Chem. Phys., 97, 7944 (1992)
[122] G. J. Moro, P. L. Nordio; Mol. Phys., 56, 255 (1985)
[123] G. J. Moro, P. L. Nordio; Mol. Phys., 57, 947 (1986)
[124] A. Ferrarini, G. Moro, P. L. Nordio; Mol. Phys., 63, 225 (1988)
[125] G. J. Moro; J. Chem. Phys., 94, 8577 (1991)
[126] G. J. Moro; J. Chem. Phys., 97, 5749 (1992)
[127] H. D. Noether; J. Polym. Sci. Part C 16, 725 (1967)
[128] P. Corradini, P. Ganis; J. Polym. Sci. 43, 311 (1960)
[129] H. G. Kilian, K. Bouekes; J. Polym. Sci. 58, 311 (1962)
[130] M. Hutnik, F. T. Gentile, P. J. Ludovice, U. W. Suter, A. S.Argon;Macromolecules, 24, 5962 (1991)
[131] Y. Li, W. L. Mattice; Macromolecules, 25, 4942 (1992)
[132] J. P. Hansen, I. R. McDonald; „ Theory of Simple Liquids“ , Academic Press,New York (1986)
[133] D. A. McQuarrie; „ Statistical Mechanics“ , Harper and Row, New York (1976)
[134] S. F. Sun; „ Physical Chemistry of Macromolecules“ ; J. Wiley & Sons, NewYork (1994)
[135] J. H. Hildebrand, R. L. Scott, „ The Solubility of Non-Electrolytes“ , Reinhold,New York (1959)
[136] H. Lechner, „ Einführung in die Kontaktwinkelmessung“ , Krüss GmbH Süd
[137] D. K. Owens, R. C. Wendt; J. Appl. Polym. Sci., 13, 1741 (1969)

Anhang 179
[138] H. Kobayashi, M. J. Owen; Macromolecules, 23, 4929 (1990)
[139] L. Börger; Diplomarbeit Marburg 1997
[140] D. N. Theodorou, U. W. Suter; Macromolecules, 19, 139 (1986)
[141] W. Voigt, „ Lehrbuch der Kristallphysik“ , Teubner, Leipzig (1928)
[142] G. Schreyer, „ Konstruieren mit Kunststoffen“ , Carl Hanser, München (1972)
[143] Prof. Dr. J. Krüger, Universität Saarbrücken, persönliche Mitteilung
[144] R. H. Boundy, R. F. Boyer, „ Styrene, Ist Polymers, Copolymers andDerivatives“ , Reinhold Publishing Corp., New York (1952)
[145] A. M. North, R. A. Pethrick, D. W. Philips; Polymer, 18, 324 (1954)
[146] H. Oberst; in „ Kunststoffe: Struktur, Physikalisches Verhalten und Prüfung“ ,(Ed. R. Nitsche, K. A. Wolf) Springer Verlag, Berlin (1962)
[147] L. D. Landau, E. M. Lifschitz; „ Lehrbuch der Theoretischen Physik“ Band 7,Akademie Verlag Berlin (1991)
[148] J. Wendling; Dissertation Marburg (1995)
[149] S. Trohalaki, A. Kloczkowski, J. E. Mark, D, Rigby, R. J. Roe in „ ComputerSimulation of Polymers“ (Ed. R. J. Roe), Prentice Hall, Englewwod Cliffs, NJ,(1991)
[150] H. Takeuchi, K. Okazaki; J. Chem. Phys., 92, 5643 (1990)
[151] H. Takeuchi; J. Chem. Phys., 93, 2062 (1990)
[152] H. Takeuchi, R. J. Roe, J.E. Mark; J. Chem. Phys. 93, 9042 (1990)
[153] F. Müller-Plathe; J. Chem. Phys., 96, 3200 (1992)
[154] F. Müller-Plathe, S. C. Rogers, W. F. van Gunsteren; Macromolecules, 25,6722 (1992)
[155] F. Müller-Plathe, S. C. Rogers, W. F. van Gunsteren; J. Chem. Phys., 98, 9895(1993)
[156] J. Han, R. H. Boyd; Macromolecules, 27, 5365 (1994)
[157] A. S. Michaels, H. J. Bixler; J. Polym. Sci., 50, 413 (1961)
[158] R. A. Pasternak, M. V. Christenson, J. Heller; Macromolecules, 3, 366 (1970)

Anhang 180
[159] L. A. Castonguay, A. K. Rappé; J. Am. Chem. Soc., 114, 5832 (1992)
[160] H.-H. Brintzinger, D. Fischer, R. Mülhaupt, B. Rieger, R. Waymouth; Angew.Chem., 107, 1255 (1995)
[161] W. Kaminsky, K. Kulper, H. H. Brintzinger, F. R. W. P. Wild; Angew. Chem.,Int. Ed. Engl., 24, 507 (1985)
[162] W. Roll, H. H. Brintzinger, B. Rieger, R. Zolk; Angew. Chem., Int. Ed. Engl.,29, 279 (1990)
[163] P. Corradini, V. Barone, R. Fusco, G. Guerra; Eur. Polym. J., 15, 1133 (1979)
[164] K. Ziegler; Angew. Chem., 76, 545 (1964)
[165] H. Cherdron; Angew. Makromol. Chem. 223, 121 (1994)
[166] M. Arndt, „ Grundlagen und Mechanismen der Polymerisation vonCycloolefinen unter Verwendung homogener Ziegler-Natta-Katalysatoren“ ,Verlag Shaker, Aachen (1994)
[167] T. Gebauer; Dissertation Marburg (1994)
[168] V. R. Jensen, K. J. Borve; Organometallics, 16, 2514 (1997)
[169] B. J. Thomas, S. K. Noh, G. K. Schulte, S. C. Sendlinger, K. H. Theopold; J.Am. Chem. Soc., 113, 893 (1991)
[170] P. J. Cossee, Catal., 3, 80 (1964)
[171] C. Mehler, W. Risse; Macromolecules, 25, 4226 (1992)
[172] T. F. A. Haselwander; Dissertation Marburg (1996)
[173] A. Sen, T.-W. Lai, R. R. Thomas; J. Organomet. Chem. 358, 567 (1988)
[174] U. Peucker; Diplomarbeit Marburg (1996)
[175] U. Peucker; Dissertation in Vorbereitung
[176] D. K. Remler, P. A. Madden; Mol. Phys., 70, 921 (1990)
[177] R. Car, M. Parrinello; Phys. Rev. Lett., 55, 2471 (1985)
[178] C. R. Landis, D. M. Root, T. Cleveland in „ Reviews in ComputationalChemistry“ , Vol. 6, (Eds. K. B. Lipkowitz, D. B. Boyd), VCH Publishers,New York (1995)

Anhang 181
[179] B. S. Heinz, W. Heitz, S. A. Krügel, F. Raubacher, J. H. Wendorff; ActaPolym. 48, 385 (1997)
[180] B. S. Heinz, F. P. Alt, W. Heitz; Macromol. Rapid Commun. 19 251 (1998)
[181] F. Raubacher; Diplomarbeit Marburg (1997)
[182] Y. Ishida, K. Yamafuji; Kolloid-Z., 177, 97 (1961)
[183] Y. Ishida; Kolloid-Z., 174, 124 (1960)
[184] W. Heitz, S. A. Krügel, R. Madan, J. H. Wendorff; Makromol. Phys. Chem.eingereicht
[185] T. A. Orofino, A. Ciferri; J. Phys. Chem., 68, 3136 (1964)
[186] J. E. Mark, P. J. Flory; J. Am. Chem. Soc., 86, 138 (1964)
[187] M. Flugel; Diplomarbeit Marburg (1995)
[188] H.-J. Zimmermann; Dissertation Darmstadt 1987
[189] M. Flugel; Dissertation in Vorbereitung
[190] F. A. Carey, R. J. Sundberg, „ Organische Chemie“ , Verrlag Chemie,Weinheim (1996)

Anhang 182
Teile dieser Arbeit wurden bereits in folgenden Tagungen und Publikationen
veröffentlicht:
Tagungsbeiträge:
• Neue Katalysatoren für die Polymerisation von Norbornensystemen bzw.
Copolymerisation von Norbornen und Ethen: Computersimulationen und
Experimente
L. Börger, T. F. A. Haselwander, W. Heitz, S. A. Krügel, F. Raubacher, U. Peucker,
J. H. Wendorff; 12. Frühjahrs-Workshop Molecular Modeling, Technische
Universität Darmstadt, 19.-20. Mai 1998
• Polynorbornene as a Model Compound for Rigid Random Coils: Simulations and
Experiments
W. Heitz, S. A. Krügel, F. Raubacher, J. H. Wendorff; CCP5/SMTG Spring School
1998, Methods in Computer Simulation, University of Bristol, 24.-31. März 1998
• Starre Knäuelmoleküle: Computersimulation und Experiment
W. Heitz, T. F. A. Haselwander, S. A. Krügel, F. Raubacher, J. H. Wendorff;
Frühjahrstagung der Deutschen Physikalischen Gesellschaft, Universität Bayreuth,
9.-13. März 1998
• Starre knäuelförmige Polymere: Computersimulationen und Experimente
W. Heitz, S. A. Krügel, F. Raubacher, J. H. Wendorff; Höchstleistungsrechnen in der
Chemie, Forschungszentrum Jülich, 16.-18. Februar 1998
• Neue Katalysatoren für die Polymerisation von Norbornen bzw. Copolymerisation
von Norbornen und Ethen: Computersimulationen und Experimente
L. Börger, T. F. A. Haselwander, W. Heitz, S. A. Krügel, U. Peucker, J. H.
Wendorff, Höchstleistungsrechnen in der Chemie, Forschungszentrum Jülich, 16.-18.
Februar 1998

Anhang 183
• Starre Knäuelmoleküle: Simulation und Experiment
W. Heitz, S. A. Krügel, F. Raubacher, J. H. Wendorff; Makromolekulares
Kolloquium Freiburg 1998
• Homogene Katalyse mit Cr(III)Katalysatoren
W. Heitz, S. A. Krügel, U. Peucker, J. H. Wendorff; Makromolekulares Kolloquium
Freiburg 1998
• Rotationsbehinderte Polymere: Einzelkettengläser - Computersimulationen und
Experimente,
S. A. Krügel, J. H. Wendorff, 11. Darmstädter Molecular Modelling Workshop,
Technische Hochschule Darmstadt, 06.-07. Mai 1997
• Synthese und Eigenschaften von rotationsbehinderten Polymeren
T. F. A. Haselwander, B. Heinz, W. Heitz, G. Herth, S. A. Krügel, J. H. Wendorff,
DECHEMA Vortragstagung Frankfurt/Main 1997
• Konformationsbehinderte Polymere: „Einzelkettengläser„
T. F. A. Haselwander, B. Heinz, W. Heitz, S. A. Krügel, F. Raubacher, J.
H.Wendorff, Makromolekulares Kolloquium Freiburg, 1997
• Structure and Properties of Cyclooolefinic Polymers: Simulation and Experiment
T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff, Molecular Modeling
of Chemicals and Materials, Molecular Graphics and Modelling Society (MGMS),
Universität Amsterdam, 09.-10. September 1996
• Eigenschaften von Polynorbornenen: Kombination von Computersimulation und
Experiment
T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff, Deutsche
Physikalische Gesellschaft Frühjahrstagung 1996, Universität Marburg, 18.-20. März
1996

Anhang 184
• Vinylische Polymerisation von Polynorbornenen mit Hilfe eines Pd(II)Katalysators:
Experimentelle Charakterisierung und Computersimulationen
T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff, Gesellschaft deutscher
Chemiker, Vortragstagung Klassische Polymere im Aufwind, Technische Universität
Dresden, 25.-26. März 1996
• Polycycloolefins: Molecular Structure and Macroscopic Properties
S. A. Krügel, J. H. Wendorff, EPF (6th European Polymer Federation) Symposium
on Polymeric Materials, Aghia Pelaghia, Kreta, 1996
• Polynorbornenes: A Combination of Computer Simulation and Experiment
T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff; BIOSYM/MSI
International Materials Science Symposium, Cambridge, 1995;
Publikationsliste:
• Polynorbornene: Synthesis, Properties and Simulations
T. F. A. Haselwander, W. Heitz, S. A. Krügel, J. H. Wendorff,
Macromol. Chem. Phys. 197, 3435 (1996)
• Poly(norbornene carboxylic acid ester)s: Synthesis and Properties,
B. Heinz, W. Heitz, S. A. Krügel, F. Raubacher, J. H. Wendorff,
Acta Polymer. 48, 385 (1997)
• Rotationally Confined Polymers: „ Single Chain Glasses“
T. F. A. Haselwander, W. Heitz. S. A. Krügel, J. H. Wendorff,
Macromolecules 30, 5345 (1997)
• Polynorbornene in the condensed state: Simulations on chain conformations and
dynamics,
S. A. Krügel, F. Raubacher, J. H. Wendorff,
Macromol. Chem. Phys. 199 757 (1998)

Anhang 185
• Polynorbornene-imides: Synthesis and Simulations,
W. Heitz, S. A. Krügel, R. Madan, J. H. Wendorff,
Macromol. Chem. Phys. eingereicht

Anhang 186
Danksagung
Herrn Prof. Dr. J. H. Wendorff danke ich für die Übernahme des Hauptreferates.
Außerdem danke ich ihm für die Betreuung und Förderung sowie für den Freiraum bei
der Gestaltung meiner Arbeit und seine stets vorhandene Diskussionsbereitschaft.
Herrn Prof. Dr. W. Heitz danke ich für die Übernahme des Koreferates und die
anregenden Diskussionen zu den Polynorbornenen.
Mein Dank gilt Frank P. Alt, Michael Flugel, Thomas F. A. Haselwander, Beate S.
Heinz, Rajni Madan und Uwe Peucker aus der Arbeitsgruppe von Herrn Prof. Dr. W.
Heitz, ohne deren präparative Tätigkeit diese Arbeit nicht möglich gewesen wäre.
Ganz besonderen Dank gilt Florian Raubacher für die intensive Zusammenarbeit und
die Diskussionen über Computersimulationen.
Weiterhin danke ich Wyneken Fimmen und Lars Börger für die Zusammenarbeit und
die stete Diskussionsbereitschaft.
Ich danke allen Mitgliedern der Arbeitsgruppe und allen Mitarbeitern des Fachbereichs,
die zum Gelingen dieser Arbeit beigetragen haben.
Meiner Mutter danke ich für die Unterstützung und Förderung während des Studiums.
Meiner Freundin Meike Schüddemage danke ich besonders für die liebevolle
Unterstützung, Geduld und das Verständnis während der letzten Jahre.