Embed Size (px)
Citation preview

Polymyositis
OA. Dr. Maya Thun,
Die Polymyositis leitet sich vom griechischen poly=viel , myo-=Muskel und -itis= entzündlich ab und würde frei übersetzt „entzündliche Muskelerkrankung mehrerer Muskelgruppen“ bedeuten. Diese Beschreibung umfasst aber eine große Anzahl an entzündlichen Muskelerkrankungen, immunologischer und nicht immunologischer Natur, wobei die Polymyositis im speziellen nur einen geringen Teil ausmacht. Die Polymyositis zählt mit der Dermatomyositis und der Einschlusskörpermyositis zur Gruppe der idiopatischen inflammatorischen Myopathien. (siehe Tabelle 1: klinische Einteilung idiopathischer Myositiden nach Sontheimer) Aufgrund neuer Erkenntnisse in der Pathogenese dieser Erkrankungen wird die derzeit noch gültige „grobe“ Einteilung mehrfach in Frage gestellt.1 Unter Polymyositis subsummiert man eine heterogene Gruppe an entzündlichen Muskelerkrankungen, immunologischer Pathogenese mit unterschiedlicher Prognose, klinischen Symptomen, differentem Krankheitsverlauf und unterschiedlichen Autoantikörpern. Die Polymyositis ist eine seltene Erkrankung. Die Prävalenz ist mit 6-7/100.000 gering. In der kaukasischen Bevölkerung liegt die Inzidenz jährlicher Neuerkrankungen zwischen 1,9 und 7,7 pro 1 Million. Die Polymyositis ist bei Frauen 2-3 mal häufiger als bei Männern.
Ätiologie und Pathogenese Die Ätiologie der Polymyositis ist weitgehend unbekannt. Mehrfacherkrankungen in Familien mit HLA-DR3 (HLA-DRB1*0301), HLA-DRw52 und HLA-DQA1*0501 Assoziation, weisen auf eine genetische Komponente in der Ätiologie der Polymyositis hin. Saisonales Auftreten von speziellen Untergruppen der Polymyositis lassen auch vermuten, dass Umwelteinflüsse, insbesondere Virusinfektionen, in der Entstehung dieser Erkrankung eine Rolle spielen. z.B. entwickeln Patienten mit anti-Jo-Autoantikörpern die ersten klinischen Beschwerden im Frühling, wohingegen Patienten mit Anti-signal-recognition-particle-Autoantikörpern den Beginn ihrer Beschwerden im Herbst angeben.2 Diese und noch weitere Auffälligkeiten legen den Einfluss von Umweltfaktoren in der Ätiologie der Polymyositis nahe. Zusätzlich erhärten diese Unterschiede zwischen den verschiedenen serologischen Untergruppen die Vermutung einer Heterogenität der Polymyositis. Zu bemerken ist auch, dass Patienten die D-penicillamin erhielten, ein Syndrom entwickelten, das weder klinisch noch serologisch von einer Polymyositis unterscheidbar war. Dieses Syndrom war nach Absetzten des Medikamentes vollständig reversibel. Dieses Phänomen lässt vermuten das einige von den „idiopathischen inflammatorischen Myopathien“ ursächlich mit einer Exposition eines bis dato nicht i-dentifizierten Agens zusammenhängt. In einzelnen Berichten wurden auch Impfungen, Virusinfektionen (-HIV) und Silikon-oder Kollagen-Implantate wie auch Silikatexposition als mögliche Auslöser einer Polymyositis beschrieben. Zwischen der Polymyositis und der Dermatomyositis gibt es pathogenetische Unterschiede. Während bei der Dermatomyositis eine humorale Immunantwort für die Muskelschädigung verantwortlich gemacht wird, steht bei der Polymyositis hingegen ein zellulärer, T-Zell

vermittelter Schädigungsmechanismus im Vordergrund. Charakteristisches histologisches Merkmal sind endomysiale T-Zellinfiltrate, die vorwiegend aus CD8-positiven T-Lymphozyten bestehen. Diese zytotoxischen T-Zellen führen zu einer Zerstörung der Muskelfasern indem sie diese bildlich umzingeln und in diese eindringen. ( siehe Abb.1) Die Nekrose der Muskelzelle wird durch Perforin Granula, sezerniert von den T-Zellen, verursacht, wobei direkt myotoxische Effekte durch Interferon-gamma, Interleukin-1- und Tumornekrosis-Faktor eine zusätzliche Rolle zu spielen scheinen. Diese charakteristische Invasion von Muskelfasern durch CD8+T-Lymphozyten fehlt bei der Dermatomyositis vollständig.3 Abb.1 Schematische Darstellung der für die Polymyositis typischen Gewebsläsion
autoaggressive T-Zellen attackieren eine Muskelfaser und verursachen eine Nekrose dieser Muskelfaser ( bei dem Anti-SRP- Syndrom sind diese Gewebsläsionen nicht nachweisbar)
Diagnose und Differentialdiagnose Die Diagnose der Polymyositis erfolgt durch den laborchemischen und histologischen Nachweis einer Myositis wenn klinisch der Verdacht einer Myositis besteht. Zur Diagnostik werden zusätzlich Hilfsmittel wie die Elektromyographie und das MRT hinzugezogen. Weiters sollten infektiöse, toxisch, metabolisch, endokrin und genetisch bedingte Myopathien ausgeschlossen werden. In 50% der Patienten mit Polymyositis können Autoantikörper nachgewiesen werden, man unterscheidet dabei „Myositis spezifische Autoantikörper“ (MSA) und „Myositis assoziierte Autoantikörper“ (MAA). (siehe Tabelle 2.) Myositis spezifische Autoantikörper sind oft früh und meist persistierend im Krankheitsverlauf nachweisbar. Diese Autoantikörper sind bei der Diagnostik hilfreich, jedoch nicht ausschlaggebend, da sie, bezogen auf die klinisch-phänomenologisch definierten Krankheitsbilder, wenig sensitiv aber dafür aber sehr spezifisch sind. Aufgrund der engen Korrelation von MSA mit relativ einheitlichen Krankheitsbildern, lassen sich unter der Polymyositis spezifische Syndrome abgrenzen, wie das Anti-synthetase Syndrom und das Anti-signal recognition particle (Anti-SRP) Syndrom. (siehe Tabelle 3.) Die Diagnosekriterien der Polymyositis von Bohan und Peter (siehe Tabelle 4.), unterscheiden jedoch nicht zwischen diesen Krankheitsentitäten, daher werden neue diagnostische Kriterien für die Polymyositis seit kurzem diskutiert (siehe Tabelle 5.). Klinisches Leitsymptom der Polymyositis ist die stammnahe Muskelschwäche, oft aber nicht immer symmetrisch . Die Patienten haben Schwierigkeiten die Arme über den Kopf zu heben oder den Kopf von der Unterlage zu heben, zusätzlich können sie Schwierigkeiten beim Schlucken entwickeln. Muskelschmerzen sind beim akuten Verlauf zusätzlich zur Muskelschwäche vorhanden, stehen aber nicht im Vordergrund.

Auch im klinischen Verlauf zeigt sich die Heterogenität der Polymyositis. Während Patienten mit Antisynthetase-Antikörper einen schubhaften, am Beginn eher schleichenden Krankheitsverlauf zeigen und klinisch eine Multiorganbeteiligung, mit Polymyositis, Polysynovitis, fibrosierender Alveolitis und „mechanic Hands, aufweisen, haben Patienten mit SRP-Antikörper einen meist akut beginnenden Verlauf und eine fulminant schwer verlaufende und oft auch therapieresistente Polymyositisform ohne Haut oder Lungenbeteiligung aber dagegen häufig mit Herzbeteiligung. Laborchemisch ist für die Polymyositis eine Erhöhung der Creatinkinase (CK) im Serum, insbesondere der CK-MM, als Hinweis einer Skelettmuskelschädigung, bezeichnend, jedoch nicht spezifisch. Differentialdiagnostisch muss daher eine CK-Erhöhung anderen Ursprungs (z.B. bei Traumen, Überbelastung, Medikamente, Makro-CK Typ 1+2) abgeklärt werden. Das MRT und die Elektromyographie, im Bereich der klinisch betroffenen Muskelgruppen, dienen zur weiteren Diagnosefindung, zusätzlich kann durch diese Methoden eine geeignete Stelle zur Muskelbiopsie ermitteln werden. Andere Ursachen eines Muskelödems im MRT (physische Überbeanspruchung, metabolische Störungen u.a.) müssen ausgeschlossen werden. Um Artefakte zu vermeiden, soll die Muskelbiopsie nicht an der Einstichstelle der EMG-Nadeln erfolgen. Eine Muskelbiopsie (histochemische + immunhistologische Untersuchung notwendig) sollte in jedem Fall angestrebt werden, um einerseits die Diagnose zu sichern und andererseits andere Muskelkrankheiten auszuschließen. Elektronenmikroskopische Untersuchungen können im speziellen zum Ausschluss einer Einschlusskörpermyositis die histologische Diagnostik ergänzen.
Medikamentöse Therapie
Die Therapie orientiert sich insbesondere nach dem Schweregrad der Krankheitsaktivität, dem Ausmaß des muskulären Befalls und an den extramuskulären Manifestationen. Als Medikamente sind derzeit die Glukokortikoide und Immunsuppressiva wie z.B. Methotrexat, Azathioprin, Ciclosporin und Cyclophosphamid in Verwendung. Mittel der ersten Wahl sind die Glukokortikoide, ihre Dosierung und Applikationsform sind vom Schweregrad der Myositis abhängig. Bei schwerer Myositis mit rasch progredientem Verlauf empfiehlt sich schon zu Therapiebeginn der zusätzliche Einsatz von Immunsuppressiva wie z.b Cyclophosphamid. Methotrexat scheint beim Antisynthetase-Syndrom besser wirksam zu sein. Cyclophosphamid ist effektiv bei fibrosierender Alveolitis und Vaskulitis. Bei refraktären Fällen ist die Kombination von Methotrexat und Azathioprin zu empfehlen. In Pivotal-Studien konnten positive Effekte der TNF-blocker auf den Krankheitsverlauf einer Polymyositis gezeigt werden
Zusammenfassung
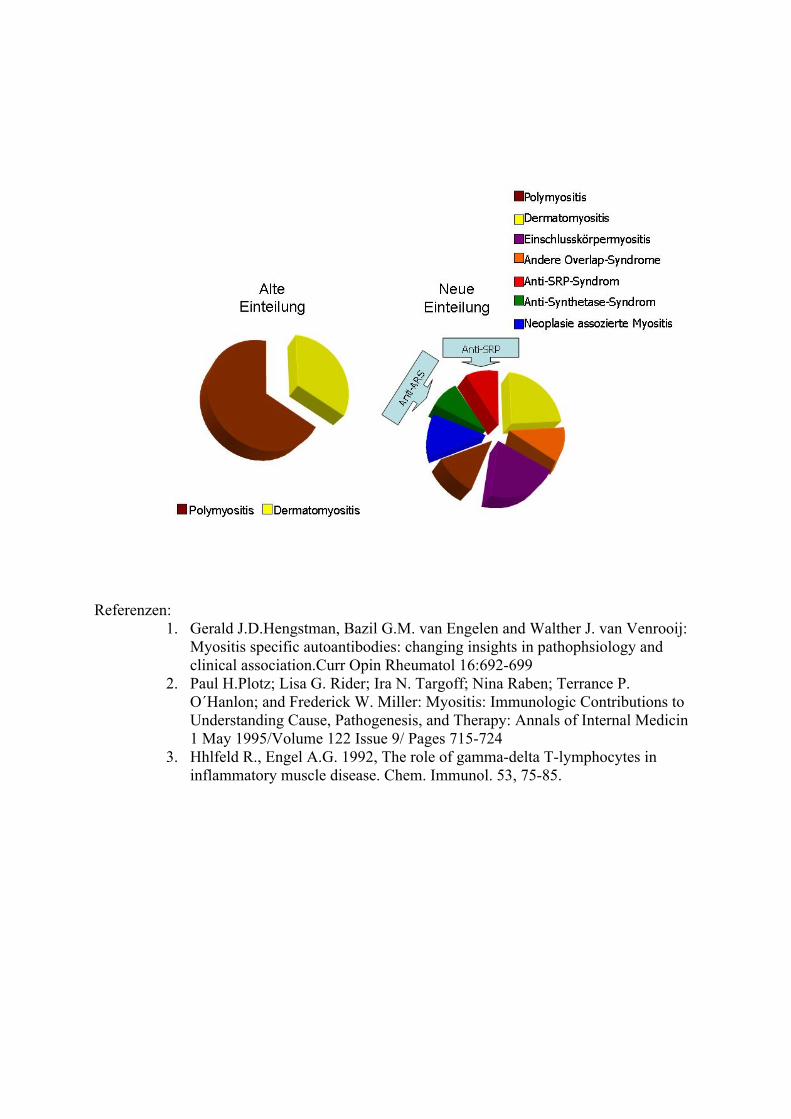
Die Polymyositis gehört zu den idipoathischen inflammatorischen Myopathien . Aufgrund der in letzter Zeit gewonnenen Erkenntnisse in der Pathogenese dieser Muskelerkrankung ist diese Krankheitsentität in einem Wandel begriffen. Derzeit kann man, durch den Nachweis von spezifischen Autoantikörper, mehrere eigenständigen Myositis-assoziierten Syndromen von der Polymyositis abgrenzen. Eine neue Klassifikation der Polymyositis ist in zu diskutieren. ( Siehe Abbildung 2)
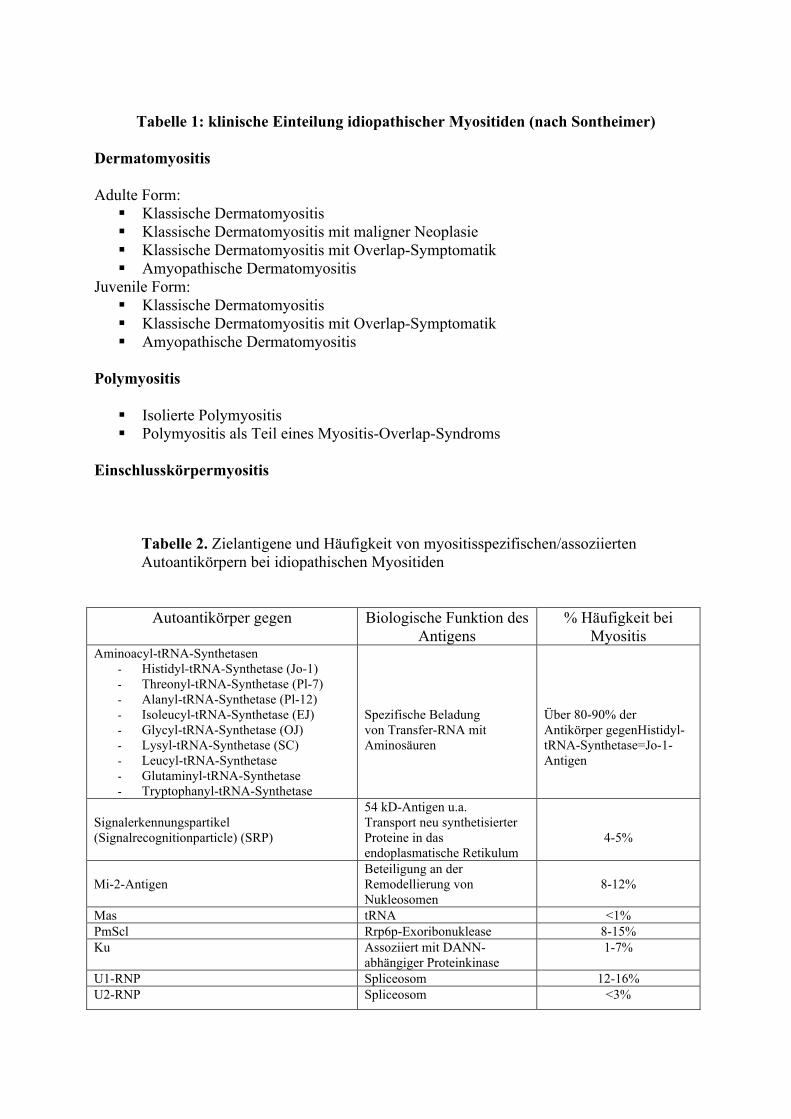
Tabelle 1: klinische Einteilung idiopathischer Myositiden (nach Sontheimer)
Dermatomyositis Adulte Form:
Klassische Dermatomyositis Klassische Dermatomyositis mit maligner Neoplasie Klassische Dermatomyositis mit Overlap-Symptomatik Amyopathische Dermatomyositis
Juvenile Form: Klassische Dermatomyositis Klassische Dermatomyositis mit Overlap-Symptomatik Amyopathische Dermatomyositis
Polymyositis
Isolierte Polymyositis Polymyositis als Teil eines Myositis-Overlap-Syndroms
Einschlusskörpermyositis
Tabelle 2. Zielantigene und Häufigkeit von myositisspezifischen/assoziierten Autoantikörpern bei idiopathischen Myositiden
Autoantikörper gegen Biologische Funktion des Antigens
% Häufigkeit bei Myositis
Aminoacyl-tRNA-Synthetasen - Histidyl-tRNA-Synthetase (Jo-1) - Threonyl-tRNA-Synthetase (Pl-7) - Alanyl-tRNA-Synthetase (Pl-12) - Isoleucyl-tRNA-Synthetase (EJ) - Glycyl-tRNA-Synthetase (OJ) - Lysyl-tRNA-Synthetase (SC) - Leucyl-tRNA-Synthetase - Glutaminyl-tRNA-Synthetase - Tryptophanyl-tRNA-Synthetase
Spezifische Beladung von Transfer-RNA mit Aminosäuren
Über 80-90% der Antikörper gegenHistidyl-tRNA-Synthetase=Jo-1-Antigen
Signalerkennungspartikel (Signalrecognitionparticle) (SRP)
54 kD-Antigen u.a. Transport neu synthetisierter Proteine in das endoplasmatische Retikulum
4-5%
Mi-2-Antigen
Beteiligung an der Remodellierung von Nukleosomen
8-12%
Mas tRNA <1% PmScl Rrp6p-Exoribonuklease 8-15% Ku Assoziiert mit DANN-
abhängiger Proteinkinase 1-7%
U1-RNP Spliceosom 12-16% U2-RNP Spliceosom <3%

Tabelle 3: klinische Syndrome und deren Autoantikörper (serologische Gruppen von Autoantikörper definieren klinische Syndrome) Autoantikörper
Charakteristische Klinisches Bild
Anti-Jo 1 und andere Antisynthetase Antikörper
Subaktuer Beginn, bevorzugt Lungenmitbeteiligung,(fibrosierende Alveolitis) Fieber, Raynaud Phänomen, PolySynovitis, „mechanic´s hands“, moderater Response auf Immunsupressive Therapie, schubweiser chronischer Verlauf
Anti-signal recognition particle (Anti-SRP)
Hoch akuter Beginn, , Beginn oft saisonal im Herbst, stark ausgeprägte Muskelschwäche, schwer verlaufende Polymyositis, 40% Herzbeteiligung, keine Hautveränderungen, Frauen häufiger betroffen als Männer, oft therapierefraktär, schlechte Überlebensprognose
Mi-2-Antigen
Chronisch rezidivierender Verlauf, klassische Dermatomyositis mit V-Zeichen,Gottronsche Pappeln, gutes Therapieansprechen, günstige Prognose
Tabelle 4. Diagnosekriterien der Poly-und Dermatomyositis (nach Bohan u. Peter 1975)
1. Symmetrische Schwäche der Schulter- / Hüftgürtelmuskulatur und der Halsflexoren, fortschreitend über Wochen und Monate mit oder ohne Dysphagie oder Beteiligung der Atemmuskulatur
2. Nachweis von Muskelfasernekrosen, Phagozytose, Regeneration mit basophilen, großen vesikulären sarkolemmalen Kernen und prominenten Nukleoli, Atrophie in perifaszikulärer Verteilung, Variabilität im Muskelfaserdurchmesser und einem entzündlichen Exsudat, oft perivaskulär
3. Erhöhung von Skelettmuskelenzymen im Serum, insbesondere der CK und oft der Aldolase, Aspartataminotransferase, Alaninaminotransferase und Laktatdehydrogenase
4. Elektromyographische Trias von kurzen, kleinen, polyphasischen Motoreinheiten, Fibrillationen, positiven, scharfen Wellen und insertionaler Irritabiltität sowie bizarren hochfrequenten wiederholten Entladungen
5. Eine der charakteristischen Hauterscheinungen der Dermatomyositis (heliotropes Gesichtexanthem, Gottron’s Papeln oder Zeichen)
Bewertung
· Ausschluss infektiöser, toxischer, metabolischer, endokriner, genetisch bedingter und anderer Myopathien
· Gesicherte Polymyositis: 4 Kriterien erfüllt
· Wahrscheinliche Polymyositis: 3 Kriterien erfüllt

· Mögliche Polymyositis: 2 Kriterien erfüllt
· Gesicherte Dermatomyositis: 3 Kriterien erfüllt plus DM-typisches Exanthem
· Wahrscheinliche Dermatomyositis: 2 Kriterien erfüllt plus DM-typisches Exanthem
· Mögliche Dermatomyositis: 1 Kriterium erfüllt plus DM-typisches Exanthem Tabelle 5. Revised diagnostic criteria of Polymyositis (Dalakas Mc Lancet 2003)
Diagnostic criteria of polymyositis
1. Myopathic weakness, which: envolves over weeks to months spares facial + eye
muscles is manifested in patients above the age of 18 years 2. Patient DOES NOT HAVE: - rash, characteristic of dermatomyositis , - a familiar history of neuromuscular disease, - exposure to myotoxic drugs (D-penicillamine, zidovudine, statins), - endocrine disease (hypothyroid, hyperthyroid, hyperparathyroid, hypercortisolism), - neurogenic disease, - dystrophies and metabolic myopathies, - IBM (excluded by clinical examination +muscle biopsy) 3. Disease can be associated with: another autoimmune disease or viral infection 4. Polymyositis is rare, as a stand alone entity 5. reconsider polymyositis if:
- disease onset is below the age of 18 years - myopathy has a slow onset and evolves over months to years (in such case think
of IBM or dystrophy) - patient has a fatigue and myalgia, without muscle weekness, even if a transient
creatine kinase elevation is seen (such patients may have fibromyalgia or fasciitis and their muscle biopsy is either normal or non-specific
- there are no typical histiological features of polymyositis, especially when there is an absence of MHC-1 or MHC/CD8 complex
Abbildung 2. Spektrum der Myositis: Gegenüberstellung alte und neuere Einteilungsmöglichkeit
Referenzen:
1. Gerald J.D.Hengstman, Bazil G.M. van Engelen and Walther J. van Venrooij: Myositis specific autoantibodies: changing insights in pathophsiology and clinical association.Curr Opin Rheumatol 16:692-699
2. Paul H.Plotz; Lisa G. Rider; Ira N. Targoff; Nina Raben; Terrance P. O´Hanlon; and Frederick W. Miller: Myositis: Immunologic Contributions to Understanding Cause, Pathogenesis, and Therapy: Annals of Internal Medicin 1 May 1995/Volume 122 Issue 9/ Pages 715-724
3. Hhlfeld R., Engel A.G. 1992, The role of gamma-delta T-lymphocytes in inflammatory muscle disease. Chem. Immunol. 53, 75-85.






























