Embed Size (px)
Citation preview

Prävalenz von Gastroschisis, Omphalozele, Spina bifida und orofazialen
Spaltbildungen bei Neugeborenen im Zeitraum Januar 2000 bis Dezember
2010 in Leipzig, Sachsen, Sachsen-Anhalt und Deutschland
Dissertation
zur Erlangung des akademischen Grades
Dr. med.
an der Medizinischen Fakultät
der Universität Leipzig
eingereicht von:
Sophia Bremer
geb. am 13.11.1989 in Leipzig
angefertigt in:
Klinik und Poliklinik für Kinder- und Jugendmedizin der Universität Leipzig
Betreuer:
Prof. Dr. med. Wieland Kiess
Beschluss über die Verleihung des Doktorgrades vom: 24.01.2017

Widmung
2
Gewidmet all denen, die mich unterstützt haben und bis zum Schluss an mich
geglaubt haben, allen voran meinem Freund Martin Hirschfeld und meinen
Eltern.

Inhaltsverzeichnis
3
Inhaltsverzeichnis
Bibliografische Beschreibung .......................................................................................... 4
I. Abkürzungsverzeichnis ............................................................................................. 6
1. Einleitung .................................................................................................................. 7
1.1 Hintergrund ........................................................................................................ 7
1.2 Gastroschisis ...................................................................................................... 8
1.3 Omphalozele .................................................................................................... 12
1.4 Orofaziale Spaltbildungen ............................................................................... 15
1.5 Spina bifida ...................................................................................................... 19
1.6 Fragestellung der Studie .................................................................................. 22
2. Publikation .............................................................................................................. 25
3. Zusammenfassung der Arbeit ................................................................................ 33
4. Literaturverzeichnis ................................................................................................ 40
II. Erklärung über die Eigenständigkeit der Arbeit ..................................................... 49
III. Lebenslauf .............................................................................................................. 50
IV. Danksagung ............................................................................................................ 52

Bibliografische Beschreibung
4
Bibliografische Beschreibung
Bremer, Sophia
Prävalenz von Gastroschisis, Omphalozele, Spina bifida und orofazialen
Spaltbildungen bei Neugeborenen im Zeitraum Januar 2000 bis Dezember 2010 in
Leipzig, Sachsen, Sachsen-Anhalt und Deutschland
Universität Leipzig, Dissertation
52 S. 1, 62 Lit. 2, 1 Artikel, 1 Abb., 2 Tab.
Referat:
Das Ziel der vorliegenden Arbeit war es, die Prävalenzen der angeborenen
Fehlbildungen Gastroschisis, Omphalozele, Spina bifida und orofazialer
Spaltbildungen bei Neugeborenen im Zeitraum Januar 2000 bis Dezember 2010 in
Leipzig, Sachsen, Sachsen-Anhalt und Deutschland zu ermitteln und auf mögliche
Trends zu untersuchen. Im Rahmen dieser retrospektiven Untersuchung erfolgte die
Datenerhebung unter Verwendung von Krankenakten des Universitätsklinikums
Leipzig, der Neonatalstatistik der Sächsischen Landesärztekammer, Vergleichsdaten
des Fehlbildungsregisters aus Sachsen-Anhalt und der Diagnosestatistik des
Forschungsdatenzentrums der Statistischen Ämter des Bundes und der Länder. Die
Prävalenz der vier Fehlbildungen betrug im Untersuchungszeitraum in Deutschland
beziehungsweise in Sachsen für Gastroschisis 1,97 bzw. 2,12, für Omphalozele 1,63
bzw. 1,48, für orofaziale Spaltbildungen 5,80 bzw. 8,11 und für Spina bifida 2,92 bzw.
2,50 je 10 000 Lebendgeborene. In Sachsen zeigte sich für alle vier Fehlbildungen eine
Zunahme der Prävalenz, allerdings mit sehr geringen Effektstärken. Auch in
Deutschland konnte eine signifikante Zunahme der Fehlbildungen beobachtet

Bibliografische Beschreibung
5
werden, ausgenommen davon war die Lebendgeborenenprävalenz der Fehlbildung
Spina bifida, die im beobachteten Zeitraum abzunehmen schien. Ob allerdings ein
tatsächlicher Anstieg der Prävalenzen besteht oder lediglich Artefakte einen Anstieg
vortäuschen, ist unklar. Änderungen in der Erfassungs- und Verschlüsselungspraxis,
Fehlcodierungen sowie Doppel- und/oder unvollständige Erfassung der Fehlbildungen
könnten die Daten verfälschen.
1 Seitenzahl insgesamt
2 Zahl der im Literaturverzeichnis ausgewiesenen Literaturangaben ohne Publikation

I. Abkürzungsverzeichnis
6
I. Abkürzungsverzeichnis
DGE Deutsche Gesellschaft für Ernährung e.V.
EUROCAT European Surveillance Systems of Congenital Anomalies
FDZ Forschungsdatenzentrum der Statistischen Ämter des Bundes und der
Länder
ICBDSR International Clearinghouse for Birth Defects Surveillance and Research
IPDTOC International Perinatal Database of Typical Orofacial Clefts
ICD-10-GM International Statistical Classification of Diseases and Related Health
Problems
KI Konfidenzintervall
LB Lebendgeborene
OR Odds Ratio
SLÄK Sächsische Landesärztekammer
SSW Schwangerschaftswoche
WHO Weltgesundheitsorganisation

1. Einleitung
7
1. Einleitung
1.1 Hintergrund
Angeborene Fehlbildungen werden als strukturelle oder funktionelle Auffälligkeiten
definiert, die zum Zeitpunkt der Geburt vorliegen. Sie entstehen während des
intrauterinen Lebens und werden pränatal, während der Geburt oder teilweise erst
im späteren Leben diagnostiziert [1]. Kongenitale Malformationen können sich
entweder als eine einzelne isolierte Anomalie oder multiple Organsysteme
betreffend, darstellen. In den Industrieländern sind sie die häufigste Ursache für die
Mortalität und Morbidität im Kindesalter [1, 2]. Jedes Jahr werden etwa 135
Millionen Kinder weltweit geboren. Ein Kind von 40, also annähernd 2,5 % aller
Neugeborenen weltweit sind von einer Fehlbildung betroffen [1, 3]. Das Auftreten
von Fehlbildungen unterscheidet sich dabei zwischen den Ländern und Regionen der
Welt, unter anderem bedingt durch unterschiedliche regionale Risikofaktoren. Die
Weltgesundheitsorganisation (WHO) schätzt die Häufigkeit angeborener
Fehlbildungen auf 40 bis 50 ‰ in den Industrienationen [2]. Pränatale Fehlbildungen
sind somit ein globales Gesundheitsproblem. In vielen Fällen sind die Ursachen für
diese Erkrankungen noch weitgehend ungeklärt. Meist ist die Ätiologie
multifaktoriell, resultierend aus verschiedenen Faktoren, die sich gegenseitig
beeinflussen [1, 4, 5]. Man geht davon aus, dass mehr als die Hälfte aller
Fehlbildungen nicht auf einen einzelnen auslösenden Faktor zurückzuführen ist [2].
Fortschritte in der Gesundheitsversorgung verbessern die Prognose vieler dieser
schweren Erkrankungen [5].
Zahlreiche Studien unter anderem aus Europa, Kanada und den USA berichten, dass
die Prävalenz von Fehlbildungen bei Neugeborenen in den letzten Jahrzehnten
zugenommen hat [48]. Das beschriebene zunehmende Auftreten von Fehlbildungen

1. Einleitung
8
ist besorgniserregend. Die Ursachen dafür sind noch weitestgehend ungeklärt [9]. Die
Zunahme von Mehrlingsgeburten in den letzten 30 Jahren, die häufiger mit
angeborenen Fehlbildungen assoziiert sind, wird als eine mögliche Ursache diskutiert
[10]. Angeborene Fehlbildungen stellen insgesamt eine erhebliche medizinisch-
ethische und gesundheitsökonomische Herausforderung dar. Sie können zu
ungünstigen Auswirkungen auf die Gesundheit und Entwicklung des Kindes bis hin
zum Tod des Kindes führen [1]. Aber auch für die Familien der Betroffenen und die
damit verbundene intensive Betreuung der Kinder sowie für die Gesellschaft stellen
Fehlbildungen eine große Herausforderung dar [11]. Die WHO meldete, dass
kongenitale Anomalien Platz 17 für die Ausgaben im Gesundheitswesen einnehmen
[1].
1.2 Gastroschisis
Gastroschisis ist definiert als eine extraumbilikale Ausstülpung der Bauchorgane
durch eine Bauchwandspalte [12]. Gastroschisis zählt mit der Omphalozele zu den
häufigsten Defekten der Bauchwand [13, 14, 15]. Diese beiden angeborenen
Fehlbildungen sind nur selten miteinander assoziiert [14]. Der Bauchwanddefekt kann
unterschiedlicher Größe sein und misst meist 3 bis 4 cm [16]. Meistens treten
Dünndarmschlingen durch den ventralen Defekt hindurch, seltener sind Magen,
Colon, Milz, Leber oder Urogenitaltrakt betroffen. Die hinausgetretenen Organe sind
von keiner schützenden membranösen Hülle, also keinem Bruchsack umgeben, was
die Gastroschisis von der Omphalozele abgrenzt [9, 14, 15]. Der Defekt ist
normalerweise rechts vom Bauchnabel gelegen. Nur sehr wenige Fälle von
linksseitigen Bauchwandspalten sind beschrieben wurden [17].

1. Einleitung
9
Pathogenese:
Die Bauchwand entsteht durch die Fusionierung von vier Faltungen, nämlich aus der
Schädelabfaltung, welche die Brustwand und den epigastrischen Teil des Abdomens
formt, dem kaudalen Blatt, welches das Perineum, die Harnblase und die
hypogastrische Region formt und zwei lateralen Blättern, die die Seitenwand des
Abdomens bilden. Diese vier Anteile treffen sich im Zentrum und vereinigen sich zum
Nabelring. In der vierten Woche der Embryonalzeit ist dieser Vorgang vervollständigt
[16]. Über die Entstehung der Gastroschisis existieren diverse Thesen: Zum einen soll
die Ruptur der Amnionmembran am Nabelschnuransatz vor vollendetem
Nabelringverschluss zum Hinausstülpen der Bauchorgane führen [16]. Andererseits
wird die abnorme Involution der rechten Vena umbilicalis, was zu einer verdünnten
Bauchwand und Herniation führt, für die Entstehung der Gastroschisis verantwortlich
gemacht. Eine geschädigte Amnionhülle wird ebenfalls als Ursache der Gastroschisis
diskutiert, welche aufgrund bisher noch nicht identifizierter Toxine resultieren soll.
Die am meisten akzeptierte Theorie besteht darin, dass sich die rechte
Mesenterialarterie intrauterin verschließt und infarziert, daraufhin die
Nabelschnurbasis nekrotisiert und die abdominalen Organe durch die entstandene
Bauchwandspalte hindurchtreten [12, 18]. Diese These wird durch die starke
Assoziation zwischen dem Auftreten der Gastroschisis und der Einnahme vasoaktiver
Substanzen durch die Mutter unterstützt [9].
Risikofaktoren:
Zu den Risikofaktoren zählen ein junges Alter der Mutter (< 20 Jahren), Infektionen
während der Gestation, Alkoholkonsum und Nikotinabusus der Mutter, ein niedriger
sozio-ökonomischer Status, ein niedriger Body-Mass-Index und weitere teratogene
Einflüsse wie zum Beispiel die Einnahme vasoaktiver Substanzen wie Aspirin,
Ibuprofen und Methylendioxymethylamphetamin (MDMA). Weiterhin geht die
verminderte Einnahme von Glutathion und alpha-Carotin sowie die vermehrte

1. Einleitung
10
Aufnahme von Nitrosaminen mit einem erhöhten Risiko für Gastroschisis einher [4, 9,
15, 19]. Einige wenige Fälle von familiärem Auftreten der Gastroschisis wurden
beobachtet, so dass eine genetische Komponente diskutiert wird [9].
Diagnostik, Therapie:
Gastroschisis wird häufig bereits im mütterlichen Serum-Screening oder in der
pränatalen Ultraschalldiagnostik detektiert. Nahezu 90 % aller Feten mit Gastroschisis
werden bereits pränatal erkannt [20]. Ein erhöhtes alpha-Fetoprotein im Serum
dürfte dabei ein erster Indikator für die Präsenz der Fehlbildung sein [21]. Eine
operative Versorgung ist frühzeitig erforderlich und besteht im Verschluss des
abdominalen Wanddefektes, um die Risiken der möglichen Verletzung innerer
Organe durch stumpfe Traumen oder durch eine Erhöhung des intraabdominellen
Druckes zu reduzieren [13]. Eine langfristige postoperative Versorgung der Patienten
ist darüber hinaus erforderlich [9]. Intestinale Dysfunktionen können aufgrund der
Gastroschisis resultieren und ursächlich für Morbidität und Mortalität des Kindes sein
[13].
Prognose:
Die Überlebenswahrscheinlichkeit von Neugeborenen mit Gastroschisis liegt
zwischen 90 und 96 % [15, 19, 22]. Die Letalität konnte in den letzten Jahren deutlich
gesunken werden, begründet mit der Entwicklung der pränatalen Diagnostik sowie
Fortschritte kinderchirurgischer Operationsmöglichkeiten und der neonatologischen
Intensivmedizin [12, 22]. Kinder mit Gastroschisis werden häufig frühzeitig und mit
intrauterinen Wachstumsrestriktionen geboren. Faktoren, die zur Morbidität der
Kinder beitragen [9]. Prognostisch entscheidend sind das Ausmaß der abdominalen
Organschädigung wie Darmatresien oder Nekrosen, ein gelegentlich auftretender
postoperativer Ileus sowie eine möglichst schnelle postoperative Genesung der
Patienten [9, 15]. Im Gegensatz zur Omphalozele ist die Gastroschisis seltener mit
weiteren Anomalien assoziiert, allerdings treten begleitend zur Gastroschisis häufig

1. Einleitung
11
intestinale Atresien oder Stenosen auf, resultierend aus der unzureichenden
Blutversorgung der betroffenen Organabschnitte [16, 21]. Verschiedene Studien
berichteten über ein gemeinsames Auftreten von Gastroschisis und
gastrointestinalen Atresien in etwa 5 bis 15 % der Fälle [23]. Zu den eher selten
auftretenden Begleitanomalien zählen ein Maldeszensus testis, das Meckeldivertikel
oder intestinale Duplikationen [16].
Epidemiologie:
Die Angaben zur Häufigkeit der Gastroschisis sind nicht einheitlich und reichen von
0,3 bis 5,5 je 10 000 Lebendgeborene [9, 14, 15, 24]. In Europa verzeichnete die
European Surveillance of Congenital Anomalies (EUROCAT) einen Aufwärtstrend der
Prävalenz der Gastroschisis von 0,54 je 10 000 Geborene in den Jahren 1980-1984 auf
2,12 je 10 000 Geborene in 2000-2002 [25]. Weltweit ermittelte man eine Zunahme
der Prävalenz von 0,1 je 10 000 Geborene in den 1970ern auf mehr als 5 je 10 000
Geborene in den frühen 2000ern [9]. Eine Zusammenschau verschiedener Studien
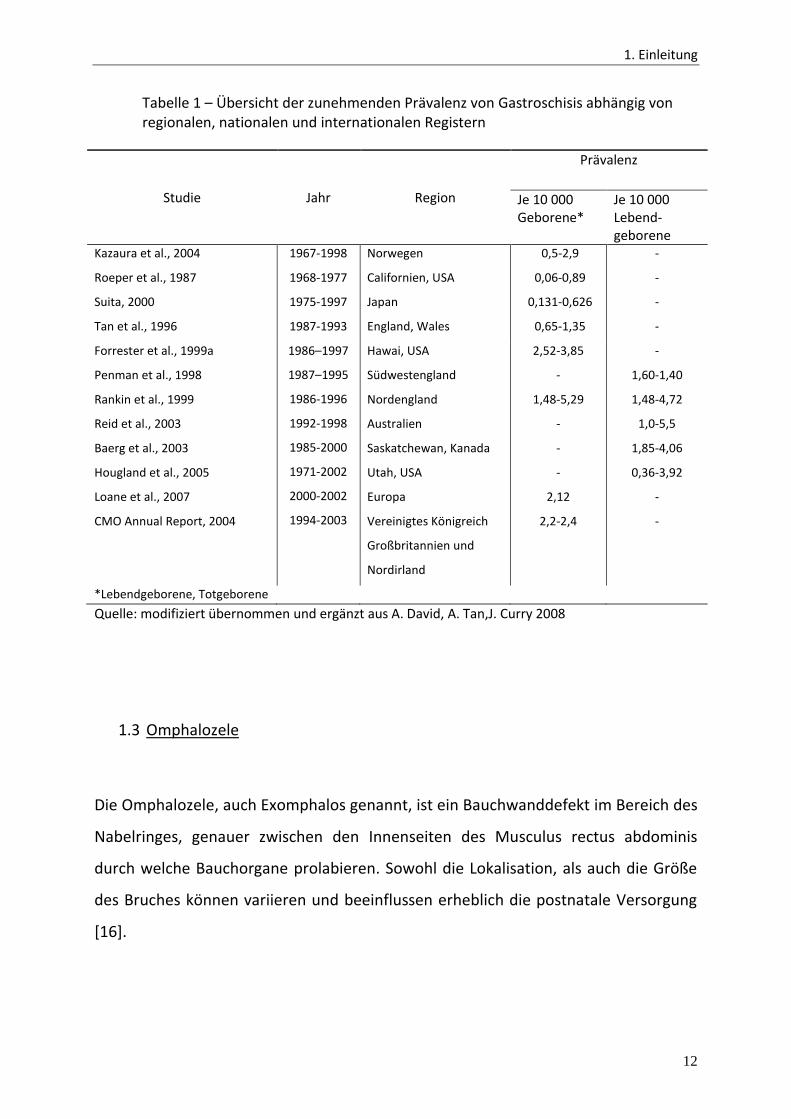
zeigt Tabelle 1. Darüber hinaus zeigen sich deutliche Unterschiede der Prävalenzen in
Herkunft und Ethnizität der Betroffenen. In weltweiten multizentrischen Studien
zeigte sich, dass häufiger Kaukasier als Kinder des afrikanischen Raumes oder des
Orients, eher Nordeuropa als Südeuropa betroffen sind [6].
1. Einleitung
12
Tabelle 1 – Übersicht der zunehmenden Prävalenz von Gastroschisis abhängig von regionalen, nationalen und internationalen Registern
Studie Jahr Region
Prävalenz
Je 10 000 Geborene*
Je 10 000 Lebend-geborene
Kazaura et al., 2004
Roeper et al., 1987
Suita, 2000
Tan et al., 1996
1967-1998
1968-1977
1975-1997
1987-1993
Norwegen
Californien, USA
Japan
England, Wales
0,5-2,9
0,06-0,89
0,131-0,626
0,65-1,35
-
-
-
-
Forrester et al., 1999a
Penman et al., 1998
Rankin et al., 1999
Reid et al., 2003
Baerg et al., 2003
Hougland et al., 2005
Loane et al., 2007
CMO Annual Report, 2004
1986–1997
1987–1995
1986-1996
1992-1998
1985-2000
1971-2002
2000-2002
1994-2003
Hawai, USA
Südwestengland
Nordengland
Australien
Saskatchewan, Kanada
Utah, USA
Europa
Vereinigtes Königreich
Großbritannien und
Nordirland
2,52-3,85
-
1,48-5,29
-
-
-
2,12
2,2-2,4
-
1,60-1,40
1,48-4,72
1,0-5,5
1,85-4,06
0,36-3,92
-
-
*Lebendgeborene, Totgeborene
Quelle: modifiziert übernommen und ergänzt aus A. David, A. Tan,J. Curry 2008
1.3 Omphalozele
Die Omphalozele, auch Exomphalos genannt, ist ein Bauchwanddefekt im Bereich des
Nabelringes, genauer zwischen den Innenseiten des Musculus rectus abdominis
durch welche Bauchorgane prolabieren. Sowohl die Lokalisation, als auch die Größe
des Bruches können variieren und beeinflussen erheblich die postnatale Versorgung
[16].

1. Einleitung
13
Pathogenese:
Bei der Omphalozele ist die Rückverlagerung der Bauchorgane in die Bauchhöhle
nach dem physiologischen Nabelbruch unzureichend oder fehlt vollständig. Diese
Rückverlagerung findet normalerweise in der 10. bis 12. Schwangerschaftswoche
(SSW) statt [16, 26]. Die Omphalozele resultiert mutmaßlich aus einer fehlerhaften
Vereinigung der lateralen mesodermalen Bauchblätter, was zu einem
Mittelliniendefekt der vorderen Bauchwand führt. Dieser Defekt manifestiert sich als
Bauchbruch von unterschiedlicher Größe und umfasst dabei zwei bis drei cm bis zu
Dimensionen, die die gesamte Bauchwand einnehmen [16]. Durch diesen Defekt
treten intraperitoneal gelegene Organe hindurch. Eine dünne Membran umhüllt die
prolabierten Organe, die unter anderem Darmschlingen, Magen, Leber, Milz,
Harnblase, Uterus oder Ovarien sein können [15]. Die innerste Schicht des
Bruchsackes ist vom Peritoneum ausgekleidet, die äußere Fläche bildet das Amnion
und dazwischen liegt die Wharton‘sche Sulze [14, 17]. Der Nabelschnuransatz
befindet sich dabei zentral auf dem Bruchsack [16].
Risikofaktoren:
Ein gehäuftes Auftreten der Omphalozele zeigte sich bei Müttern, die jünger als 20
Jahre oder älter als 35 Jahre alt waren sowie bei Auftreten von Mehrlingsgeburten
[27]. Die Lebensumstände der Mutter, im Gegensatz zur Gastroschisis, scheinen das
Risiko der Omphalozele weniger stark zu beeinflussen [16]. Allerdings zeigte sich in
einer US-amerikanischen Studie, dass Alkoholkonsum im ersten Trimenon und starker
Nikotinabusus das Risiko für das Auftreten einer Omphalozele erhöhen können [28].
Diagnostik und Therapie:
Im mütterlichen Serum zeigen sich erhöhte Werte des alpha-Fetoproteins, welche
allerdings nicht die gleichen Dimensionen wie bei der Gastroschisis erreichen. Eine
definitive sonografische Diagnose der Omphalozele kann ab der 12. SSW gestellt
werden, wenn keine Verwechslung mit dem physiologischen Nabelbruch mehr

1. Einleitung
14
besteht. Allerdings kann eine rupturierte Omphalozele im pränatalen Ultraschall
schwer von einer Gastroschisis unterschieden werden [29]. Sobald die Diagnose
Omphalozele gestellt worden ist, sollte eine umfangreiche weitere Diagnostik
erfolgen, um mögliche Begleitanomalien zu detektieren [21]. Die Entbindung erfolgt
häufig mithilfe einer Sectio caesarea, vor allem dann, wenn es sich um einen Defekt
größeren Ausmaßes handelt, um so eine mögliche Ruptur des Bruchsackes oder eine
Leberverletzung zu verhindern [16, 21]. Die Therapie beinhaltet die Stabilisation und
operative Rückverlagerung des Bruchsackinhaltes [26].
Prognose:
Bestimmender Faktor der Prognose der Omphalozele sind die häufig zusätzlich
bestehenden Begleitanomalien, die in 50 bis 70 % der Fälle auftreten. Im Gegenteil
dazu ist bei der Gastroschisis vor allem das Schädigungsausmaß der betroffenen
Bauchorgane entscheidend [13, 26]. In Tabelle 2 sind die Unterschiede und
Gemeinsamkeiten der beiden Bauchwanddefekte dargestellt (Tabelle 2).
Chromosomenaberrationen, die häufig mit der Omphalozele assoziiert sind, sind die
Trisomie 13, 18 und 21 [21, 26]. Das Risiko für chromosomale Anomalien scheint
häufiger bei Kindern mit zentraler und nicht mit epigastrischer Omphalozele
aufzutreten [21]. Man beobachtete Begleitanomalien eher bei Omphalozelen mit
kleinerem Ausmaß (<4 cm), als bei sehr großen Defekten (55% gegenüber 36%).
Begleitanomalien, die nicht den Gastrointestinaltrakt betreffen, zum Beispiel
Herzfehler oder Lungenhypoplasien, sind in bis zu 24 % der Fälle beschrieben, wobei
Herzfehler als häufigste Begleiterkrankung beobachtet wurde [21, 30].
Epidemiologie:
Die Angaben über das Auftreten der Omphalozele sind nicht einheitlich. Die Prävalenz
der Omphalozele schwankt zwischen 1,5 und 3,3 je 10 000 Geborenen [15, 24, 26, 31,
21, 12]. In einer norwegischen Studie blieb im Zeitraum von 1967 bis 1995 das
Auftreten von Omphalozele mit 2 je 10 000 Geborene stabil [55].

1. Einleitung
15
EUROCAT stellte zwischen 1998 und 2007 eine Zunahme des Auftretens von 1,77 auf
2,41 je 10 000 Geborene fest [56]. Häufiger ist dabei das männliche Geschlecht
betroffen [14].
Tabelle 2: Charakteristik von Omphalozele und Gastroschisis
Omphalozele Gastroschisis
Bruchsack Vorhanden Nicht vorhanden
Begleitanomalien Häufig Selten
Lokalisation des Defekts Nabel Rechts vom Nabel
Alter der Mutter < 20, > 35 Jahre < 20 Jahre
Entbindung Sectio caesarea, Vaginal Sectio caesarea, Vaginal
Operative Versorgung Nicht dringend Dringend
Prognostische Faktoren Assoziierte Anomalien Ausmaß der Darmschädigung
Quelle: modifiziert übernommen aus Christison-Lagay ER et al. 2011
1.4 Orofaziale Spaltbildungen
Orofaziale Spaltbildungen zählen zu den am weitesten verbreiteten angeborenen
Fehlbildungen insgesamt und stellen die häufigste Fehlbildung des Kopf- und
Halsbereiches dar [32, 33, 34]. Die Bezeichnung orofaziale Spaltbildung umfasst die
angeborenen Fehlbildungen der Lippenspalte (Q36*), der Gaumenspalte mit
Lippenspalte (Q37*) sowie der isolierten Gaumenspalte (Q35*). Am häufigsten tritt
dabei die Lippenspalte mit oder ohne Gaumenspalte (Q36*, Q37*) auf, nämlich in
mehr als 60 % der Fälle. Orofaziale Spaltbildungen werden in syndromale und nicht-
syndromale Formen unterschieden. In etwa 70 % der Fälle treten Lippenspalten mit
oder ohne Gaumenspalte als nicht-syndromale Formen auf und auch ohne andere
Geburtsschäden oder Entwicklungsstörungen [35, 36]. Hingegen werden bei

1. Einleitung
16
isolierten Gaumenspalten syndromale und nicht-syndromale Formen zu gleichen
Anteilen beobachtet [37]. Häufig zeigt sich ein gemeinsames Auftreten von
orofazialen Spaltbildungen und Chromosomenaberrationen, vor allem mit Trisomie
13 und 18 [29].
Pathogenese:
Die embryonale Entwicklung des Gesichts beginnt in der vierten SSW, die Entwicklung
der Lippen, des Gaumens und des Kiefers vollzieht sich während der vierten bis zur
zwölften SSW. Orofaziale Spaltbildungen entstehen aufgrund fehlerhaft erfolgter
Prozesse der normalen Gesichtsentwicklung [38]. Der primäre Gaumen ist in der
sechsten SSW geformt und bei der Lippenspalte betroffen. Während der sechsten bis
zwölften SSW entwickelt sich der sekundäre Gaumen, welcher bei einer
Gaumenspalte zusätzlich betroffen sein kann. Bei den Lippen- und Lippen-
Kieferspalten handelt es sich um Störungen in der Bildung der primitiven Nase
zwischen der fünften und sechsten SSW. Dabei kommt es nicht zur Fusionierung der
Nasenwülste (primäre Lippen-Kiefer-Spalte) oder die noch nicht substituierte
Epithelmauer reisst ein (sekundäre Spaltbildung). Eine Gaumenspalte entsteht durch
die unvollständige oder fehlende Vereinigung der seitlichen Gaumenfortsätze in der
achten Embryonalwoche [57].
Risikofaktoren:
Die Ursache orofazialer Spaltbildungen ist multifaktoriell. Zu den genetischen
Ursachen zählen chromosomale Veränderungen, insbesondere auf Chromosom 9
gelegen, genetische Störanfälligkeiten auf teratogene Expositionen und komplexe
genetische Mitwirkung multipler Gene. Für die Entstehung orofazialer Spaltbildungen
gehen exogene Einflüsse wie ein hohes Alter der Mutter, die Ethnizität, Nikotin-,
Alkoholabusus, übermäßiger Koffeingenuss der Mutter, die Einnahme von
Benzodiazepinen, Kortikosteroiden, Antikonvulsiva, Vitamin A, als auch andere
berufliche Expositionen mit einem erhöhten Risiko einher. Auch eine unzureichende

1. Einleitung
17
Folsäuresupplementation soll in einem kausalen Zusammenhang mit der Entstehung
orofazialer Spaltbildungen stehen [32, 39].
Komplikationen:
Orofaziale Spaltbildungen führen zu einer Vielzahl von Komplikationen. Abgesehen
von kosmetischen Problemen, können unter anderem Ohrinfektionen, Schluck- oder
Hörprobleme und Sprachentwicklungsverzögerungen auftreten, so dass eine
interdisziplinäre Versorgung der Patienten notwendig ist [32, 40]. Aber auch
psychologische, soziale und sozioökologische Auswirkungen auf das betroffene Kind
werden häufig beobachtet [39]. Neugeborene mit einer orofazialen Spaltbildung
haben lebenslang eine erhöhte Inzidenz für psychische Gesundheitsprobleme sowie
eine erhöhte Mortalitäts- und Morbiditätsrate in allen Altersstufen, vor allem in
weniger entwickelten Ländern [36, 38].
Therapie:
Eine interdisziplinäre Behandlung, bestehend aus operativer Versorgung, Logopädie,
zahnärztliche Betreuung und psychologischer Unterstützung, stellt die Basistherapie
orofazialer Spaltbildungen dar [36].
Epidemiologie:
Die weltweite Prävalenz orofazialer Spaltbildungen variiert zwischen 9,92 und 14,29
je 10 000 Geborene [11, 34, 35, 40]. Abhängig von geografischer Lage und Ethnizität
schwankt das Auftreten deutlich. Datenauswertungen zwischen 1993 bis 1998
reichen von 3,4 bis 22,9 je 10 000 Geborene [11]. Höhere Prävalenzen wurden in
Asien (1:700) und bei Ureinwohnern Amerikas (1:100) ermittelt, hingegen in Afrika
(1:2500) und bei Kaukasiern (1:1000) eher niedrigere Prävalenzen beobachtet
wurden [29, 41].

1. Einleitung
18
Die WHO und das „National Institute for Dental and Craniofacial Research“ gründeten
2003 die „International Perinatal Database of Typical Orofacial Clefts“ (IPDTOC), um
Daten über das Auftreten orofazialer Spaltbildungen systematisch zu sammeln und so
mögliche Trends erkennen zu können [34]. Dafür stehen Datenerhebungen von drei
Organisationen zur Verfügung: EUROCAT von Europa, das „National Birth Defects
Prevention Network“ (NBDPN) aus den USA sowie das „International Clearinghouse
for Birth Defects Surveillance and Research“ (ICBDSR), welches weltweit Daten über
Fehlbildungen sammelt [34]. Durch die IPDTOC wird das Auftreten der Lippenspalte
mit oder ohne Gaumenspalte (Q36*, Q37*) mit 9,92 je 10 000 Geborene angegeben.
Dabei teilt sich die Häufigkeit wie folgt auf: für die isolierte Lippenspalte (Q36*) wird
eine Prävalenz von 3,28 und für die Lippenspalte mit Gaumenspalte (Q37*) eine
Prävalenz von 6,64 je 10 000 Geborene angegeben. Mithilfe der IPDTOC konnte im
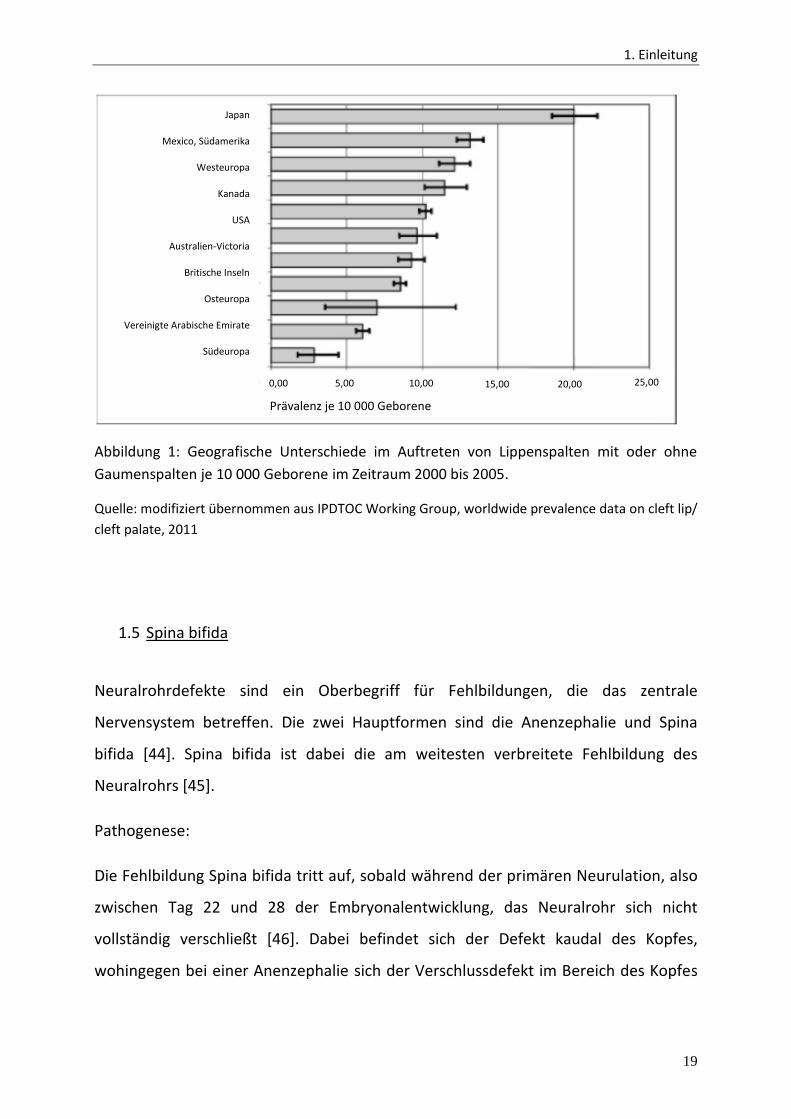
Zeitraum von 2000 bis 2005 die Prävalenzen von 54 Registern aus 30 verschiedenen
Ländern mit stark auseinanderweichenden Häufigkeiten verglichen werden
(Abbildung 1) [42].
Die Lippenspalte mit oder ohne Gaumenspalte tritt doppelt so oft bei Jungen wie bei
Mädchen auf. Hingegen ist bei isolierten Gaumenspalten das weibliche Geschlecht
häufiger betroffen [41, 43]. Nur auf eine Gesichtshälfte begrenzte Spaltbildungen
treten gehäuft links auf (2:1) [35, 41].
1. Einleitung
19
Abbildung 1: Geografische Unterschiede im Auftreten von Lippenspalten mit oder ohne
Gaumenspalten je 10 000 Geborene im Zeitraum 2000 bis 2005.
Quelle: modifiziert übernommen aus IPDTOC Working Group, worldwide prevalence data on cleft lip/
cleft palate, 2011
1.5 Spina bifida
Neuralrohrdefekte sind ein Oberbegriff für Fehlbildungen, die das zentrale
Nervensystem betreffen. Die zwei Hauptformen sind die Anenzephalie und Spina
bifida [44]. Spina bifida ist dabei die am weitesten verbreitete Fehlbildung des
Neuralrohrs [45].
Pathogenese:
Die Fehlbildung Spina bifida tritt auf, sobald während der primären Neurulation, also
zwischen Tag 22 und 28 der Embryonalentwicklung, das Neuralrohr sich nicht
vollständig verschließt [46]. Dabei befindet sich der Defekt kaudal des Kopfes,
wohingegen bei einer Anenzephalie sich der Verschlussdefekt im Bereich des Kopfes
Prävalenz je 10 000 Geborene
Japan
Mexico, Südamerika
Westeuropa
Kanada
USA
Australien-Victoria
Britische Inseln
Osteuropa
Vereinigte Arabische Emirate
Südeuropa
Südafrika 0,00 5,00 10,00 15,00 20,00 25,00

1. Einleitung
20
befindet [44]. Die Bezeichnung Spina bifida umfasst verschiedene Ausprägungen
eines Neuralrohrdefekts [44, 47]. Die Menigomyelozele ist dabei die am weitesten
verbreitete Unterform, die bei mehr als 90 % aller Spina bifida-Fälle auftritt. Diese
beschreibt eine Protrusion des Myelons und der Meningen durch einen
fehlgespaltenen Wirbel [44].
Diagnostik, Therapie:
Screeninguntersuchungen des mütterlichen Serums und pränatale
Ultraschalldiagnostik ermöglichen es, frühzeitig die Diagnose der Spina bifida zu
stellen. Hochauflösende Ultraschalluntersuchungen erlauben dabei Aussagen über
die genaue Lokalisation des knöchernen Defektes und des Weichgewebes,
einschließlich einer Beurteilung des fetalen Zentralen Nervensystems [48].
Angeborene Fehlbildungen des Neuralrohrs können entweder als geschlossene Form,
Spina bifida occulta (Q76.0), oder als offene Form, Spina bifida aperta (Q05.*)
auftreten. Die Therapie dieser Ausprägungen ist sehr unterschiedlich und reicht von
lediglich konservativen Nachsorgeuntersuchungen der unter anderem oft zufällig
entdeckten Spina bifida occulta bis zur operativen Versorgungen, die vereinzelt
bereits pränatal bei der Spina bifida aperta durchgeführt werden [48].
Prognose:
Neugeborene mit einer Anenzephalie sterben meist innerhalb der ersten Lebenstage
oder Lebenswochen. Aufgrund einer verbesserten medizinischen Versorgung ist die
Mortalität von Neugeborenen mit Spina bifida gesunken. Seit 1960 ist die
Überlebensrate über 50 % [49]. Fast ausnahmslos bestehen bei den Betroffenen
langfristig medizinische Probleme [50]. Oftmals resultiert aus dieser Malformation
eine Lähmung der Beine abhängig davon auf welcher Höhe der Wirbelsäule sich die
Läsion befindet. Nicht selten ist die Spina bifida mit körperlichen und/oder kognitiven
Beeinträchtigungen assoziiert [49]. Irreversible neurologische, urologische und

1. Einleitung
21
orthopädische Schädigungen führen zu lebenslangen Einschränkungen der Patienten
[51]. In einer französischen Studie beobachtete man über einen Zeitraum von fast 30
Jahren, dass in circa 24 % weitere Anomalien mit der Spina bifida assoziiert sind, dazu
zählten Chromosomenaberrationen, orofaziale Spaltbildungen, Klumpfuß und renale
Anomalien [52]. In einigen Fällen ist die Fehlbildung von der Arnold-Chiari-
Malformation vom Typ 2 begleitet [50].
Risikofaktoren:
Die exakte Ursache der Spina bifida ist bis heute ungeklärt. Zu den etablierten
Risikofaktoren zählen eine positive Familienanamnese, ein bereits vor der
Schwangerschaft bestehender maternaler Diabetes mellitus, Adipositas der Mutter,
das Alter der Mutter (< 19 Jahre und > 40 Jahre) sowie eine unzureichende
Folsäuresupplementation vor und während der Schwangerschaft [44, 49].
Epidemiologische Studien aus den frühen 1990er zeigten erstmals den
Zusammenhang zwischen maternaler Folsäureeinnahme und dem vollständigen
Neuralrohrverschluss des Kindes auf. Denn die perikonzeptionelle Einnahme von
Folsäure kann das Risiko für die Entstehung von Neuralrohrdefekten reduzieren. Der
dabei genau unterliegende biologische Mechanismus bei der Prävention von
Neuralrohrdefekten durch die Folsäuresupplementation ist nicht eindeutig geklärt
[44]. Aufgrund dieser Erkenntnisse wurde 1998 in den USA und Kanada begonnen
Getreideprodukte mit Folsäure anzureichern [46]. Daraufhin beobachtete man auch
eine Zunahme des Folsäuregehalts im Blut der Bevölkerung sowie einen Rückgang der
Prävalenz dieser angeborenen Malformation. Es ist bisher allerdings nicht möglich die
Fehlbildung vollständig zu verhindern [44]. Laut einer US-Studie von 2003 bis 2006
supplementieren nicht alle Frauen im gebärfähigen Alter die empfohlene
Folsäuretagesdosis von 400 Mikrogramm pro Tag [58]. Die Deutsche Gesellschaft für
Ernährung e.V. (DGE) empfiehlt ebenfalls täglich 400 Mikrogramm Folsäure in Form

1. Einleitung
22
von Supplementen bereits vier Wochen vor der Schwangerschaft und weiterführend
im ersten Trimenon einzunehmen [59].
Epidemiologie:
Die weltweite Prävalenz für Fehlbildungen des Zentralen Nervensystems reicht von
10 bis 100 je 10 000 Geburten [44, 47]. EUROCAT ermittelte im Zeitraum von 2000 bis
2008 eine Prävalenz für Neuralrohrdefekte, chromosomale Anomalien
ausgeschlossen, von 9,43 je 10 000 Geborene [60]. Die Prävalenz für Spina bifida
beträgt laut EUROCAT im Zeitraum von 2008 bis 2012 4,02 je 10 000 Geborene [61].
Einen signifikanten Rückgang der Prävalenz registrierte man in Ungarn zwischen den
Jahren 1994-2005 gegenüber dem Zeitraum 1980-1990, nachdem ein
Folsäuresupplementations-Programm für Schwangere eingeführt worden war [51].
Innerhalb des Zeitraumes 1995-1996 und 2003-2004 stellte man in den USA eine
Abnahme um 23 % fest. Begründet wurde diese Abnahme ebenfalls mit dem
positiven Effekt von Folsäureanreicherung in Getreideprodukten [44]. Hingegen
ermittelte eine andere US-amerikanische Studie eine stabil gebliebene Prävalenz der
Fehlbildung zwischen 1997-2009 in den USA [53].
1.6 Fragestellung der Studie
Das Ziel dieser Studie war es, das Auftreten von den Fehlbildungen Gastroschisis,
Omphalozele, Spina bifida und den orofazialen Spaltbildungen im Zeitraum Januar
2000 bis Dezember 2010 in Leipzig, Sachsen, Sachsen-Anhalt und Deutschland zu
untersuchen.

1. Einleitung
23
Folgende Fragen wurden dabei bearbeitet:
1. Ist aus diesen Untersuchungen ein Trend für das Auftreten von Gastroschisis,
Omphalozele, Spina bifida und orofazialen Spaltbildungen für die untersuchten
Regionen abzulesen?
2. Wie sind diese Erhebungen im internationalen Vergleich und verglichen mit
den ermittelten Prävalenzen durch das Fehlbildungsregister in Sachsen-Anhalt
einzuordnen?
3. Welche Konsequenzen sollten aus dieser Datenerhebung resultieren? Müssen
infolgedessen Präventionsmaßnahmen, wie zum Beispiel eine umfassendere
perikonzeptionelle Folsäuresupplementation in Deutschland, noch besser
implementiert werden?
Die vier Fehlbildungen Gastroschisis, Omphalozele, Spina bifida und orofaziale
Spaltbildungen wurden ausgewählt, da für sie international unterschiedliche
Prävalenzen beschrieben sind. Außerdem stellen sie nach angeborenen Herzfehlern
die häufigsten Fehlbildungen dar. Teilweise wären diese Fehlbildungen verhinderbar,
wie zum Beispiel durch Prävention mittels Folsäuresupplementation vor und während
der Schwangerschaft.
Für die Datenerhebung standen vier Datenquellen zur Verwendung bereit.
Krankenakten des Universitätsklinikums Leipzig, die Neonatalstatistik der Sächsischen
Landesärztekammer (SLÄK), das Fehlbildungsregister Sachsen-Anhalts sowie die
Diagnosestatistik des Forschungsdatenzentrums der Statistischen Ämter des Bundes
und der Länder (FDZ) konnten für die Datenerhebung herangezogen werden. Die
Codierung der Fehlbildungen erfolgte anhand der vom Deutschen Institut für
Medizinische Dokumentation und Information (DIMDI) herausgegebenen ICD-10-GM.
Untersucht wurden die Bauchwanddefekte Gastroschisis, verschlüsselt als Q79.3 und
die Omphalozele, die mit Q79.2 codiert wird. Als orofaziale Spaltbildungen wurden

1. Einleitung
24
die isolierte Lippenspalte (Q36.*) und die Lippenspalte mit Gaumenspalte (Q37.*)
zusammengefasst. Für die Neuralrohrdefekte wurde die Codierung Q05.* betrachtet,
welche der Spina bifida aperta entspricht.

2.Publikation
25
2. Publikation
Die Ergebnisse dieser Studie wurden in der Zeitschrift „Das Gesundheitswesen“ des
Thieme Verlages veröffentlicht.
Prävalenz von Gastroschisis, Omphalozele, Spina bifida und orofazialen Spaltbildungen
bei Neugeborenen im Zeitraum Januar 2000 bis Dezember 2010 in Leipzig, Sachsen,
Sachsen-Anhalt und Deutschland
Sophia Bremer, Mandy Vogel, Dr. rer. pol. Urban Janisch, Annette Friedrich, Dr. med.
Anke Rißmann, Prof. Dr. med. Ulrich Thome, PD Dr. med. Matthias Knüpfer, PD Dr. med.
Ulf Bühligen, Prof. Dr. med. Wieland Kiess

2.Publikation
26

2.Publikation
27

2.Publikation
28
2.Publikation
29

2.Publikation
30

2.Publikation
31

2.Publikation
32

3. Zusammenfassung
33
3. Zusammenfassung der Arbeit
Dissertation zur Erlangung des akademischen Grades
Dr. med.
Prävalenz von Gastroschisis, Omphalozele, Spina bifida und orofazialen Spaltbildungen
bei Neugeborenen im Zeitraum Januar 2000 bis Dezember 2010 in Leipzig, Sachsen,
Sachsen-Anhalt und Deutschland
Eingereicht von
Sophia Bremer
Angefertigt in der
Klinik und Poliklinik für Kinder- und Jugendmedizin der Universität Leipzig
Betreut von
Prof. Dr. med. Wieland Kiess
Angeborene Fehlbildungen sind, vor allem in den Industrieländern, die häufigste Ursache
für die Mortalität und Morbidität im frühen Kindesalter. Das Auftreten verschiedener
Fehlbildungen scheint in den letzten Jahren zugenommen zu haben. Diese Dissertation
möchte einen Beitrag zu der Klärung dieses Verdachts leisten.

3. Zusammenfassung
34
Ausgangspunkt dieser Arbeit waren Aussagen zahlreicher weltweiter und
multizentrischer Studien über die Zunahme angeborener Fehlbildungen [4, 5, 6, 7, 8].
Kongenitale Malformationen sind die häufigste Todesursache im frühen Kindesalter in
den Industrienationen [54]. Diese stellen insgesamt eine erhebliche medizinisch-
ethisches und gesundheitsökonomische Herausforderung dar. Die vorliegende Studie
untersuchte lokale und nationale Trends der Prävalenzen von Gastroschisis,
Omphalozele, Spina bifida und orofazialen Spaltbildungen im Zeitraum von 2000 bis
2010 in Leipzig, Sachsen, Sachsen-Anhalt und Deutschland. Im beschriebenen Zeitraum
wurden 7 702 035 Kinder in Deutschland geboren. In unserer Studie registrierten wir
insgesamt 9 443 Kinder, die eine der vier untersuchten Fehlbildungen in Deutschland
aufwiesen.
Für die Datenerhebung konnten Krankenakten des Universitätsklinikums Leipzig, die
Neonatalstatistik der Sächsischen Landesärztekammer, das Fehlbildungsregister
Sachsen-Anhalts sowie die Diagnosestatistik des Forschungsdatenzentrums der
Statistischen Ämter des Bundes und der Länder herangezogen werden. Mithilfe des
Fehlbildungsregisters EUROCAT, einer populationsbezogenen Fehlbildungserfassung, die
mit 43 Registern aus 23 Ländern insgesamt 29 % aller Geburten in Europa überblickt und
Fehlbildungen zusammenfasst, war ein internationaler Vergleich der untersuchten
Prävalenzen möglich.
Folgende Fragen wurden in dieser Studie bearbeitet:
Frage 1: Ist aus diesen Untersuchungen ein Trend für das Auftreten von Gastroschisis,
Omphalozele, Spina bifida und orofazialen Spaltbildungen für die untersuchten Regionen
abzulesen?

3. Zusammenfassung
35
Die Prävalenz der Fehlbildungen betrug im Untersuchungszeitraum in Deutschland
beziehungsweise in Sachsen 1,97/2,12 (Gastroschisis), 1,63/1,48 (Omphalozele),
5,80/8,11 (orofaziale Spaltbildungen) und 2,92/2,50 (Spina bifida) je 10 000 LB. In
Sachsen zeigte sich ein Trendanstieg, dessen Effektstärken jedoch sehr gering sind
(OR/Jahr zwischen 1,01-1,09). Bei den gemeldeten Fällen von Omphalozele,
Gastroschisis und orofazialen Spaltbildungen beobachteten wir in Deutschland einen
statistisch signifikanten Anstieg über den betrachteten Zeitraum, allerdings mit sehr
geringen Effektgrößen wie z.B. für die Omphalozele (OR 1,04, KI 1,02 - 1,05, p-korrigiert
< 0,05) und Gastroschisis (OR 1,025, KI 1,0 - 1,04, p-korrigiert < 0,05). Wir führen diesen
statistisch signifikanten Anstieg vielmehr auf die hohe Fallzahl zurück, da die Gesamtzahl
der Geburten in Deutschland in die Berechnungen einging. Darüber hinaus befürchten
wir, dass über die Jahre während der Datenerfassung Fehlcodierungen und Doppel- oder
lückenhafte Erfassungen der Fehlbildungen sowie möglicherweise durch intrauterine
Verlegungen über Landesgrenzen hinweg in unterschiedlichem Ausmaß auftraten. Die
Häufigkeit der Fehlbildung Spina bifida nahm dagegen im betrachteten Zeitraum in
Deutschland signifikant ab.
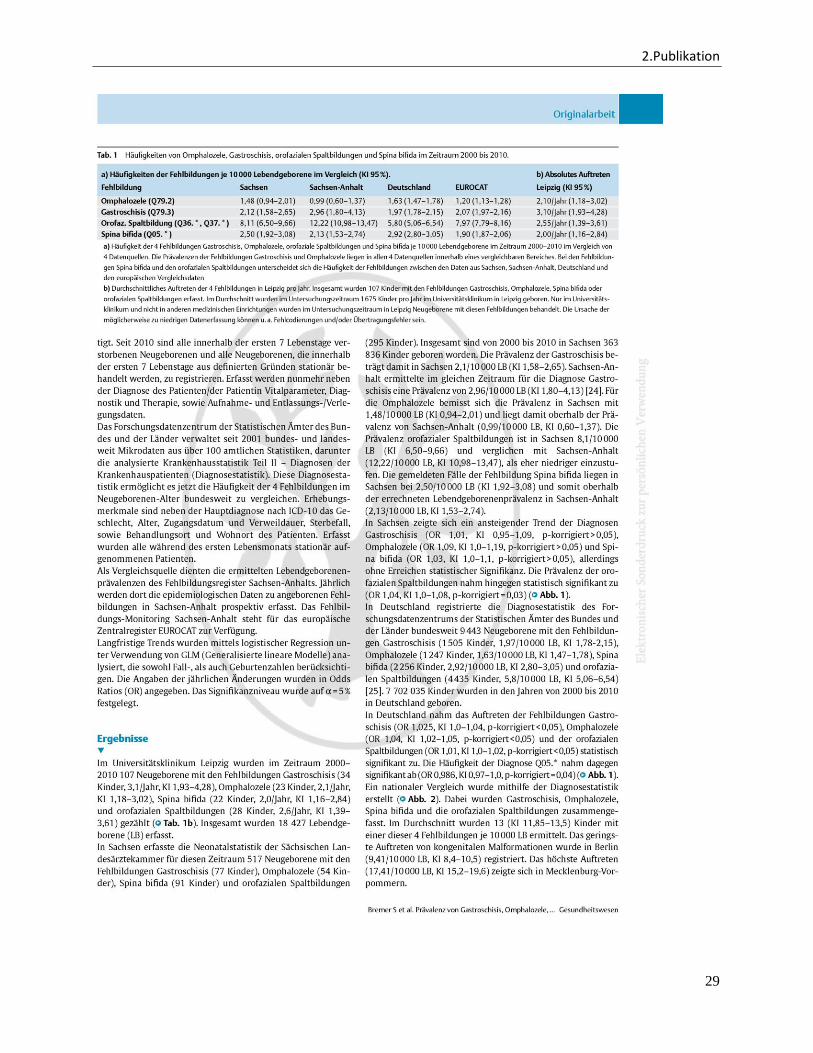
Im Universitätsklinikum Leipzig wurden im Zeitraum 2000 bis 2010 107 Neugeborene mit
den Fehlbildungen Gastroschisis (3,1/Jahr, KI 1,93-4,28), Omphalozele (2,1/Jahr, KI 1,18-
3,02), Spina bifida (2,0/Jahr, KI 1,16-2,84) und orofazialen Spaltbildungen (2,6/Jahr, KI
1,39-3,61) gezählt.
Im nationalen Vergleich zwischen den 16 Bundesländern zeigten sich erfreulicherweise
keine erheblichen Unterschiede im Auftreten der vier Fehlbildungen. Im Durchschnitt
wurden 13 Kinder je 10 000 LB mit einer dieser vier Fehlbildungen je Bundesland
ermittelt. In Berlin zeigte sich dabei die niedrigste Prävalenz (9,41/10 000 LB). In
Mecklenburg-Vorpommern traten die Fehlbindungen scheinbar häufiger auf (17,41/10

3. Zusammenfassung
36
000 LB). Wir vermuten, dass der Fehler der kleinen Zahlen, also eine Verfälschung des
Ergebnisses aufgrund einer geringen Gesamtmenge dafür verantwortlich ist. Eine
sorgfältigere Meldung und Registrierung von kongenitalen Anomalien in Mecklenburg-
Vorpommern oder ein tatsächlich gehäuftes Auftreten von Fehlbildungen in einer Region
sind nicht auszuschließen.
Frage 2: Wie sind diese Erhebungen im internationalen Vergleich und verglichen mit den
ermittelten Prävalenzen durch das Fehlbildungsregister in Sachsen-Anhalt einzuordnen?
Als Vergleichsquelle dienten die epidemiologischen Daten zu angeborenen
Fehlbildungen, die in Sachsen-Anhalt prospektiv erfasst und jährlich veröffentlicht
werden. Die Datensammlung von EUROCAT ermöglichte es uns, einen internationalen
Vergleich mit den erhobenen Daten dieser Studie durchzuführen. Es zeigen sich teilweise
Unterschiede im Auftreten der vier kongenitalen Malformationen, die in dieser Arbeit
tabellarisch visualisiert wurden. Die Häufigkeit der Fehlbildung Gastroschisis liegt in
Sachsen, Sachsen-Anhalt und Deutschland im europäischen Vergleichsrahmen. Das
Auftreten der Omphalozele ist in Sachsen und Deutschland geringfügig oberhalb der
Lebendgeborenenprävalenz von Sachsen-Anhalt und der europäischen Vergleichsdaten.
Die ermittelten Prävalenzen der orofazialen Spaltbildungen liegen in Sachsen,
Deutschland und in den europäischen Vergleichsdaten in einer vergleichbaren
Größenordnung. In Sachsen-Anhalt ist das Auftreten der orofazialen Spaltbildungen
oberhalb dieser Vergleichswerte. Das Auftreten der Fehlbildung Spina bifida ist in
Sachsen, Sachsen-Anhalt und den europäischen Vergleichsdaten in einer
Größenordnung, hingegen die gemeldeten Fälle in Deutschland über diesen liegen. Wir
vermuten, dass eine ungleiche Datenerfassung für lokale und nationale Unterschiede

3. Zusammenfassung
37
verantwortlich ist, so dass die Vergleichbarkeit und Interpretation der erhobenen Daten
erschwert ist.
Frage 3: Welche Konsequenzen sollten aus dieser Datenerhebung resultieren? Müssen
infolgedessen Präventionsmaßnahmen, wie zum Beispiel eine umfassendere
perikonzeptionelle Folsäuresupplementation in Deutschland, noch besser implementiert
werden?
Die Datenanalyse lässt es nicht mit Sicherheit zu, über das Auftreten der vier
untersuchten Fehlbildungen zu urteilen. Um einen nicht zu wünschenden Aufwärtstrend
von Fehlbildungen zu detektieren, damit infolgedessen zum Beispiel
Präventionsmaßnahmen erweitert werden können, wären valide Daten erforderlich. Der
Erfolg von Präventionsmaßnahmen, wie zum Beispiel der Folsäuresupplementation,
kann infolge der unvollständigen Datenerhebung ebenfalls nur eingeschränkt gemessen
werden. Eine prospektive Erfassung von Fehlbildungen wäre wünschenswert.
Stärken und Schwächen der Studienarbeit
Bei dieser Studie handelt es sich um eine retrospektive Analyse. Die Stärken dieser
Dissertation liegen in der umfangreichen Datenanalyse von vier unterschiedlichen
Datenquellen aus Leipzig, Sachsen, Sachsen-Anhalt und Deutschland. Somit war ein
regionaler Vergleich der Prävalenzen der vier Fehlbildungen durchführbar. Teilweise war
es uns möglich die Datenerhebungen zu kontrollieren und zu korrigieren. Die SLÄK erhält
zur Erstellung der sächsischen Neonatalstatistik Daten aus dem Universitätsklinikum
Leipzig. Durch das Heranziehen der Krankenakten der Uniklinik Leipzig fiel auf, dass die

3. Zusammenfassung
38
Diagnosen Omphalozele und Gastroschisis in Sachsen teilweise zusammengefasst und als
eine Diagnose an die SLÄK übermittelt wurden. Nachträglich ließen sich die Daten exakt
zuordnen. Nahezu 60 % aller Fälle im Universitätsklinikum Leipzig wurden nicht korrekt
als Gastroschisis, sondern als Omphalozele an die SLÄK gemeldet. Eine umfassende und
flächendeckende Daten-Kuratierung war im Rahmen dieser Arbeit nicht möglich und
wird auch in keinem der genutzten Register systematisch durchgeführt. Eine
Datenkorrektur oder ein systematisches Qualitätsmanagement bei der Datenerhebung
könnten während des Beobachtungszeitraums an den unterschiedlichen
Datenerfassungsstandorten unterschiedlich ausgeprägt gewesen sein. Wir vermuten,
dass Routineerfassungen ohne Datenbereinigung im alltäglichen Klinikbetrieb zu
Erfassungsfehlern führen.
Die Aussagekraft der Ergebnisse ist eingeschränkt, da für die untersuchten Fehlbildungen
insgesamt geringe Fallzahlen ermittelt wurden. Bei der Interpretation der Daten könnte
der Fehler der kleinen Zahlen zu falschen Schlussfolgerungen geführt haben. Außerdem
bleibt unklar, ob Änderungen in der Erfassungs- und Verschlüsselungspraxis,
Fehlcodierungen, Übertragungsfehler, Doppel- und/oder lückenhafte Erfassung der
Fehlbildungen die Daten verfälscht haben könnten. Ob also ein tatsächlicher Anstieg der
Prävalenzen besteht oder lediglich Artefakte einen Anstieg vortäuschen, ist ungewiss.
Perspektive
1964 wurde das „National Congenital Anomalies System“ im Zuge der Contergan-
Katastrophe in den USA gegründet. Dieses soll es unter anderem ermöglichen,
teratogene Gefahren, wie zum Beispiel den Wirkstoff Thalidomid frühzeitig zu
identifizieren [1]. Mit der Gründung von EUROCAT im Jahre 1979, existiert in Europa ein

3. Zusammenfassung
39
Fehlbildungsregister, welches eine schnelle Auskunft über das Auftreten von mehr als 80
Fehlbildungen ermöglicht. Die kontinuierliche Erstellung von gegenwärtigen
Prävalenzübersichten erlaubt unter anderem die Wirksamkeit primärer Prävention und
pränataler Diagnostik einzuschätzen [8, 62]. Alle Gebiete, die nicht in diesem Register
vorkommen, sind schwer zugänglich, um Informationen über angeborene Fehlbildungen
zu erhalten. Es ist nur mit sehr hohem Aufwand möglich, Fehlbildungs-Prävalenzen
zeitnah zu erfassen: auf Grund der dezentralen Daten-Erfassung, der nur über durchaus
langwierige und bürokratische Wege zu erlangenden Zugangserlaubnis zu den Daten und
der aufwendigen Überprüfung der Originaldaten zur Validierung (im Rahmen der
vorliegenden Arbeit waren mehrfach Reisen zum jeweiligen Standort der
Datenquellen/Register erfolgt) ist eine Datenauswertung in Deutschland nur mit großem
Zeitaufwand und mit Jahre dauernder Verzögerung möglich. Nur in Sachsen-Anhalt und
Rheinland-Pfalz wird das Auftreten von Fehlbildungen prospektiv erfasst und auch nur in
diesen Bundesländern können zeitnah Veränderungen der Fehlbildungsprävalenz
erkannt werden. Signifikante und besonders nur regional auftretende Änderungen im
Auftreten von kongenitalen Malformationen bleiben mit großer Wahrscheinlichkeit in
Deutschland unbemerkt. Angesichts der anscheinenden oder scheinbaren Zunahme von
Fehlbildungen und der offensichtlich fehlerhaften Datenlage ist ein bundesweites oder
sind weitere regionale Register für eine bessere und zeitnahe Erkennung und Erfassung
von Fehlbildungen in Deutschland notwendig.

4. Literaturverzeichnis
40
4. Literaturverzeichnis
1. Singh S, Chukwunyere DN, Omembelede J, Onankpa B: Foetal congenital anomalies:
An experience from a tertiary health institution in north-west nigeria (2011-2013).
The Nigerian postgraduate medical journal 2015; 22(3): 174–8.
2. Zhang X: Prevalence of birth defects and risk-factor analysis from a population-based
survey in Inner Mongolia, China.
3. Czeizel AE: The primary prevention of birth defects: Multivitamins or folic acid?
International journal of medical sciences 2004; 1(1): 50–61.
4. Alvarez SM, Burd RS: Increasing prevalence of gastroschis repairs in the United
States: 1996-2003. Journal of Pediatric Surgery 2007; 42(6): 943–6.
5. Gianicolo, Emilio Antonio Luca, Mangia C, Cervino M, Bruni A, Andreassi MG, Latini G:
Congenital anomalies among live births in a high environmental risk area—A case-
control study in Brindisi (southern Italy). Environmental Research 2014; 128: 9–14.
6. Castilla EE, Mastroiacovo P, Orioli IM: Gastroschisis: International epidemiology and
public health perspectives. Am. J. Med. Genet. 2008; 148C(3): 162–79.
7. Baerg J, Kaban G, Tonita J, Pahwa P, Reid D: Gastroschisis: A sixteen-year review. J.
Pediatr. Surg. 2003; 38(5): 771–4.

4. Literaturverzeichnis
41
8. Dolk H: EUROCAT: 25 years of European surveillance of congenital anomalies.
Archives of Disease in Childhood - Fetal and Neonatal Edition 2005; 90(5): F355–F358.
9. David AL, Tan A, Curry J: Gastroschisis: sonographic diagnosis, associations,
management and outcome. Prenatal diagnosis 2008; 28(7): 633–44.
10. Boyle B, McConkey R, Garne E, et al.: Trends in the prevalence, risk and pregnancy
outcome of multiple births with congenital anomaly: a registry-based study in 14
European countries 1984-2007. BJOG 2013; 120(6): 707–16.
11. Mossey PA, Modell B: Epidemiology of oral clefts 2012: an international perspective.
Front Oral Biol 2012; 16: 1–18.
12. Marseglia L, Manti S, D'Angelo G, et al.: Gastroesophageal reflux and congenital
gastrointestinal malformations. World journal of gastroenterology WJG 2015; 21(28):
8508–15.
13. Christison-Lagay ER, Kelleher CM, Langer JC: Neonatal abdominal wall defects. Semin
Fetal Neonatal Med 2011; 16(3): 164–72.
14. Hardy D, Bhalla VK, Parkhurst C, Pipkin WL, Howell CG, Hatley RM: Gastroschisis
associated with an omphalocele and intestinal atresia. Pediatr Surg Int 2014; 30(3):
353–5.

4. Literaturverzeichnis
42
15. Corey KM, Hornik CP, Laughon MM, McHutchison K, Clark RH, Smith PB: Frequency of
anomalies and hospital outcomes in infants with gastroschisis and omphalocele. Early
human development 2014; 90(8): 421–4.
16. S. Ionescu, D. Stanescu, M. Mocanu, et al.: Differential Diagnosis of Abdominal Wall
Defects - Omphalocele versus Gastroschisis. Chirurgia (2014) 109: 7-14
17. Ledbetter DJ: Gastroschisis and Omphalocele. Surgical Clinics of North America 2006;
86(2): 249–60.
18. Feldkamp ML, Carey JC, Sadler TW: Development of gastroschisis: review of
hypotheses, a novel hypothesis, and implications for research. American journal of
medical genetics. Part A 2007; 143A(7): 639–52.
19. Fillingham A, Rankin J: Prevalence, prenatal diagnosis and survival of gastroschisis.
Prenatal diagnosis 2008; 28(13): 1232–7.
20. Weichert J, Kahl FO, Schröer A, Bohlmann MK, Diedrich K, Hartge DR: Kongenitale
Gastroschisis--pränatale Diagnose und perinatales Management. Z Geburtshilfe
Neonatol 2010; 214(4): 135–44.
21. Christison-Lagay ER, Kelleher CM, Langer JC: Neonatal abdominal wall defects.
Seminars in Fetal and Neonatal Medicine 2011; 16(3): 164–72.

4. Literaturverzeichnis
43
22. Krause H, Pötzsch S, Hass H, et al.: Ventrale Bauchwanddefekte--Darstellung der
Entwicklung in Prävalenz und operativem Vorgehen anhand von Gastroschisis und
Omphalozele. Zentralbl Chir 2009; 134(6): 524–31.
23. Bauman Z, Nanagas V: The Combination of Gastroschisis, Jejunal Atresia, and Colonic
Atresia in a Newborn. Case reports in pediatrics 2015; 2015: 129098.
24. Askarpour S, Ostadian N, Javaherizadeh H, Chabi S: Omphalocele, Gastroschisis:
Epidemiology, Survival, and Mortality in Imam Khomeini Hospital, Ahvaz-Iran. Polish
Journal of Surgery 2012; 84(2).
25. Loane M, Dolk H, Bradbury I: Increasing prevalence of gastroschisis in Europe 1980-
2002: a phenomenon restricted to younger mothers? Paediatr Perinat Epidemiol
2007; 21(4): 363–9.
26. Sharma D, Murki S, Pratap T: A newborn with omphalocele and umbilical cord cyst:
an interesting entity. Iranian journal of pediatrics 2014; 24(4): 449–50.
27. Marshall J, Salemi JL, Tanner JP, et al.: Prevalence, Correlates, and Outcomes of
Omphalocele in the United States, 1995-2005. Obstetrics and gynecology 2015;
126(2): 284–93.
28. Mac Bird T, Robbins JM, Druschel C, Cleves MA, Yang S, Hobbs CA: Demographic and
environmental risk factors for gastroschisis and omphalocele in the National Birth
Defects Prevention Study. Journal of Pediatric Surgery 2009; 44(8): 1546–51.

4. Literaturverzeichnis
44
29. Achermann S, Addor M-C, Schinzel A: 2000-38 Der Anteil pränatal erfasster Fälle von
ausgewählten Fehlbildungen in der EUROCAT-Studie.
30. Corey KM, Hornik CP, Laughon MM, McHutchison K, Clark RH, Smith PB: Frequency of
anomalies and hospital outcomes in infants with gastroschisis and omphalocele. Early
human development 2014; 90(8): 421–4.
31. Calvert N, Damiani S, Sunario J, Bower C, Dickinson JE: The outcomes of pregnancies
following a prenatal diagnosis of fetal exomphalos in Western Australia. The
Australian & New Zealand journal of obstetrics & gynaecology 2009; 49(4): 371–5.
32. Brooklyin S, Jana R, Aravinthan S, Adhisivam B, Chand P: Assessment of folic Acid and
DNA damage in cleft lip and cleft palate. Clin Pract 2014; 4(1): 608.
33. Shkoukani MA, Chen M, Vong A: Cleft lip - a comprehensive review. Frontiers in
pediatrics 2013; 1: 53.
34. Prevalence at birth of cleft lip with or without cleft palate: data from the
International Perinatal Database of Typical Oral Clefts (IPDTOC). Cleft Palate
Craniofac. J. 2011; 48(1): 66–81.
35. Shkoukani MA, Chen M, Vong A: Cleft Lip – A Comprehensive Review. Front. Pediatr.
2013; 1.
36. Wehby GL, Cassell CH: The impact of orofacial clefts on quality of life and healthcare
use and costs. Oral diseases 2010; 16(1): 3–10.

4. Literaturverzeichnis
45
37. Parada C, Chai Y: Roles of BMP signaling pathway in lip and palate development.
Frontiers of oral biology 2012; 16: 60–70.
38. Leslie EJ, Marazita ML: Genetics of cleft lip and cleft palate. American journal of
medical genetics. Part C, Seminars in medical genetics 2013; 163C(4): 246–58.
39. Mehrotra D: Genomic expression in non syndromic cleft lip and palate patients: A
review. Journal of oral biology and craniofacial research 2015; 5(2): 86–91.
40. Reiter R, Haase S, Brosch S: Orofaziale Spaltbildungen. Laryngorhinootologie 2012;
91(2): 84–95.
41. Dixon MJ, Marazita ML, Beaty TH, Murray JC: Cleft lip and palate: understanding
genetic and environmental influences. Nature reviews. Genetics 2011; 12(3): 167–78.
42. Prevalence at Birth of Cleft Lip With or Without Cleft Palate: Data From the
International Perinatal Database of Typical Oral Clefts (IPDTOC). The Cleft Palate-
Craniofacial Journal 2011; 48(1): 66–81.
43. Calzolari E, Bianchi F, Rubini M, Ritvanen A, Neville AJ: Epidemiology of cleft palate in
Europe: implications for genetic research. Cleft Palate Craniofac. J. 2004; 41(3): 244–
9.
44. Au KS, Ashley-Koch A, Northrup H: Epidemiologic and genetic aspects of spina bifida
and other neural tube defects. Dev Disabil Res Revs 2010; 16(1): 6–15.

4. Literaturverzeichnis
46
45. Sandler AD: Children with spina bifida: key clinical issues. Pediatr. Clin. North Am.
2010; 57(4): 879–92.
46. Benedum C, Yazdy M, Mitchell A, Werler M: Risk of Spina Bifida and Maternal
Cigarette, Alcohol, and Coffee Use during the First Month of Pregnancy. IJERPH 2013;
10(8): 3263–81.
47. Clemmensen D, Thygesen M, Rasmussen MM, Fenger-Grøn M, Petersen OB, Mosdal
C: Decreased incidence of myelomeningocele at birth: effect of folic acid
recommendations or prenatal diagnostics? Childs Nerv Syst 2011; 27(11): 1951–5.
48. Coleman BG, Langer JE, Horii SC: The diagnostic features of spina bifida: the role of
ultrasound. Fetal diagnosis and therapy 2015; 37(3): 179–96.
49. Parker SE, Yazdy MM, Mitchell AA, Demmer LA, Werler MM: A description of spina
bifida cases and co-occurring malformations, 1976-2011. Am. J. Med. Genet. 2014;
164(2): 432–40.
50. Fleischman AR, Oinuma M: Fortification of corn masa flour with folic acid in the
United States. American journal of public health 2011; 101(8): 1360–4.
51. Szabó N, Gergev G, Valek A, Eller J, Kaizer L, Sztriha L: Birth prevalence of neural tube
defects: a population-based study in South-Eastern Hungary. Childs Nerv Syst 2013;
29(4): 621–7.

4. Literaturverzeichnis
47
52. Stoll C, Dott B, Alembik Y, Roth M: Associated malformations among infants with
neural tube defects. Am. J. Med. Genet. 2011; 155(3): 565–8.
53. Lloyd JC, Wiener JS, Gargollo PC, Inman BA, Ross SS, Routh JC: Contemporary
epidemiological trends in complex congenital genitourinary anomalies. J. Urol. 2013;
190(4 Suppl): 1590–5.
54.Tennant, Peter W G, Pearce MS, Bythell M, Rankin J: 20-year survival of children born
with congenital anomalies: a population-based study. Lancet 2010; 375(9715): 649–
56.
55.Didriksen A KM, Irgens LM, Lie RT, Bjerkedal T, Egenaes J. Gastrochisis in Norway
1967–1995. In: EUROCAT-ICBDMS International symposium on registration and
prevention of congenital anomalies; 1998. Florence; 1998.
56.http://www.eurocat-network.eu/content/Stat-Mon-Report-2007.pdf, 12.03.2016
57. Dt Ärztebl 1998; 95: A-2262–2267 [Heft 37]
58.Tinker, S.C.; Cogswell, M.E.; Devine, O.; Berry, R.J. Folic acid intake among US women
aged 15–44 years, National Health and Nutrition Examination Survey, 2003–2006.
Am. J. Prev. Med. 2010, 38, 534–542.

4. Literaturverzeichnis
48
59. https://www.dge.de/presse/pm/folsaeure-in-der-praevention/, 12.03.2016
60. EUROCAT (2014). EUROCAT Special Report: Prevalence of Neural Tube Defects in
Younger Mothers in Europe 2000-2008: Analysis of the EUROCAT Database.
EUROCAT Central Registry, University of Ulster.
61. www.eurocat-network.eu/ACCESSPREVALENCEDATA/PrevalenceTables, 12.03.2016
62. www.eurocat-network.eu/aboutus/whatiseurocat/whatiseurocat, 12.03.2016

II. Erklärung über die Eigenständigkeit der Arbeit
49
II. Erklärung über die Eigenständigkeit der Arbeit
Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne unzulässige
Hilfe oder Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Ich
versichere, dass Dritte von mir weder unmittelbar noch mittelbar geldwerte Leistungen
für Arbeiten erhalten haben, die im Zusammenhang mit dem Inhalt der vorgelegten
Dissertation stehen, und dass die vorgelegte Arbeit weder im Inland noch im Ausland in
gleicher oder ähnlicher Form einer anderen Prüfungsbehörde zum Zweck einer
Promotion oder eines anderen Prüfungsverfahrens vorgelegt wurde. Alles aus anderen
Quellen und von anderen Personen übernommene Material, das in der Arbeit
verwendet wurde oder auf das direkt Bezug genommen wird, wurde als solches
kenntlich gemacht. Insbesondere wurden alle Personen genannt, die direkt an der
Entstehung der vorliegenden Arbeit beteiligt waren.
Leipzig, den 29.03.2016 S.Bremer
Datum Unterschrift

III. Lebenslauf
50
III. Lebenslauf
Sophia Bremer, geboren am 13.11.1989 in Leipzig
Beruf
Seit 04/2016 Ärztin in Weiterbildung für Innere Medizin an der Helios
Klinik Leisnig
Akademische und schulische Ausbildung
2008 – 2015 Medizinstudium an der Universität Leipzig
Abschlussnote gut (2,16)
2014, 2015 2. und 3. Ärztliche Prüfung: Note gut
2014 - 2015 Praktisches Jahr an der Universität Leipzig, Sana Klinikum
Borna, Kantonspital Luzern, Sankt Georg Leipzig
2012 – 2013 Auslandssemester in Valencia, Spanien
2010 1. Ärztliche Prüfung: Note gut
2008 Abitur an der Friedrich-Schiller-Schule, Leipzig

III. Lebenslauf
51
Veröffentlichung
Sophia Bremer, Mandy Vogel, Dr. rer. pol. Urban Janisch, Annette Friedrich, Dr. med.
Anke Rißmann, Prof. Dr. med. Ulrich Thome, PD Dr. med. Matthias Knüpfer, PD Dr. med.
Ulf Bühligen, Prof. Dr. med. Wieland Kiess
Prävalenz von Gastroschisis, Omphalozele, Spina bifida und orofazialen Spaltbildungen
bei Neugeborenen im Zeitraum Januar 2000 bis Dezember 2010 in Leipzig, Sachsen,
Sachsen-Anhalt und Deutschland. Paper publiziert in „Das Gesundheitswesen“

IV. Danksagung
52
IV. Danksagung
Ich bedanke mich herzlichst bei meinem Betreuer Herrn Professor Dr. med. Kieß für die
Überlassung des Dissertationsthemas und die kontinuierliche Unterstützung sowie das
stetige kritische Hinterfragen der Ergebnisse und deren Interpretation.
Ein weiterer besonderer Dank gilt Mandy Vogel für die immerwährende freundliche und
fachliche Unterstützung bei der Bewältigung aller statistischen Herausforderungen sowie
für die gestalterische Ausarbeitung aller Daten.
Ich danke weiterhin Frau Dr. med. Anke Rißmann für die objektive Betrachtung aller
Ergebnisse und die stetige Zustimmung für dieses Dissertationsprojekt.
Außerdem gilt mein Dank allen Partner-Autoren für deren Unterstützung und
fortlaufenden Zuspruch.













