Embed Size (px)
Citation preview

Dr. Stephanie BlumAlexandra Fürst Dr. Christine OechsleinCornelia Wawretschek
SOP-Sammlung für die Pharmaindustrie
Standardarbeitsanweisungen für GMP-Bereiche
n Downloadn veränderbare Word-Dateien

Inhaltsverzeichnis
(1)(EL16) © Maas & Peither AG – GMP-Verlag
IVZINHALTSVERZEICHNIS
Zu Ihrer Information: Ausführlicher als diese Übersicht sind die Inhaltsverzeichnisse vorden einzelnen Kapiteln.
BAND 1
Kapitel AEinleitungA.1 VorwortA.2 Die AutorenA.3 Tipps für die Erstellung und den Umgang mit SOPs
Kapitel BAllgemeine DokumenteB.1 OrganigrammB.2.1 Leiter der Qualitätskontrolle, Sachkundige PersonB.2.2 Leiter der HerstellungB.2.3 Leiter der QualitätssicherungB.2.4 Verantwortliche Person gemäß § 2 AM-HandelsVB.3 Site Master File
Kapitel 1Qualitätsmanagement1.0 SOP-100-01
Qualitätsmanagement und Managementverantwortung1.1 SOP-101-02
Überprüfung der Chargendokumentation und Marktfreigabe1.2 SOP-102-02
Lieferantenqualifizierung einschließlich Dienstleister – Outsourced Activities1.3 SOP-103-01
Qualitätsrisikomanagement1.4 SOP-104-01
Umgang mit Abweichungen und Änderungen1.5 SOP-105-01
Maßnahmen zur Fehlerkorrektur und Vorbeugung (CAPA) einschließlich Ursachen-analyse
1.6 SOP-106-01Audits und Selbstinspektionen
1.7 SOP-107-02Herstellung und Analytik im Auftrag – Verträge und Management
SOP-BuchIVZ.fm Seite 1

SOP-Sammlung für die Pharmaindustrie
(2) (EL16) © Maas & Peither AG – GMP-Verlag
IVZ 1.8 SOP-108-01Product Quality Review (PQR)
1.9 SOP-109-01Beanstandungen (Complaints)
1.10 SOP-110-01Produktrückruf (Alarmplan)
1.15 SOP-115-01Technologietransfer
1.30 SOP-130-01Umgang mit Fälschungen
BAND 2
Kapitel 2Qualitätskontrolle2.1 SOP-201-01
Stabilitätsuntersuchungen2.2 SOP-202-01
Umgang mit OOS-, OOT- und OOE-Ergebnissen2.5 SOP-205-01
Umgang mit Proben, Reagenzien und Referenzstandards im Labor2.7 SOP-207-02
Validierung analytischer Methoden und Methodentransfer
Kapitel 3Personal3.1 SOP-301-01
Qualifizierung von Mitarbeitern3.2 SOP-302-01
Gesundheitsüberwachung
Kapitel 4Dokumentation4.0 SOP-400-01
Datenintegrität und Data Governance4.1 SOP-401-01
Erstellen, Verteilen und Aktualisieren von SOPs4.2 SOP-402-01
Unterschriften und Unterschriftsberechtigung4.3 SOP-403-02
Protokollführung, Logbücher und Laborjournale4.4 SOP-404-01
Archivierung von GMP-relevanten Dokumenten und Aufzeichnungen4.6 SOP-406-01
Erstellung von Spezifikationen und Prüfanweisungen
SOP-BuchIVZ.fm Seite 2

Inhaltsverzeichnis
(3)(EL16) © Maas & Peither AG – GMP-Verlag
IVZ4.7 SOP-407-01Erstellung von Herstellungs- und Verpackungsanweisungen
4.8 SOP-408-01Technische Dokumentation im Pharmabetrieb (Räume, Geräte und Anlagen)
BAND 3
Kapitel 5Gebäude und Anlagen5.0 SOP-500-01
Hygieneplan5.1 SOP-501-02
Qualifizierung von Anlagen und Geräten5.2 SOP-502-01
Schädlingsüberwachung und -bekämpfung5.3 SOP-503-01
Reinigung von Räumen und Einrichtungen5.5 SOP-505-02
Reinigung und Reinigungsvalidierung5.17 SOP-517-02
Life Cycle computergestützter Systeme (CS)
Kapitel 6Material und Produkt6.0 SOP-600-02
Validierungsmasterplan6.1 SOP-601-02
Prozessvalidierung6.2 SOP-602-01
Bereitstellung und Aufbewahrung von Rückstellmustern6.4 SOP-604-01
Wareneingang und Wareneingangskontrollen 6.5 SOP-605-01
Vernichtung von Ausgangsstoffen, Packmitteln und Arzneimitteln6.40 SOP-640-01
Beschaffung (Sourcing) von Wirk- und Hilfsstoffen6.41 SOP-641-01
Risikobewertung von Hilfsstoffen
SOP-BuchIVZ.fm Seite 3

SOP-Sammlung für die Pharmaindustrie
(4) (EL16) © Maas & Peither AG – GMP-Verlag
IVZ Kapitel 7Transport7.0 SOP-700-01
GDP Masterplan7.2 SOP-702-01
Qualifizierung von Transportunternehmen7.3 SOP-703-01
Qualifizierung von Transportverpackungen 7.70 SOP-770-1
Transport von Arzneimitteln7.80 SOP-780-01
Transportvalidierung
SOP-BuchIVZ.fm Seite 4

Maas & Peither Pharma GmbH
Standardarbeitsanweisung Seite 2 von 21
SOP-501-02 Qualifizierung von Anlagen und
Geräten
Gültig ab:
01.09.2016
© 2016 Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 2
Inhalt
1 Geltungsbereich ................................................................................................................. 1
2 Ziel/Zweck ............................................................................................................................ 3
3 Anwendungsbereich .......................................................................................................... 3
4 Definitionen/Abkürzungen ................................................................................................. 4
5 Grundlagen, mitgeltende Unterlagen ............................................................................... 5
5.1 Grundlagen .................................................................................................................. 5 5.2 Mitgeltende Unterlagen ................................................................................................ 5
6 Arbeitsablauf und Verantwortlichkeiten ........................................................................... 5
6.1 Grundsätze ................................................................................................................... 5 6.2 Qualifizierungsauftrag .................................................................................................. 6 6.3 Einteilung der Ausrüstung in Risikoklassen ................................................................. 7 6.4 Vorbereitung der Qualifizierung ................................................................................... 7
6.4.1 Beschaffung .................................................................................................... 7 6.4.2 Lastenheft (User Requirements Specification (URS)) .................................... 8 6.4.3 Qualifizierungsplan .......................................................................................... 9
6.5 Designqualifizierung und Pflichtenheft ......................................................................... 9 6.5.1 Risikoklasse 1 ................................................................................................. 9 6.5.2 Risikoklassen 2 und 3 ..................................................................................... 9
6.6 Abnahmeprüfungen (FAT/SAT) ................................................................................. 10 6.6.1 Werksabnahmeprüfung (Factory Acceptance Test (FAT)) ........................... 11 6.6.2 Standortabnahmeprüfung (Site Acceptance Test, SAT) ............................... 11 6.6.3 FAT/SAT-Testpläne/-protokolle .................................................................... 12
6.7 Prüfpläne IQ/OQ/PQ .................................................................................................. 13 6.8 Geräteinstallation und Installationsqualifizierung (IQ) ............................................... 13
6.8.1 Risikoklasse 1 ............................................................................................... 13 6.8.2 Risikoklasse 2 und 3 ..................................................................................... 14
6.9 Funktionsqualifizierung (OQ), Gerätefunktionsprüfung ............................................. 15 6.9.1 Risikoklasse 1 ............................................................................................... 15 6.9.2 Risikoklasse 2 und 3 ..................................................................................... 15
6.10 Leistungsqualifizierung (PQ) ...................................................................................... 15 6.11 Qualifizierungsbericht ................................................................................................ 16 6.12 Übergabe an den Nutzer ............................................................................................ 17 6.13 Requalifizierung und Review ..................................................................................... 17 6.14 Zuständigkeiten und Verantwortlichkeiten ................................................................. 18
6.14.1 Verantwortung des Leiters der Herstellung, des Leiters der Qualitätskontrolle bzw. des Leiters Forschung & Entwicklung .................................................. 18
6.14.2 Verantwortung des Geräteverantwortlichen.................................................. 18 6.14.3 Verantwortung des Qualifizierungsbeauftragten ........................................... 18 6.14.4 Verantwortung der Qualitätssicherung .......................................................... 19 6.14.5 Verantwortung des Qualitätszirkels .............................................................. 19 6.14.6 Verantwortung der Sachkundigen Person (QP) ........................................... 20 6.14.7 Verantwortung des Einkaufs ......................................................................... 20 6.14.8 Verantwortung jedes Mitarbeiters ................................................................. 20
7 Führung und Ablage von Dokumenten .......................................................................... 20
7.1 Dokumentation zu Geräten der Risikoklasse 1 .......................................................... 20 7.2 Dokumentation zu Geräten der Risikoklasse 2 und 3................................................ 21
8 Anlagen .............................................................................................................................. 21

A.3 Tipps für die Erstellung und den Umgang mit SOPs
(1)C. Oechslein © Maas & Peither AG – GMP-Verlag
A.3
A.3 Tipps für die Erstellung und den Umgang mit SOPs
Der Begriff der Standardarbeitsanweisungen (Standard Opera-ting Procedures) ist aus dem GxP-regulierten Bereich nicht mehr wegzudenken: Alle internationalen Regelwerke fordern, dass qualitätsrelevante Arbeiten und Abläufe in schriftlichen Verfah-rensbeschreibungen festgelegt sind.
Was sind SOPs?
Anstelle des Begriffs „SOP“ werden in Regelwerken und in GxP-pflichtigen Betrieben auch andere Begrifflichkeiten gleichbe-deutend verwendet, beispielsweise Arbeitsanweisung, Ver-fahrensanweisung, Verfahrensbeschreibung, Betriebsan-weisung. Unabhängig von der jeweiligen Bezeichnung handelt es sich in jedem Falle um gelenkte Dokumente, die integrale Bestandteile des dokumentierten Qualitätssiche-rungssystems sind, welches der EU-GMP-Leitfaden und die AMWHV (Arzneimittel- und Wirkstoffherstellungsverordnung) fordern.
Definitionen
„Standardarbeitsanweisung SOP ist eine schriftliche Anwei-sung zur Beschreibung der einzelnen Schritte wiederkehren-der Arbeitsgänge (Standardarbeitsverfahren), einschließlich der zu verwendenden Materialien und Methoden.“ (AMWHV §2, Abs. 15)
„Eine genehmigte, schriftliche Verfahrensanweisung zur Durchführung von Arbeiten, die nicht unbedingt ein bestimmtes Produkt oder Material betreffen, sondern mehr von allgemeiner Art sind (z. B. Bedienungsanweisungen für Geräte und Materialien mit Vorschriften zur Wartung und Reinigung, Reinigungen von Räumlichkeiten und Umge-bungskontrollen, Probenahme und Inspektion)“ (nach WHO, sinngemäß übersetzt)
Arbeitsanweisung,
Verfahrensanweisung,
Verfahrensbeschreibung,
Betriebsanweisung

SOP-Sammlung für die Pharmaindustrie
(2) C. Oechslein © Maas & Peither AG – GMP-Verlag
A.3
Qualitätssicherungssysteme nach ISO 9001:2000ff beschrei-ben Abläufe üblicherweise auf verschiedenen Dokumentati-onsebenen: ¬ Übergeordnete Ebene: Richtlinien, Policies, Master-SOPs,
„übergeordnete Verfahrensanweisungen” oder Qualitätssi-cherungsverfahrensanweisungen (QVA). Diese legen grundsätzliche Verfahrensweisen fest (z. B. zur Validierung) oder beschreiben abteilungs-/bereichsübergreifende Abläufe (z. B. Umgang mit Beanstandungen)
¬ Die Basis des Qualitätssicherungssystems besteht aus vie-len, detaillierten Arbeitsanweisungen: Prüfanweisungen (PA), Arbeitsanweisungen (AA) oder SOPs, in denen ganz konkrete Handlungen beschrieben sind (z. B. Reinigung einer Maschine).
Im Gegensatz dazu unterscheidet der EU-GMP-Leitfaden keine unterschiedlichen Ebenen an Qualitätssicherungsdoku-menten, sondern bezeichnet sie einheitlich als „Verfahrensbe-schreibungen” bzw. „Procedures“. Der Begriff „Standard Opera-ting Procedure” oder SOP taucht dagegen im EU-GMP-Leitfaden nicht auf.
Für die betriebliche Praxis bedeutet das, dass ein Unterneh-men sein System der Qualitätsvorgabedokumente gestalten kann, wie es für das Unternehmen am besten passt (d. h. eine oder mehrere Dokumentationsebenen) und auch die Begriff-lichkeiten für die Dokumentationsebenen selbst festlegen kann. Entscheidend aus GMP-Sicht ist, dass das Dokumentati-onssystem und die Bezeichnung der Dokumente schriftlich de-finiert sind (z. B. in einer Verfahrensanweisung oder SOP) und dieses System in der Praxis funktioniert.
Die in diesem Buch enthaltenen Musterdokumente folgen dem Ansatz des EU-GMP-Leitfadens und unterscheiden nicht in verschiedene Hierarchieebenen. So ist beispielsweise der Validierungsmasterplan als einfache SOP im Dokumentensys-tem integriert.
Qualitätssicherungssystemenach ISO 9001:2000ff

A.3 Tipps für die Erstellung und den Umgang mit SOPs
(3)C. Oechslein © Maas & Peither AG – GMP-Verlag
A.3
Wozu SOPs?
Um Arzneimittel von einheitlicher Qualität herzustellen, darf es für die einzelnen Mitarbeiter bei der Durchführung einer Tätigkeit keine Interpretationsspielräume, individuelle Freiheit oder Kreativität geben. Damit jeder seine Aufgabe auf Anhieb richtig macht, müssen alle wiederkehrenden Tätigkeiten detailliert in SOPs beschrieben sein.
Dabei gelten folgende GrundsätzeSOPs ¬ sind keine Absichtserklärungen, ¬ sollen praxisgerecht und detailliert beschreiben, wie eine
Arbeit ausgeführt werden muss, ¬ müssen unmissverständlich und klar formuliert sein, ¬ müssen in der aktuellen Version vor Ort am Arbeitsplatz
verfügbar sein und allen Betroffenen bekannt sein,¬ müssen von jedem Mitarbeiter (im Geltungsbereich) täglich
strikt eingehalten werden – egal ob Anfänger oder erfahre-ner Praktiker, Chef oder Lehrling.
¬ Es muss immer nach der aktuellsten, genehmigten Version gearbeitet werden.
¬ Abweichungen von SOPs müssen in jedem Falle dokumen-tiert werden.
¬ Gewünschte Änderungen müssen dem Vorgesetzen zur Kenntnis gebracht werden, damit eine sorgfältige Beurtei-lung dieser Änderung erfolgen kann.
¬ Alle gewünschten Änderungen dürfen erst umgesetzt wer-den, wenn sie von hierfür benannten Personen genehmigt wurden.
¬ SOPs müssen regelmäßig überprüft und gegebenenfalls aktualisiert werden, damit sie jederzeit den tatsächlich durchführbaren Arbeitsablauf praxisgerecht beschreiben.
Gut verfasste SOPs haben einen dreifachen Nutzen
1. Für den Mitarbeiter, welcher im Idealfall an der Erstellung seiner Vorschriften beteiligt ist, denn er kennt die real durchführbaren Abläufe am besten. Für ihn ist die SOP wie ein Kochrezept oder eine Art Checkliste: Wenn er seine Ar-beit genau gemäß genehmigter SOP ausführt, hat er seine Pflicht erfüllt.
2. Für den Vorgesetzten: Er prüft die Vorschrift (und lässt sie prüfen, z. B. von den Anschlussabteilungen, die auch von der SOP betroffen sind) und legt mit seiner Unterschrift den Ablauf wie beschrieben fest. Er kann sich fortan darauf verlassen (denn seine Mitarbeiter im GMP-Bereich sind ja zuverlässig!), dass alle Mitarbeiter gemäß SOP arbeiten und jede Abweichung gemeldet wird. Juristisch gesehen
keine Interpretatations-spielräume
Zum Zwecke der besseren Les-barkeit wird auf geschlechtsspe-zifische Formulierungen verzich-tet. In allen Fällen sind Frauen und Männer gleichermaßen ge-meint.
Nutzen für die Mitarbeiter
Nutzen für Vorgesetzte

SOP-Sammlung für die Pharmaindustrie
(4) C. Oechslein © Maas & Peither AG – GMP-Verlag
A.3
entlastet die SOP den Vorgesetzten: Er kann damit bewei-sen, dass er eine Aufgabe verbindlich und schriftlich be-stimmten Mitarbeitern übertragen hat. Deswegen ist es wichtig, in SOPs genau die zuständigen Mitarbeiter zu nen-nen und die SOPs zu schulen.
3. Für die Qualitätssicherung: Sie prüft die Übereinstim-mung der SOP mit den gültigen GMP-Regeln (firmeninter-nes QS-System und nationale/internationale Gesetze und Richtlinien), bevor sie die Arbeitsanweisung genehmigt und in Kraft setzt. Damit ist sichergestellt, dass der Inhalt der SOPs den aktuellen regulatorischen Vorgaben ent-spricht.
Wie sollen SOPs geschrieben sein?
Jedes Unternehmen hat die Möglichkeit und die Pflicht, seine eigenen Standards (z. B. einheitliche Gliederung) zu setzen. Diese sollten am besten in einer Verfahrensanweisung („SOP der SOPs“) beschrieben sein.
Wenn es in einem Unternehmen für sinnvoll erachtet wird, ver-schiedene Ebenen an Verfahrensanweisungen zu definieren, dann können für die unterschiedlichen Dokumentenebenen auch unterschiedliche Vorgaben bezüglich Struktur und Inhalt definiert werden, beispielsweise:¬ technische SOPs (z. B. Bedienungs-, Reinigungs-, Wartungs-
und Kalibrier-SOPs): gekürztes Inhaltsverzeichnis, anderes Review-Intervall, der Genehmiger ist nicht in der Qualitäts-sicherung angesiedelt, sondern in der Technik
¬ übergeordnete SOPs (z. B. Masterpläne, bereichsübergrei-fende Regelungen, internationale Qualitätsstrategien): spezielles Inhaltsverzeichnis, englische Sprache (da die Anwender dieser Dokumentenkategorie im Berufsalltag täglich Englisch verwenden)
Die in dieser SOP-Sammlung vorliegenden Verfahrensanwei-sungen unterscheiden nicht verschiedene Dokumentene-benen und haben daher alle ein einheitliches Format, wie es in kleinen und mittelständischen Unternehmen vielfach üblich ist.
Worauf es bei GMP-Vorgabedokumenten im Allgemeinen – also auch SOPs – ankommt, ist im EU-GMP-Leitfaden beschrieben.
Nutzen für dieQualitätssicherung
Für die Inhalte und den Umfangvon Verfahrensanweisungen
gibt es keine präzisen gesetzli-chen Vorgaben oder verbindliche
Standards.

Maas & Peither Pharma GmbH
Standardarbeitsanweisung Seite 9 von 30
SOP-400-01 Datenintegrität und Data G overnanc e Gültig ab: 1.5.2018
2018 © Maas & Peither AG – GMP-Verlag Musterdokumente – SOP-Sammlung für die Pharmaindustrie 9
• SOP-532 Umgang mit Abweichungen und Änderungen bei computergestützten Systemen • SOP-533 Konzept zur Datenmigration • SOP-534 Stilllegung und Außerbetriebnahme von computergestützten Systemen
6 Arbeitsablauf und Verantwortlichkeiten
6.1 G runds ätze 1. Die Integrität von Daten gehört zu den obersten Unternehmenszielen und darf keinem
anderen Managementziel untergeordnet werden. „Data Governance“ und „Datenintegrität“ sind daher „Chefsache“.
2. Die Beherrschung von Daten resultiert aus der Summe vieler Einzelmaßnahmen, von denen keine die andere ersetzt – dennoch darf sie nie auf punktuelle oder isolierte Maßnahmen beschränkt sein, sondern ist als ganzheitlicher Ansatz über den gesamten Daten-Lebenszyklus und den gesamten Lebenszyklus des zugehörigen Produktes bzw. der Anlage zu betrachten.
3. Mangelhafte Datenintegrität wird grundsätzlich nicht als „zufälliger Fehler“ oder „persönlicher Mitarbeiterfehler“ eingestuft, sondern muss stets auf die tatsächliche Ursache hin untersucht werden (gemäß SOP-105 „Maßnahmen zu Fehlerkorrektur und Vorbeugung (CAPA)“).
4. Der Umfang der notwendigen Maßnahmen zur Erzielung von Datenintegrität ist abhängig von der Kritikalität der Daten. Letztere wird vom Dateneigner in Zusammenarbeit mit der Qualitätssicherung risikobasiert bestimmt, bevor das zugehörige System bzw. der zugehörige Arbeitsablauf für GxP-Zwecke in Betrieb genommen wird (siehe Punkt 5.2.5).
Die Anforderungen an „integre Daten“ werden oftmals mit dem Akronym „ALCOA“ bzw. „ALCOA Plus“ subsummiert. Im Folgenden ist beschrieben, wie diese Anforderungen im Einzelnen im Unternehmen umgesetzt werden.
Oftmals sind systematische Fehler oder konkurrierende Ziele (z. B. Lieferfähigkeit ist der Unternehmensleitung wichtiger als ehrlicher Umgang mit Daten) Ursache für Datenfälschungen. Es wäre grundfalsch, in so einem Falle „Mitarbeiterfehlverhalten“ auf der operativen Ebene als Ursache zu diagnostizieren. Weitere Datenintegritäts-Verstöße sind damit vorprogrammiert.
Die virtuelle Firma Maas & Peither Pharma GmbH hat sich entschieden, die Beherrschung von Daten (Data Governance) als Voraussetzung für „Datenintegrität“ ausdrücklich zur Verantwortung der Geschäftsführung zu machen und für alle Mitarbeiter sichtbar zur „Chefsache“ zu erklären. Das ist auch der Grund, weshalb diese SOP von der Geschäftsführung ausdrücklich gutgeheißen – d. h. unterschrieben werden sollte. Nur so ist gewährleistet, dass die Geschäftsleitung tatsächlich keine konkurrierenden Ziele verfolgt, die es dem Mitarbeiter schwer machen, bei der Wahrheit zu bleiben. Ganz falsch wäre es, „Datenintegrität“ ausschließlich durch Maßnahmenpakete für die operative Ebene erreichen zu wollen, während in der Managementebene keine Bewusstseinsänderung stattfindet. Psychologisch wichtig ist auch die Signalwirkung für die Mitarbeiter, wenn sie wissen, dass korrekter Umgang mit Daten von alleroberster Ebene gefordert und honoriert wird.
6.2 Herrs chaft über Daten – B eherrs chung von Daten – Data G overnance Verglichen mit der einfachen Protokollierung von Daten auf Papier erfordert die Erzeugung komplexer Daten in elektronischen Systemen umfangreichere Maßnahmen, um Schwachstellen, die zu Datenveränderung führen könnten, vorzubeugen, zu entdecken und zu korrigieren. Die mehrfache Weiterleitung von Daten über mehrere Schnittstellen verschiedener Computersysteme oder vom/zum Dienstleister macht die Situation unübersichtlich und erleichtert zufällige oder systematische Datenverluste.
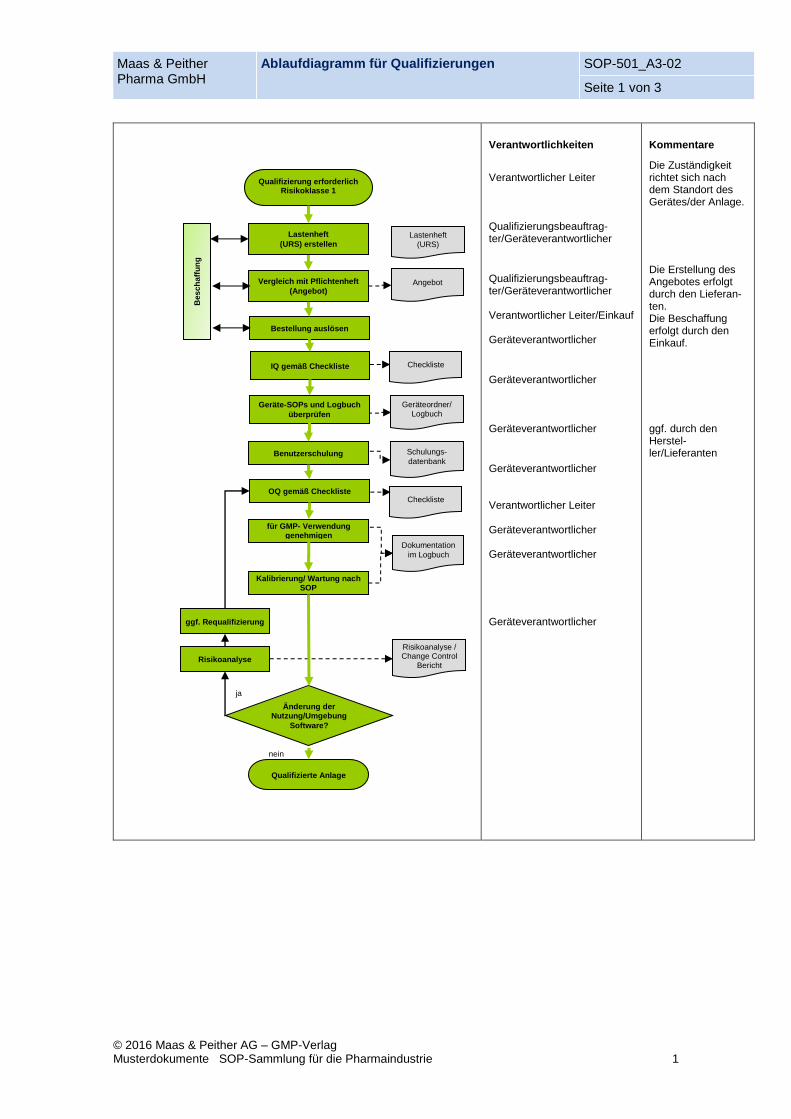
Maas & Peither Pharma GmbH
Ablaufdiagramm für Qualifizierungen SOP-501_A3-02
Seite 1 von 3
© 2016 Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 1
Verantwortlichkeiten
Verantwortlicher Leiter Qualifizierungsbeauftrag-ter/Geräteverantwortlicher
Qualifizierungsbeauftrag-ter/Geräteverantwortlicher Verantwortlicher Leiter/Einkauf Geräteverantwortlicher
Geräteverantwortlicher Geräteverantwortlicher
Geräteverantwortlicher Verantwortlicher Leiter Geräteverantwortlicher Geräteverantwortlicher
Geräteverantwortlicher
Kommentare
Die Zuständigkeit richtet sich nach dem Standort des Gerätes/der Anlage.
Die Erstellung des Angebotes erfolgt durch den Lieferan-ten. Die Beschaffung erfolgt durch den Einkauf. ggf. durch den Herstel-ler/Lieferanten
Risikoanalyse
Vergleich mit Pflichtenheft
(Angebot)
Bestellung auslösen
Checkliste IQ gemäß Checkliste
Geräteordner/ Logbuch
Geräte-SOPs und Logbuch
überprüfen
Dokumentation
im Logbuch
Änderung der Nutzung/Umgebung
Software?
Qualifizierte Anlage
Angebot
Lastenheft
(URS) erstellen Lastenheft
(URS)
Benutzerschulung
OQ gemäß Checkliste
Schulungs-
datenbank
für GMP- Verwendung genehmigen
Kalibrierung/ Wartung nach SOP
nein
ja
ggf. Requalifizierung
Risikoanalyse / Change Control
Bericht
Qualifizierung erforderlich Risikoklasse 1
Checkliste
Bes
ch
aff
un
g

Maas & Peither Pharma GmbH
Checkliste für Qualifizierungen SOP-501_A4-02
Seite 1 von 3
© 2016 Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 1
Allgemeine Angaben
Bezeichnung Inventar-Nr. Hersteller/ Lieferant
Gerätetyp-Nr., Serien-Nr.
(… Text …) (… Text …) (… Text …) (… Text …)
Bitte vollständig ausfüllen.
Das Gerät/die Anlage
wird isoliert betrieben □
ist/sind Teil einer Anlage □ (Bezeichnung d. A. …)
Standort des Geräts (Gebäude, Raum-Nr. …)
Der Betrieb erfolgt im:
Reinraum □ Analysenlabor □ F & E □
Lager □ Technikbereich □ Sonstige
Räume □
Bereitstellung Klin. PP □
Ermittlung der Risikoklasse – Teil 1
Risikoklasse 0 Geräte aus serienmäßiger Fertigung ohne jegliche Messeinheit und Kalibrierfunktion
Es ist keine Qualifizierung erforderlich. □
Risikoklasse 1 einfache Geräte aus serienmäßiger Ferti-gung mit Messeinheit und Kalibrierfunktion
Die Qualifizierung schließt mit der Funkti-onsqualifizierung (OQ) ab.
□
Risikoklasse 2 Geräte und Anlagen von geringer bis mittlerer Komplexität
Die Qualifizierung erfolgt vollständig. □
Risikoklasse 3 Geräte und Anlagen von hoher Komplexität Die Qualifizierung erfolgt vollständig ein-schließlich FAT/SAT.
□
Risikoklasse ermittelt: (Geräteverantwortlicher)
(Datum/Unterschrift)
Risikoklasse geprüft: (Qualitätssicherung)
(Datum/Unterschrift)
Risikoklasse genehmigt: (Verantwortlicher Leiter)
(Datum/Unterschrift)
Formblatt erstellt: (Datum/Unterschrift)
überprüft: genehmigt:

Maas & Peither Pharma GmbH
Standardarbeitsanweisung Seite 1 von 29
SOP-505-02 Reinigung und Reinigungsvalidierung
Gültig ab:
01.09.2016
2016 © Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 1
SOP-505-02 Reinigung und Reinigungsvalidierung
1 Geltungsbereich
Diese Standardarbeitsanweisung gilt am Standort Pharmahausen der Maas & Peither Pharma GmbH für sämtliche GMP-relevanten Bereiche aus:
Betreuung Lohnherstellung und Außenlager
Herstellung Solida
Verpackung
Qualifizierung/Validierung
Produktionsplanung
Technik
Prüfung Ausgangsstoffe/Packmittel
Analytische Entwicklung
Betreuung Auftragslabore
Probenahme
Pharmazeutische Technologie und Scale-up
Bereitstellung klinischer Prüfpräparate
Einkauf
Qualitätssicherung
Leitung Herstellung
Leiter der Qualitätskontrolle
Leiter Forschung und Entwicklung
Dieses Dokument ist für alle Mitarbeiter der genannten Bereiche verbindlich, sofern sie in Rei-nigungsvalidierungsprojekte eingebunden sind, sowie für ggf. beauftragte externe Dienstleister oder Berater.
erstellt:
(Datum/Unterschrift)
überprüft: (Datum/Unterschrift)
genehmigt: (Datum/Unterschrift)

Maas & Peither Pharma GmbH
Standardarbeitsanweisung Seite 2 von 29
SOP-505-02 Reinigung und Reinigungsvalidierung
Gültig ab:
01.09.2016
2016 © Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 2
Inhalt
1 Geltungsbereich .................................................................................................................. 1
2 Ziel/Zweck ............................................................................................................................. 4
3 Anwendungsbereich ........................................................................................................... 4
4 Definitionen/Abkürzungen .................................................................................................. 4
5 Grundlagen, mitgeltende Unterlagen ................................................................................ 6
5.1 Grundlagen .................................................................................................................. 6 5.2 Mitgeltende Unterlagen ................................................................................................ 6
6 Arbeitsablauf und Verantwortlichkeiten............................................................................ 7
6.1 Reinigung ..................................................................................................................... 7 6.1.1 Reinigungsanweisungen ................................................................................. 7 6.1.2 Reinigungsmittel .............................................................................................. 8 6.1.3 Kennzeichnung und Aufbewahrung ................................................................ 9 6.1.4 Reinigungsarten .............................................................................................. 9 6.1.5 Reinigung bei Kampagnenfertigung oder von Dedicated Equipment ........... 10 6.1.6 Außerplanmäßige Reinigungen .................................................................... 10
6.2 Festlegung zulässiger Standzeiten ............................................................................ 10 6.3 Reinigungsvalidierung ................................................................................................ 10
6.3.1 Grundsätze .................................................................................................... 11 6.3.2 Auswahl der Leitsubstanzen ......................................................................... 14 6.3.3 Grenzwerte und Akzeptanzkriterien .............................................................. 15 6.3.4 Grenzwerte und Akzeptanzkriterien für mikrobiologische Kontamination .... 19 6.3.5 Auswahl eines oder mehrerer Probenahmeverfahren .................................. 19 6.3.6 Auswahl der Probenahmematerialien und -hilfsmittel ................................... 20 6.3.7 Auswahl der zu beprobenden Ausrüstungsteile und Festlegung
repräsentativer Probenahmestellen .............................................................. 20 6.3.8 Auswahl der analytischen Methode .............................................................. 21 6.3.9 Probenahme .................................................................................................. 22 6.3.10 Dokumentation .............................................................................................. 22
6.4 Vorgehensweise bei bestandener/nicht bestandener Reinigungsvalidierung ........... 23 6.5 Aufrechterhaltung des Validierungsstatus ................................................................. 23
6.5.1 Revalidierung ................................................................................................ 23 6.5.2 Monitoring ..................................................................................................... 24 6.5.3 Vorgehensweise bei bestandenem/nicht bestandenem Monitoring ............. 24 6.5.4 Review........................................................................................................... 24
6.6 Zuständigkeiten und Verantwortlichkeiten ................................................................. 25 6.6.1 Verantwortung der Qualitätssicherung .......................................................... 25 6.6.2 Verantwortung des Leiters der Forschung & Entwicklung, Herstellung bzw.
Technik, Einkauf ............................................................................................ 25 6.6.3 Verantwortung des Leiters der Forschung & Entwicklung ............................ 25 6.6.4 Verantwortung des Leiters der Herstellung................................................... 25 6.6.5 Verantwortung des Leiters der Qualitätskontrolle ......................................... 26 6.6.6 Verantwortung der Sachkundigen Person (QP) ........................................... 26 6.6.7 Verantwortung der analytischen Probenahme .............................................. 26 6.6.8 Verantwortung der Analytischen Entwicklung ............................................... 26 6.6.9 Verantwortung der Betreuung Auftragslabore .............................................. 26 6.6.10 Verantwortung des Dienstleisters Labor D GmbH ........................................ 26 6.6.11 Verantwortung des Dienstleisters Labor TOX GmbH ................................... 27 6.6.12 Verantwortung des Validierungskoordinators ............................................... 27

Maas & Peither Pharma GmbH
Standardarbeitsanweisung Seite 3 von 29
SOP-505-02 Reinigung und Reinigungsvalidierung
Gültig ab:
01.09.2016
2016 © Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 3
6.6.13 Verantwortung des Geräteverantwortlichen.................................................. 27 6.6.14 Verantwortung jedes Mitarbeiters ................................................................. 27 6.6.15 Verantwortung des RV-Probenehmers der Produktion ................................ 27
7 Führung und Ablage von Dokumenten ........................................................................... 28
8 Anlagen ............................................................................................................................... 28
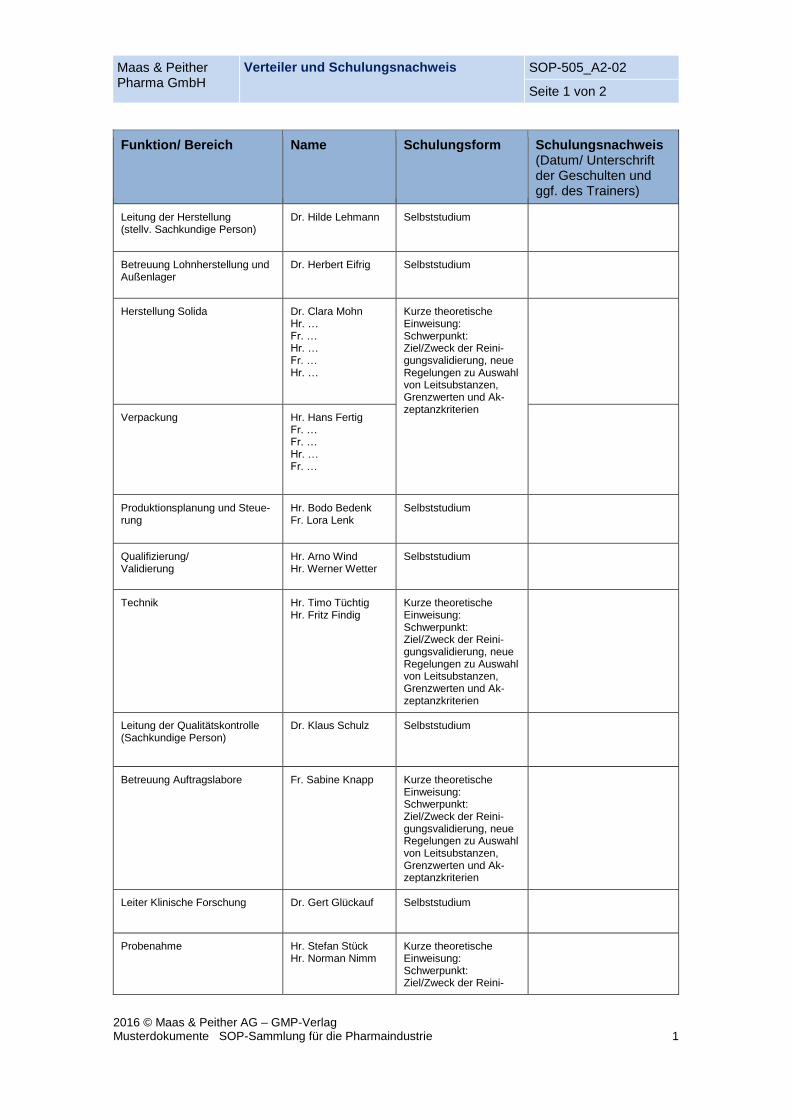
Maas & Peither Pharma GmbH
Verteiler und Schulungsnachweis SOP-505_A2-02
Seite 1 von 2
2016 © Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 1
Funktion/ Bereich Name Schulungsform Schulungsnachweis (Datum/ Unterschrift der Geschulten und ggf. des Trainers)
Leitung der Herstellung (stellv. Sachkundige Person)
Dr. Hilde Lehmann Selbststudium
Betreuung Lohnherstellung und Außenlager
Dr. Herbert Eifrig Selbststudium
Herstellung Solida Dr. Clara Mohn Hr. … Fr. … Hr. … Fr. … Hr. …
Kurze theoretische Einweisung: Schwerpunkt: Ziel/Zweck der Reini-gungsvalidierung, neue Regelungen zu Auswahl von Leitsubstanzen, Grenzwerten und Ak-zeptanzkriterien
Verpackung Hr. Hans Fertig Fr. … Fr. … Hr. … Fr. …
Produktionsplanung und Steue-rung
Hr. Bodo Bedenk Fr. Lora Lenk
Selbststudium
Qualifizierung/ Validierung
Hr. Arno Wind Hr. Werner Wetter
Selbststudium
Technik Hr. Timo Tüchtig Hr. Fritz Findig
Kurze theoretische Einweisung: Schwerpunkt: Ziel/Zweck der Reini-gungsvalidierung, neue Regelungen zu Auswahl von Leitsubstanzen, Grenzwerten und Ak-zeptanzkriterien
Leitung der Qualitätskontrolle (Sachkundige Person)
Dr. Klaus Schulz Selbststudium
Betreuung Auftragslabore Fr. Sabine Knapp Kurze theoretische Einweisung: Schwerpunkt: Ziel/Zweck der Reini-gungsvalidierung, neue Regelungen zu Auswahl von Leitsubstanzen, Grenzwerten und Ak-zeptanzkriterien
Leiter Klinische Forschung Dr. Gert Glückauf Selbststudium
Probenahme Hr. Stefan Stück Hr. Norman Nimm
Kurze theoretische Einweisung: Schwerpunkt: Ziel/Zweck der Reini-
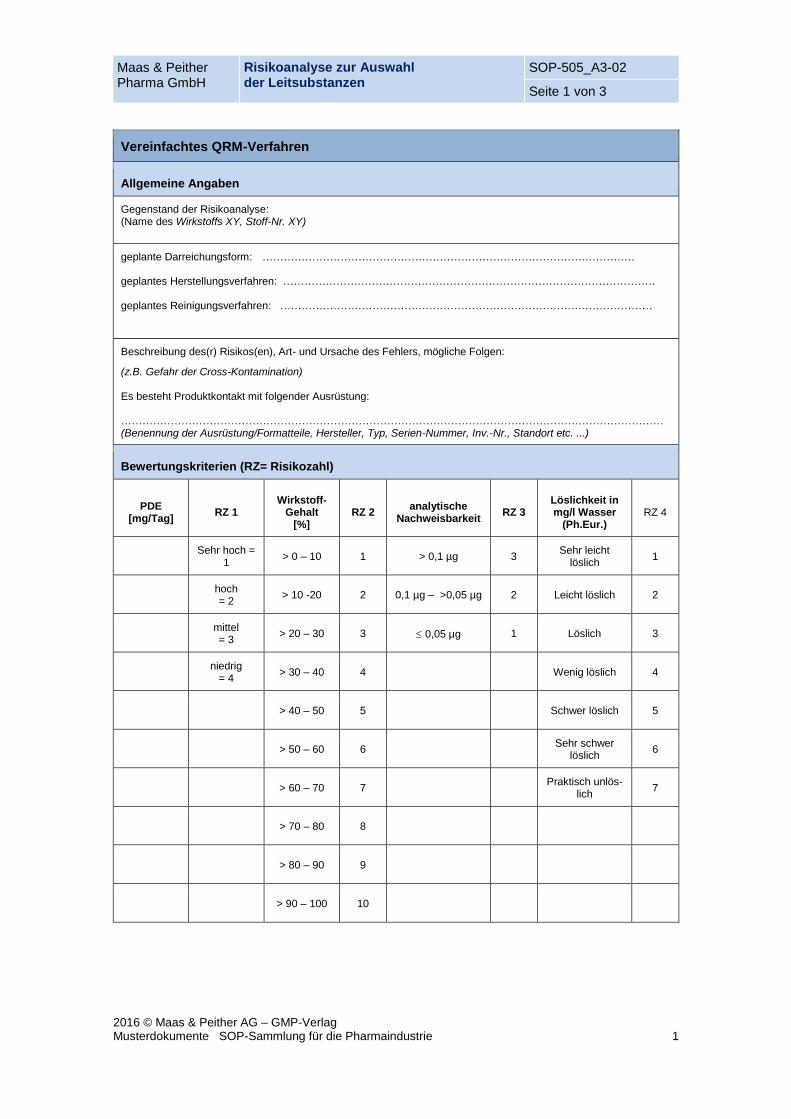
Maas & Peither Pharma GmbH
Risikoanalyse zur Auswahl der Leitsubstanzen
SOP-505_A3-02
Seite 1 von 3
2016 © Maas & Peither AG – GMP-Verlag Musterdokumente SOP-Sammlung für die Pharmaindustrie 1
Vereinfachtes QRM-Verfahren
Allgemeine Angaben
Gegenstand der Risikoanalyse: (Name des Wirkstoffs XY, Stoff-Nr. XY)
geplante Darreichungsform: …………………………………………………………………………………………… geplantes Herstellungsverfahren: …………………………………………………………………………………………… geplantes Reinigungsverfahren: ……………………………………………………………………………………………
Beschreibung des(r) Risikos(en), Art- und Ursache des Fehlers, mögliche Folgen:
(z.B. Gefahr der Cross-Kontamination) Es besteht Produktkontakt mit folgender Ausrüstung: ……………………………………………………………………………………………………………………………………… (Benennung der Ausrüstung/Formatteile, Hersteller, Typ, Serien-Nummer, Inv.-Nr., Standort etc. ...)
Bewertungskriterien (RZ= Risikozahl)
PDE [mg/Tag]
RZ 1 Wirkstoff-
Gehalt [%]
RZ 2 analytische
Nachweisbarkeit RZ 3
Löslichkeit in mg/l Wasser
(Ph.Eur.) RZ 4
Sehr hoch =
1 > 0 – 10 1 > 0,1 µg 3
Sehr leicht löslich
1
hoch = 2
> 10 -20 2 0,1 µg – >0,05 µg 2 Leicht löslich 2
mittel = 3
> 20 – 30 3 0,05 µg 1 Löslich 3
niedrig
= 4 > 30 – 40 4 Wenig löslich 4
> 40 – 50 5 Schwer löslich 5
> 50 – 60 6 Sehr schwer
löslich 6
> 60 – 70 7 Praktisch unlös-
lich 7
> 70 – 80 8
> 80 – 90 9
> 90 – 100 10

Maas & Peither Pharma GmbH
Standardarbeitsanweisung Seite 3 von 15
SOP-601-02 P rozes s validierung
Gültig ab: 01.10.2018
2018 © Maas & Peither AG – GMP-Verlag Musterdokumente – SOP-Sammlung für die Pharmaindustrie 3
2 Ziel/Zweck Ziel dieser SOP ist es, das Vorgehen bei der Prozessvalidierung fester Formen so weit wie möglich zu standardisieren, damit die Durchführung dieser Projekte effizient und auf die projekt-spezifisch kritischen Aspekte fokussiert ablaufen kann. Während der Validierungsmasterplan (SOP-600) das allgemeine Vorgehen bei Validierungsprojekten, die verantwortlichen Personen und die erforderlichen Dokumente definiert, beschreibt diese SOP die inhaltlichen Aspekte der Prozessvalidierung fester Formen. Der hier beschriebene Ansatz setzt das zeitgemäße „Life-Cycle-Modell der Validierung“ um: basierend auf den Erkenntnissen der Prozessentwicklung wird eine „Control Strategy“ definiert, anschließend in der sog. traditionellen Form die Eignung von Prozess und Control Strategy anhand von Konformitätschargen überprüft und schließlich über den gesamten Produktlebenszyklus im Sinne einer „Ongoing Process Verification“ (OPV) verifiziert.
3 Anwendungsbereich Diese SOP legt fest,
• wie Herstellungsverfahren für feste Formen prospektiv validiert werden, • in welcher Weise sichergestellt wird, dass der Prozess über den gesamten Produktlebens-
zyklus in validem Zustand bleibt, • welche Akzeptanzkriterien für feste Formen generell zu setzen sind • wie eine Control Strategy festzulegen ist.
Diese SOP befasst sich nicht mit prozessbegleitender Validierung, der Validierung von Verpa-ckungsprozessen oder Reinigungsverfahren. Das Vorgehen in diesen Fällen ist in eigenen SOPs festgelegt. Auch die Vorgehensweise für eine Kontinuierliche Prozessverifizierung (CPV) ist nicht Gegenstand dieser SOP.
In kleineren Unternehmen kann der Anwendungsbereich auch auf die prozessbegleitende (con-current) Validierung ausgedehnt werden, d. h. beide Validierungsformen werden in einer einzi-gen SOP beschrieben. Eine Aufteilung in zwei SOPs, wie im vorliegenden Fall, ist immer dann vorteilhaft, wenn viele unterschiedliche Mitarbeiter in den Prozess involviert sind und die Abläu-fe (prospektiv/concurrent) deutlich verschieden sind. In diesem Falle würde eine Zusammenfas-sung in einer einzigen SOP zu einer zu umfangreichen SOP führen.
Auf eine Erwähnung der retrospektiven Validierung wurde hier bewusst verzichtet, da dieses Vorgehen nicht mehr dem Stand der Technik entspricht.
4 Definitionen/Abkürzungen
Bitte hier nur diejenigen Abkürzungen und Fachbegriffe aufführen, die im eigenen Betrieb tat-sächlich geläufig sind!
Begriff/ Abkürzung Definition bei Maas & Peither Pharma GmbH
Akzeptanzkriterium Vorab festgelegte Anforderung, die erfüllt sein muss, damit eine Validierung erfolgreich abgeschlossen werden kann.
Chargenprotokoll Ein Chargenprotokoll enthält alle chargenbezogenen Vorgaben und wird während der Herstellung handschriftlich mit den aktuell anfal-lenden Ist-Werten ergänzt (entspricht dem Begriff des Herstel-

A.2 Die Autoren
(1) © Maas & Peither AG – GMP-Verlag (EL12)
A.2
A.2 Die Autoren
A.2.1 Die Autorinnen
Dr. Stephanie [email protected]
Unternehmensberaterin, GeschäftsführerincirQum, Frankfurt am Main
Dr. Stephanie Blum gründete 2008 das Beratungsunternehmen cirQum in Frank-furt und ist dessen Inhaberin und Geschäftsführerin. Zuvor war die promovierteMolekularbiologin 10 Jahre in leitenden Positionen in der biotechnologischen undpharmazeutischen Industrie tätig. Frau Dr. Blum arbeitet als Auditorin, Beraterinund Referentin und ist Autorin zahlreicher Fachpublikationen.
cirQum ist aktiv als Dienstleister für pharmazeutisches Qualitätsmanagement (GMP, GCP, GLP, GCLP, GDP, GACP) mit den Schwerpunkten Auftrags-Audits, Quali-tätsmanagement-Beratung und Schulung. Im Mittelpunkt stehen dabei GMP-Au-dits bei Lohnherstellern und Auftragslaboren sowie bei Herstellern von Wirkstof-fen, Hilfsstoffen und Packmitteln. Hinzu kommen für die Hersteller pflanzlicher Arzneimittel GACP-Audits bei Anbauern und Lieferanten pflanzlicher Rohstoffe. Im GDP-Bereich stehen Audits bei Spediteuren, Lägern und Großhändlern im Fokus und in den Bereichen GCP, GCLP und GLP werden CROs, Prüfzentren, Labore und Prüfeinrichtungen auditiert. Ein weiterer Schwerpunkt sind Schulungen und Semi-nare zu GXP-Themen sowie die Beratung rund um das pharmazeutische Qualitäts-management.
Alexandra Fü[email protected]
Dipl. Ing (FH) ChemieQA Specialist Pharma, Ludwigshafen
Alexandra Fürst bietet seit 2012 freiberufliche Unterstützung in der Pharmazeuti-schen Industrie an.
Ihre Projekte umfassten bisher QA-Aufgaben wie das Erstellen von SOPs, PQRs, Schulungen, Abweichungen und Änderungsmanagement. Zusätzliche Tätigkeiten wie das Erstellen von Quality Manual und SMF und die Durchführung von internen Audits sowie Lieferantenaudits ergänzen das Leistungsspektrum. Des Weiteren kommen Überarbeitung von Herstellungsanweisungen sowie Prozessentwicklung und -optimierung für Pharma-Firmen, Wirkstoffhersteller und Tierarzneimittelher-steller dazu.
Nach ihrem Studium lebte Frau Fürst 9 Jahre in England und arbeitete dort u.a. für Eli Lilly. Hier war sie anfangs im Bereich Health and Safety für den Research Cen-ter in Windlesham tätig. Später wechselte sie an den Herstellungsstandort in Ba-singstoke und betreute zuerst die Qualitätskontrolle als SOP- und Schulungs-Coor-dinator und anschließend als Stabilitäts-Coordinatorin.
A-Autoren.fm Seite 1 Donnerstag, 13. April 2017 12:51 12

SOP-Sammlung für die Pharmaindustrie
(2) © Maas & Peither AG – GMP-Verlag (EL12)
A.2
Dr. Christine [email protected]
ApothekerinGMP-Praxis, Bad Säckingen
Dr. Christine Oechslein führt als freiberufliche GMP-Trainerin interne GMP-Trainingsbei Pharmafirmen, Wirkstoffherstellern und Zuliefererbetrieben durch. Zuvor warsie viele Jahre in der Pharmaindustrie tätig. Als Referentin bei verschiedenen Ver-anstaltern und Autorin vermittelt sie GMP-Wissen mit den Schwerpunkten Prozes-svalidierung, GMP-Training und GMP in der Entwicklung.
Nach dem Studium der Pharmazie begann Frau Dr. Oechslein ihre Berufslauf-bahn 1987 bei Kettelhack-Riker als Projektleiterin. Später wechselte sie zu Sandoz Pharma, wo sie als Laborleiterin für die Entwicklung neuartiger Drug Delivery Sys-tems für Peptid-Wirkstoffe verantwortlich war. Außerdem gehörten Rezeptur- und Prozessentwicklung, Prozessvalidierung und Klinikmusterbereitstellung zu ihren Aufgaben. Parallel dazu fertigte sie ihre Dissertation an. Nach ihrem Wechsel in die Qualitätssicherung konzipierte sie ein Qualitätssicherungshandbuch für die phar-mazeutische Entwicklung, verwaltete das SOP-System und begleitete Prozessvali-dierungen.
Seit 1997 war sie als freiberufliche Mitarbeiterin im Bereich Qualitätssysteme bei Novartis Pharma AG beschäftigt. Parallel dazu qualifizierte sie sich durch ein Kon-taktstudium an der Pädagogischen Hochschule Freiburg zur GMP-Trainerin. Sie ist Autorin zahlreicher Fachpublikationen.
Cornelia [email protected]
Pharmazeutisch-technische AssistentinGxP-Services Cornelia Wawretschek, Berlin
Cornelia Wawretschek arbeitet seit 15 Jahren freiberuflich als Unternehmensbera-terin für Qualitätssicherung. Sie sammelte langjährig praktische Erfahrungen aufden Gebieten Pharmazeutische Verfahren und Analytik mit den Kompetenz-schwerpunkten Entwicklung fester, halbfester und flüssiger Arzneiformen, Steril-fertigung sowie Klinische Prüfpräparate.
Seit 1986 bei der Schering AG, Berlin (jetzt: Bayer HealthCare Pharmaceuticals) tätig, unterstützte sie als Referentin die pharmazeutischen, chemischen und analy-tischen Entwicklungsfunktionen des Standorts Berlin auf dem Gebiet der Quali-tätssicherung. Dazu gehörten vor allem die Vorbereitung und Begleitung behörd-licher Inspektionen und Qualitätsaudits.
2001 gründete Frau Wawretschek das Unternehmen GxP-Services, Berlin. Sie be-rät Pharma- und Biotechnologieunternehmen beim Aufbau und der Aktualisie-rung Pharmazeutischer Qualitätssysteme und erstellt für sie SOPs. Weitere Schwer-punkte sind die Vor- und Nachbereitung von behördlichen Inspektionen, die Erstellung von Modulen zur Planung und Verwaltung von Schulungen und die Durchführung von Trainingsmaßnahmen.
A-Autoren.fm Seite 2 Donnerstag, 13. April 2017 12:51 12





























