Embed Size (px)
Citation preview

Stereoselektive Synthese tea 2,6-Methano-2-ben~n-5-ole 177
Stereoselektive Synthese tertiarer 2,6-Methano-2-benzazocin-5-ole Ulrike Holzgrabe', Barthold Piening und Rolf Haller t
Pharmazeutisches Institut der Universitiit Kiel, Gutenbergstr. 76-78, D-2300 E e l
Eingegangen am 28. Miirz 1989
Der 3.1 1-dipyridylsubstituiene 5-0xc~2,6-methano-2-benzazocin-~n- s;iureeSter 1 wird rnit Gngmrd- bzw. lithiumorganischen Reagentien zu ter- tiiiren Akoholen umgesetzt Auf diesem Wege gelingt die stereoselektive Einfiihrung von Methyl-, Phenyl-, Ropargyl- und Ethinylsubstituenten an C-
tion der so erhaltenen &ohole erfolgt mit Hilfe der 'H- und l3c-m- Spektroskopie.
Stereoselective Syntheses of kdmy 2 , 6 - M e t h a n u - Z b e n z i n - h I ~
The conversion of the 3.11-dipyridine substituted methyl 5-hydmxy-2.6 m e t h a n o - 2 - b e ~ e ~ x y l a 1 e s 1 with Crignurd- and lithium
des Methanobenzazocingeriistes. Die der Konfigura- organic compounds stereoselectively yields terriory alcohols with methyl-, phenyl-. propargyl- and efhinylsubstituents at C-5. The relative configuration of the alcohols so obtained is determined by means of 'H-NMR and I3C- NMR spektroscopic data
Die oxidative Cyclisierung substituierter 1 -Benyl4-piperidon-3-carbon- siiureester mit Cer(IV)sulfat in schwefelsaurer Liisung fiihrt zu 5-0x0-2.6- m e t h a n o - 2 - b e ~ i n a c a r b o n s a ~ s t e ~ ' ) , deren Grundgeriist sich von den stark analgetisch wirkenden Benzomorphanen nur in der Stellung des Stickstoffs im Methanobenzazocingeriist unterscheide?.
Gegenstand dieser Untersuchungen ist die Stereoselektivi- tat der Umsetzung der Carbonylgruppe des in guten Aus- beuten zughglichen 3.1 1-dipyridylsubstituierten 5-0x0- 2 ,6-methano-2-benins 1 mit Methylmagnesiumiodid, Phenyl- und Propargyl-magnesiumbromid sowie Methylli- thium und Lithiumacetylid-Ethylendiamin-Komplex. Die sich aus dieser Synthese ergebenden ter t ihn Alkohole sollten staker wirksamere Derivate sein als das Ausgangs- keton 12).
Tab. 1: Ergebnisse der Umsetzung des Ketons 1 mit metallorganischen Reagentien (Epimerenverhiiltnis: a = axiale Hydroxylgruppe und e = equa- toriale Hydroxylgruppe)
Reagens a : e
CHjMgI 80: 20 CH3Li 100: 0 C&MgBr loo: 0 HCdXH2MgBr *) LiCrCHxH2NCHzCHzNH2 loo: 0
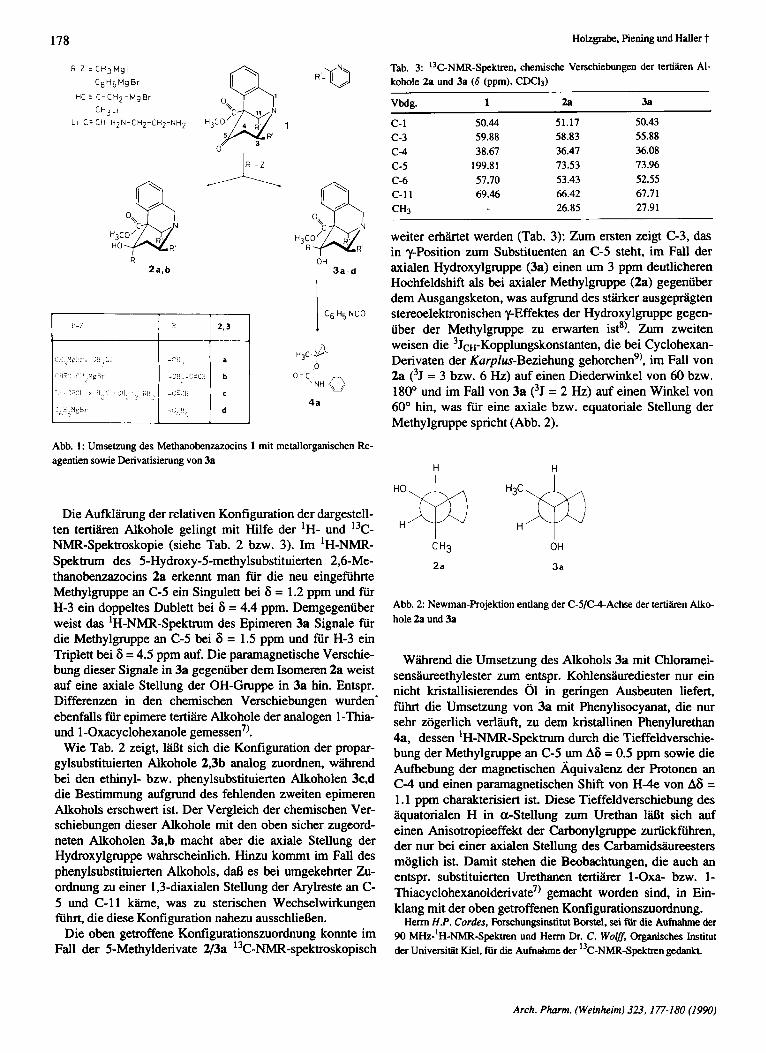
Die Umsetzung des 5-0~0-2,6-rnethano-2-be~ocins 1 rnit Alkylmagnesiumhalogeniden gelingt ausschlieSlich in trockenem Dichlormethan als Lijsungsmittel (vergl. Lit.3)), w h n d die Reaktion mit lithiumorganischen Verbindun- gen sowie mit Phenylmagnesiumbromid in Tetrahydrofuran durchgefiihrt werden kann (Exp. Teil). Bei allen Reaktionen zeigt sich, daR lediglich die Ketogruppe und nie die Ester- funktion angegriffen wird (Abb. 1). Bei der Umsetzung rnit den Alkylmagnesiumhalogeniden werden beide moglichen epimeren Alkohole gebildet (Tab. 1). Wiihrend sich im Fall der Reaktion rnit Methylmagnesiumiodid beide Isomere
isolieren l iekn und sich ein Epimerenverhdtnis von 80% Alkohol mit axialer Hydroxylgruppe (3) und 20% rnit equa- torialer OH-Gruppe (2) bestimmen lieS (Integration des Signals der Methylgruppe an C-5 im 'H-NMR-Spektrum), erhdt man bei der Reaktion mit Propargylmagnesiumbro- mid ein Epimerengemisch, dessen Zusammensetzung sich mit 'H-NMR-spektroskopischen oder chromatographischen Methoden nicht quantitativ bestimmen l a t , und dessen isomere Alkohole sich nur schwer getrennt isolieren lassen. Bei der Umsetzung von 1 mit Phenylmagnesiumbromid, Methyllithium sowie Lithiumacetylid-Ethylendiamin- Komplex wird iiberwiegend, d.h. zu mehr als 95% (genaue- re Angaben 1 a t die 'H-NMR-spektroskopische Bestim- mung nicht zu) der Alkohol mit axialer Hydroxylgruppe ge- bildet. Damit greift das Nukleophil also mit hoher Stereose- lektivitat equatorial (exocyclisch) die Ketofunktion an C-5 an. Diese Lmkung des Reaktionsverlaufes l a t sich durch den Pyridinring an C-11, der eine P-syn-axiale Stellung zur Ketogruppe einnimmt4), erkliiren: Er blockiert den Angnff der Grignard-Verbindung von der Unterseite des Molekiils fast vollstihdig. Diese Beobachtungen stehen im Einklang mit Untersuchungen von Kamemitzky et al.') und Cherest und Felkin6), die bei der Umsetzung von cyclischen Ketonen mit bstshdigen, syn-axialen Substituenten mit me- tallorganischen Verbindungen analoge Stereoselektivitliten gefunden haben.
Tab. 2 'H-NMR-Spektren, chemische Verschiebungen der tertiiiren Al- kohole 2 und 3 (6 (ppm), CDCl3)
Verb. H-3 H-4e H 4 a H-11 OH CH3 an C-5 ~~ ~~
2a 4.4 1.7 1.8 5.2 3.6 1.2 3a 4.5 1.9 1.9 5.1 5.6 1.5 2b 4.2 2.2 3.1 5.2 3.5 3b 4.4 1.8 2.4 5.1 6.1 k 4.5 2.2 2.2 5.1 5.6 3d 4.4 2.5 3.0 5.5 1.6
Arch. Phorm. (Weinheim) 323,177-180 (1990) OVCH Verlagsgesellschaft mbH, D-6940 Weinheim, 1990 0365-6233/90/0303-0177 $02.50/0
Holzgrabe. Piening und Haller t 178
R Z = CH3Mgl
CbHgMgBr
HC I C-CHZ-MgBr
C H ~ L I
L I - C ~ C H HzN-CHZ-CHZ-NHZ
R'
R 2a ,b
OH 3 a d
L I 1 I
Abb. 1: Umsetzung des Methanobenzazocins 1 mit metallorganischen Re- agentien sowie Derivatisierung von 3a
Die Aufldhng der relativen Konfiguration der dargestell- ten ter t ihn Alkohole gelingt rnit Hilfe der 'H- und 13C- NMR-Spektroskopie (siehe Tab. 2 bzw. 3). Im 'H-NMR- Spektrum des 5-Hydroxy-5-methylsubstituierten 2,6-Me- thanobenzazocins 2a erkennt man fiir die neu eingefuhrte Methylgruppe an C-5 ein Singulett bei 6 = 1.2 ppm und fur H-3 ein doppeltes Dublett bei 6 = 4.4 ppm. Demgegenuber weist das 'H-NMR-Spektrum des Epimeren 3a Signale fur die Methylgruppe an C-5 bei 6 = 1.5 ppm und fiir H-3 ein Triplett bei 6 = 4.5 ppm auf. Die paramagnetische Verschie- bung dieser Signale in 3a gegenuber dem Isomeren 2a weist auf eine axiale Stellung der OH-Gruppe in 3a hin. Entspr. Differenzen in den chemischen Verschiebungen wurden' ebenfalls fiir epimere tertiiire Alkohole der analogen 1-Thia- und 1 -0xacyclohexanole gemessen7).
Wie Tab. 2 zeigt, lN3t sich die Konfiguration der propar- gylsubstituierten Alkohole 2,3b analog zuordnen, wiihrend bei den ethinyl- bzw. phenylsubstituierten Alkoholen 3c,d die Bestimmung aufgrund des fehlenden zweiten epimeren Alkohols erschwert ist. Der Vergleich der chemischen Ver- schiebungen dieser Alkohole rnit den oben sicher zugeord- neten Alkoholen 3a,b macht aber die axiale Stellung der Hydroxylgruppe wahrscheinlich. Hinzu kommt im Fall des phenylsubstituierten Alkohols, daf3 es bei umgekehrter Zu- ordnung zu einer 1,3-diaxialen Stellung der Arylreste an C- 5 und C-11 k h e , was zu sterischen Wechselwirkungen fiihrt, die diese Konfiguration nahezu ausschliekn.
Die oben getroffene Konfgurationszuordnung konnte im Fall der 5-Methylderivate 2/3a '3C-NMR-spektroskopisch
Tab. 3: '3C-NMR-Speklren, chemische Verschiebungen der t e r t i k n Al- kohole 2a und 3a (6 (pprn), cDc13)
vbdg. 1 2a 3a
c-1 50.44 51.17 50.43 c-3 59.88 58.83 55.88 C 4 38.67 36.47 36.08 c - 5 199.81 73.53 73.96 C-6 57.70 53.43 52.55 c-11 69.46 66.42 67.71 CH3 26.85 27.91
weiter erhwet werden (Tab. 3): Zum ersten zeigt C-3, das in y-Position zum Substituenten an C-5 steht, im Fall der axialen Hydroxylgruppe (3a) einen um 3 ppm deutlicheren Hochfeldshift als bei axialer Methylgruppe (2a) gegenuber dem Ausgangsketon, was aufgrund des stiirker ausgeprilgten stereoelektronischen y-Effektes der Hydroxylgruppe gegen- uber der Methylgruppe zu erwarten is?. Zum zweiten weisen die 3J~~-Kopplungskonstanten, die bei Cyclohexan- Derivaten der Kurplus-Beziehung gehorchen'), im Fall von 2a (3J = 3 bzw. 6 Hz) auf einen Diederwinkel von 60 bzw. 180" und im Fall von 3a (3J = 2 Hz) auf einen Winkel von 60" hin, was fur eine axiale bzw. equatoriale Stellung der Methylgruppe spricht (Abb. 2).
H H
CH3 OH
2 a 3 a
Abb. 2: Newman-Projektion entlang der C-S/C4-Achse der terti2ren Ako- hole 2a und 3a
Wiihrend die Umsetzung des Alkohols 3a rnit Chloramei- sensaureethylester zum entspr. Kohlensaurediester nur ein nicht kristallisierendes 01 in geringen Ausbeuten liefert, fiihrt die Umsetzung von 3a mit Phenylisocyanat, die nur sehr zogerlich verlauft, zu dem kristallinen Phenylurethan 4a, dessen 'H-NMR-Spektrum durch die Tieffeldverschie- bung der Methylgruppe an C-5 um A6 = 0.5 ppm sowie die Aufhebung der magnetischen Aquivalenz der Protonen an C-4 und einen paramagnetischen Shift von H-4e von A6 = 1.1 ppm charakterisiert ist. Diese Tieffeldverschiebung des aquatorialen H in a-Stellung zum Urethan l a t sich auf einen Anisotropieeffekt der Carbonylgruppe zuriickfiihren, der nur bei einer axialen Stellung des Carbamidsiiureesters moglich ist. Damit stehen die Beobachtungen. die auch an entspr. substituierten Urethanen te r t ih r 1-Oxa- bzw. 1- Thiacyclohexanolderivate7) gemacht worden sind, in Ein- klang mit der oben getroffenen Konfigurationszuordnung.
Herm H.P. Cordes, Forschungsinstitut Borstel. sei fib die Aufnahme der 90 MHz-'H-NMR-Spe.ktren und Herm Dr. C. W O E Organisches Institut der Universitit Gel , fiir die Auhahme der '3C-NMR-Spekeen gedankt.
Arch. Pharm. (Weinheim) 323,177-180 (1990)

Stereoselektive Synthese tert 2,6-Methano-2-benzaz~cin-5-ole 179
Experimenteller Teil
Allg. Angaben: Schmelzpunktsapparatur nach Dr. Tottoli (BUchi), unkorr.- Elementaranalysen: Mirkoanalyt Labor. I. Beetz, Kronach.- IR: Beckman Acculab 10.- 'H-NMR-Spektren: Bruker WH 90 (90 MHz).- I3C- NMR-Spektren: Bruker AM 300 (75.47 MHz).- Kieselgel: Kieselgel60 (70 - 230 mesh ASTM, Merck 7734).
Die DarsteUung des N-Benzyl-2,6-di-2-pyridyl4-piperidon-3-carbon~u- re-methylesters erfolgt nach ') und dessen Oxidation zum 5-0x0-3L1 lanti- di-2-pyridyI-4,5-dihydro- 1H,3H-2r,6c-methanobenz[c]azwin-6-carbonsau- remethyl-ester 1 nachlb'.
5-Hydroxy-5-methyl-3t.l lanti-di-2-pyridyl4,S-dihydro-IH3H-2r,6c- methanobenr[c]azocin-6-carbo~~~emethylester 2&3a
1. Darstellung unter Grignardbedingungen: Aus 120 mg ( 5 mmol) Mg und 700 mg (5 mmol) CH31 wird in 50 ml
EtzO H3CMgI hergestellt. EtzO wird bei Raumtemp. i.Vak. abdestilliert, die Suspension gut gekiihlt und 20 ml absol. CHzClz zugetropft. Zu diesem frisch hergestellten Crignard-Reagens wird unter Eiskiihlung und Argon eine Usung von 1 g (2.5 mmol) 1 in 20 ml absol. CHzC12 zugetropft. Nach 15 min wird mit gesattigter NHQLiisung unter Eiskiihlung hydrolysiert und die wurige Phase 3 mal mit CHzClz extrahiert. Die vereinigten org. Phasen werden iiber Na2SO4 getrocknet und i.Vak. eingeengt.- Gesamt- ausb. 0.8 g (80%).- Epimerentrennung durch sc an 100 g Kieselgel (Ethyl- acetat 9.0, Toluol 1.0, konz. NH3 0.1). 2a: Rf = 0.8.- Kristallisation aus Et0H.- Schmp. 217OC.- C ~ H z N 3 0 3
(415.5).- Ber. C 72.3 H 6.06 N 10.1 Gef. 72.4 H 6.08 N 9.5.- IR (KBr):
3a: Rf = 0.7.- Kristallisation aus Ethylacetat, Umkristallisation aus MeOH.- Schmp. 195°C.- CzHzN303 (415.5).- Ber. C 72.3 H 6.06 N 10.1 Gef. 72.3 H 6.13 N 10.2.- IR (KBr): 3560 (OH); 1730 cm-' (C=O Ester).
3400 (OH); 1740 m-' (C=O Ester).
2. Darstellung mit Methyllithium: 500 mg (1.25 mmol) 1 werden in absol. THF gelbst. Unter Eiskiihlung
werden 2 ml (2.5 mmol) einer 1.6 molaren ujsung von H3CLi in EtzO unter Argon zugetropft. Nach DC Kontrolle wird mit gesattigter NH&L Usung und Eiswasser hydrolysiert, mit Citronenssure neutralisiert und i.Vak. eingeengt Die Usung wird mit HzO versetzt und mit CHzClz mehr- mals extrahiert. Die org. Phasen werden iiber Na2SO4 getrocknet und i.Vak. eingeengt. Als Riickstand bleibt ein rotes 61, das sc (s.o.) gereinigt wird. Ausb. 0.3 g (60%) 3a.
5-Hydroxy-S-prop-2-inyl-3t,ll -anti-di-2-pyridyl-$J-dihydro-I H3H- 2r.6c-methanobenz[c]azocin-6-carbo~~~emethylester 2,3b
Aus 120 mg (5 mmol) Mg und 600 mg (5 mmol) Propargylbromid wird unter Zugabe einer Spatelspitze H g c 1 ~ ~ ~ ) Ropargylmagnesiumbromid in EtzO unter Argon hergestellt. Anschlieknd werden unter Eiskiihlung 20 ml absol. CHzClz zugetropft, dann 500 mg (1.25 mmol) 1 in absol. CHzC12. Man Nhrt In h und hydrolysiert mit gesiittigter NH&I-Liisung und Eis- wasser. Die warige Phase wird mehrmals mit CHzClz extrahiert, die verei- nigten organischen Phasen iiber NazS04 getrocknet und i.Vak. eingeengt.- Gesamtausb. 0.05 g (lo%).- Epimerentrennung durch SC an 100 g Kiesel- gel (Ethylacetat 4.0, MeOH 3.0, Cyclohexan 6.0, konz NHJ 0.1). 2b Rf = 0.8.- Kristallisation aus EtOwHz0.- Schmp. 192OC.-
CnHzN303 (439.5).- Ber. C 73.8 H 5.79 N 9.6 Gef. C 73.8 H 5.80 N 9.6.- IR (KBr): 3540 (OH); 3260 (&H); 2120 (CsC); 1715 cm.' (C=O Ester). 3 b Rf = 0.7.- Kristallisation aus EtOH/HZO.- Schmp. 21lOC.-
CnHzN303 (439.5).- Ber. C 73.7 H 5.79 N 9.6 Gef. C 73.8 H 5.80 N 9.6.- IR (KBr): 3400 (OH); 32% (=CH); 2120 (W): 1735 cm-' (0 Ester).
Si-Ethinyl-Sc-hydroxy-3t.l I -anti-di-2-pyridyl4 5-dihydro-IH3H-2r.6~- methanobenz[c]azocin-6-carbo~~~e~thylester(3c)
Zu einer Suspension aus 1 g (10 mmol) Lithiumacetylid-Ethylendiamin- Komplex in absol. THF werden 2 g (5 mmol) 1, geltist in absol. THF, in- nerhalb von I5 min bei Raumtemp. getropft. Anschlieknd wird 1 h bei 35°C geriihrt, dann unter Eiskiihlung vorsichtig mit Eiswasser hydrolysiert, rnit Citronensiiure neutralisiert und die L&ung i.Vak. eingeengt. Der erhal- tene Riickstand wird mit HzO versetzt und mehrmals mit CHzClz extra- hiert. Die vereinigten org. Phasen werden iiber NaZSO4 getrocknet, das Lii- sungsmittel wird i.Vak. entfemt. A l s Riickstand bleibt ein rotes 01, aus dem sich nach sc-Reinigung an 200 g Kieselgel (Ethylacetat 10.0, konz. NH3 0.1, Rf = 0.9) 3c isolieren und aus MeOH kristallisieren IUt. Ausb. 1.4 g (70%)- Schmp. 174OC.- CS23N303 (425.5).- Ber. C 73.4 H 5.45 N 9.9 Gef. C 73.3 H 5.50 N 9.8.- IR (KBr): 3560 (OH); 3350 (=CH); 2140 (w); 1745 an-' (C=O Ester).
Sc-Hydroxy-St-phenyl-3t.I 1 -anti-di-2-pyridyl4,5-dihydro-l H3H-2r.6~- methanoberu[c~azocin-6-carbonsciuremethyr (3d)
Aus 120 mg (5 mmol) Mg und 730 mg (5 mmol) Brombenzol wird Phe- nylmagnesiumbromid in Etz0 hergestellt Zu diesem Grignard-Reagens wird unter Eiskiihlung 1 g (2.5 mmol) 1, gelbst in 50 ml absol. THF, ge- tropft. Der sich gelb verfrirbende Reaktionsansatz wird nach l stdg. RIUlren bei Raumtemp. mit geattigter NH.,Cl-Usung und Eiswasser hydrolysiert und die warige Phase mit CHzClz extrahiert. Die vereinigten org. Phasen werden iiber NazS04 getrocknet, das Usungsmittel wird entfemt Als Riickstand bleibt ein rotes 61, aus dem nach Abtrennung von nicht umge- setztem Ausgangsprodukt durch SC an 100 g Kieselgel (Ethylacetat 9.0, Cyclohexan 1.0, Rf = 0.5 (l), Rf = 0.8 (3d)) 3d in geringer Menge isoliert werden kann. Kristallisation aus EtOH/HzO (3:1).- Schmp. 141OC.- C3&&03 (477.6).- Ber. C 75.5 H 5.70 N 8.8 Gef. 75.5 H 5.68 N 8.8.- IR (KBr): 3500 (OH); 1750 cm-' (C=O Ester).
St-Methyl-Sc-N-phenylcarbamoyl-3tJ I -anti-di-2-pyridy1-4 S-dihydro- 1H3H-2r,6c-methano~nz[c]azocim-6-carbo~~~emethylester (4)
Zu 0.5 g Phenylisocyanat in 10 ml absol. THF werden 0.3 g des Alke hols 3a. gel6st in 5 ml absol. THF. getropft. Anschlieknd wird unter Riihren 5 h zum Ruckflu6 erhitzt, danach das Usungsmittel i.Vak. abde- stilliert und der 6lige Riickstand mit wenig Ethylacetat angerieben. Bei Raumtemp. scheidet sich 4a kristallm ab. Umkristallisation aus EtOH. Ausb. 220 mg (60%).- Schmp. 235'C.- C3zH@404 (521.3).- Ber. C 71.9 H 5.65 N 10.5 Gef. C 72.8 H 5.56 N 10.5.- IR (KBr): 3300 (NH); 1745 cm" (C=O).- 'H-NMR (8 (ppm) CDC13): 1.80 (m. lH, H A ) , 1.96 (s, 3H, CH3). 2.98 (dd, 1H. J = 14 bzw. 3.5 HZ, H 4 ) , 3.71 (s. 3H, OCH3). 3.88.4.32 (AB, 2H, J,, = 18 Hz, H-1). 4.56 (dd. 1H. J = 12 b m . 2.5 HZ, H-3), 5.05 (s, lH, H-11). 5.20 (breif 1H. NH). 6.95 - 8.83 (m. 17H. aromat.).
Literatur
l a R.Haller, R.Kohlmorgen und W.Hiinsel, Tetrahedron Lett. 1973,1205. 1 b U.Holzgrabe, B.Piening. RKohlmorgen und E.Stoll, Arch.Pharm.
(Weinheim)32I, 917 (1988). 2 A.F.Casy und R.F.Parfin, Opioid Analgesics, Chemistry and Recep
tors, Plenum Press, New York 1986. 3 H.G.Viehe und M.Reinstein, Chem.Ber. 95,2557 (1962). 4 K.-F.Hesse, U.Holzgrabe und B.Piening, ZNaturforsch. 436, 901
(1988). 5 A.V.Kamemitzky und A.A.Akhrem, Tetrahedron 1962.705. 6 M.Cherest und H.Felkin, Tetrahedron Lett. 1968,2205. 7 R.Haller und J.Ebersberg, ArcbPharm. (Weinheim)302,677 (1%9).
Arch. Pharm. (Weinheim) 323, 177-180 (1990)

180 Holzgrabe, Piening und Haller t
8 J.R.Lambertund A.R.Vagenas, 0rg.Magn.Res. 17,265 (1981). 1 1 R.Haller und R.Kohlmorgen, Arch.Phann. (Weinheim) 309, 206 9 A.H.Haines und M.S.Shandiz, J.Chem.Soc., Perkin I, 1981,1671. (1976). 10 U.Ashauer-Holzgrabe und R.Haller, Arch.Pham. (Weinheirn) 319, 12 J.H.Wotiz. J.Am.Chem.Soc. 73,5503 (1951).
1079 (1986). m6601
Arch. Pharm. (Weinheirn) 323,177-180 (1990)















![Stereoselektive [3,3]-sigmatrope Umlagerung von 3-Aza-1 ...elpub.bib.uni-wuppertal.de/servlets/DerivateServlet/...Stereoselektive [3,3]-sigmatrope Umlagerung von 3-Aza-1-oxa-Cope-Systemen](https://img.pdfslide.org/doc/110x75/60ec87bd5fdb122b4548ccbe/stereoselektive-33-sigmatrope-umlagerung-von-3-aza-1-elpubbibuni-stereoselektive.jpg)













