Embed Size (px)
DESCRIPTION
Das technisch-wissenschaftliche Journal TechnoPharm richtet sich an Experten und Entscheider, die in Pharmaunternehmen und Zulieferbetrieben für Planung, Installation, Betrieb und Wartung von Produktionsanlagen und nicht zuletzt für F&E zuständig sind.
Citation preview

APV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V.
23890 ISSN 2191- 8341
www.ecv.de
Auf die Dosierung kommt es anKleinstmengenverarbeitung von Pulvern fordert Hersteller und Anlagenbauer
Design ist keine Frage der SchönheitRäume ausstatten, gestalten und qualifizieren
Unsichtbare GefahrenSterilfilter in Abgasleitungen von Autoklavieranlagen als Waffe gegen Bakterien, Viren und Co.
Tuning für die FeuchtemessungMikrowellenresonanz-Sensoren messen erstmals in Wirbelschichtgeräten im 2-Frequenz-Bereich
06 ∙ 2013

Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-06\Anzeigensatz-keine-Druck-PDFs\fette-TP-2013-06_216x303.indd Zuletzt gesichert: 14.11.13 (11:05:24 Uhr)
NEXT GENERATION TABLETING TECHNOLOGYNEW FE SERIES
+ Design – Simplicity as a key to efficiency
+ 360° accessibility+ Guaranteed easy and safe operation
+ FE55 red dot design award 2012+ FE35 IF product design award 2013
and red dot design award 2013
www.fette-compacting.com

Von Pillendrehern undPlätzchenbäckern
Medizin und Pharmazie leisten im-mer noch Großes für die Menschheit.Es ist erstaunlich und faszinierendzugleich, was hier in den vergange-nen Jahrhunderten alles passiert ist.Vor knapp 150 Jahren wurden –wennauch mit ganz anderen Maßstäbenals heute – erstmals „maschinell“Tabletten hergestellt. Die kleinen Ap-paraturen verdienten kaum den Na-men „Maschine“, die Apotheker wur-den im Volksmund nur „Pillendre-her“ genannt. Ob des großen Ge-schicks bei der Pillenherstellung ge-nossen sie allerdings dasselbe Anse-hen wie kunstfertige Handwerker.Denn die kleinen runden Pillen – da-mals Pilulae genannt – wurden nichtnur von Hand gedreht, sondern – fürdie besonders gut situierte Kund-schaft – auf Wunsch sogar vergoldet.Doch bis der Kranke seine Arznei fürein paar Taler kaufen konnte, hatteder Apotheker eine Menge zu tun.Und das erinnerte gar nicht immeran Arznei, sondern eher an eineBackstube. Denn zuerst musste –einfach ausgedrückt – eine Masseaus Hefe, Glyzerin und destilliertemWasser oder aber aus Süßholzsaftund -wurzel hergestellt werden. AlsWirkstoff gab man dann beispiels-weise Baldrian dazu, was der Beruhi-gung diente. Jetzt musste der Teignur noch in Form gebracht werden,womit wir uns wieder in der Analogieder Backstube und der Plätzchen be-finden. Zuerst wurde die Masse aus-gerollt. Bevor ein findiger Apothekerzur Herstellung der Pilulae das sogenannte Pillenbrett erfand, wurdevon Hand gerollt, geschnitten undgeformt. Neben den Pilulae kannteman noch die Tabulae, Rotuli, Tro-chisci und die Pastili, wobei die nach
deutscher Methode hergestellten Pa-stili als Vorläufer der Tablette be-trachtet werden können. Auch hierwurden Pulvermischungen befeuch-tet, geknetet, ausgestrichen, getrock-net und meist rautenförmig zuge-schnitten oder in Form gepresst. Eineerste offizielle Monographie für Tab-letten gab es im DAB VI von 1926,eine Erwähnung war bereits imDAB V von 1910 in der MonographiePastili enthalten.
Wenn Sie jetzt kurz vor Weihnach-ten Ihre ersten Plätzchen backen,versuchen Sie es doch selbst einmalmit dem Formen. Sie werden feststel-len: Es ist gar nicht so leicht undbraucht Zeit und Geduld! Vorteilbeim Backen: Schmeckt das Ganzenicht wie gewünscht, noch eine PriseZimt oder Zucker dazu und schonkönnen größere Schäden vermiedenwerden.
Bei den ersten handgefertigtenTabletten hingegen war eine gleich-mäßige Dosierung kaum möglich,was unweigerlich zu Schwankungendes Wirkstoffgehaltes führen musste.Alles in allem eine unwissenschaftli-che, ineffiziente und vor allem hygie-nisch fragwürdige Angelegenheit, dienoch einmal einen ganz anderen, be-sonderen Blick auf die Erfindung derersten Tablettenpresse und die In-dustrialisierung mit ihren hohen Hy-gienestandards verschafft. Man führesich bildlich vor Augen: ModerneTablettenpressen der Gegenwart ha-ben einem Output von ca. 200000Stück pro Stunde.
So viele Plätzchen werden wir inder heimischen Küche nicht herstel-len, weshalb eine 100 prozentige Au-tomatisierung – von modernenHightech-Geräten abgesehen – we-
der erforderlich noch gewünscht ist.Schon gar nicht zu Weihnachten, wodie Plätzchenherstellung traditionellmehr zelebriert als automatisiertwird. Für viele ist es die erste Zeitdes Jahres, in der sie ihre Geschwin-digkeit drosseln und die persönlicheTaktzahl verringern. Wo beispiels-weise das Wort Qualitätskontrollenur im Zusammenhang mit Plätz-chengeschmack fällt. Auch Naschengenannt. Eine Zeit, wo Herstellungund Themen wie z.B. Primärpack-mittel keine Rolle spielen. Wo Sekun-därverpackungen in Form von bun-ten Blechdosen durch die Kinderausgesucht werden. Wo Beipackzet-tel einsprachig als nette Weihnachts-grüße dazugelegt werden. Und wodie einzige Regularie, die es zwin-gend einzuhalten gilt, die ist, ja nie-manden bei seiner Aussendung zuvergessen.
Einen gravierenden Fehler aller-dings können Sie machen: Sollte sicham Ende des Herstellungszyklus daskomplette Gebäck gut verpackt aufdemWeg zu Freunden und Verwand-ten befinden, haben Sie ein Problemmit der Lagerhaltung. Denn Vorrats-haltung empfiehlt sich bei Plätzchendefinitiv mehr als bei Tabletten. Indiesem Sinne bleiben Sie gesund und
Fröhliche WeihnachtenIhreKerstin Jarosch
Editorial

TERMINE 302
FOKUS: HERSTELLUNGFESTER ARZNEIFORMEN
Seyfang, Karlheinz; Steckel, Hartwig 304PulverabfüllungKleinste Mengen richtig dosieren
Germer, Katharina; Wolf, Bertram 312Übertragung der Granulierung in zweigetrennten Schritten auf einen Ein-Schritt-WirbelschichtprozessTeil 2: Wirbelschichtagglomeration und-trocknung in einem Schritt und Vergleich mitZwei-Schritt-Prozessen
Kollar, Benjamin; Breitkreutz, Jörg;Wiedey, Wolfgang; Bartscher, Kathrin;Döscher, Claas 317Mikrowellenresonanz-SensorenInnovation für die Feuchtemessung inWirbelschichtgeräten
PROZESS- UND VERFAHRENSTECHNIK
Grumbach, Carsten; Czermak, Peter 322Abluftfiltration unter der LupeRisiken bei der Sterilfiltration derAutoklavenabluft
Heuwes, Guido 328Ausstattung, Gestaltung undQualifizierung von RäumenRäume, Luft, Technik
MESSEN/STEUERN/REGELN
Hoffmann, Christina 334Inline Leitfähigkeitsmessung fürpharmazeutische Prozesse
Schwarzkopf, Danica 337Basiswissen Kalibriermanagement
MASCHINEN- UND ANLAGENBAU
Wuhrmann, Daniel; Reusch, Philipp 342ProjektmanagementRechtssicherheit für Pharma-Ingenieure: Teil 1
AUTOMATION
Schade, Markus 346Jahre vergehen, Funktionen bestehen
IT
Bauer, Claus 349Cloud Computing
PRODUKTE 354
IMPRESSUM 356
Inhaltsverzeichnis
TechnoPharm 3, Nr. 6, 301 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 301Inhaltsverzeichnis
BeiratsgremiumKonstantin Clevermann, Dortmund · Prof. Dr. Jürgen Hannemann, Biberach · Dr. Udo Janske, Darmstadt · Prof. Dr. Gerd Kutz,Detmold · Heinz Kudernatsch, Nürnberg · Hans Ulrich Petereit, Darmstadt · Dr. Elke Sternberger-Rützel, Freiberg a.N. · Dr. MikeSchäfers, Eschweiler · Prof. Dr. Hartwig Steckel, Kiel · Dr. Frank Stieneker, Hofheim · Roland Szymoniak, Frankfurt am Main · Dr.Jochen Thies, Warendorf · Dipl. Ing. Frank Wilde, Basel · Prof. Dr. Ing. Dominik Rabus, Forchtenberg · Dipl. Ing. Frank Lehmann,Allschwil (Schweiz)
APVnewsNachrichten und Mitteilungen von der Arbeitsgemeinschaft fürPharmazeutische Verfahrenstechnik e.V.(Ausgabe 06/13, nach S. 356)
Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-07\Anzeigensatz-keine-Druck-PDFs\letzner-pi-2013-07-216x303.indd Zuletzt gesichert: 11.07.13 (12:53:19 Uhr)
Enthärtung im „sanitary design“
L Komplett in EdelstahlL Heisswasser- Sanitisierung
Umkehrosmose / Elektrodeionisation
L Letzner-Kohlensäure-PatentL Wassereinsparung bis zu 50 %
Mehrstufen Druckkolonnendestillation
L Anti-Rouging KonzeptL Vollisolierung L Leistungsregelung
LetzTOC / Mehrkanal online TOC Messung
L gemäß Ph.Eur. USP. JP16 complianceL Automatischer Systemseignungstest (SST)
Letzner Pharmawasseraufbereitung GmbH · Robert-Koch-Str. 1 · 42499 HückeswagenTel. +49(0)2192/83883 · Telefax +49(0)2192/921733 · www.letzner.de

TERMINE 302
FOKUS: HERSTELLUNGFESTER ARZNEIFORMEN
Seyfang, Karlheinz; Steckel, Hartwig 304PulverabfüllungKleinste Mengen richtig dosieren
Germer, Katharina; Wolf, Bertram 312Übertragung der Granulierung in zweigetrennten Schritten auf einen Ein-Schritt-WirbelschichtprozessTeil 2: Wirbelschichtagglomeration und-trocknung in einem Schritt und Vergleich mitZwei-Schritt-Prozessen
Kollar, Benjamin; Breitkreutz, Jörg;Wiedey, Wolfgang; Bartscher, Kathrin;Döscher, Claas 317Mikrowellenresonanz-SensorenInnovation für die Feuchtemessung inWirbelschichtgeräten
PROZESS- UND VERFAHRENSTECHNIK
Grumbach, Carsten; Czermak, Peter 322Abluftfiltration unter der LupeRisiken bei der Sterilfiltration derAutoklavenabluft
Heuwes, Guido 328Ausstattung, Gestaltung undQualifizierung von RäumenRäume, Luft, Technik
MESSEN/STEUERN/REGELN
Hoffmann, Christina 334Inline Leitfähigkeitsmessung fürpharmazeutische Prozesse
Schwarzkopf, Danica 337Basiswissen Kalibriermanagement
MASCHINEN- UND ANLAGENBAU
Wuhrmann, Daniel; Reusch, Philipp 342ProjektmanagementRechtssicherheit für Pharma-Ingenieure: Teil 1
AUTOMATION
Schade, Markus 346Jahre vergehen, Funktionen bestehen
IT
Bauer, Claus 349Cloud Computing
PRODUKTE 354
IMPRESSUM 356
Inhaltsverzeichnis
TechnoPharm 3, Nr. 6, 301 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 301Inhaltsverzeichnis
BeiratsgremiumKonstantin Clevermann, Dortmund · Prof. Dr. Jürgen Hannemann, Biberach · Dr. Udo Janske, Darmstadt · Prof. Dr. Gerd Kutz,Detmold · Heinz Kudernatsch, Nürnberg · Hans Ulrich Petereit, Darmstadt · Dr. Elke Sternberger-Rützel, Freiberg a.N. · Dr. MikeSchäfers, Eschweiler · Prof. Dr. Hartwig Steckel, Kiel · Dr. Frank Stieneker, Hofheim · Roland Szymoniak, Frankfurt am Main · Dr.Jochen Thies, Warendorf · Dipl. Ing. Frank Wilde, Basel · Prof. Dr. Ing. Dominik Rabus, Forchtenberg · Dipl. Ing. Frank Lehmann,Allschwil (Schweiz)
APVnewsNachrichten und Mitteilungen von der Arbeitsgemeinschaft fürPharmazeutische Verfahrenstechnik e.V.(Ausgabe 06/13, nach S. 356)
Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-07\Anzeigensatz-keine-Druck-PDFs\letzner-pi-2013-07-216x303.indd Zuletzt gesichert: 11.07.13 (12:53:19 Uhr)
Enthärtung im „sanitary design“
L Komplett in EdelstahlL Heisswasser- Sanitisierung
Umkehrosmose / Elektrodeionisation
L Letzner-Kohlensäure-PatentL Wassereinsparung bis zu 50 %
Mehrstufen Druckkolonnendestillation
L Anti-Rouging KonzeptL Vollisolierung L Leistungsregelung
LetzTOC / Mehrkanal online TOC Messung
L gemäß Ph.Eur. USP. JP16 complianceL Automatischer Systemseignungstest (SST)
Letzner Pharmawasseraufbereitung GmbH · Robert-Koch-Str. 1 · 42499 HückeswagenTel. +49(0)2192/83883 · Telefax +49(0)2192/921733 · www.letzner.de

APVAPV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik, Kurfürstenstr. 59, 55118, Mainz,Tel. + 49 (0) 6131-9769-0, Fax +49 (0) 6131-9769-69, e-mail: [email protected]
04. / 05.12.2013 BERLIN 2nd APV-Workshop on Quality by Design Driven Development of Coated Dosage Forms
09. / 10.12.2013 MÜNCHEN Praxis der Entwicklung, Stabilitätsprüfung und Herstellung von Betäubungsmitteln
11. / 12.12.2013 WIESBADEN Analyse komplexer technischer Störungen / Die hohe Schule des Trouble Shooting
CONCEPT HEIDELBERGCONCEPT HEIDELBERG GmbH, Rischerstr. 8, 69123 Heidelberg, Tel. + 49 (0) 6221-84440, Fax +49 (0) 6221-844434,e-mail: [email protected]. / 24.01.2014 MANNHEIM Die Leitung der Qualitätskontrolle / Pflichten und Verantwortlichkeiten nach AMWHVund EU- GMP Leitfaden
28. / 29.01.2014 HEIDELBERG Die Leitung der Herstellung
28. / 29.01.2014 HEIDELBERG Granulierung / Grundlagen, Optimierung, Trouble-Shooting
29.–31.01.2014 KARLSRUHE Der Hygienebeauftragte / Block 1Grundlagen der Betriebs- und Personalhygiene
29.–31.01.2014 HEIDELBERG Der QS-/GMP-Beauftragte in der pharmazeutischen Industrie / Block I
19.–30.01.2014 HEIDELBERG Tablettierung / Grundlagen, Optimierung, Trouble-Shooting
04. / 05.02.2014 MANNHEIM Pharma-Technik für Nicht-Techniker – Einführung in die pharmazeutische Anlagentechnik
04. / 05.02.2014 HEIDELBERG Kalibrierung / Qualifizierung / Validierung in der Packmittelprüfung
04. / 05.02.2014 HEIDELBERG GMP-/GDP-Anforderungen an Lager und Transport
04. / 05.02.2014 HEIDELBERG GMP-Grundlagen der Sterilproduktion
11. / 12.02.2014 HEIDELBERG GMP-gerechte Medientechnik
12. / 13.02.2014 HEIDELBERG Rohrleitungen für Pharma-Wasser und Reindampf
11.–13.02.2014 HEIDELBERG GMP-gerechte Medientechnik und Rohrleitungen für Pharmawasser und Reindampf
18. / 19.02.2014 HEIDELBERG FDA-/GMP-gerechter Prozess-Transfer – Zulassung, Projektmanagement, Technologie
18. / 19.02.2014 MANNHEIM Schlanke GMP-Systeme – Effizienz in der Qualitätssicherung
19.–21.02.2014 MANNHEIM Der Validierungsbeauftragte in der pharmazeutischen Industrie
20.–21.02.2014 MANNHEIM SPS in der Pharmaindustrie / Validierung, aktuelle GAMP®- und Part 11-Anforderungen
25.02.2014 BASEL (CH) GMP-Basis-/Einstiegsschulung – Schweiz –
26.–28.02.2014 HEIDELBERG Der QS-/GMP-Beauftragte in der pharm. Industrie – Block II
EUROPEAN COMPLIANCE ACADEMYEuropean Compliance Academy, P.O. Box 10 21 68, 69011 Heidelberg, e-mail: [email protected]. / 29.01.2014 BERLIN Protective Packaging Solutions for Pharmaceutical Product Stability
05. / 06.02.2014 WIEN (AT) Radiopharmaceuticals – Quality, Safety and GMP Requirements
27. / 28.02.2014 BERLIN Pharmaceutical Contracts / GMP and Legal Compliance and pre-course session Contracting in China
FORUM INSTITUTFORUM Institut für Management GmbH, Postfach 10 50 60, 69040 Heidelberg, Tel + 49 (0) 6221-500 500,Fax +49 (0) 6221-500 505, e-mail: [email protected].–12.02.2014 BONN Labeling, Packungsbeilage und QRD-Templates
PTS TRAINING SERVICEPTS Training Service, Postfach 4308, 59737 Arnsberg, Tel. + 49 (0 )2932-51477, Fax +49 (0) 2932-51674, e-mail: [email protected] UNNA Basistraining Qualifizierung / Modul 1
18.02.2014 OLTEN (CH) GMP Basistraining in der Schweiz
19.02.2014 UNNA Basistraining Validierung: /Modul 2
26.02.2014 OLTEN (CH) GMP / The Basics
11. / 12.02.2014 UNNA OE Strategien / Operational Excellence mit vier Fallbeispielen und Betriebsbesichtigung
11. / 12.03.2014 BADEN-BADEN PQS Pharmaceutical Quality System / Experte für Qualitätskontrolle Modul 1
18.03.2014 KOBLENZ Räume, Luft und Technik – Modul 1 / Gestaltung und Qualifizierung von Räumen
19.03.2014 KOBLENZ Räume, Luft und Technik – Modul 2 / Qualifizierung von Lüftungsanlagen
20.03.2014 KOBLENZ Räume, Luft und Technik – Modul 3 / Messtechnik: Umsetzung gemäß Annex 1,DIN ISO 14644
25.03.2014 OLTEN (CH) GDP Gute Vertriebspraxis
TTCTTC (Technology Training Center), Werner-Glatt-Straße 1, 79589 Binzen, Tel. + 49 (0) 7621-664-535, e-mail: [email protected]
04. / 05.02.2014 BINZEN Explosionsschutz in der Feststofffertigung
11.–13.03.2014 BINZEN Fluidized bed processing
Termine
TechnoPharm 3, Nr. 6, 302 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)302 Termine
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-06\Anzeigensatz-keine-Druck-PDFs\phast-TP-2013-06_216x303.indd Zuletzt gesichert: 19.11.13 (06:58:44 Uhr)
SETTING STANDARDS
FROM BENCH TO MARKET
• Contract(cGMP)qualitycontrol -smallmolecules,biologics,highly
potentcompounds -ICHstabilitystudies -batchreleasebyQualifiedPersons
• Development -developmentandvalidationof
analyticalmethods -productdevelopment
• Qualityservice -clinicaltrialsupply/manufacturing
-on-sitetechnicalandanalyticalsupply
-certifiedtrainingcourses
• ReferenceStandardSubstances -USP,BP,EP -QualificationNMR,MS
FDAinspected2013
PHAST_AZ_Technopharm-RZ.indd 1 18.11.13 15:37

APVAPV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik, Kurfürstenstr. 59, 55118, Mainz,Tel. + 49 (0) 6131-9769-0, Fax +49 (0) 6131-9769-69, e-mail: [email protected]
04. / 05.12.2013 BERLIN 2nd APV-Workshop on Quality by Design Driven Development of Coated Dosage Forms
09. / 10.12.2013 MÜNCHEN Praxis der Entwicklung, Stabilitätsprüfung und Herstellung von Betäubungsmitteln
11. / 12.12.2013 WIESBADEN Analyse komplexer technischer Störungen / Die hohe Schule des Trouble Shooting
CONCEPT HEIDELBERGCONCEPT HEIDELBERG GmbH, Rischerstr. 8, 69123 Heidelberg, Tel. + 49 (0) 6221-84440, Fax +49 (0) 6221-844434,e-mail: [email protected]. / 24.01.2014 MANNHEIM Die Leitung der Qualitätskontrolle / Pflichten und Verantwortlichkeiten nach AMWHVund EU- GMP Leitfaden
28. / 29.01.2014 HEIDELBERG Die Leitung der Herstellung
28. / 29.01.2014 HEIDELBERG Granulierung / Grundlagen, Optimierung, Trouble-Shooting
29.–31.01.2014 KARLSRUHE Der Hygienebeauftragte / Block 1Grundlagen der Betriebs- und Personalhygiene
29.–31.01.2014 HEIDELBERG Der QS-/GMP-Beauftragte in der pharmazeutischen Industrie / Block I
19.–30.01.2014 HEIDELBERG Tablettierung / Grundlagen, Optimierung, Trouble-Shooting
04. / 05.02.2014 MANNHEIM Pharma-Technik für Nicht-Techniker – Einführung in die pharmazeutische Anlagentechnik
04. / 05.02.2014 HEIDELBERG Kalibrierung / Qualifizierung / Validierung in der Packmittelprüfung
04. / 05.02.2014 HEIDELBERG GMP-/GDP-Anforderungen an Lager und Transport
04. / 05.02.2014 HEIDELBERG GMP-Grundlagen der Sterilproduktion
11. / 12.02.2014 HEIDELBERG GMP-gerechte Medientechnik
12. / 13.02.2014 HEIDELBERG Rohrleitungen für Pharma-Wasser und Reindampf
11.–13.02.2014 HEIDELBERG GMP-gerechte Medientechnik und Rohrleitungen für Pharmawasser und Reindampf
18. / 19.02.2014 HEIDELBERG FDA-/GMP-gerechter Prozess-Transfer – Zulassung, Projektmanagement, Technologie
18. / 19.02.2014 MANNHEIM Schlanke GMP-Systeme – Effizienz in der Qualitätssicherung
19.–21.02.2014 MANNHEIM Der Validierungsbeauftragte in der pharmazeutischen Industrie
20.–21.02.2014 MANNHEIM SPS in der Pharmaindustrie / Validierung, aktuelle GAMP®- und Part 11-Anforderungen
25.02.2014 BASEL (CH) GMP-Basis-/Einstiegsschulung – Schweiz –
26.–28.02.2014 HEIDELBERG Der QS-/GMP-Beauftragte in der pharm. Industrie – Block II
EUROPEAN COMPLIANCE ACADEMYEuropean Compliance Academy, P.O. Box 10 21 68, 69011 Heidelberg, e-mail: [email protected]. / 29.01.2014 BERLIN Protective Packaging Solutions for Pharmaceutical Product Stability
05. / 06.02.2014 WIEN (AT) Radiopharmaceuticals – Quality, Safety and GMP Requirements
27. / 28.02.2014 BERLIN Pharmaceutical Contracts / GMP and Legal Compliance and pre-course session Contracting in China
FORUM INSTITUTFORUM Institut für Management GmbH, Postfach 10 50 60, 69040 Heidelberg, Tel + 49 (0) 6221-500 500,Fax +49 (0) 6221-500 505, e-mail: [email protected].–12.02.2014 BONN Labeling, Packungsbeilage und QRD-Templates
PTS TRAINING SERVICEPTS Training Service, Postfach 4308, 59737 Arnsberg, Tel. + 49 (0 )2932-51477, Fax +49 (0) 2932-51674, e-mail: [email protected] UNNA Basistraining Qualifizierung / Modul 1
18.02.2014 OLTEN (CH) GMP Basistraining in der Schweiz
19.02.2014 UNNA Basistraining Validierung: /Modul 2
26.02.2014 OLTEN (CH) GMP / The Basics
11. / 12.02.2014 UNNA OE Strategien / Operational Excellence mit vier Fallbeispielen und Betriebsbesichtigung
11. / 12.03.2014 BADEN-BADEN PQS Pharmaceutical Quality System / Experte für Qualitätskontrolle Modul 1
18.03.2014 KOBLENZ Räume, Luft und Technik – Modul 1 / Gestaltung und Qualifizierung von Räumen
19.03.2014 KOBLENZ Räume, Luft und Technik – Modul 2 / Qualifizierung von Lüftungsanlagen
20.03.2014 KOBLENZ Räume, Luft und Technik – Modul 3 / Messtechnik: Umsetzung gemäß Annex 1,DIN ISO 14644
25.03.2014 OLTEN (CH) GDP Gute Vertriebspraxis
TTCTTC (Technology Training Center), Werner-Glatt-Straße 1, 79589 Binzen, Tel. + 49 (0) 7621-664-535, e-mail: [email protected]
04. / 05.02.2014 BINZEN Explosionsschutz in der Feststofffertigung
11.–13.03.2014 BINZEN Fluidized bed processing
Termine
TechnoPharm 3, Nr. 6, 302 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)302 Termine
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-06\Anzeigensatz-keine-Druck-PDFs\phast-TP-2013-06_216x303.indd Zuletzt gesichert: 19.11.13 (06:58:44 Uhr)
SETTING STANDARDS
FROM BENCH TO MARKET
• Contract(cGMP)qualitycontrol -smallmolecules,biologics,highly
potentcompounds -ICHstabilitystudies -batchreleasebyQualifiedPersons
• Development -developmentandvalidationof
analyticalmethods -productdevelopment
• Qualityservice -clinicaltrialsupply/manufacturing
-on-sitetechnicalandanalyticalsupply
-certifiedtrainingcourses
• ReferenceStandardSubstances -USP,BP,EP -QualificationNMR,MS
FDAinspected2013
PHAST_AZ_Technopharm-RZ.indd 1 18.11.13 15:37

und haben erst kürzlich die Zulas-sung für die Therapie der ZystischenFibrose erhalten [4].
In Blister-basierten Mehrdosenin-halatoren (z.B. Diskus Inhalator, El-penhaler, sowie weitere in der Ent-wicklung) werden ebenfalls Pulver-mengen im unteren mg-Bereich zwi-schen 5 mg und 15 mg gefüllt.
Sogenannte „Disposable Devices“,also Inhalationspulver incl. Inhalatorzur einmaligen Verwendung, machenebenfalls die Dosierung von Klein-mengen, entweder in einen Blister(z.B. Monohaler, Twincer) oder inden Inhalator direkt (z.B. TwinCaps)notwendig [5, 6].
Entscheidendes Qualitätskrite-rium für den Füllprozess ist, dasssich das Pulver während des gesam-ten Füllprozesses nicht verändert(physikalische Veränderung, Ände-rung der Schüttdichte, Entmischung)und dass die Gehaltseinheitlichkeits-anforderungen der Arzneibücher so-wie der Zulassungsbehörden erfülltwerden. Dies beginnt bereits bei derHomogenität der Pulvermischung,die mit einem geeigneten Verfahrenbestimmt werden sollte und unter-halb von 1.5 % (relative Standard-abweichung, RSD) liegen sollte. Fürdie daraus abgeteilten Einzeldosensollte eine Füllgenauigkeit von < 5 %RSD erreicht werden, um schließlicheine einheitliche und den Anforde-rungen entsprechende „freigegebeneDosis“ aus dem Inhalationsgerät er-reichen zu können. Die Erfordernissefür die Dose Content Uniformity(DCU) von Inhalationsprodukten istin Arzneibüchern monografiert und
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 305Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 1: Zusammenhang zwischen Mischungsgüte,Füllgenauigkeit und Freigegebener Dosis bei Pul-verinhalativa (Quelle: Pharm. Institut, AU Kiel).
PulverabfüllungKleinste Mengen richtig dosieren
Dr. Karlheinz Seyfang . Harro Höfliger GmbH, Allmersbach im TalProf. Dr. Hartwig Steckel . Pharmazeutisches Institut der Universität Kiel, Kiel
Korrespondenz: Prof. Dr. Hartwig Steckel, Christian-Albrechts-Universität zu Kiel, Pharmazeutisches Institut, Gutenbergstraße 76,24118 Kiel; e-mail: [email protected]
ZusammenfassungDie Kleinstmengendosierung von Pulvern mit geringer Füllgewichtsschwankung stellteine besondere Herausforderung für den pharmazeutischen Hersteller, aber auch denAnlagenbauer dar. Die Kenntnis der Pulvereigenschaften ist eine essentielle Voraus-setzung für die Auswahl des richtigen Füll- bzw. Dosiersystems. Zur Verfügung stehenMaschinen, die eine 100 %-Befüllung einer Blisterkavität oder einer Kapsel bewerkstel-ligen, sowie solche bei denen Pulver über eine Dosiereinheit mittels Druck- oder Vaku-umanwendung in eine Vorlage dosiert werden kann. Als Sonderfall wird die Dosierungüber eine vibrierende Kapillare vorgestellt. Ein zunehmend wichtiger Aspekt ist die Füll-mengen-Kontrolle, die sowohl stichprobenartig als auch als 100 %-Kontrolle durchgeführtwerden kann.
Einleitung
Die Kleinstmengendosierung vonPulvern im Pharmabereich stellt einebesondere Herausforderung für denpharmazeutischen Hersteller dar. Un-ter Kleinstmengendosierung soll indiesem Zusammenhang eine Füll-menge von 1 mg –max. 100 mg einesPulvers verstanden werden. Derartigkleine Mengen sind vor allem im Be-reich der Inhalationspulver in vor-abgeteilter Form, also abgefüllt inBlister oder Hartkapseln, zu finden[1]. Andere Verwendungen für dieKleinstmengendosierung sind dassterile Befüllen von Vials mit Zytosta-tika-Pulver, das Dosieren reiner Wirk-stoffe in Kapseln für frühe klinischeStudien oder das Abfüllen von Rea-genzien in Mikrotiterplatten oderKartuschen für die Serienanalyse [2].
Dieser Artikel beschränkt sich aufdie Darstellung der verschiedenenFülltechniken zur Befüllung von Kap-seln oder Blisterkavitäten mit Pul-vern zur Inhalation. Für die Auswahleines Verfahrens zur Abfüllung oderDosierung von Pulvern sind folgendeAspekte zu beachten:. Einzelmasse der Dosis
. Fließeigenschaften des Pulvers
. Schüttdichte und Verdichtung desPulvers
. Geometrische Eigenschaften
. Feuchteempfindlichkeit
. Temperaturempfindlichkeit
. Segregationsneigung
. Elektrostatische Aufladung undHaftung an Maschinenoberflächen
Eine sorgfältige Charakterisierungdes zu verarbeitenden Pulvers solltealso jeder Füllaktivität vorausgehen,da das Ergebnis einer solchen Ana-lyse bereits bestimmte Fülltechnikenausschließt oder andere bevorzugterscheinen lässt [z.B. 3].
Definition 1Unter „Füllen“ oder „Befüllen“ ei-ner Kapsel oder einer Blisterka-vität versteht man das „randvollfüllen“ der gesamten Vorlage. Esist also kein Dosierprozess imVorfeld erforderlich, sonderndas Pulver wird gravimetrisch,mit Vakuum oder aufgrund derKohäsivität des Pulvers in dieKavität hineingedrückt. Die zubefüllende Kavität funktioniertin diesem Fall als die eigentlicheDosierkammer.
Definition 2Unter Dosierung versteht mandas Abteilen einer Pulverporti-on, entweder gravimetrisch odervolumetrisch, so dass eine genauabgemessene Menge in das Kap-selunterteil bzw. den Blisterüberführt wird und diese nichtzu 100 % ausfüllt.
Im Bereich der Pulverinhalativakommen überwiegend (volumetri-sche) Dosierverfahren zum Einsatz,bei bestimmten Blisterpackungenist auch die „Randvoll“-Befüllung an-zutreffen.
In gängigen Kapsel-basierten In-halationsprodukten wird eine Füll-menge zwischen 5 mg und 25 mg ei-ner Pulvermischung in Hartkapselnoder bei neueren Entwicklungenauch in HPMC (Hydroxypropyl-methylcellulose)-Kapseln dosiert.Produkte mit darüber hinausgehen-den Füllmengen (z.B. Tobi® Inhalati-onspulver: ca. 50 mg einer sprüh-getrockneten Formulierung; Colo-breathe® Inhalationskapseln: 125 mgreines Wirkstoffmikronisat, Bronchi-tol® Kapseln: 40 mg mikronisiertesMannitol) stellen die Ausnahme dar
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)304 Steckel und Seyfang . Kleinstmengendosierung von Pulvern

und haben erst kürzlich die Zulas-sung für die Therapie der ZystischenFibrose erhalten [4].
In Blister-basierten Mehrdosenin-halatoren (z.B. Diskus Inhalator, El-penhaler, sowie weitere in der Ent-wicklung) werden ebenfalls Pulver-mengen im unteren mg-Bereich zwi-schen 5 mg und 15 mg gefüllt.
Sogenannte „Disposable Devices“,also Inhalationspulver incl. Inhalatorzur einmaligen Verwendung, machenebenfalls die Dosierung von Klein-mengen, entweder in einen Blister(z.B. Monohaler, Twincer) oder inden Inhalator direkt (z.B. TwinCaps)notwendig [5, 6].
Entscheidendes Qualitätskrite-rium für den Füllprozess ist, dasssich das Pulver während des gesam-ten Füllprozesses nicht verändert(physikalische Veränderung, Ände-rung der Schüttdichte, Entmischung)und dass die Gehaltseinheitlichkeits-anforderungen der Arzneibücher so-wie der Zulassungsbehörden erfülltwerden. Dies beginnt bereits bei derHomogenität der Pulvermischung,die mit einem geeigneten Verfahrenbestimmt werden sollte und unter-halb von 1.5 % (relative Standard-abweichung, RSD) liegen sollte. Fürdie daraus abgeteilten Einzeldosensollte eine Füllgenauigkeit von < 5 %RSD erreicht werden, um schließlicheine einheitliche und den Anforde-rungen entsprechende „freigegebeneDosis“ aus dem Inhalationsgerät er-reichen zu können. Die Erfordernissefür die Dose Content Uniformity(DCU) von Inhalationsprodukten istin Arzneibüchern monografiert und
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 305Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 1: Zusammenhang zwischen Mischungsgüte,Füllgenauigkeit und Freigegebener Dosis bei Pul-verinhalativa (Quelle: Pharm. Institut, AU Kiel).
Die perfekte Lösung für Tablettierwerkzeuge
Reinigung und Konservierung in 1 Prozess
Ihr Nutzen
9 wässrige Reinigung - ohne Lösungsmittel
9 im 1-Schritt-Verfahren - bis zu 80 % schneller!
9 perfekter Korrosionsschutz - längere Lebensdauer
9 analytisch rein - höhere Sicherheit
Borer Chemie AGGewerbestrasse 13, 4528 Zuchwil / SwitzerlandTel. +41 32 686 56 00, Fax +41 32 686 56 90www.borer.ch, [email protected]
Kontaktieren Sie uns! [email protected]
PulverabfüllungKleinste Mengen richtig dosieren
Dr. Karlheinz Seyfang . Harro Höfliger GmbH, Allmersbach im TalProf. Dr. Hartwig Steckel . Pharmazeutisches Institut der Universität Kiel, Kiel
Korrespondenz: Prof. Dr. Hartwig Steckel, Christian-Albrechts-Universität zu Kiel, Pharmazeutisches Institut, Gutenbergstraße 76,24118 Kiel; e-mail: [email protected]
ZusammenfassungDie Kleinstmengendosierung von Pulvern mit geringer Füllgewichtsschwankung stellteine besondere Herausforderung für den pharmazeutischen Hersteller, aber auch denAnlagenbauer dar. Die Kenntnis der Pulvereigenschaften ist eine essentielle Voraus-setzung für die Auswahl des richtigen Füll- bzw. Dosiersystems. Zur Verfügung stehenMaschinen, die eine 100 %-Befüllung einer Blisterkavität oder einer Kapsel bewerkstel-ligen, sowie solche bei denen Pulver über eine Dosiereinheit mittels Druck- oder Vaku-umanwendung in eine Vorlage dosiert werden kann. Als Sonderfall wird die Dosierungüber eine vibrierende Kapillare vorgestellt. Ein zunehmend wichtiger Aspekt ist die Füll-mengen-Kontrolle, die sowohl stichprobenartig als auch als 100 %-Kontrolle durchgeführtwerden kann.
Einleitung
Die Kleinstmengendosierung vonPulvern im Pharmabereich stellt einebesondere Herausforderung für denpharmazeutischen Hersteller dar. Un-ter Kleinstmengendosierung soll indiesem Zusammenhang eine Füll-menge von 1 mg –max. 100 mg einesPulvers verstanden werden. Derartigkleine Mengen sind vor allem im Be-reich der Inhalationspulver in vor-abgeteilter Form, also abgefüllt inBlister oder Hartkapseln, zu finden[1]. Andere Verwendungen für dieKleinstmengendosierung sind dassterile Befüllen von Vials mit Zytosta-tika-Pulver, das Dosieren reiner Wirk-stoffe in Kapseln für frühe klinischeStudien oder das Abfüllen von Rea-genzien in Mikrotiterplatten oderKartuschen für die Serienanalyse [2].
Dieser Artikel beschränkt sich aufdie Darstellung der verschiedenenFülltechniken zur Befüllung von Kap-seln oder Blisterkavitäten mit Pul-vern zur Inhalation. Für die Auswahleines Verfahrens zur Abfüllung oderDosierung von Pulvern sind folgendeAspekte zu beachten:. Einzelmasse der Dosis
. Fließeigenschaften des Pulvers
. Schüttdichte und Verdichtung desPulvers
. Geometrische Eigenschaften
. Feuchteempfindlichkeit
. Temperaturempfindlichkeit
. Segregationsneigung
. Elektrostatische Aufladung undHaftung an Maschinenoberflächen
Eine sorgfältige Charakterisierungdes zu verarbeitenden Pulvers solltealso jeder Füllaktivität vorausgehen,da das Ergebnis einer solchen Ana-lyse bereits bestimmte Fülltechnikenausschließt oder andere bevorzugterscheinen lässt [z.B. 3].
Definition 1Unter „Füllen“ oder „Befüllen“ ei-ner Kapsel oder einer Blisterka-vität versteht man das „randvollfüllen“ der gesamten Vorlage. Esist also kein Dosierprozess imVorfeld erforderlich, sonderndas Pulver wird gravimetrisch,mit Vakuum oder aufgrund derKohäsivität des Pulvers in dieKavität hineingedrückt. Die zubefüllende Kavität funktioniertin diesem Fall als die eigentlicheDosierkammer.
Definition 2Unter Dosierung versteht mandas Abteilen einer Pulverporti-on, entweder gravimetrisch odervolumetrisch, so dass eine genauabgemessene Menge in das Kap-selunterteil bzw. den Blisterüberführt wird und diese nichtzu 100 % ausfüllt.
Im Bereich der Pulverinhalativakommen überwiegend (volumetri-sche) Dosierverfahren zum Einsatz,bei bestimmten Blisterpackungenist auch die „Randvoll“-Befüllung an-zutreffen.
In gängigen Kapsel-basierten In-halationsprodukten wird eine Füll-menge zwischen 5 mg und 25 mg ei-ner Pulvermischung in Hartkapselnoder bei neueren Entwicklungenauch in HPMC (Hydroxypropyl-methylcellulose)-Kapseln dosiert.Produkte mit darüber hinausgehen-den Füllmengen (z.B. Tobi® Inhalati-onspulver: ca. 50 mg einer sprüh-getrockneten Formulierung; Colo-breathe® Inhalationskapseln: 125 mgreines Wirkstoffmikronisat, Bronchi-tol® Kapseln: 40 mg mikronisiertesMannitol) stellen die Ausnahme dar
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)304 Steckel und Seyfang . Kleinstmengendosierung von Pulvern

Den Verschluss bildet ein Membran-filter, der nach Aufsetzen des Füllkop-fes auf den Blister den Dosierraumbegrenzt. Durch die pulverundurch-lässige Membran hindurch kann imBlisternapf ein Unterdruck erzeugtwerden, der das Pulver zum Fließendurch die Kapillare bringt, bis derHohlraum komplett befüllt ist [8].Das Füllen von freifließenden Pulvern
ist im Gegensatz zum zuvor beschrie-benen Füllprinzip mit diesem Systemdurch die Anpassung des Kapillar-durchmessers bedingt möglich, je-doch ist zu beachten, dass beim Ab-heben des Füllkopfes bei zu geringerKohäsivität Pulver aus dem Pulver-vorrat nachrieseln kann. Andererseitsbesteht durch die Abdeckung der Sie-gelfläche auch bei klebrigen Pulvern
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 307Steckel und Seyfang . Kleinstmengendosierung von Pulvern
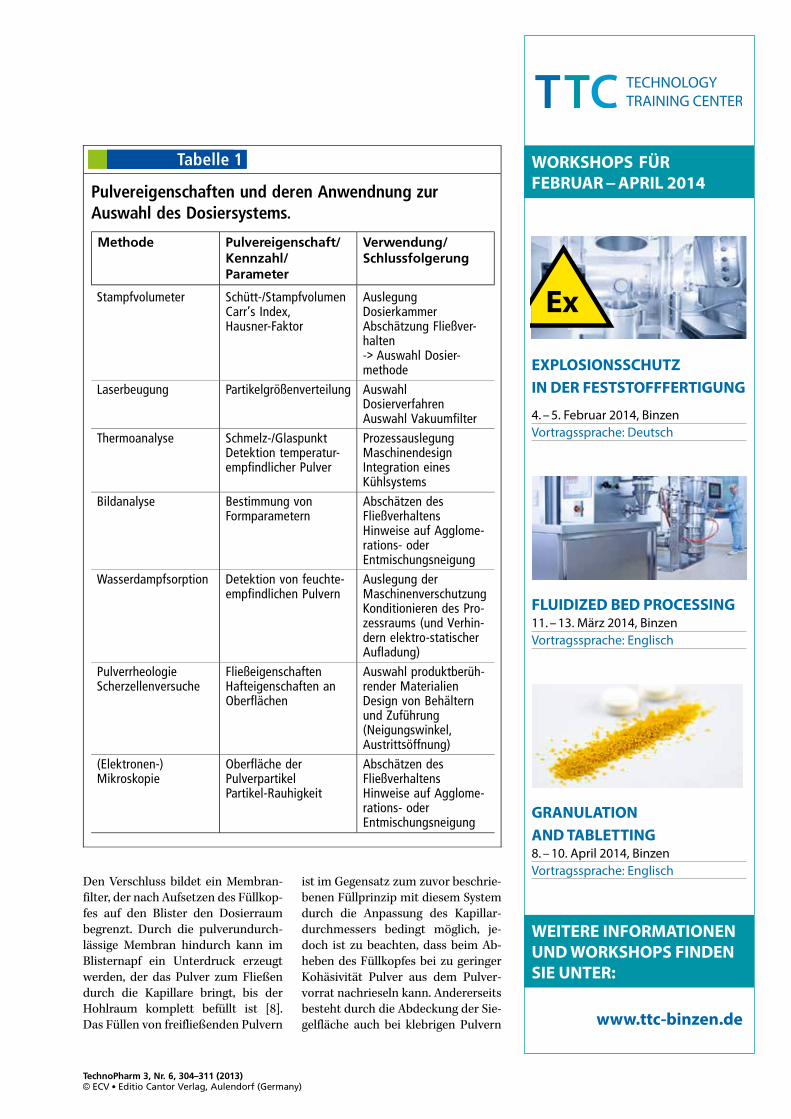
Tabelle 1
Pulvereigenschaften und deren Anwendnung zurAuswahl des Dosiersystems.
Methode Pulvereigenschaft/Kennzahl/Parameter
Verwendung/Schlussfolgerung
Stampfvolumeter Schütt-/StampfvolumenCarr’s Index,Hausner-Faktor
AuslegungDosierkammerAbschätzung Fließver-halten-> Auswahl Dosier-methode
Laserbeugung Partikelgrößenverteilung AuswahlDosierverfahrenAuswahl Vakuumfilter
Thermoanalyse Schmelz-/GlaspunktDetektion temperatur-empfindlicher Pulver
ProzessauslegungMaschinendesignIntegration einesKühlsystems
Bildanalyse Bestimmung vonFormparametern
Abschätzen desFließverhaltensHinweise auf Agglome-rations- oderEntmischungsneigung
Wasserdampfsorption Detektion von feuchte-empfindlichen Pulvern
Auslegung derMaschinenverschutzungKonditionieren des Pro-zessraums (und Verhin-dern elektro-statischerAufladung)
PulverrheologieScherzellenversuche
FließeigenschaftenHafteigenschaften anOberflächen
Auswahl produktberüh-render MaterialienDesign von Behälternund Zuführung(Neigungswinkel,Austrittsöffnung)
(Elektronen-)Mikroskopie
Oberfläche derPulverpartikelPartikel-Rauhigkeit
Abschätzen desFließverhaltensHinweise auf Agglome-rations- oderEntmischungsneigung
derzeit nicht global harmonisiert(USA z.B. mit anderen Anforderun-gen als EU). Die entsprechenden Zu-sammenhänge sind in Abb. 1 ver-deutlicht.
Methoden zur Pulver-charakterisierung
Für frei-fließende, vor allem aber fürkohäsive Pulver sollten einige Pulver-parameter bekannt sein, um eineerste Einschätzung über das zu ver-wendende Füllsystem gewinnen zukönnen. Dazu zählen Kenngrößen,die mittels der im Arzneibuch be-schriebenen Standardtests ermitteltwerden, wie Böschungswinkel,Stampf- zu Schüttdichte-Verhältnis(Hausner-Faktor), Fließzeiten, wahreund scheinbare Dichte sowie die spe-zifische Oberfläche. Darüber hinausgibt es eine Reihe von ergänzendenMessverfahren, mit denen die Sensi-tivität des abzufüllenden Pulvers ge-genüber Temperatur und Feuchte(Thermoanalyse und dynamischeWasserdampfsorption) untersuchtwerden kann. Auch pulverrheologi-sche Messungen sowie Messungenmit einer Scherzelle geben Auf-schluss über die Fließfähigkeit, Flui-disierbarkeit und Adhäsivität einesPulvers. Tabelle 1 listet die üblichenVerfahren zur Pulvercharakterisie-rung auf und beschreibt deren Rele-vanz für das auszuwählende Pulver-system.
Für die Konstruktion und denBau von Dosiersystemen müssengeeignete Fertigungsmaterialienfür alle produktberührenden Teileausgewählt werden, um Klebenund Anhaftungen der Pulver odergar einzelner Mischungsbestand-teile zu vermeiden. In Abb. 2a sindbeispielhaft pulverrheologischeMessungen (durchgeführt mit ei-nem Freeman FT4 Powder Rheo-meter, London, UK) zur Grenzflä-chenreibung (Haftung verschiede-ner Pulver an Maschinenmateria-lien) dargestellt, mit deren Hilfedie Entscheidung für einen bevor-zugten Werkstoff der produktbe-rührenden Teile getroffen werden
kann. Der Messablauf ist in Abb. 2bdargestellt: Das Pulver wird in ei-nem 50 mm × 85 ml Gefäß mit ei-nem Rührflügel vorkonditioniert,bevor die Pulver dann mit einem48 mm Stempel komprimiert wer-den; dabei werden unterschiedlicheWerkstoffe für den Stempel verwen-det. Mit diesen Materialien wirddann die Scherkraft nach unter-schiedlicher Vorkomprimierung ge-messen, Abb. 2c [7].
FüllverfahrenfürPulver inBlister
Eine randvoll-Befüllung von Kapselnist heute – abgesehen von der manu-ellen Befüllung im Labor oder in derRezeptur – eher unüblich. Auf moder-nen Kapselfüllmaschinen wird dasPulver in einem geeigneten Dosiersys-tem separat vordosiert und die abge-teilte Portion anschließend in die Kap-selunterteile übergeben. Um eine stö-rungsfreie Übergabe der Dosis und einkorrektes Verschließen zu gewährleis-ten, sind die Kapseln bei diesem Ver-fahren nur teilgefüllt. Bei einigen spe-ziellen Inhalationssystemen bestehtjedoch die Aufgabe, einen Blisternapfrandvoll mit Pulver zu befüllen. Hierzustehen derzeit zwei verschiedeneTechnologien zur Verfügung (Abb. 3und 4): In einem speziell entwickeltenVerfahren wird das vorgeformte Blis-terband kopfüber, also mit der geöff-neten Blisterkavität nach unten, mitvorgegebenem Anpressdruck in ein ni-velliertes Pulverbett gedrückt. Dabeifüllt das Pulver den Blisternapf unddie aus Druck und Pulvereigenschaf-ten resultierende scheinbare Dichtebestimmt die dosierte Masse. Nachdem Austritt des Blisters aus dem Pul-verbett wird mit einem Rakel über-schüssiges Pulver abgestreift, an-schließend der Blister um 180° ge-dreht, die Siegelflächen gereinigt undmit einer zweiten Aluminiumfolie ver-siegelt (Abb. 3). Die Voraussetzung zurBefüllung eines Blisterstreifens aufdiese Art ist eine gewisse Kohäsivitätdes Pulvers, da das einmal gefülltePulver ansonsten nicht in der Kavitätdes Blisters gehalten werden kann.Andererseits kann bei extrem kohäsi-ven Pulvern die Reinigung der Siegel-fläche stark erschwert sein. Die er-reichbare Füllgewichtsgenauigkeitmit diesem Verfahren beträgt ca. 1,5bis 2,5 % (RSD des Füllgewichts); dieLeistung der Maschine liegt bei 3600Kavitäten pro Minute.
In einem neueren Ansatz wird dasPulver aus dem Pulvervorrat mittelsUnterdruck durch eine Kapillare mit1,5 mm bis 2 mm Innendurchmesserin die Blisterkavität gezogen (Abb. 4).
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)306 Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 2:) Messung der Grenzflächenreibung mitverschiedenen Materialproben und LactoseMonohydrat (Respitose ML001). Zifferncode:01-Edelstahl 316L, spiegelpoliert;02-Edelstahl 316L Referenz; 06-Aluminium,eloxiert; 09-Chromnitrid; 10-Aluminium elo-xiert mit PTFE-Einbettung; 16-PEEK; DLC =Diamond-like carbon (Quelle 2a: Pharm. In-stitut, AU Kiel, Abb. 2b, c: Freeman Technology,London, UK.).
Den Verschluss bildet ein Membran-filter, der nach Aufsetzen des Füllkop-fes auf den Blister den Dosierraumbegrenzt. Durch die pulverundurch-lässige Membran hindurch kann imBlisternapf ein Unterdruck erzeugtwerden, der das Pulver zum Fließendurch die Kapillare bringt, bis derHohlraum komplett befüllt ist [8].Das Füllen von freifließenden Pulvern
ist im Gegensatz zum zuvor beschrie-benen Füllprinzip mit diesem Systemdurch die Anpassung des Kapillar-durchmessers bedingt möglich, je-doch ist zu beachten, dass beim Ab-heben des Füllkopfes bei zu geringerKohäsivität Pulver aus dem Pulver-vorrat nachrieseln kann. Andererseitsbesteht durch die Abdeckung der Sie-gelfläche auch bei klebrigen Pulvern
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 307Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Tabelle 1
Pulvereigenschaften und deren Anwendnung zurAuswahl des Dosiersystems.
Methode Pulvereigenschaft/Kennzahl/Parameter
Verwendung/Schlussfolgerung
Stampfvolumeter Schütt-/StampfvolumenCarr’s Index,Hausner-Faktor
AuslegungDosierkammerAbschätzung Fließver-halten-> Auswahl Dosier-methode
Laserbeugung Partikelgrößenverteilung AuswahlDosierverfahrenAuswahl Vakuumfilter
Thermoanalyse Schmelz-/GlaspunktDetektion temperatur-empfindlicher Pulver
ProzessauslegungMaschinendesignIntegration einesKühlsystems
Bildanalyse Bestimmung vonFormparametern
Abschätzen desFließverhaltensHinweise auf Agglome-rations- oderEntmischungsneigung
Wasserdampfsorption Detektion von feuchte-empfindlichen Pulvern
Auslegung derMaschinenverschutzungKonditionieren des Pro-zessraums (und Verhin-dern elektro-statischerAufladung)
PulverrheologieScherzellenversuche
FließeigenschaftenHafteigenschaften anOberflächen
Auswahl produktberüh-render MaterialienDesign von Behälternund Zuführung(Neigungswinkel,Austrittsöffnung)
(Elektronen-)Mikroskopie
Oberfläche derPulverpartikelPartikel-Rauhigkeit
Abschätzen desFließverhaltensHinweise auf Agglome-rations- oderEntmischungsneigung
WORKSHOPS FÜR FEBRUAR − APRIL 2014
WEITERE INFORMATIONEN UND WORKSHOPS FINDEN SIE UNTER:
www.ttc-binzen.de
EXPLOSIONSSCHUTZ IN DER FESTSTOFFFERTIGUNG
4. – 5. Februar 2014, BinzenVortragssprache: Deutsch
FLUIDIZED BED PROCESSING11. – 13. März 2014, BinzenVortragssprache: Englisch
GRANULATION AND TABLETTING8. – 10. April 2014, BinzenVortragssprache: Englisch
derzeit nicht global harmonisiert(USA z.B. mit anderen Anforderun-gen als EU). Die entsprechenden Zu-sammenhänge sind in Abb. 1 ver-deutlicht.
Methoden zur Pulver-charakterisierung
Für frei-fließende, vor allem aber fürkohäsive Pulver sollten einige Pulver-parameter bekannt sein, um eineerste Einschätzung über das zu ver-wendende Füllsystem gewinnen zukönnen. Dazu zählen Kenngrößen,die mittels der im Arzneibuch be-schriebenen Standardtests ermitteltwerden, wie Böschungswinkel,Stampf- zu Schüttdichte-Verhältnis(Hausner-Faktor), Fließzeiten, wahreund scheinbare Dichte sowie die spe-zifische Oberfläche. Darüber hinausgibt es eine Reihe von ergänzendenMessverfahren, mit denen die Sensi-tivität des abzufüllenden Pulvers ge-genüber Temperatur und Feuchte(Thermoanalyse und dynamischeWasserdampfsorption) untersuchtwerden kann. Auch pulverrheologi-sche Messungen sowie Messungenmit einer Scherzelle geben Auf-schluss über die Fließfähigkeit, Flui-disierbarkeit und Adhäsivität einesPulvers. Tabelle 1 listet die üblichenVerfahren zur Pulvercharakterisie-rung auf und beschreibt deren Rele-vanz für das auszuwählende Pulver-system.
Für die Konstruktion und denBau von Dosiersystemen müssengeeignete Fertigungsmaterialienfür alle produktberührenden Teileausgewählt werden, um Klebenund Anhaftungen der Pulver odergar einzelner Mischungsbestand-teile zu vermeiden. In Abb. 2a sindbeispielhaft pulverrheologischeMessungen (durchgeführt mit ei-nem Freeman FT4 Powder Rheo-meter, London, UK) zur Grenzflä-chenreibung (Haftung verschiede-ner Pulver an Maschinenmateria-lien) dargestellt, mit deren Hilfedie Entscheidung für einen bevor-zugten Werkstoff der produktbe-rührenden Teile getroffen werden
kann. Der Messablauf ist in Abb. 2bdargestellt: Das Pulver wird in ei-nem 50 mm × 85 ml Gefäß mit ei-nem Rührflügel vorkonditioniert,bevor die Pulver dann mit einem48 mm Stempel komprimiert wer-den; dabei werden unterschiedlicheWerkstoffe für den Stempel verwen-det. Mit diesen Materialien wirddann die Scherkraft nach unter-schiedlicher Vorkomprimierung ge-messen, Abb. 2c [7].
FüllverfahrenfürPulver inBlister
Eine randvoll-Befüllung von Kapselnist heute – abgesehen von der manu-ellen Befüllung im Labor oder in derRezeptur – eher unüblich. Auf moder-nen Kapselfüllmaschinen wird dasPulver in einem geeigneten Dosiersys-tem separat vordosiert und die abge-teilte Portion anschließend in die Kap-selunterteile übergeben. Um eine stö-rungsfreie Übergabe der Dosis und einkorrektes Verschließen zu gewährleis-ten, sind die Kapseln bei diesem Ver-fahren nur teilgefüllt. Bei einigen spe-ziellen Inhalationssystemen bestehtjedoch die Aufgabe, einen Blisternapfrandvoll mit Pulver zu befüllen. Hierzustehen derzeit zwei verschiedeneTechnologien zur Verfügung (Abb. 3und 4): In einem speziell entwickeltenVerfahren wird das vorgeformte Blis-terband kopfüber, also mit der geöff-neten Blisterkavität nach unten, mitvorgegebenem Anpressdruck in ein ni-velliertes Pulverbett gedrückt. Dabeifüllt das Pulver den Blisternapf unddie aus Druck und Pulvereigenschaf-ten resultierende scheinbare Dichtebestimmt die dosierte Masse. Nachdem Austritt des Blisters aus dem Pul-verbett wird mit einem Rakel über-schüssiges Pulver abgestreift, an-schließend der Blister um 180° ge-dreht, die Siegelflächen gereinigt undmit einer zweiten Aluminiumfolie ver-siegelt (Abb. 3). Die Voraussetzung zurBefüllung eines Blisterstreifens aufdiese Art ist eine gewisse Kohäsivitätdes Pulvers, da das einmal gefülltePulver ansonsten nicht in der Kavitätdes Blisters gehalten werden kann.Andererseits kann bei extrem kohäsi-ven Pulvern die Reinigung der Siegel-fläche stark erschwert sein. Die er-reichbare Füllgewichtsgenauigkeitmit diesem Verfahren beträgt ca. 1,5bis 2,5 % (RSD des Füllgewichts); dieLeistung der Maschine liegt bei 3600Kavitäten pro Minute.
In einem neueren Ansatz wird dasPulver aus dem Pulvervorrat mittelsUnterdruck durch eine Kapillare mit1,5 mm bis 2 mm Innendurchmesserin die Blisterkavität gezogen (Abb. 4).
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)306 Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 2:) Messung der Grenzflächenreibung mitverschiedenen Materialproben und LactoseMonohydrat (Respitose ML001). Zifferncode:01-Edelstahl 316L, spiegelpoliert;02-Edelstahl 316L Referenz; 06-Aluminium,eloxiert; 09-Chromnitrid; 10-Aluminium elo-xiert mit PTFE-Einbettung; 16-PEEK; DLC =Diamond-like carbon (Quelle 2a: Pharm. In-stitut, AU Kiel, Abb. 2b, c: Freeman Technology,London, UK.).

Pulver im Bereich von 0.5 mg bis10 mg mit einer Genauigkeit von 1-2 % RSD füllen.
Hier befinden sich die relativ klei-nen, konischen Dosierkammern (Boh-rungen mit Innendurchmesser 1 mmbis ca. 6 mm) auf einem Edelstahlrohr(= Dosierwalze) mit eng tolerierterWandstärke. Nach innen werden dieseBohrungen von einem Filter mit ca.1μm Porengröße begrenzt, der Luftdurchlässt aber Pulverteilchen zurück-hält. Über entsprechende Kanäle, Ver-teiler und Ventile kann in den Bohrun-gen im Wechsel ein definierter Unter-oder Überdruck erzeugt werden. Wiein Abb. 6 dargestellt, befindet sich überder Dosierwalze das Pulverbett, undein geeigneter Rührer sorgt dafür, dassdas meist kohäsive Füllgut im Verlauf
der Zeit keine Brücken bildet. Der Do-siervorgang beginnt „in der zwölf-Uhr-Position“ mit dem Einsaugen des Pul-vers, unterstützt durch den Rührer.Danach dreht sich die Walze mit denbefüllten Bohrungen um 180° (im Bildim Uhrzeigersinn), wobei die Dosier-kammern ein Rakel passieren, dasüberschüssiges Pulver sehr präzise ab-streift. Über einen kurzen Druckimpulswird dann das vordosierte Pulver andas Zielbehältnis (Kapseln, Blisternäp-fe, etc.) übergeben. Schließlich erfolgtin der „neun-Uhr-Position“ eine in-lineReinigung der Dosierbohrungen, umanhaftende Pulverpartikel von Filteroder Innenwand zu entfernen. Diesist erforderlich, um auch bei stark an-haftenden Pulvern, wie beispielsweisesprühgetrockneten Proteinen oder
Peptiden, einen stabilenAbfüllprozess zu gewähr-leisten. Optional kann in„drei-Uhr-Position“ einKamera- oder spektro-skopisches System zuFüllkontrolle oder In-Prozess-Analytik inte-griert werden.
Kommerzielle Systemeverfügen über bis zu 6 Do-sierwalzen, die pro Ma-schinentakt jeweils biszu 16 Dosierungen erzeu-gen, so dass sich ein ent-sprechend hoher Durch-satz ergibt (Ausstoßleis-tung derzeit bis zu 250Blisternäpfe/min und beiKapselfüllmaschinen biszu 140 Takten/min, ent-sprechend > 1500 Dosie-
rungen pro Minute).Eine neuere Entwicklungmacht sich
das vibrationsgesteuerte Fließen vonPulvern in Kapillaren zunutze [9, 10].Pulver wird dabei aus dem Pulvervor-rat in eine Kapillare mit sich verjüngen-dem Auslauf überführt. Wird eine Vi-bration auf die Kapillare aufgebracht,beginnt das Pulver zu fließen und kannin Kapseln, Flaschen oder Blisterkavi-täten dosiert werden. Die dosierteMenge pro Zeiteinheit ist eine Funk-tion der Pulvereigenschaften sowieder Frequenz und Amplitude der auf-gebrachten Schwingung [11]. Um auchbei irregulärem Fließverhalten eineausreichende Dosiergenauigkeit zu ge-währleisten, können solche Dosier-aggregate mit einem System zur in-lineMessung der Dosiermenge rückgekop-
Abb. 5: links: Schema der Stechheberdosierung; rechts: Anwendungen und technische Daten zur Stechheberdosierung.
nicht die Gefahr, dass die Siegelflächeverschmutzt und so die Dichtigkeitder Blister gefährdet wird. Derzeitiggefertigte Maschinen (Füllköpfe mitje 60 bis 80 Kapillaren) erlauben eineArbeitsgeschwindigkeit von 900 –2400 Kavitäten/min und eine Füll-gewichtsgenauigkeit von < 2 % RSD.
Beiden Verfahren ist gemeinsam,dass der zu befüllende Blisternapfzugleich die Dosierkammer darstellt,weshalb besonders hohe Anforde-rungen an die Gleichförmigkeit derNäpfe und somit an die Präzisionund Reproduzierbarkeit der Tiefzieh-prozesse gestellt werden.
Dosierverfahren für Pulver
Es gibt eine Reihe von Füllverfahrenzur Befüllung von Hartkapseln mitPulvermaterial, Granulaten, Pelletsoder sogar Minitabletten Die Dosie-rung von Kleinstmengen eines Pul-vers, also die exakte Dosierung im Be-reich zwischen 1 und 100 mg, ist nurmit wenigen Füllmaschinen zu errei-chen: Diese arbeiten entweder nachdem Stechheber/Vakuumstechheber-Prinzip, dem Prinzip der Vakuumwal-zendosierung oder der Mikrovibrati-onsdosierung. In allen Fällen, in de-nen das dosierte Pulver über eine freieStrecke in die Kapsel/den Blister fällt,ist zudem die Integration einer 100 %Füllgewichtskontrolle möglich.
Die genannten Dosierverfahrenwerden mittlerweile nicht nur in Kap-selfüllmaschinen eingesetzt, sondernauch in Sondermaschinen, mit denenBlister, Kartuschen oder spezielleringförmige Mehrdosenbehältnisse(Disks) befüllt und verschlossen wer-den. Die Dosierung von kleinen Pul-vermengen mit dem Stechheber(Abb. 5) mit oder ohne Vakuum er-laubt die Dosierung von mäßig bisschlechtfließenden Pulvern im Be-reich von > 10 mg bis 600 mg.
Bei diesem volumetrischen Dosier-system wird die Dosierkammer von ei-nem Dosierrohr und einem Stößel ge-bildet, die unabhängig voneinandervertikal bewegt werden können und inihrem Durchmesser dem Zielbehältnis,z.B. der Kapsel, angepasst sind. Durch
Änderung der relati-ven Höhe des Stö-ßels kann das Kam-mervolumen stufen-los verstellt werden.Der Pulvervorrat be-findet sich bei die-sem Verfahren inder Regel in einemrotierenden Behäl-ter, in welchem dasPulver entlüftet undmit einem Rakel aufein definiertes Ni-veau gebracht wer-den kann. Die Stech-heber tauchen vonoben ins Pulverbett ein und grenzendie Dosis ab, indem sich das Dosierrohrbis zum Boden absenkt und dabei mitPulver füllt. Um beim Ausheben undder (raschen) Transferbewegung zumZielbehältnis kein Pulver zu verlieren,wird die Pulversäule innerhalb des Do-sierrohrs zuvor durch leichtes Absen-ken des Stößels mit 40 bis 80 N kom-primiert. Dies führt zu einer Brücken-bildung im Dosierrohr und verhindertweitgehend das Ausfließen des Pulvers.Die Übergabe der Pulverdosis erfolgtdurch Herausschieben der Pulversäulemit dem Stößel.
Es sei hier angemerkt, dass extremgut fließende Pulver und Granulate,wie sie in der Tablettierung er-wünscht sind, wegen der relativ ge-ringen Verdichtungskräfte mit die-sem Verfahren schlecht zu dosierensind. Die Korngrößen der Partikelmüssen – wie bei anderen Verfahrenauch – der absoluten Dosiermenge
angepasst werden. Stark klebendePulver können an der Außenseiteder Stechheber anhaften und durchanschließend unkontrolliertes Ablö-sen den Prozess stören. Als Alterna-tive kann die Stirnseite des Stößelsporös konstruiert sein (Siebgewebe,Sintermaterial, Membranfilter), sodass durch Verbindung mit einempneumatischen System das Pulverschonend mittels Unterdruck im Do-sierrohr gehalten und mittels Druck-impuls übergeben werden kann.
Pulvereigenschaften wie Kompri-mierbarkeit, Kohäsivität und Par-tikelgrößenverteilung geben hierden Ausschlag für die Verwendungdes Stechhebers mit Komprimierenoder des Vakuumsystems. Die Do-siergenauigkeit liegt pulverabhängigzwischen 1 und 3 % RSD.
Mit dem Vakuum-Walzendosier-system (Abb. 6) lassen sich hingegenauch schlecht bis gar nicht fließende
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)308 Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 3: Überkopfbefüllen von Blisterstreifen. (Quelle Abb 3-8: HarroHäfliger GmbH, Allmersbach i.T.).
Abb. 4 : Membranfüller zur 100 %-Befüllung beliebig geformter Kavitäten; links: Schema derMembranbefüllung; rechts: Anwendungen und technische Daten zum Membranfüller.

Pulver im Bereich von 0.5 mg bis10 mg mit einer Genauigkeit von 1-2 % RSD füllen.
Hier befinden sich die relativ klei-nen, konischen Dosierkammern (Boh-rungen mit Innendurchmesser 1 mmbis ca. 6 mm) auf einem Edelstahlrohr(= Dosierwalze) mit eng tolerierterWandstärke. Nach innen werden dieseBohrungen von einem Filter mit ca.1μm Porengröße begrenzt, der Luftdurchlässt aber Pulverteilchen zurück-hält. Über entsprechende Kanäle, Ver-teiler und Ventile kann in den Bohrun-gen im Wechsel ein definierter Unter-oder Überdruck erzeugt werden. Wiein Abb. 6 dargestellt, befindet sich überder Dosierwalze das Pulverbett, undein geeigneter Rührer sorgt dafür, dassdas meist kohäsive Füllgut im Verlauf
der Zeit keine Brücken bildet. Der Do-siervorgang beginnt „in der zwölf-Uhr-Position“ mit dem Einsaugen des Pul-vers, unterstützt durch den Rührer.Danach dreht sich die Walze mit denbefüllten Bohrungen um 180° (im Bildim Uhrzeigersinn), wobei die Dosier-kammern ein Rakel passieren, dasüberschüssiges Pulver sehr präzise ab-streift. Über einen kurzen Druckimpulswird dann das vordosierte Pulver andas Zielbehältnis (Kapseln, Blisternäp-fe, etc.) übergeben. Schließlich erfolgtin der „neun-Uhr-Position“ eine in-lineReinigung der Dosierbohrungen, umanhaftende Pulverpartikel von Filteroder Innenwand zu entfernen. Diesist erforderlich, um auch bei stark an-haftenden Pulvern, wie beispielsweisesprühgetrockneten Proteinen oder
Peptiden, einen stabilenAbfüllprozess zu gewähr-leisten. Optional kann in„drei-Uhr-Position“ einKamera- oder spektro-skopisches System zuFüllkontrolle oder In-Prozess-Analytik inte-griert werden.
Kommerzielle Systemeverfügen über bis zu 6 Do-sierwalzen, die pro Ma-schinentakt jeweils biszu 16 Dosierungen erzeu-gen, so dass sich ein ent-sprechend hoher Durch-satz ergibt (Ausstoßleis-tung derzeit bis zu 250Blisternäpfe/min und beiKapselfüllmaschinen biszu 140 Takten/min, ent-sprechend > 1500 Dosie-
rungen pro Minute).Eine neuere Entwicklungmacht sich
das vibrationsgesteuerte Fließen vonPulvern in Kapillaren zunutze [9, 10].Pulver wird dabei aus dem Pulvervor-rat in eine Kapillare mit sich verjüngen-dem Auslauf überführt. Wird eine Vi-bration auf die Kapillare aufgebracht,beginnt das Pulver zu fließen und kannin Kapseln, Flaschen oder Blisterkavi-täten dosiert werden. Die dosierteMenge pro Zeiteinheit ist eine Funk-tion der Pulvereigenschaften sowieder Frequenz und Amplitude der auf-gebrachten Schwingung [11]. Um auchbei irregulärem Fließverhalten eineausreichende Dosiergenauigkeit zu ge-währleisten, können solche Dosier-aggregate mit einem System zur in-lineMessung der Dosiermenge rückgekop-
Abb. 5: links: Schema der Stechheberdosierung; rechts: Anwendungen und technische Daten zur Stechheberdosierung.
nicht die Gefahr, dass die Siegelflächeverschmutzt und so die Dichtigkeitder Blister gefährdet wird. Derzeitiggefertigte Maschinen (Füllköpfe mitje 60 bis 80 Kapillaren) erlauben eineArbeitsgeschwindigkeit von 900 –2400 Kavitäten/min und eine Füll-gewichtsgenauigkeit von < 2 % RSD.
Beiden Verfahren ist gemeinsam,dass der zu befüllende Blisternapfzugleich die Dosierkammer darstellt,weshalb besonders hohe Anforde-rungen an die Gleichförmigkeit derNäpfe und somit an die Präzisionund Reproduzierbarkeit der Tiefzieh-prozesse gestellt werden.
Dosierverfahren für Pulver
Es gibt eine Reihe von Füllverfahrenzur Befüllung von Hartkapseln mitPulvermaterial, Granulaten, Pelletsoder sogar Minitabletten Die Dosie-rung von Kleinstmengen eines Pul-vers, also die exakte Dosierung im Be-reich zwischen 1 und 100 mg, ist nurmit wenigen Füllmaschinen zu errei-chen: Diese arbeiten entweder nachdem Stechheber/Vakuumstechheber-Prinzip, dem Prinzip der Vakuumwal-zendosierung oder der Mikrovibrati-onsdosierung. In allen Fällen, in de-nen das dosierte Pulver über eine freieStrecke in die Kapsel/den Blister fällt,ist zudem die Integration einer 100 %Füllgewichtskontrolle möglich.
Die genannten Dosierverfahrenwerden mittlerweile nicht nur in Kap-selfüllmaschinen eingesetzt, sondernauch in Sondermaschinen, mit denenBlister, Kartuschen oder spezielleringförmige Mehrdosenbehältnisse(Disks) befüllt und verschlossen wer-den. Die Dosierung von kleinen Pul-vermengen mit dem Stechheber(Abb. 5) mit oder ohne Vakuum er-laubt die Dosierung von mäßig bisschlechtfließenden Pulvern im Be-reich von > 10 mg bis 600 mg.
Bei diesem volumetrischen Dosier-system wird die Dosierkammer von ei-nem Dosierrohr und einem Stößel ge-bildet, die unabhängig voneinandervertikal bewegt werden können und inihrem Durchmesser dem Zielbehältnis,z.B. der Kapsel, angepasst sind. Durch
Änderung der relati-ven Höhe des Stö-ßels kann das Kam-mervolumen stufen-los verstellt werden.Der Pulvervorrat be-findet sich bei die-sem Verfahren inder Regel in einemrotierenden Behäl-ter, in welchem dasPulver entlüftet undmit einem Rakel aufein definiertes Ni-veau gebracht wer-den kann. Die Stech-heber tauchen vonoben ins Pulverbett ein und grenzendie Dosis ab, indem sich das Dosierrohrbis zum Boden absenkt und dabei mitPulver füllt. Um beim Ausheben undder (raschen) Transferbewegung zumZielbehältnis kein Pulver zu verlieren,wird die Pulversäule innerhalb des Do-sierrohrs zuvor durch leichtes Absen-ken des Stößels mit 40 bis 80 N kom-primiert. Dies führt zu einer Brücken-bildung im Dosierrohr und verhindertweitgehend das Ausfließen des Pulvers.Die Übergabe der Pulverdosis erfolgtdurch Herausschieben der Pulversäulemit dem Stößel.
Es sei hier angemerkt, dass extremgut fließende Pulver und Granulate,wie sie in der Tablettierung er-wünscht sind, wegen der relativ ge-ringen Verdichtungskräfte mit die-sem Verfahren schlecht zu dosierensind. Die Korngrößen der Partikelmüssen – wie bei anderen Verfahrenauch – der absoluten Dosiermenge
angepasst werden. Stark klebendePulver können an der Außenseiteder Stechheber anhaften und durchanschließend unkontrolliertes Ablö-sen den Prozess stören. Als Alterna-tive kann die Stirnseite des Stößelsporös konstruiert sein (Siebgewebe,Sintermaterial, Membranfilter), sodass durch Verbindung mit einempneumatischen System das Pulverschonend mittels Unterdruck im Do-sierrohr gehalten und mittels Druck-impuls übergeben werden kann.
Pulvereigenschaften wie Kompri-mierbarkeit, Kohäsivität und Par-tikelgrößenverteilung geben hierden Ausschlag für die Verwendungdes Stechhebers mit Komprimierenoder des Vakuumsystems. Die Do-siergenauigkeit liegt pulverabhängigzwischen 1 und 3 % RSD.
Mit dem Vakuum-Walzendosier-system (Abb. 6) lassen sich hingegenauch schlecht bis gar nicht fließende
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)308 Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 3: Überkopfbefüllen von Blisterstreifen. (Quelle Abb 3-8: HarroHäfliger GmbH, Allmersbach i.T.).
Abb. 4 : Membranfüller zur 100 %-Befüllung beliebig geformter Kavitäten; links: Schema derMembranbefüllung; rechts: Anwendungen und technische Daten zum Membranfüller.
LYNX-SPECTRA 3D
▪ Inspection of Inhaler Powders
▪ Precise Volume Measurement
▪ Laser Technology
NEW APPLICATION!
Dry Powder Inspection

pelt werden. Das Messsystem steuertdann den Vibrationsantrieb, wobeizur Erhöhung der Leistung eine Unter-scheidung in Grob- und Feindosierungmöglich ist (Abb. 7). Das Nachrieselnvon Pulver nach Abschalten derSchwingung kann zum einen über dieGeometrie und den Innendurchmesserder Kapillarenöffnung verhindert wer-den, zum anderen ermöglicht eine in-tegrierte Spursperre die Vergrößerungdes Fließkanals und damit die Erhö-hung der Durchsatzleistung. Der Do-sierbereich für dieses System liegt zwi-schen 10 mg und > 2000 mg. Alle hierdiskutierten Systeme sind mittlerweilesowohl als „Table Top“, d. h., als kleineLaborfüllmaschine sowie als vollinte-grierte Füllanlagen erhältlich und de-cken dabei den Bedarf der typischenFüllaufgaben ab.
Füllgewichtskontrolle
Während die stichprobenartige Über-prüfung des Füllgewichts bis heute ei-nen festen Platz in der In-Prozess-Kon-trolle hat, stellen pharmazeutischeHersteller heute höhere Anforderungenbis hin zu einer 100 % Füllgewichts-kontrolle.
GravimetrischeFüllgewichtskontrolleDie gravimetrische Füllgewichtskon-trolle von Hartkapseln lässt sich bei-spielsweise durch die 100 % Verwie-gung aller gefüllten Kapseln erreichen.Da die Varianz des Kapselleergewichtszur Gesamttoleranz beiträgt, ist diesjedoch eher für Füllmassen > 200 mg
bzw. Kapseln Größe 3 bis00 geeignet und sinnvoll,da in diesen Fällen die Va-rianz weniger als 2 % derFüllmenge entspricht.
Eine verlässliche gravi-metrische Stichproben-Kontrolle ist in der Regelnur durch Wiegen der be-füllten Kapseln bzw. desBlisters sowie eine erneuteWägung nach Entleerungzu erreichen.
Lediglich für die Kap-selbefüllung im unterenLeistungsbereich ist eineMaschine am Markt, dievollautomatisch jede Leer-kapsel wiegt, anschlie-ßend der Füllstation zu-führt und nach dem Befül-len/Verschließen erneutverwiegt. Bei der Dosie-rung in Blister sind auto-matische gravimetrischeKontrollen nur als (zerstö-rende) statistische In-Pro-zess-Kontrollen verfügbar.Beispielsweise gibt es Wiegeauto-maten, die einzelne Blisterstreifen Napffür Napf aufziehen und deren Inhalt inWaagschalen entleeren undwiegen. Beianderen Systemen werden Streifenoder einzelne Näpfe ausgestanzt, zu-nächst brutto gewogen, danach mittelsHohlnadeln leergesaugt und anschlie-ßend erneut gewogen.
Für den Entwicklungsbereich undzum Befüllen von Kleinmengen (z.B.für frühe präklinische Studien oderPhase 1 Studien) sind gravimetrisch-
arbeitende Dosiersysteme verfügbar;hierbei wird eine Waage an das Dosier-system (Vibrationsprinzip, Schnecken-dosierung) gekoppelt und die Ein-waage als Regelgröße für die Dosierungverwendet, z.B. Xcelodose [12], Quan-tos, Powdernium [13]. Diese Systemearbeiten extrem langsam und sind des-halb nur für Kleinmengen geeignet.
Andere Prinzipien der Füll-gewichtskontrolleIn den letzten Jahren wurden unter-
schiedliche Verfahrenbeschrieben, um in-linedie dosierte Masse zuüberprüfen. Neben derIntegration kapazitiverSensoren in die Kapsel-füllmaschine [14] wur-den Mikrowellenreso-natoren [15, 16], Rönt-gensysteme [17] undoptische Verfahren [18]beschrieben.
Einen innovativen An-satz zur 100 % Kontrolledes Füllgewichts stellt
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)310 Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 7: Dosiersystem nach dem Prinzip „Vibrierende Kapillare“.
Abb. 8: Funktionsweise des kapazitiven Sensors zur 100 % in-line Kontrolle der Dosiermenge.
Abb. 6: links: Schema Vakuum-Walzendosierer; rechts: Anwendungen und technische Daten zur Vakuumwalzendosierung.

die Integration eines kapazitiven Sen-sors in das Dosiersystem dar (Abb. 8).Die Kapazität des Kondensators wirddurch das durchströmende Pulver ge-ändert, so dass nach Kalibrierung mitdem zu füllenden Pulver eine 100 % Ge-wichtskontrolle ermöglicht wird [19].
Mit einem Kamerasystem kann dieAnwesenheit des gefüllten Pulvers über-prüft werden. Die Verwendung vonspektroskopischen Verfahren (IR, NIRoder Raman-Sensoren) erlaubt zudemdie Quantifizierung einer Pulverkom-ponente im Gemisch und stellt damiteinen Parameter für die Homogenitätdes gefüllten Pulvers sowie der Verän-derung des Pulvers währende des Füll-vorgangs, z.B. durch Entmischung, dar.
Röntgenstrahlung kann ebenfallszu einer „Ja/Nein“-Kontrolle des Füll-prozesses eingesetzt werden. Eineabsolute Quantifizierung des Füll-gutes ist bei Kleinstmengen damitallerdings derzeit nicht zu erreichen.
Zusammenfassung
Es gibt heute kaum eine Abfüllaufgabefür Pulver, die nicht mit den vorhande-nen Technologien gelöst werden kann.Ein solides Wissen über das Pulver,dessen Eigenschaften und Sensitivitätgegenüber äußeren Einflüssen ist dabeierforderlich, um für das zu füllendeoder zu dosierende Produkt das rich-tige Abfüllprinzip und die dazugehöri-gen Einstellungen auszuwählen. DieFüllgewichtsgenauigkeit liegt in der Re-gel unterhalb von 3 %, dies kann auchbei sehr kleinen Dosiermengen (1 mg)und sehr schlecht fließenden Pulvernerreicht werden. Eine quantitative Füll-mengenkontrolle lässt sich heute be-reits in viele der vorgestellten Abfüll-verfahren integrieren.
Fachliteratur[1] Eskandar F, Lejeune M, Edge S., Low
powder mass filling of dry powder inha-lation formulations, Drug Dev Ind Pharm37:1 (2011) 24-32.
[2] Kane NR, Broce B, Gonzalez-Zugasti J,Lewis WP, LeQuesne M and Lemmo AV, ASystem for DispensingSub-Milligram Do-ses of Active Pharmaceutical Powders forEarly Stage Solubility Assays , J Lab Au-tomation 9:4 (2004) 218-227
[3] Tan SB and Newton JM, Powder flowabilityas an indication of capsule filling perfor-mance, Int J Pharm 61:1–2 (1990) 145-155
[4] Rote Liste 2013, Editio Cantor Verlag,Aulendorf, Deutschland
[5] Smith IJ, Parry-Billings M, The inhalers ofthe future? A review of dry powder devi-ces on the market today, Pulm PharmacolTher 16(2): (2003) 79-95.
[6] Friebel C and Steckel H, Single-use dis-osable dry powder inhalers for pulmonarydrug delivery, Expert Opin Drug Deliv 7:12(2010) 1359-1372
[7] Freeman R, Measuring the flow propertiesof consolidated, conditioned and aeratedpowders – A comparative study using apowder rheometer and a rotational shearcell, Powder Technology 174:1-2 (2007)Pages 25-33
[8] Weigel M, Fülleinrichtung zum volume-trischen Dosieren von Pulver, EP 2 195 244
[9] Yang S and J.R.G. Evans JRG, Acousticinitiation of powder flow in capillaries,Chem Engineering Sci 60 (2005) 413-421
[10] Yang S and Li X, Experimental and analyticalstudy of ultrasonic micro powder feeding, J.Phys. D: Appl. Phys. 36 (2003) 1349-1354
[11] Chen X, Seyfang K and Steckel H, Deve-lopment of a micro dosing system for finepowder using a vibrating capillary. Part 1:the investigation of factors influencing onthe dosing performance, Int J Pharm 433(2012) 34-41
[12] Bi M, Sun CC, Alvarez F and Alvarez-Nu-nez F, The Manufacture of Low-Dose OralSolid Dosage Form to Support Early Cli-nical Studies Using an Automated Micro-Filing System, AAPS PharmSciTech. 12:1(2011) 88-95
[13] NN, Microdosing equipment fills niche inR&D, clinical trial, Tablets & Capsules 3(2009) 37-39
[14] S. De Caris, A. Ansaloni, Verfahren zumAuswiegen von Inhaltsstoffen in Kapselnund Vorrichtung zum Dosieren von In-haltsstoffen, DE 44 37 597
[15] R. Trebbi, Method for monitoring theproperties of pharmaceutical articles,US 7,042,231
[16] H.-W. Müller, Messeinrichtung zur zer-störungsfreien Bestimmung der Einwaagein Kapseln,EP 1 634 041
[17] F. Bessler, W. Bauer, X-Ray Net WeightControl of Pharmaceutical Products,17th World Conference on NondestructiveTesting, 25-28 Oct 2008, Shanghai, China
[18] P. Stöckel, Optische 100 %-Inspektion aufKapselfüllmaschinen, http://sine.ni.com/cs/app/doc/p/id/cs-11377
[19] Chen X, Seyfang K and Steckel H, Deve-lopment of a micro-dosing system for finepowder using a vibrating capillary, part 2:The implementation of a process analyticaltechnology tool in a closed-loop dosingsystem, Int J Pharm 433 (2012) 42-50
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 311Steckel und Seyfang . Kleinstmengendosierung von Pulvern
pelt werden. Das Messsystem steuertdann den Vibrationsantrieb, wobeizur Erhöhung der Leistung eine Unter-scheidung in Grob- und Feindosierungmöglich ist (Abb. 7). Das Nachrieselnvon Pulver nach Abschalten derSchwingung kann zum einen über dieGeometrie und den Innendurchmesserder Kapillarenöffnung verhindert wer-den, zum anderen ermöglicht eine in-tegrierte Spursperre die Vergrößerungdes Fließkanals und damit die Erhö-hung der Durchsatzleistung. Der Do-sierbereich für dieses System liegt zwi-schen 10 mg und > 2000 mg. Alle hierdiskutierten Systeme sind mittlerweilesowohl als „Table Top“, d. h., als kleineLaborfüllmaschine sowie als vollinte-grierte Füllanlagen erhältlich und de-cken dabei den Bedarf der typischenFüllaufgaben ab.
Füllgewichtskontrolle
Während die stichprobenartige Über-prüfung des Füllgewichts bis heute ei-nen festen Platz in der In-Prozess-Kon-trolle hat, stellen pharmazeutischeHersteller heute höhere Anforderungenbis hin zu einer 100 % Füllgewichts-kontrolle.
GravimetrischeFüllgewichtskontrolleDie gravimetrische Füllgewichtskon-trolle von Hartkapseln lässt sich bei-spielsweise durch die 100 % Verwie-gung aller gefüllten Kapseln erreichen.Da die Varianz des Kapselleergewichtszur Gesamttoleranz beiträgt, ist diesjedoch eher für Füllmassen > 200 mg
bzw. Kapseln Größe 3 bis00 geeignet und sinnvoll,da in diesen Fällen die Va-rianz weniger als 2 % derFüllmenge entspricht.
Eine verlässliche gravi-metrische Stichproben-Kontrolle ist in der Regelnur durch Wiegen der be-füllten Kapseln bzw. desBlisters sowie eine erneuteWägung nach Entleerungzu erreichen.
Lediglich für die Kap-selbefüllung im unterenLeistungsbereich ist eineMaschine am Markt, dievollautomatisch jede Leer-kapsel wiegt, anschlie-ßend der Füllstation zu-führt und nach dem Befül-len/Verschließen erneutverwiegt. Bei der Dosie-rung in Blister sind auto-matische gravimetrischeKontrollen nur als (zerstö-rende) statistische In-Pro-zess-Kontrollen verfügbar.Beispielsweise gibt es Wiegeauto-maten, die einzelne Blisterstreifen Napffür Napf aufziehen und deren Inhalt inWaagschalen entleeren undwiegen. Beianderen Systemen werden Streifenoder einzelne Näpfe ausgestanzt, zu-nächst brutto gewogen, danach mittelsHohlnadeln leergesaugt und anschlie-ßend erneut gewogen.
Für den Entwicklungsbereich undzum Befüllen von Kleinmengen (z.B.für frühe präklinische Studien oderPhase 1 Studien) sind gravimetrisch-
arbeitende Dosiersysteme verfügbar;hierbei wird eine Waage an das Dosier-system (Vibrationsprinzip, Schnecken-dosierung) gekoppelt und die Ein-waage als Regelgröße für die Dosierungverwendet, z.B. Xcelodose [12], Quan-tos, Powdernium [13]. Diese Systemearbeiten extrem langsam und sind des-halb nur für Kleinmengen geeignet.
Andere Prinzipien der Füll-gewichtskontrolleIn den letzten Jahren wurden unter-
schiedliche Verfahrenbeschrieben, um in-linedie dosierte Masse zuüberprüfen. Neben derIntegration kapazitiverSensoren in die Kapsel-füllmaschine [14] wur-den Mikrowellenreso-natoren [15, 16], Rönt-gensysteme [17] undoptische Verfahren [18]beschrieben.
Einen innovativen An-satz zur 100 % Kontrolledes Füllgewichts stellt
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 304–311 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)310 Steckel und Seyfang . Kleinstmengendosierung von Pulvern
Abb. 7: Dosiersystem nach dem Prinzip „Vibrierende Kapillare“.
Abb. 8: Funktionsweise des kapazitiven Sensors zur 100 % in-line Kontrolle der Dosiermenge.
Abb. 6: links: Schema Vakuum-Walzendosierer; rechts: Anwendungen und technische Daten zur Vakuumwalzendosierung.

wogen (300 g) und ohne vorherigesMischen in den Wirbelschichtappa-rat überführt. Der Ventilator wurdeeingeschaltet und das Wirbelbett er-zeugt. Nach 3 min wurde der Sprüh-vorgang gestartet. Die Zweistoffdüse(Durchmesser 1,0 mm, Position derDüsenkappe 2,5 Skalenteile, Düsen-Schlick, Untersiemau/Coburg, BRD)wurde im Topspraymodus betriebenund befand sich im Zentrum des Pro-zessbehälters. Wie bei den vorher-gehenden Untersuchungen wurden300 g Pulver vorgelegt und 400 g Gra-nulierlösung versprüht. An dieSprüh- und Agglomerationsphaseschloss sich die Trocknungsphaseunmittelbar an. Während des Prozes-ses wurden im Abstand von 4 minProben gezogen, um die Agglomerat-bildung zu bewerten und besondersgegen Ende der Trocknungsphase dieRestfeuchte zu messen. Bei einerRestfeuchte kleiner 5 % wurde dieTrocknung beendet.
3. Ergebnisse undDiskussion Wirbelschicht-agglomeration und-trocknung (WW)
Für die Zulufttemperaturen 50, 60und 70 °C und die Sprühraten 10 bis20 g/min verliefen die Wirbelschicht-prozesse stabil, lediglich der Zuluft-strom musste bei zunehmender Ag-glomeration geringfügig nachregu-liert werden. In keinem Fall kam eszum Zusammenbruch des Wirbel-bettes oder zu größeren Produkt-ablagerungen an der Behälterwandoder im Filter. Sprührate 22 g/minführte zu Überfeuchtung währenddes Prozesses unabhängig von derZulufttemperatur, bei Sprührate klei-ner 10 g/min erfolgte keine Agglome-ratbildung aufgrund eines zu gerin-gen Angebotes an Bindemittellösungpro Zeiteinheit. Bei Zulufttemperaturkleiner 50 °C war die Trocknungsratezu gering, das Produkt wurde schonbei niedrigen Sprühraten von 10 und12 g/min aufgrund einer zu großenMenge an Flüssigkeit pro Zeiteinheitüberfeuchtet, und das Wirbelbettbrach zusammen. Zulufttemperatu-
ren größer 70 °C sind ap-parativ möglich, aber dieProdukttemperaturkönnte während des Pro-zesses für temperatur-empfindliche Arznei-stoffe inakzeptableWerte erreichen. Bei denZulufttemperaturen 50-70 °C blieb die Produkt-temperatur unter 40 °C.Mit zunehmender Zu-lufttemperatur verrin-gerte sich die Gesamt-prozesszeit aufgrundder erhöhten Trock-nungsrate, aber dafürmusste mit einer gerin-geren Agglomerationund letztendlich mit ei-ner geringeren Korn-größe gerechnet werden.
Mit zunehmenderSprührate steigt bei Rezeptur 1(Abb. 1) die Korngröße an, die stär-kere Befeuchtung in kurzer Zeit lässterwartungsgemäß größere Agglome-rate entstehen, bei Rezeptur 2(Abb. 2) ist diese Tendenz jedochnicht zu beobachten, die d50-Werte
streuen im Bereich 250-400μm. Trotzder erhöhten Sprührate werden hieroffenbar keine größeren Granulat-körner gebildet, beim Trocknen ent-stehen starke Feststoffbrücken ausNatriumcitrat, und in der Endphaseder Trocknung tritt erhöhter Abriebauf.
Für beide Rezepturen ergeben sichbei der Erhöhung der Sprührate von10 auf 20 g/min keine Trends bezüg-lich Schütt- und Stampfdichte, Kom-pressibilitätsindex, Böschungswinkelund Fließzeit, jedoch streuen dieWerte (Tab. 2). Alle Granulate zeigenzumindest zufriedenstellendes Fließ-verhalten und fließen bei der Mes-sung der Fließzeit gleichmäßig ausdem Trichter aus. Die niedrigerenKompressibilitätsindizes bei Rezep-tur 2 resultieren aus der bereits dis-kutierten größeren Härte und Dichteder Granulatkörner und der damitverbundenen geringeren Verdich-tung bei der mechanischen Belas-tung im Stampfvolumeter.
Damit entsprechen die Eigen-schaften der Wirbelschichtgranulateder Rezepturen 1 und 2 für die Zu-lufttemperaturen 50-70 °C und dieSprühraten 10-20 g/min den Granu-laten aus Mischeragglomerationund Wirbelschichttrocknung [4],
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 313Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2
Tabelle 1
Prozessparameter Wirbelschicht-agglomeration und -trocknung
Ansatzmenge Pulver [g] 300
AnsatzmengeGranulierlösung [g]
400
KonzentrationGranulierlösung [%]
10
Zuluftstrom [m3/h] 40-60
Zulufttemperatur [ºC] 50, 60, 70
Stellung Düsenkappe[Skalenteile]
2,5
Sprühdruck [bar] 2
Sprührate [g/min] 10, 12, 14, 16,18, 20
Abb. 1: Mittlere Korngröße der Rezeptur 1 inAbhängigkeit von Zulufttemperatur undSprührate.
Abb. 2: Mittlere Korngröße der Rezeptur 2 inAbhängigkeit von Zulufttemperatur undSprührate.
Übertragung der Granulierungin zwei getrennten Schrittenauf einen Ein-Schritt-WirbelschichtprozessTeil 2: Wirbelschichtagglomeration und -trocknung in einem Schrittund Vergleich mit Zwei-Schritt-Prozessen
Katharina Germer und Prof. Dr. Bertram Wolf . Hochschule Anhalt, 06406 Bernburg
Korrespondenz: Prof. Dr. Bertram Wolf, Hochschule Anhalt, Fachbereich 7, Pharmatechnik, Neues Labor, Strenzfelder Allee 28,06406 Bernburg; e-mail: [email protected]
1. Einleitung
Die Zielstellung der vorliegendenStudie bestand im Nachweis, dassim Labormaßstab Granulate in derWirbelschicht im Ein-Schritt-Ver-fahren (Agglomeration und Trock-nung) hergestellt werden könnenohne signifikante Veränderung derQualität der Produkte im Vergleichzu Mischergranulaten [1–3]. In einerersten Studie wurden zwei unter-schiedliche Modellrezepturen imMischer agglomeriert, mit demFeuchtgranulierer granuliert undauf Horden an der Luft getrocknet,in einer zweiten Studie wurde dasFeuchtagglomerat genauso her-gestellt, aber in der Wirbelschichtgetrocknet [4], und in der drittenStudie wurden Feuchtagglomerationund Trocknung ohne Unterbre-chung des Prozesses in der Wirbel-schicht durchgeführt. In den dreiStudien wurden bestimmte Prozess-parameter verändert und deren Ein-
fluss auf die Granulateigenschaftenuntersucht, um die Robustheit derProzesse und den Arbeitsbereichzu ermitteln. Für die beiden Rezep-turen wurden für Granulate undTabletten typische Füllmittel einge-setzt, wobei eine Rezeptur überwie-gend unlöslich, die andere dagegeneinen hohen Anteil einer löslichenKomponente enthält. Damit sollteder Einfluss der Rezeptureigenschaf-ten auf den Granulierprozess unddie Granulateigenschaften unter-sucht werden. Die hier vorgestelltenUntersuchungen im Labormaßstabbilden die Grundlage für das eigent-liche Ziel, den Ein-Schritt-Prozessder Wirbelschichtagglomerationund -trocknung auf eine Tech-nikums-Wirbelschichtanlage zuüberführen, die Granulate sowohlim Batchprozess als auch in kon-tinuierlicher Verfahrensweise herzu-stellen und letztendlich die Wirbel-schichtgranulation auf einer kon-tinuierlichen Anlage zu realisieren.
2. Material und Methoden
Die Rezepturen wurden in der vor-hergehenden Publikation [4] be-schrieben, ebenso die Prüfmethodenfür die Granulateigenschaften [4, 5].Bei der Wirbelschichtagglomerationund -trocknung wurden die Prozess-parameter Zulufttemperatur undSprührate variiert, um die Robustheitund Reproduzierbarkeit des Prozes-ses zu untersuchen (Tab. 1). DerSprühdruck wurde in dieser Studiekonstant bei 2 bar gewählt, über Er-gebnisse aus Untersuchungen zumEinfluss des Sprühdruckes auf denGranulierprozess und die Granulat-eigenschaften wird in einer späterenPublikation berichtet.
Zur Herstellung der Granulatewurde der Batch-Labor-Wirbel-schichtgranulierer und -trockner(GPCG 1.1, Glatt GmbH, Binzen)ohne Material bei 60 °C Zulufttem-peratur 10 min vorgewärmt. Die Pul-verkomponenten wurden einge-
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)312 Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2

wogen (300 g) und ohne vorherigesMischen in den Wirbelschichtappa-rat überführt. Der Ventilator wurdeeingeschaltet und das Wirbelbett er-zeugt. Nach 3 min wurde der Sprüh-vorgang gestartet. Die Zweistoffdüse(Durchmesser 1,0 mm, Position derDüsenkappe 2,5 Skalenteile, Düsen-Schlick, Untersiemau/Coburg, BRD)wurde im Topspraymodus betriebenund befand sich im Zentrum des Pro-zessbehälters. Wie bei den vorher-gehenden Untersuchungen wurden300 g Pulver vorgelegt und 400 g Gra-nulierlösung versprüht. An dieSprüh- und Agglomerationsphaseschloss sich die Trocknungsphaseunmittelbar an. Während des Prozes-ses wurden im Abstand von 4 minProben gezogen, um die Agglomerat-bildung zu bewerten und besondersgegen Ende der Trocknungsphase dieRestfeuchte zu messen. Bei einerRestfeuchte kleiner 5 % wurde dieTrocknung beendet.
3. Ergebnisse undDiskussion Wirbelschicht-agglomeration und-trocknung (WW)
Für die Zulufttemperaturen 50, 60und 70 °C und die Sprühraten 10 bis20 g/min verliefen die Wirbelschicht-prozesse stabil, lediglich der Zuluft-strom musste bei zunehmender Ag-glomeration geringfügig nachregu-liert werden. In keinem Fall kam eszum Zusammenbruch des Wirbel-bettes oder zu größeren Produkt-ablagerungen an der Behälterwandoder im Filter. Sprührate 22 g/minführte zu Überfeuchtung währenddes Prozesses unabhängig von derZulufttemperatur, bei Sprührate klei-ner 10 g/min erfolgte keine Agglome-ratbildung aufgrund eines zu gerin-gen Angebotes an Bindemittellösungpro Zeiteinheit. Bei Zulufttemperaturkleiner 50 °C war die Trocknungsratezu gering, das Produkt wurde schonbei niedrigen Sprühraten von 10 und12 g/min aufgrund einer zu großenMenge an Flüssigkeit pro Zeiteinheitüberfeuchtet, und das Wirbelbettbrach zusammen. Zulufttemperatu-
ren größer 70 °C sind ap-parativ möglich, aber dieProdukttemperaturkönnte während des Pro-zesses für temperatur-empfindliche Arznei-stoffe inakzeptableWerte erreichen. Bei denZulufttemperaturen 50-70 °C blieb die Produkt-temperatur unter 40 °C.Mit zunehmender Zu-lufttemperatur verrin-gerte sich die Gesamt-prozesszeit aufgrundder erhöhten Trock-nungsrate, aber dafürmusste mit einer gerin-geren Agglomerationund letztendlich mit ei-ner geringeren Korn-größe gerechnet werden.
Mit zunehmenderSprührate steigt bei Rezeptur 1(Abb. 1) die Korngröße an, die stär-kere Befeuchtung in kurzer Zeit lässterwartungsgemäß größere Agglome-rate entstehen, bei Rezeptur 2(Abb. 2) ist diese Tendenz jedochnicht zu beobachten, die d50-Werte
streuen im Bereich 250-400μm. Trotzder erhöhten Sprührate werden hieroffenbar keine größeren Granulat-körner gebildet, beim Trocknen ent-stehen starke Feststoffbrücken ausNatriumcitrat, und in der Endphaseder Trocknung tritt erhöhter Abriebauf.
Für beide Rezepturen ergeben sichbei der Erhöhung der Sprührate von10 auf 20 g/min keine Trends bezüg-lich Schütt- und Stampfdichte, Kom-pressibilitätsindex, Böschungswinkelund Fließzeit, jedoch streuen dieWerte (Tab. 2). Alle Granulate zeigenzumindest zufriedenstellendes Fließ-verhalten und fließen bei der Mes-sung der Fließzeit gleichmäßig ausdem Trichter aus. Die niedrigerenKompressibilitätsindizes bei Rezep-tur 2 resultieren aus der bereits dis-kutierten größeren Härte und Dichteder Granulatkörner und der damitverbundenen geringeren Verdich-tung bei der mechanischen Belas-tung im Stampfvolumeter.
Damit entsprechen die Eigen-schaften der Wirbelschichtgranulateder Rezepturen 1 und 2 für die Zu-lufttemperaturen 50-70 °C und dieSprühraten 10-20 g/min den Granu-laten aus Mischeragglomerationund Wirbelschichttrocknung [4],
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 313Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2
Tabelle 1
Prozessparameter Wirbelschicht-agglomeration und -trocknung
Ansatzmenge Pulver [g] 300
AnsatzmengeGranulierlösung [g]
400
KonzentrationGranulierlösung [%]
10
Zuluftstrom [m3/h] 40-60
Zulufttemperatur [ºC] 50, 60, 70
Stellung Düsenkappe[Skalenteile]
2,5
Sprühdruck [bar] 2
Sprührate [g/min] 10, 12, 14, 16,18, 20
Abb. 1: Mittlere Korngröße der Rezeptur 1 inAbhängigkeit von Zulufttemperatur undSprührate.
Abb. 2: Mittlere Korngröße der Rezeptur 2 inAbhängigkeit von Zulufttemperatur undSprührate.
Übertragung der Granulierungin zwei getrennten Schrittenauf einen Ein-Schritt-WirbelschichtprozessTeil 2: Wirbelschichtagglomeration und -trocknung in einem Schrittund Vergleich mit Zwei-Schritt-Prozessen
Katharina Germer und Prof. Dr. Bertram Wolf . Hochschule Anhalt, 06406 Bernburg
Korrespondenz: Prof. Dr. Bertram Wolf, Hochschule Anhalt, Fachbereich 7, Pharmatechnik, Neues Labor, Strenzfelder Allee 28,06406 Bernburg; e-mail: [email protected]
1. Einleitung
Die Zielstellung der vorliegendenStudie bestand im Nachweis, dassim Labormaßstab Granulate in derWirbelschicht im Ein-Schritt-Ver-fahren (Agglomeration und Trock-nung) hergestellt werden könnenohne signifikante Veränderung derQualität der Produkte im Vergleichzu Mischergranulaten [1–3]. In einerersten Studie wurden zwei unter-schiedliche Modellrezepturen imMischer agglomeriert, mit demFeuchtgranulierer granuliert undauf Horden an der Luft getrocknet,in einer zweiten Studie wurde dasFeuchtagglomerat genauso her-gestellt, aber in der Wirbelschichtgetrocknet [4], und in der drittenStudie wurden Feuchtagglomerationund Trocknung ohne Unterbre-chung des Prozesses in der Wirbel-schicht durchgeführt. In den dreiStudien wurden bestimmte Prozess-parameter verändert und deren Ein-
fluss auf die Granulateigenschaftenuntersucht, um die Robustheit derProzesse und den Arbeitsbereichzu ermitteln. Für die beiden Rezep-turen wurden für Granulate undTabletten typische Füllmittel einge-setzt, wobei eine Rezeptur überwie-gend unlöslich, die andere dagegeneinen hohen Anteil einer löslichenKomponente enthält. Damit sollteder Einfluss der Rezeptureigenschaf-ten auf den Granulierprozess unddie Granulateigenschaften unter-sucht werden. Die hier vorgestelltenUntersuchungen im Labormaßstabbilden die Grundlage für das eigent-liche Ziel, den Ein-Schritt-Prozessder Wirbelschichtagglomerationund -trocknung auf eine Tech-nikums-Wirbelschichtanlage zuüberführen, die Granulate sowohlim Batchprozess als auch in kon-tinuierlicher Verfahrensweise herzu-stellen und letztendlich die Wirbel-schichtgranulation auf einer kon-tinuierlichen Anlage zu realisieren.
2. Material und Methoden
Die Rezepturen wurden in der vor-hergehenden Publikation [4] be-schrieben, ebenso die Prüfmethodenfür die Granulateigenschaften [4, 5].Bei der Wirbelschichtagglomerationund -trocknung wurden die Prozess-parameter Zulufttemperatur undSprührate variiert, um die Robustheitund Reproduzierbarkeit des Prozes-ses zu untersuchen (Tab. 1). DerSprühdruck wurde in dieser Studiekonstant bei 2 bar gewählt, über Er-gebnisse aus Untersuchungen zumEinfluss des Sprühdruckes auf denGranulierprozess und die Granulat-eigenschaften wird in einer späterenPublikation berichtet.
Zur Herstellung der Granulatewurde der Batch-Labor-Wirbel-schichtgranulierer und -trockner(GPCG 1.1, Glatt GmbH, Binzen)ohne Material bei 60 °C Zulufttem-peratur 10 min vorgewärmt. Die Pul-verkomponenten wurden einge-
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)312 Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2

Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-11\Anzeigensatz-keine-Druck-PDFs\bohle-pi-2013-11-216x303.indd Zuletzt gesichert: 08.11.13 (08:52:02 Uhr)
DIE TECHNOLOGISCHE ZUKUNFT HAT BEGONNEN:
Bohle Uni Cone BUC® produktiver, praktischer, präziser
Ihre Vorteile:
• Hohe Ausbeute und Vermeidung von Zwillingsbildung beim Coaten
• Trocknen, Granulieren und Coaten ohne Umbau im selben Gerät
• Bequeme Bedienung durch seitlich angebrachte Düsen
• Optimierte Prozessführung mittels innovativer PAT-Methoden (Partikelgröße und Feuchte)www.lbbohle.de
Herkömmliches Wurster-Verfahren
Wegweisende Bohle Uni Cone BUC®-Technologie
Die Alternative
zum traditionellen
Wurster-Verfahren
und die Wirbelschichtagglomera-tion und -trocknung als Ein-Schritt-Prozess erweist sich als ge-eignete Methode zur Granulather-stellung im Labormaßstab.
4. Vergleich der dreiMethoden
Mischergranulierung / Luft-trocknung, Mischergranu-lierung / Wirbelschicht-trocknung und Wirbelschicht-agglomeration / -trocknungDie drei unterschiedlichenMethodenzur Granulatherstellung Mischer-agglomeration / Lufttrocknung (ML),Mischeragglomeration / Wirbel-schichttrocknung (MW) und Wirbel-schichtagglomeration und -trock-nung (WW) führten in allen Fällenzu akzeptablen Produkten, die Eigen-schaften der Granulate liegen in Be-reichen, die sie zur Verpressung zuTabletten empfehlen.
Bei Rezeptur 1 streuen die Werteder mittleren Korngröße d50 im Be-reich 270-480μm (Abb. 3), und eineAbhängigkeit von der Herstellungs-methode ist nicht erkennbar. DieSchüttdichte liegt bei der Mischer-agglomeration (Tab. 3, Methoden MLund MW: 0,40-0,57 g/ml) deutlich hö-
her als bei der Wirbelschichtagglome-ration (Chargen WW: 0,18-0,27 g/ml),weil die Agglomerate im feuchten Zu-stand wesentlich stärker verdichtetwerden. Das Verdichtungsverhaltender trockenen Granulate bei der Be-handlung im Stampfvolumeter ist da-gegen wieder ähnlich: die aus Schütt-und Stampfdichte berechneten Kom-pressibilitätsindizes liegen unabhän-gig von der Herstellungsmethode bei10-16 %, was geringer Verdichtungund guten Fließeigenschaften ent-spricht. Der Böschungswinkel für Mi-schergranulierung / Wirbelschicht-trocknung (MW) ist zwar niedrig (16°)und zeigt gute Fließeigenschaften an,jedoch erfolgte kein freiwilliges Flie-ßen aufgrund elektrostatischer Auf-ladung (Fließzeit nicht messbar). DieGranulate aus der Mischergranulie-rung / Lufttrocknung (ML) fließengut, was auch mit dem Böschungs-winkel 27° belegt wird. Die Wirbel-schichtgranulate (WW) weisen dage-
gen weniger gute Fließeigenschaftenbezüglich Böschungswinkel (34°) undFließzeit (18-23 s) auf, was auf elek-trostatische Aufladung der Produktebei der Wirbelschichttrocknung zu-rückgeführt werden kann, jedoch ent-sprechen die geringe Verdichtung undein Kompressibilitätsindex von 10 %sehr gutem Fließen [5].
Die mittlere Korngröße der Char-gen Rezeptur 2 streut zwischen 260und 515μm, wie bei Rezeptur 1 istkeine Abhängigkeit von der Herstel-lungsmethode erkennbar (Abb. 4).Der Trend bei der Schüttdichte(Tab. 4) entspricht der Rezeptur 1:niedrige Werte bei Wirbelschichtgra-nulaten (WW), deutlich höhereWerte bei Mischeragglomeraten(ML und MW). Die Verdichtung dertrockenen Mischeragglomerate istinfolge der hohen Dichte und derstarken Feststoffbrücken (auskristal-lisiertes Natriumcitrat) bei Stampfensehr gering, demzufolge sind auchdie Kompressibilitätsindizes niedrig(5-10 %) und damit die Fließeigen-schaften exzellent. Die lockeren Wir-belschichtgranulate (WW) werdendagegen im Stampfvolumeter starkverdichtet und weisen Kompressibi-litätsindizes von 24 % und damit we-niger gute Fließeigenschaften auf.Die Werte für Böschungswinkel undFließzeit belegen diesen Trend: ver-gleichsweise hohe Werte für Wirbel-schichtgranulate WW (37°, 12-18 s)und niedrigere für MischergranulateML und MW (20-26°, 5-8 s). Die Gra-nulate Rezeptur 2 erfüllen die Forde-rungen nach Fließzeiten unter 25 s,Böschungswinkeln unter 40° und ge-ringer oder mittlerer Verdichtung dertrockenen Produkte bei der Unter-suchung mit dem Stampfvolumeter
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)314 Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2
Tabelle 2
Eigenschaften der Granulatrezepturen 1 und 2und Bewertung der Fließeigenschaften
Rezeptur 1 Rezeptur 2
Bereich Fließeigen-schaft
Bereich Fließeigen-schaft
d50 [m] 300-500 250-400
Schüttdichte [g/ml] 0,21-0,27 0,21-0,33
KI [%] 22-24 mäßig 7-13 ausgezeich-net bis gut
Böschungswinkel[Grad]
33-39 gut biszufrieden-stellend
32-36 gut biszufrieden-stellend
Fließzeit (100 g) [s] 10-15 zufrieden-stellend
7-20 gut bis mäßig
Abb. 3: Vergleich der mittleren KorngrößeRezeptur 1.
Abb. 4: Vergleich der mittlere KorngrößenRezeptur 2.

Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-11\Anzeigensatz-keine-Druck-PDFs\bohle-pi-2013-11-216x303.indd Zuletzt gesichert: 08.11.13 (08:52:02 Uhr)
DIE TECHNOLOGISCHE ZUKUNFT HAT BEGONNEN:
Bohle Uni Cone BUC® produktiver, praktischer, präziser
Ihre Vorteile:
• Hohe Ausbeute und Vermeidung von Zwillingsbildung beim Coaten
• Trocknen, Granulieren und Coaten ohne Umbau im selben Gerät
• Bequeme Bedienung durch seitlich angebrachte Düsen
• Optimierte Prozessführung mittels innovativer PAT-Methoden (Partikelgröße und Feuchte)www.lbbohle.de
Herkömmliches Wurster-Verfahren
Wegweisende Bohle Uni Cone BUC®-Technologie
Die Alternative
zum traditionellen
Wurster-Verfahren
und die Wirbelschichtagglomera-tion und -trocknung als Ein-Schritt-Prozess erweist sich als ge-eignete Methode zur Granulather-stellung im Labormaßstab.
4. Vergleich der dreiMethoden
Mischergranulierung / Luft-trocknung, Mischergranu-lierung / Wirbelschicht-trocknung und Wirbelschicht-agglomeration / -trocknungDie drei unterschiedlichenMethodenzur Granulatherstellung Mischer-agglomeration / Lufttrocknung (ML),Mischeragglomeration / Wirbel-schichttrocknung (MW) und Wirbel-schichtagglomeration und -trock-nung (WW) führten in allen Fällenzu akzeptablen Produkten, die Eigen-schaften der Granulate liegen in Be-reichen, die sie zur Verpressung zuTabletten empfehlen.
Bei Rezeptur 1 streuen die Werteder mittleren Korngröße d50 im Be-reich 270-480μm (Abb. 3), und eineAbhängigkeit von der Herstellungs-methode ist nicht erkennbar. DieSchüttdichte liegt bei der Mischer-agglomeration (Tab. 3, Methoden MLund MW: 0,40-0,57 g/ml) deutlich hö-
her als bei der Wirbelschichtagglome-ration (Chargen WW: 0,18-0,27 g/ml),weil die Agglomerate im feuchten Zu-stand wesentlich stärker verdichtetwerden. Das Verdichtungsverhaltender trockenen Granulate bei der Be-handlung im Stampfvolumeter ist da-gegen wieder ähnlich: die aus Schütt-und Stampfdichte berechneten Kom-pressibilitätsindizes liegen unabhän-gig von der Herstellungsmethode bei10-16 %, was geringer Verdichtungund guten Fließeigenschaften ent-spricht. Der Böschungswinkel für Mi-schergranulierung / Wirbelschicht-trocknung (MW) ist zwar niedrig (16°)und zeigt gute Fließeigenschaften an,jedoch erfolgte kein freiwilliges Flie-ßen aufgrund elektrostatischer Auf-ladung (Fließzeit nicht messbar). DieGranulate aus der Mischergranulie-rung / Lufttrocknung (ML) fließengut, was auch mit dem Böschungs-winkel 27° belegt wird. Die Wirbel-schichtgranulate (WW) weisen dage-
gen weniger gute Fließeigenschaftenbezüglich Böschungswinkel (34°) undFließzeit (18-23 s) auf, was auf elek-trostatische Aufladung der Produktebei der Wirbelschichttrocknung zu-rückgeführt werden kann, jedoch ent-sprechen die geringe Verdichtung undein Kompressibilitätsindex von 10 %sehr gutem Fließen [5].
Die mittlere Korngröße der Char-gen Rezeptur 2 streut zwischen 260und 515μm, wie bei Rezeptur 1 istkeine Abhängigkeit von der Herstel-lungsmethode erkennbar (Abb. 4).Der Trend bei der Schüttdichte(Tab. 4) entspricht der Rezeptur 1:niedrige Werte bei Wirbelschichtgra-nulaten (WW), deutlich höhereWerte bei Mischeragglomeraten(ML und MW). Die Verdichtung dertrockenen Mischeragglomerate istinfolge der hohen Dichte und derstarken Feststoffbrücken (auskristal-lisiertes Natriumcitrat) bei Stampfensehr gering, demzufolge sind auchdie Kompressibilitätsindizes niedrig(5-10 %) und damit die Fließeigen-schaften exzellent. Die lockeren Wir-belschichtgranulate (WW) werdendagegen im Stampfvolumeter starkverdichtet und weisen Kompressibi-litätsindizes von 24 % und damit we-niger gute Fließeigenschaften auf.Die Werte für Böschungswinkel undFließzeit belegen diesen Trend: ver-gleichsweise hohe Werte für Wirbel-schichtgranulate WW (37°, 12-18 s)und niedrigere für MischergranulateML und MW (20-26°, 5-8 s). Die Gra-nulate Rezeptur 2 erfüllen die Forde-rungen nach Fließzeiten unter 25 s,Böschungswinkeln unter 40° und ge-ringer oder mittlerer Verdichtung dertrockenen Produkte bei der Unter-suchung mit dem Stampfvolumeter
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)314 Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2
Tabelle 2
Eigenschaften der Granulatrezepturen 1 und 2und Bewertung der Fließeigenschaften
Rezeptur 1 Rezeptur 2
Bereich Fließeigen-schaft
Bereich Fließeigen-schaft
d50 [m] 300-500 250-400
Schüttdichte [g/ml] 0,21-0,27 0,21-0,33
KI [%] 22-24 mäßig 7-13 ausgezeich-net bis gut
Böschungswinkel[Grad]
33-39 gut biszufrieden-stellend
32-36 gut biszufrieden-stellend
Fließzeit (100 g) [s] 10-15 zufrieden-stellend
7-20 gut bis mäßig
Abb. 3: Vergleich der mittleren KorngrößeRezeptur 1.
Abb. 4: Vergleich der mittlere KorngrößenRezeptur 2.
Mikrowellenresonanz-SensorenInnovation für die Feuchtemessung in Wirbelschichtgeräten
Benjamin Kollar, Jörg Breitkreutz . Institut für Pharmazeutische Technologie und Biopharmazie, Heinrich-Heine-Universität, DüsseldorfWolfgang Wiedey, Kathrin Bartscher . NextPharma, Pharbil Waltrop GmbH, WaltropClaas Döscher . AMS Advanced Microwave Systems GmbH, Hamburg
Korrespondenz: Benjamin Kollar, Institut für Pharmazeutische Technologie und Biopharmazie, Heinrich-Heine-Universität,Universitätsstr. 1, 40225 Düsseldorf, e-mail: [email protected]
ZusammenfassungMikrowellenresonanz-Sensoren ermöglichen eine Messung der Produktfeuchte in Wirbel-schichtgeräten. Die bisher verfügbaren Sensoren haben jedoch nur einen eingeschränktenAnwendungsbereich bis etwa 10 % relativer Feuchte im festen Produkt. Erstmals wirdhier ein neuartiger Sensortyp vorgestellt, der durch die Messung bei zwei Frequenzenauch in höheren Feuchtebereichen verlässliche Messwerte liefert. Anhand der beispiel-haften Auswertung einer Messreihe werden die Vorteile der 2-Frequenz-Messunggegenüber den bisherigen 1-Frequenz-Sensoren dargestellt. Final wird das Potential desSensors bei der Anwendung in einem Wirbelschichtgerät der Firma Glatt verdeutlicht.
Einleitung
Bei der Entwicklung und Herstel-lung von festen pharmazeutischenArzneiformen ist der Wassergehaltein wichtiges Qualitätsmerkmal.Von den Ausgangsstoffen über dieZwischenprodukte bis zu den fer-tigen Arzneimitteln können durchden Wassergehalt sowohl die physi-kalischen als auch die chemischenEigenschaften beeinflusst werden.Zu den physikalischen Eigenschaf-ten, die sich in Abhängigkeit vomWassergehalt ändern, gehören zumBeispiel die Fließfähigkeit und Ver-pressbarkeit von Pulvern und Gra-nulaten [1]. Die Stabilität der Wirk-stoffe und die Reaktivität von Wirk-und Hilfsstoffen zählen zu den vomWassergehalt geprägten che-mischen Eigenschaften [2]. Somitist die Bestimmung des Wasser-gehalts eine grundlegende Prüf-und Kontroll-Operation im Verlaufdes pharmazeutischen Herstel-lungsprozesses.
Je nach Anforderungen und Ein-satzgebieten sind dafür verschiedeneMethoden verfügbar, die sich in ih-rem Messprinzip und den damit ver-bundenen Vor- und Nachteilen deut-lich unterscheiden.
Wird eine Probe zur Bestimmungdem Prozessfluss entzogen (Off-line-Messung), wird für die Feuchte-bestimmung meist der Trocknungs-verlust mittels thermogravimetri-scher Methode (bevorzugt automati-siert mit Hilfe von Trocknungswaa-gen) und die Titration nach demKarl-Fischer-Verfahren angewandt.
Diese Feuchtemessmethoden kön-nen nicht direkt in Wirbelschicht-Pro-zessen (in-line-Messung) angewandtwerden. Die Nah-Infrarot Spektrosko-pie ist das derzeit häufigste ange-wandte Verfahren zur in-line Feuchte-bestimmung in der Wirbelschicht. Alsnachteilhaft erwiesen sich dabei dieDichteabhängigkeit des Messsignals,oberflächliche Messung aufgrundvon geringer Eindringtiefe und Pro-bleme durch Verschmutzung des
Messfensters. Burggraeve et al. gibtin dem aktuellen Review [3] einenbreiten Überblick über die verfügbareTechnik. Die Mikrowellenresonanz-Technologie konnte sich in den letz-ten Jahren gerade für diesen Anwen-dungsbereich als vielversprechend er-weisen [4, 5].
Mittelfristiges Ziel ist es, Mikro-wellenresonanz-Sensoren entspre-chend der Vorgaben der FDA Guide-line Process Analytical Technologies(PAT) einzusetzen [6]. Durch das da-mit gewonnene Prozessverständnisund die kontinuierliche Produktkon-trolle könnten zukünftige Ansprücheeines Quality-by-Design Ansatzes er-füllt werden [7].
Messprinzip der Mikro-wellenresonanz-Sensoren
Die starke Wechselwirkung der DipolevonWassermolekülen mit demMikro-wellenfeld bildet die Grundlage für dieFeuchtemessung mittels Mikrowellen-resonanz-Technologie. Mikrowellen-
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 317Kollar et al. . Mikrowellenresonanz-Sensoren
mit Kompressibilitätsindizes unter25 %.
5. Zusammenfassung
Im Labormaßstab wurde gezeigt,dass zwei unterschiedliche Granu-latformulierungen im Wirbel-schichtprozess in einem Schritt (Ag-glomeration und Trocknung) her-gestellt werden können. Die Granu-lateigenschaften unterscheiden sichzwar von den Granulaten aus derMischeragglomeration, insbeson-dere ist die Schüttdichte deutlich ge-ringer, was zu Verminderung derFließeigenschaften führt, jedoch lie-gen die Prüfparameter Verdichtung,Kompressibilitätsindex, Böschungs-winkel und Fließzeit in akzeptablenGrenzen und erfüllen die konventio-nell aufgestellten Forderungen unddie der Bewertungsskala im Arznei-
buch. Bei einer Umstellung vomGra-nulationsprozess in zwei Schritten(Mischeragglomeration und nach-folgende separate Trocknung) aufeine Wirbelschichtagglomerationund -trocknung im Produktions-maßstab müssen auf jeden Fall diespezifischen Eigenschaften der je-weiligen Rezeptur wie Löslichkeitder Komponenten, Quellung und Fä-higkeit zurWasserretention, lipophi-le-hydrophile Eigenschaften, Korn-größe und Korngrößenverteilungberücksichtigt werden, und eineEvaluierung des Prozesses im Labor-und Technikumsmaßstab vor derEinführung in die Produktion istempfehlenswert.
6. Danksagung
Für die Durchführung der experi-mentellen Untersuchungen im Rah-
men der Bachelorarbeit wird FrauMaike Retta gedankt.
Fachliteratur[1] Lieberman, H.A., Lachman, L. and
Schwartz, J.B. (editors): PharmaceuticalDosage Forms: Tablets, Volume 2. MarcelDekker New York and Basel, 1990
[2] Gao, J.Z.H., Jain, A., Motheram, R., Gray, D.B., Hussain, M.A.: Fluid bed granulation ofa poorly water soluble, low density, mi-cronized drug: comparison with highshear granulation. Int.J.Pharm. 2002, 237(1-2), 1-14
[3] Serno, P., Kleinebudde, P., Knop, K.: Gra-nulieren. Grundlagen, Verfahren, Formu-lierungen. Editio Cantor Verlag, Aulen-dorf, 2007
[4] Germer, K, Wolf, B.: Übertragung derGranulierung in zwei getrennten Schrit-ten auf einen Ein-Schritt-Wirbelschicht-prozess. Teil 1: Mischergranulierung /Lufttrocknung und Mischergranulierung/ Wirbelschichttrocknung. TechnoPharm,in Vorbereitung
[5] Europäisches Arzneibuch, 7. Grundaus-gabe, 2012
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)316 Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2
Tabelle 3
Vergleich der Granulatparameter Rezeptur 1, k.F. kein freiwilliges Fließen.
Rezeptur 1 ML30 ML50 ML120 MW50 MW60 MW70 WW50 WW60 WW70
Schüttdichte[g/ml]
0,39 0,57 0,4 0,45 0,45 0,44 0,27 0,23 0,18
KI [%] 13 10 16 12 14 13 10 12 10
Böschungs-winkel [º]
27 26 27 17 16 16 33 33 37
Fließzeit(100 g) [s]
10 5 8 k.F. k.F. k.F. 23 18 20
Tabelle 4
Vergleich der Granulateigenschaften Rezeptur 2.
Rezeptur 2 ML30 ML50 ML120 MW50 MW60 MW70 WW50 WW60 WW70
Schüttdichte[g/ml]
0,47 0,41 0,48 0,59 0,59 0,57 0,22 0,19 0,19
KI [%] 10 9 9 5 9 9 23 24 24
Böschungs-winkel [º]
27 26 26 29 28 20 38 36 37
Fließzeit(100 g) [s]
7 8 7 7 6 5 12 18 13

Mikrowellenresonanz-SensorenInnovation für die Feuchtemessung in Wirbelschichtgeräten
Benjamin Kollar, Jörg Breitkreutz . Institut für Pharmazeutische Technologie und Biopharmazie, Heinrich-Heine-Universität, DüsseldorfWolfgang Wiedey, Kathrin Bartscher . NextPharma, Pharbil Waltrop GmbH, WaltropClaas Döscher . AMS Advanced Microwave Systems GmbH, Hamburg
Korrespondenz: Benjamin Kollar, Institut für Pharmazeutische Technologie und Biopharmazie, Heinrich-Heine-Universität,Universitätsstr. 1, 40225 Düsseldorf, e-mail: [email protected]
ZusammenfassungMikrowellenresonanz-Sensoren ermöglichen eine Messung der Produktfeuchte in Wirbel-schichtgeräten. Die bisher verfügbaren Sensoren haben jedoch nur einen eingeschränktenAnwendungsbereich bis etwa 10 % relativer Feuchte im festen Produkt. Erstmals wirdhier ein neuartiger Sensortyp vorgestellt, der durch die Messung bei zwei Frequenzenauch in höheren Feuchtebereichen verlässliche Messwerte liefert. Anhand der beispiel-haften Auswertung einer Messreihe werden die Vorteile der 2-Frequenz-Messunggegenüber den bisherigen 1-Frequenz-Sensoren dargestellt. Final wird das Potential desSensors bei der Anwendung in einem Wirbelschichtgerät der Firma Glatt verdeutlicht.
Einleitung
Bei der Entwicklung und Herstel-lung von festen pharmazeutischenArzneiformen ist der Wassergehaltein wichtiges Qualitätsmerkmal.Von den Ausgangsstoffen über dieZwischenprodukte bis zu den fer-tigen Arzneimitteln können durchden Wassergehalt sowohl die physi-kalischen als auch die chemischenEigenschaften beeinflusst werden.Zu den physikalischen Eigenschaf-ten, die sich in Abhängigkeit vomWassergehalt ändern, gehören zumBeispiel die Fließfähigkeit und Ver-pressbarkeit von Pulvern und Gra-nulaten [1]. Die Stabilität der Wirk-stoffe und die Reaktivität von Wirk-und Hilfsstoffen zählen zu den vomWassergehalt geprägten che-mischen Eigenschaften [2]. Somitist die Bestimmung des Wasser-gehalts eine grundlegende Prüf-und Kontroll-Operation im Verlaufdes pharmazeutischen Herstel-lungsprozesses.
Je nach Anforderungen und Ein-satzgebieten sind dafür verschiedeneMethoden verfügbar, die sich in ih-rem Messprinzip und den damit ver-bundenen Vor- und Nachteilen deut-lich unterscheiden.
Wird eine Probe zur Bestimmungdem Prozessfluss entzogen (Off-line-Messung), wird für die Feuchte-bestimmung meist der Trocknungs-verlust mittels thermogravimetri-scher Methode (bevorzugt automati-siert mit Hilfe von Trocknungswaa-gen) und die Titration nach demKarl-Fischer-Verfahren angewandt.
Diese Feuchtemessmethoden kön-nen nicht direkt in Wirbelschicht-Pro-zessen (in-line-Messung) angewandtwerden. Die Nah-Infrarot Spektrosko-pie ist das derzeit häufigste ange-wandte Verfahren zur in-line Feuchte-bestimmung in der Wirbelschicht. Alsnachteilhaft erwiesen sich dabei dieDichteabhängigkeit des Messsignals,oberflächliche Messung aufgrundvon geringer Eindringtiefe und Pro-bleme durch Verschmutzung des
Messfensters. Burggraeve et al. gibtin dem aktuellen Review [3] einenbreiten Überblick über die verfügbareTechnik. Die Mikrowellenresonanz-Technologie konnte sich in den letz-ten Jahren gerade für diesen Anwen-dungsbereich als vielversprechend er-weisen [4, 5].
Mittelfristiges Ziel ist es, Mikro-wellenresonanz-Sensoren entspre-chend der Vorgaben der FDA Guide-line Process Analytical Technologies(PAT) einzusetzen [6]. Durch das da-mit gewonnene Prozessverständnisund die kontinuierliche Produktkon-trolle könnten zukünftige Ansprücheeines Quality-by-Design Ansatzes er-füllt werden [7].
Messprinzip der Mikro-wellenresonanz-Sensoren
Die starke Wechselwirkung der DipolevonWassermolekülen mit demMikro-wellenfeld bildet die Grundlage für dieFeuchtemessung mittels Mikrowellen-resonanz-Technologie. Mikrowellen-
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 317Kollar et al. . Mikrowellenresonanz-Sensoren
mit Kompressibilitätsindizes unter25 %.
5. Zusammenfassung
Im Labormaßstab wurde gezeigt,dass zwei unterschiedliche Granu-latformulierungen im Wirbel-schichtprozess in einem Schritt (Ag-glomeration und Trocknung) her-gestellt werden können. Die Granu-lateigenschaften unterscheiden sichzwar von den Granulaten aus derMischeragglomeration, insbeson-dere ist die Schüttdichte deutlich ge-ringer, was zu Verminderung derFließeigenschaften führt, jedoch lie-gen die Prüfparameter Verdichtung,Kompressibilitätsindex, Böschungs-winkel und Fließzeit in akzeptablenGrenzen und erfüllen die konventio-nell aufgestellten Forderungen unddie der Bewertungsskala im Arznei-
buch. Bei einer Umstellung vomGra-nulationsprozess in zwei Schritten(Mischeragglomeration und nach-folgende separate Trocknung) aufeine Wirbelschichtagglomerationund -trocknung im Produktions-maßstab müssen auf jeden Fall diespezifischen Eigenschaften der je-weiligen Rezeptur wie Löslichkeitder Komponenten, Quellung und Fä-higkeit zurWasserretention, lipophi-le-hydrophile Eigenschaften, Korn-größe und Korngrößenverteilungberücksichtigt werden, und eineEvaluierung des Prozesses im Labor-und Technikumsmaßstab vor derEinführung in die Produktion istempfehlenswert.
6. Danksagung
Für die Durchführung der experi-mentellen Untersuchungen im Rah-
men der Bachelorarbeit wird FrauMaike Retta gedankt.
Fachliteratur[1] Lieberman, H.A., Lachman, L. and
Schwartz, J.B. (editors): PharmaceuticalDosage Forms: Tablets, Volume 2. MarcelDekker New York and Basel, 1990
[2] Gao, J.Z.H., Jain, A., Motheram, R., Gray, D.B., Hussain, M.A.: Fluid bed granulation ofa poorly water soluble, low density, mi-cronized drug: comparison with highshear granulation. Int.J.Pharm. 2002, 237(1-2), 1-14
[3] Serno, P., Kleinebudde, P., Knop, K.: Gra-nulieren. Grundlagen, Verfahren, Formu-lierungen. Editio Cantor Verlag, Aulen-dorf, 2007
[4] Germer, K, Wolf, B.: Übertragung derGranulierung in zwei getrennten Schrit-ten auf einen Ein-Schritt-Wirbelschicht-prozess. Teil 1: Mischergranulierung /Lufttrocknung und Mischergranulierung/ Wirbelschichttrocknung. TechnoPharm,in Vorbereitung
[5] Europäisches Arzneibuch, 7. Grundaus-gabe, 2012
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 312–316 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)316 Germer und Wolf . Ein-Schritt-Wirbelschichtprozess Teil 2
Tabelle 3
Vergleich der Granulatparameter Rezeptur 1, k.F. kein freiwilliges Fließen.
Rezeptur 1 ML30 ML50 ML120 MW50 MW60 MW70 WW50 WW60 WW70
Schüttdichte[g/ml]
0,39 0,57 0,4 0,45 0,45 0,44 0,27 0,23 0,18
KI [%] 13 10 16 12 14 13 10 12 10
Böschungs-winkel [º]
27 26 27 17 16 16 33 33 37
Fließzeit(100 g) [s]
10 5 8 k.F. k.F. k.F. 23 18 20
Tabelle 4
Vergleich der Granulateigenschaften Rezeptur 2.
Rezeptur 2 ML30 ML50 ML120 MW50 MW60 MW70 WW50 WW60 WW70
Schüttdichte[g/ml]
0,47 0,41 0,48 0,59 0,59 0,57 0,22 0,19 0,19
KI [%] 10 9 9 5 9 9 23 24 24
Böschungs-winkel [º]
27 26 26 29 28 20 38 36 37
Fließzeit(100 g) [s]
7 8 7 7 6 5 12 18 13

Für die Charakterisierung des Pro-totyp-Sensors wurden anfangs statio-näre Messungen mit ubiquitärenpharmazeutischen Hilfsstoffen zurGranulierung durchgeführt. Exempla-risch sind in Abb. 4 die Messwerte desPrototyp-Sensors gegenüber der Refe-renzmessung (Karl-Fischer-Titration,Moisture Meter CA-100 /VA-100 Mit-subishi Chemical Corporation, Tokyo,Japan) für natürliche Maisstärke (TypCPharmGel®, Cargill, Krefeld) auf-getragen. Zusätzlich wurde der Trock-nungsverlust (LOD/IR) mit einer auto-matischen Trocknungswaage (MA-45,Sartorius AG, Göttingen) bestimmt.Um Proben mit unterschiedlichenWassergehalten zu erlangen, wurdendie Substanzen bei definierten Tem-peratur-/Feuchtebedingungen kon-ditioniert bzw. direkt durch Wasser-zugabe befeuchtet.
Jede so vorbehandelte Probewurde mit dem jeweiligen Messver-fahren dreimal vermessen. Aus dendrei Messungen wurde der arithme-tische Mittelwert errechnet und dieStandardabweichung bestimmt.
Die Messwerte beiderFrequenzen zeigen an-fangs den gleichen Ver-lauf. Bei der niedrigenMessfrequenz zeigt sichdann ab 15 % Feuchtedie von Buschmüller etal. beschriebene Proble-matik. Der Mikrowellen-wert kann keinemFeuchtewert mehr ein-deutig zugeordnet wer-den. DieWerte der hohenFrequenz lassen jedochbis zu > 30 % Feuchte ei-nen linearen Anstieg er-kennen.
Eine Erklärung fürdieses Verhalten bei hö-heren Feuchten kann dieVeränderung der dielek-trischen Eigenschaftensein. Abhängig von demrelativen Wassergehaltist das Wasser uneinheit-lich stark an die Stärkegebunden, wodurch sichdie dielektrischen Eigen-
schaften ändern. Bandbreiten-Ver-breiterung und Resonanzfrequenz-verschiebung lassen somit nicht di-rekt auf die Produktfeuchte schlie-ßen [11]. Ein weiterer Vorteil derZwei-Frequenz-Messung ist, dasssich durch die Erfassung von denzwei unabhängigen Messwertreihendie Möglichkeiten der Auswertungerheblich erweitern.
Unterteilt man den Messverlauf inzwei Phasen mit dem Schwellenwertvon 15 % Feuchte, lässt sich der Be-reich < 15 % sehr gut durch eine Li-
nearkombination der beiden Mikro-wellenwerte entsprechend der Glei-chung erfassen (Abb. 5A).
Für den Bereich oberhalb von15 % relativer Feuchte ist kein linea-rer Zusammenhang vorhanden. Je-doch kann durch Bildung des Quo-tienten aus den beiden Mikrowellen-werten ein Feuchtewert zuverlässigprognostiziert werden (Abb. 5B).Diese Verfahren wurde von Knöchelet al. 2010 erstmals beschrieben [11].
Neben Maisstärke wurden ebensoProben aus Kartoffelstärke Ph.Eur.,(Roquette, Frankfurt) vermessen. Ta-belle 1 gibt einen Überblick der er-langten Daten in Form des Korrelati-onskoeffizients nach Pearson. Wie zu-vor beschrieben wurde jeweils der Be-reich bis 15 % und der Bereich ober-halb 15 % relativer Feuchte getrenntbetrachtet. Dabei wird der Vorteil derzweiphasigen Berechnungsmethodedeutlich. Im Bereich bis 15 % relativeFeuchte ist die Linearkombinationmit Korrelationskoeffizients größer/gleich 0,99 optimal. Die Berechnungdurch das Quotientenverfahren zeigtsich dort mit Werten von 0,8752 fürMaisstärke und 0,9074 bei Kartoffel-stärke weit unterlegen. Im hohenFeuchtebereich oberhalb 15 % relati-ver Feuchte verhalten sich die Werte
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 319Kollar et al. . Mikrowellenresonanz-Sensoren
Abb. 3: Trocknung eines Granulates mit Messungenauigkeitbei hoher Produktfeucht (≥ 10 %), (–) Messung bei 2,5 GHz,(.) LOD/IR; modifiziert nach Buschmüller et al. [10].
Abb. 4: Stationäre Messung, Maisstärke (Typ CPharmGel®,Cargill, Krefeld), n=3.
Abb. 5: AuswertungA. LinearkombinationB. Quotientverfahren
resonanz-Sensoren werden generell inzwei unterschiedlichen Bauformeneingesetzt, als Hohlraumresonatorenund den hier betrachteten Streufeld-resonatoren. Bei den Hohlraumreso-natoren muss die Probe in den Mess-hohlraum eingebracht werden, was fürdie Anwendung in Wirbelschichtgerä-ten nachteilig ist. Im Gegensatz dazuerzeugen Streufeldresonatoren dasMikrowellenfeld oberhalb ihrer glat-ten Oberfläche, sodass das Messgutnur an ihm vorbeigeführt werden
muss. Damit eignen sich die Streufeld-resonatoren besonders gut für denEinbau in Förderstrecken oder in Be-hältnissen mit Produktberührung.
Wird eine feuchte Probe in dasFeld eines Mikrowellenresonanz-Sensors eingebracht, hat dies zweiEffekte. Zum einen sinkt die Reso-nanzfrequenz fRes und zum anderenverschlechtert sich die Güte der Re-sonanzschwingung, was mit einerVerbreiterung der Bandbreite B derResonanzkurve einhergeht (Abb. 1).
Bestimmt man die beiden Para-meter B und fRes bei einem mit einerProbe beladenen Resonator (2) ge-genüber einem leeren Resonator (1)erhält man durch jeweilige Differenz-bildung die Werte für die Resonanz-frequenzverschiebung ΔfRes undBandbreiten-Verbreiterung ΔB.
Sowohl die Resonanzfrequenzver-schiebung als auch die Bandbreiten-Verbreiterung sind zu der im Mess-
feld eingebrachten Materialmengeproportional. Die eingebrachte Was-sermenge verändert die beiden Grö-ßen hingegen in unterschiedlichemAusmaß. Durch Bildung des Quotien-ten beider Differenzen ergibt sich soein dichteunabhängiger Mikrowel-len-Feuchtemesswert M(Ψ) [8].
Mit Messdaten aus einer Referenz-bestimmung, zum Beispiel der Karl-Fischer-Titration, können die Werteaus der Mikrowellenresonanzmessungin Beziehung gesetzt werden und eineKalibriergleichung erstellt werden.
Bisherige Sensoren
Die erste Beschreibung eines pharma-zeutischen Einsatzes von Mikrowel-lenresonanz-Sensoren zur Feuch-temessung in der Wirbelschicht er-folgte von Buschmüller et al. im Jahr2008 [9, 10]. Abb. 2 (links) zeigt einenSensor des dort eingesetzten Typs inausgebautem Zustand. Deutlich ist dermikrowellenleitende keramische Ringan der Sensoroberfläche zu erkennen.Oberhalb dieses Rings bildet sich dasResonanzstreufeld. Der von Buschmül-ler et al. eingesetzte Sensor und ebensodie zurzeit am Markt verfügbaren Sen-soren nutzen nur eine einzige Reso-
nanzfrequenz zur Messung. Mithilfedes am Sensorkopf eingebauten PT100Temperaturfühler liefert die zugehö-rige Mess-Software in Echtzeit nebendem Mikrowellenfeuchtewert auch einTemperaturmesswert.
Buschmüller et al. vermaßen vor-nehmlich Granulate, bei denen dieGranulierprozesse wegen der Feuch-teempfindlichkeit der Arzneistoffe ineinem relativ niedrigen Feuchte-bereich durchgeführt wurden. Durchsofortige, präzise Erkennung des spe-zifizierten Endpunktes konnten dieTrocknungszeiten bis auf 30 % derkonventionellen Methode verkürztwerden.
Wurden jedoch Granulationen undTrocknungen mit hoher Anfangs-
feuchte vermessen, zeig-ten sich Probleme bei re-lativen Feuchten oberhalbvon etwa 10 %. Die Streu-ung der Messwerte wargrößer und der Mikrowel-len-Feuchtewert wich zu-nehmend von den mittelsTrocknungsverlust be-stimmten ab (Abb. 3).
Um diese Limitierung zuumgehen undMikrowellen-resonanz-Sensoren in wei-teren Anwendungsberei-chen einsetzen zu können,wurde die Technologie vonder Firma AMS AdvancedMicrowave Systems GmbHweiterentwickelt.
Innovativer Sensor mitzwei Messfrequenzen
Der in Abb. 2 (rechts) vorgestelltePrototyp misst bei zwei unterschied-lichen Resonanzfrequenzen. Die Re-sonanzfrequenz wird innerhalb einerSekunde von 2,5 GHz auf 8,5 GHzgewechselt. Auch die Bauformkonnte verkleinert werden, wie inAbb. 2 erkennbar. Der verringerteSensorkopfdurchmesser von 5 cm er-möglicht nun den Einbau in Wirbel-schichtgeräte im Labormaßstab unddamit grundsätzlich die Anwendungin der pharmazeutischen Entwick-lung.
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)318 Kollar et al. . Mikrowellenresonanz-Sensoren
Abb. 1: Messprinzip, Wasser-Einfluss auf dieResonanz.
Abb. 2: Mikrowellenresonanzsensoren, links: hydorpharm®
fbma (AMS GmbH), Sensor-Durchmesser 12 cm,Messfrequenz 2,5 GHz rechts: Neuer Prototyp Sensor-Durchmesser 5 cm, Messfrequenz 2,5 GHz und 8,5 GHz.(Quelle: B. Kollar)
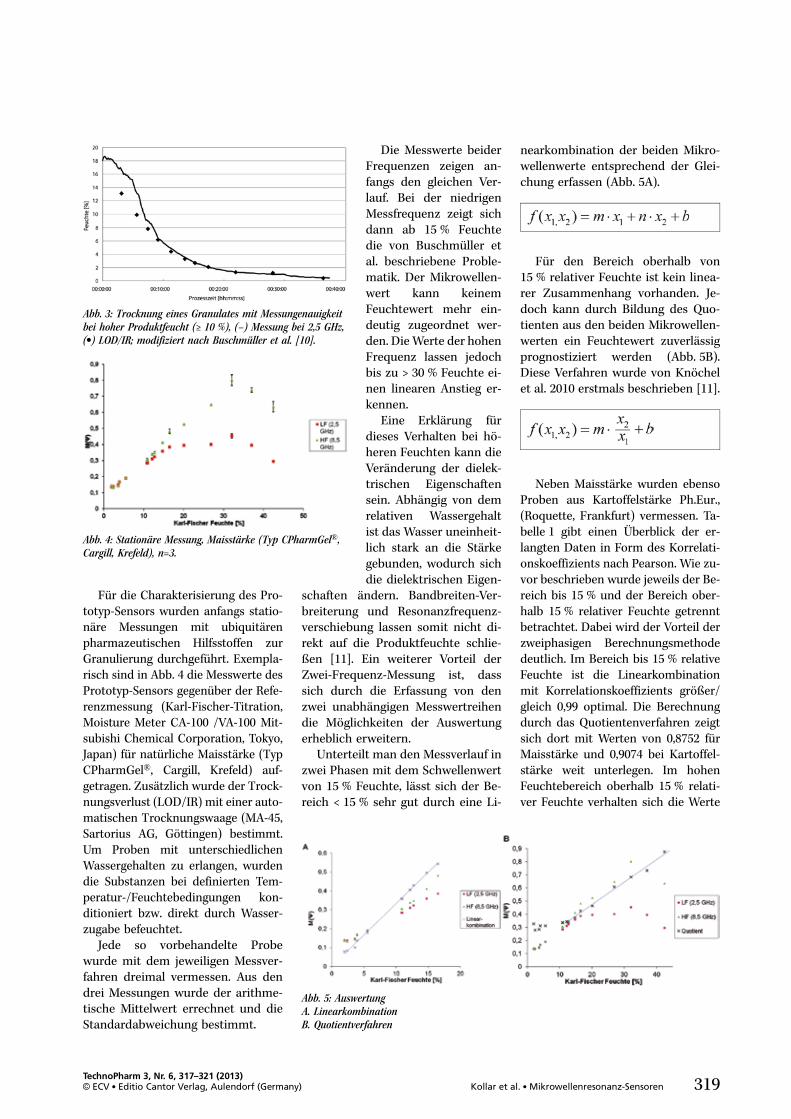
Für die Charakterisierung des Pro-totyp-Sensors wurden anfangs statio-näre Messungen mit ubiquitärenpharmazeutischen Hilfsstoffen zurGranulierung durchgeführt. Exempla-risch sind in Abb. 4 die Messwerte desPrototyp-Sensors gegenüber der Refe-renzmessung (Karl-Fischer-Titration,Moisture Meter CA-100 /VA-100 Mit-subishi Chemical Corporation, Tokyo,Japan) für natürliche Maisstärke (TypCPharmGel®, Cargill, Krefeld) auf-getragen. Zusätzlich wurde der Trock-nungsverlust (LOD/IR) mit einer auto-matischen Trocknungswaage (MA-45,Sartorius AG, Göttingen) bestimmt.Um Proben mit unterschiedlichenWassergehalten zu erlangen, wurdendie Substanzen bei definierten Tem-peratur-/Feuchtebedingungen kon-ditioniert bzw. direkt durch Wasser-zugabe befeuchtet.
Jede so vorbehandelte Probewurde mit dem jeweiligen Messver-fahren dreimal vermessen. Aus dendrei Messungen wurde der arithme-tische Mittelwert errechnet und dieStandardabweichung bestimmt.
Die Messwerte beiderFrequenzen zeigen an-fangs den gleichen Ver-lauf. Bei der niedrigenMessfrequenz zeigt sichdann ab 15 % Feuchtedie von Buschmüller etal. beschriebene Proble-matik. Der Mikrowellen-wert kann keinemFeuchtewert mehr ein-deutig zugeordnet wer-den. DieWerte der hohenFrequenz lassen jedochbis zu > 30 % Feuchte ei-nen linearen Anstieg er-kennen.
Eine Erklärung fürdieses Verhalten bei hö-heren Feuchten kann dieVeränderung der dielek-trischen Eigenschaftensein. Abhängig von demrelativen Wassergehaltist das Wasser uneinheit-lich stark an die Stärkegebunden, wodurch sichdie dielektrischen Eigen-
schaften ändern. Bandbreiten-Ver-breiterung und Resonanzfrequenz-verschiebung lassen somit nicht di-rekt auf die Produktfeuchte schlie-ßen [11]. Ein weiterer Vorteil derZwei-Frequenz-Messung ist, dasssich durch die Erfassung von denzwei unabhängigen Messwertreihendie Möglichkeiten der Auswertungerheblich erweitern.
Unterteilt man den Messverlauf inzwei Phasen mit dem Schwellenwertvon 15 % Feuchte, lässt sich der Be-reich < 15 % sehr gut durch eine Li-
nearkombination der beiden Mikro-wellenwerte entsprechend der Glei-chung erfassen (Abb. 5A).
Für den Bereich oberhalb von15 % relativer Feuchte ist kein linea-rer Zusammenhang vorhanden. Je-doch kann durch Bildung des Quo-tienten aus den beiden Mikrowellen-werten ein Feuchtewert zuverlässigprognostiziert werden (Abb. 5B).Diese Verfahren wurde von Knöchelet al. 2010 erstmals beschrieben [11].
Neben Maisstärke wurden ebensoProben aus Kartoffelstärke Ph.Eur.,(Roquette, Frankfurt) vermessen. Ta-belle 1 gibt einen Überblick der er-langten Daten in Form des Korrelati-onskoeffizients nach Pearson. Wie zu-vor beschrieben wurde jeweils der Be-reich bis 15 % und der Bereich ober-halb 15 % relativer Feuchte getrenntbetrachtet. Dabei wird der Vorteil derzweiphasigen Berechnungsmethodedeutlich. Im Bereich bis 15 % relativeFeuchte ist die Linearkombinationmit Korrelationskoeffizients größer/gleich 0,99 optimal. Die Berechnungdurch das Quotientenverfahren zeigtsich dort mit Werten von 0,8752 fürMaisstärke und 0,9074 bei Kartoffel-stärke weit unterlegen. Im hohenFeuchtebereich oberhalb 15 % relati-ver Feuchte verhalten sich die Werte
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 319Kollar et al. . Mikrowellenresonanz-Sensoren
Abb. 3: Trocknung eines Granulates mit Messungenauigkeitbei hoher Produktfeucht (≥ 10 %), (–) Messung bei 2,5 GHz,(.) LOD/IR; modifiziert nach Buschmüller et al. [10].
Abb. 4: Stationäre Messung, Maisstärke (Typ CPharmGel®,Cargill, Krefeld), n=3.
Abb. 5: AuswertungA. LinearkombinationB. Quotientverfahren
resonanz-Sensoren werden generell inzwei unterschiedlichen Bauformeneingesetzt, als Hohlraumresonatorenund den hier betrachteten Streufeld-resonatoren. Bei den Hohlraumreso-natoren muss die Probe in den Mess-hohlraum eingebracht werden, was fürdie Anwendung in Wirbelschichtgerä-ten nachteilig ist. Im Gegensatz dazuerzeugen Streufeldresonatoren dasMikrowellenfeld oberhalb ihrer glat-ten Oberfläche, sodass das Messgutnur an ihm vorbeigeführt werden
muss. Damit eignen sich die Streufeld-resonatoren besonders gut für denEinbau in Förderstrecken oder in Be-hältnissen mit Produktberührung.
Wird eine feuchte Probe in dasFeld eines Mikrowellenresonanz-Sensors eingebracht, hat dies zweiEffekte. Zum einen sinkt die Reso-nanzfrequenz fRes und zum anderenverschlechtert sich die Güte der Re-sonanzschwingung, was mit einerVerbreiterung der Bandbreite B derResonanzkurve einhergeht (Abb. 1).
Bestimmt man die beiden Para-meter B und fRes bei einem mit einerProbe beladenen Resonator (2) ge-genüber einem leeren Resonator (1)erhält man durch jeweilige Differenz-bildung die Werte für die Resonanz-frequenzverschiebung ΔfRes undBandbreiten-Verbreiterung ΔB.
Sowohl die Resonanzfrequenzver-schiebung als auch die Bandbreiten-Verbreiterung sind zu der im Mess-
feld eingebrachten Materialmengeproportional. Die eingebrachte Was-sermenge verändert die beiden Grö-ßen hingegen in unterschiedlichemAusmaß. Durch Bildung des Quotien-ten beider Differenzen ergibt sich soein dichteunabhängiger Mikrowel-len-Feuchtemesswert M(Ψ) [8].
Mit Messdaten aus einer Referenz-bestimmung, zum Beispiel der Karl-Fischer-Titration, können die Werteaus der Mikrowellenresonanzmessungin Beziehung gesetzt werden und eineKalibriergleichung erstellt werden.
Bisherige Sensoren
Die erste Beschreibung eines pharma-zeutischen Einsatzes von Mikrowel-lenresonanz-Sensoren zur Feuch-temessung in der Wirbelschicht er-folgte von Buschmüller et al. im Jahr2008 [9, 10]. Abb. 2 (links) zeigt einenSensor des dort eingesetzten Typs inausgebautem Zustand. Deutlich ist dermikrowellenleitende keramische Ringan der Sensoroberfläche zu erkennen.Oberhalb dieses Rings bildet sich dasResonanzstreufeld. Der von Buschmül-ler et al. eingesetzte Sensor und ebensodie zurzeit am Markt verfügbaren Sen-soren nutzen nur eine einzige Reso-
nanzfrequenz zur Messung. Mithilfedes am Sensorkopf eingebauten PT100Temperaturfühler liefert die zugehö-rige Mess-Software in Echtzeit nebendem Mikrowellenfeuchtewert auch einTemperaturmesswert.
Buschmüller et al. vermaßen vor-nehmlich Granulate, bei denen dieGranulierprozesse wegen der Feuch-teempfindlichkeit der Arzneistoffe ineinem relativ niedrigen Feuchte-bereich durchgeführt wurden. Durchsofortige, präzise Erkennung des spe-zifizierten Endpunktes konnten dieTrocknungszeiten bis auf 30 % derkonventionellen Methode verkürztwerden.
Wurden jedoch Granulationen undTrocknungen mit hoher Anfangs-
feuchte vermessen, zeig-ten sich Probleme bei re-lativen Feuchten oberhalbvon etwa 10 %. Die Streu-ung der Messwerte wargrößer und der Mikrowel-len-Feuchtewert wich zu-nehmend von den mittelsTrocknungsverlust be-stimmten ab (Abb. 3).
Um diese Limitierung zuumgehen undMikrowellen-resonanz-Sensoren in wei-teren Anwendungsberei-chen einsetzen zu können,wurde die Technologie vonder Firma AMS AdvancedMicrowave Systems GmbHweiterentwickelt.
Innovativer Sensor mitzwei Messfrequenzen
Der in Abb. 2 (rechts) vorgestelltePrototyp misst bei zwei unterschied-lichen Resonanzfrequenzen. Die Re-sonanzfrequenz wird innerhalb einerSekunde von 2,5 GHz auf 8,5 GHzgewechselt. Auch die Bauformkonnte verkleinert werden, wie inAbb. 2 erkennbar. Der verringerteSensorkopfdurchmesser von 5 cm er-möglicht nun den Einbau in Wirbel-schichtgeräte im Labormaßstab unddamit grundsätzlich die Anwendungin der pharmazeutischen Entwick-lung.
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)318 Kollar et al. . Mikrowellenresonanz-Sensoren
Abb. 1: Messprinzip, Wasser-Einfluss auf dieResonanz.
Abb. 2: Mikrowellenresonanzsensoren, links: hydorpharm®
fbma (AMS GmbH), Sensor-Durchmesser 12 cm,Messfrequenz 2,5 GHz rechts: Neuer Prototyp Sensor-Durchmesser 5 cm, Messfrequenz 2,5 GHz und 8,5 GHz.(Quelle: B. Kollar)

wacht werden kann, sodass bei Errei-chen des Endpunkts der Prozessohne Zeitverlust beendet werdenkann. Damit soll die Mikrowellenre-sonanztechnologie zur kontinuierli-chen Feuchtemessung im Sinne desPAT- und QbD-Ansatzes im pharma-zeutischen Umfeld etabliert werden.
Fachliteratur[1] G. E. Amidon, M. E.Houghton, The Effect of
Moisture on the Mechanical and PowderFlow Properties of Microcrystalline Cellu-lose, Pharm. Res. 12 (1995), 923–929
[2] J. T. Carstensen, Effect of Moisture on theStability of Solid Dosage Forms, Drug.Dev. Ind. Pharm. 14 (1988), 1927–1969
[3] A. Burggraeve, T. Monteyne, C. Vervaet, J.P. Remon, T. De Beer, Process analyticaltools for monitoring, understanding, and
control of pharmaceutical fluidized bedgranulation: A review, Eur. J. Pharm. Bio-pharm. 83 (2013), 2–15
[4] C. Buschmüller, W.Wiedey, C. Döscher, M.Plitzko, J. Breitkreutz, In-line Monitoringof Granule Moisture and Temperaturethroughout the entire fluidized-bed Gra-nulation Process using Microwave Reso-nance Technology, Pharm. Ind. 71 (2009),1403-1408.
[5] V. Lourenço, T. Herdling, G. Reich, J. Me-nezesa, D. Lochmann, Combining micro-wave resonance technology to multi-variate data analysis as a novel PAT toolto improve process understanding in fluidbed granulation, Eur. J. Pharm. Biopharm.78 (2011) 513–521.
[6] Guidance for industry PAT – A frameworkfor innovative pharmaceutical develop-ment, manufacturing and quality assu-rance. Sept. 2004 http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070305.pdf
[7] ICH, Guideline Q8R2: PharmaceuticalDevelopment, Technical Report, 2009.
ICH, Guideline Q9: Quality Risk Manage-ment, Technical Report, 2006.ICH, Guideline Q10: PharmaceuticalQuality System, Technical Report, 2008.http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html
[8] R. Knöchel, W. Taute, C. Döscher, Strayfield ring resonator and a novel throughguide resonator for precise microwavemoisture and density measurements,Meas. Sci. Technol. 18 (2007) 1061–1068.
[9] C. Buschmüller, W. Wiedey, C. Döscher, J.Dressler, J. Breitkreutz, In-line monitoringof granule moisture in fluidized-bed dryersusing microwave resonance technology,Eur. J. Pharm. Biopharm. 69 (2008) 380–387.
[10] C. Buschmüller, In-line monitoring ofgranule moisture in fluidized bed granu-lators using microwave resonance tech-nology as novel PAT tool, Diss. (2009),Düsseldorf, Heinrich-Heine-Universität
[11] R. Knöchel, R. Jahns, W. Taute, C. Döscher,A Resonator-based moisture meter forhigh moisture levels, KonferenzbandAQUAMETRY 2010 Weimar, Deutschland
umgekehrt. So ist die Linearkombina-tion unbrauchbar, bei der Maisstärkewird der Koeffizient sogar negativ mit-0,0536, während die Koeffizientendes Quotientenverfahrens eine guteKorrelation mit 0,9862 für Maisstärkeund 0,9747 für Kartoffelstärke wieder-geben.
Messung in der Wirbel-schicht
Bei der Entwicklung des neuen Zwei-Frequenz-Prototyps wurde die Bau-form darauf hin ausgerichtet, dasser in dem kleinvolumigen Wirbel-schichtgerät GPCG1 der Firma Glatt(Binzen, Deutschland) eingesetztwerden kann.
Wie in Abb. 6 zu sehen, wird derschmale Sensorkopf durch einenStandardflansch in den Expansions-behälter eingebracht und befestigt.Im Inneren des Behältnisses schließtder Sensorkopf bündigmit der Behälterober-fläche ab. Somit kön-nen Beeinträchtigun-gen des Wirbelschicht-betts ausgeschlossenwerden. Nachdem dasProdukt eingefüllt wur-de, befindet sich derSensor im Ruhezustandnoch oberhalb des ge-schütteten Materials.Bei Betrieb strömt undwirbelt das Material an
der Sensoroberfläche vorbei unddurchdringt so das Resonanzfeld. Bei-spielhaft ist in Abb. 7 der Messverlaufeiner Trocknung von zuvor befeuch-teter Maisstärke dargestellt.Die durch LOD/IR ermittelten Wertezeigen eine Abnahme der Feuchteausgehend von über 20 % bis auf 4,4 %rel. Feuchte. Die unterschiedlichenVerläufe der Mikrowellenwerte sinddeutlich im Diagramm erkennbar.Während die Mikrowellenwerte beihohen Feuchten größer 10 % entspre-chend der LOD/IR-Werte abnehmen,zeigen sie im niedrigen Feuchte-bereich kaum Abnahme, obwohl dieLOD/IR-Werte weiter sinken.
Bei dem Sensor älteren Typs (s.Abb. 3) trat diese Differenz bei nied-rigen Feuchten nicht auf. Gerade beiFeuchten kleiner als 6 % stimmtendie Messwerte besonders gut mitden Referenzmethoden überein.Möglicherweise sind auf der Sensor-oberfläche adhärierende Pulver- oderGranulatpartikel dafür verantwort-lich. Für den neuen Sensor, der off-line präzise und richtige Messwerteauch bei niedrigen Feuchten liefert,müsste die Oberflächenbeschaffen-heit optimiert werden, um ähnlichgute Messwerte zu erlangen.
Zukünftig soll durch weitere Mes-sungen die Auswertung so weiterent-wickelt werden, dass die Feuchte inder Wirbelschicht materialunabhän-gig über weite Feuchtebereiche ein-deutig bestimmt werden kann. Letzt-lich soll ein System entstehen, mitdem zum Beispiel eine Trocknungdurch direkte kontinuierliche Feuch-teanzeige von Beginn an stetig über-
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)320 Kollar et al. . Mikrowellenresonanz-Sensoren
Tabelle 1
Korrelationskoeffizient nach Pearson, Mikrowellenwertegegen LOD/IR-Messung.
MLF(Ψ) –LOD/IR
MHF(Ψ) –LOD/IR
Linear-komb. –LOD/IR
QuotientMHF/MLF –LOD/IR
MaisstärkeKorrelations-koeffizient0-15 % rel. F.
0,9947 0,9933 0,9961 0,8752
Korrelations-koeffizient15-40 % rel. F.
-0,5664 0,4639 -0,0536 0,9862
KartoffelstärkeKorrelations-koeffizient0-15 % rel. F.
0,9886 0,9860 0,9889 0,9074
Korrelations-koeffizient15-40 % rel. F.
0,6050 0,9187 0,8108 0,9747
Abb. 6: Glatt GPCG1 mit montiertem Prototyp-Sensor. (Quelle: B. Kollar) Abb. 7: Trocknung von Maisstärke im Glatt GPCG1, LOD/IR n=1.

wacht werden kann, sodass bei Errei-chen des Endpunkts der Prozessohne Zeitverlust beendet werdenkann. Damit soll die Mikrowellenre-sonanztechnologie zur kontinuierli-chen Feuchtemessung im Sinne desPAT- und QbD-Ansatzes im pharma-zeutischen Umfeld etabliert werden.
Fachliteratur[1] G. E. Amidon, M. E.Houghton, The Effect of
Moisture on the Mechanical and PowderFlow Properties of Microcrystalline Cellu-lose, Pharm. Res. 12 (1995), 923–929
[2] J. T. Carstensen, Effect of Moisture on theStability of Solid Dosage Forms, Drug.Dev. Ind. Pharm. 14 (1988), 1927–1969
[3] A. Burggraeve, T. Monteyne, C. Vervaet, J.P. Remon, T. De Beer, Process analyticaltools for monitoring, understanding, and
control of pharmaceutical fluidized bedgranulation: A review, Eur. J. Pharm. Bio-pharm. 83 (2013), 2–15
[4] C. Buschmüller, W.Wiedey, C. Döscher, M.Plitzko, J. Breitkreutz, In-line Monitoringof Granule Moisture and Temperaturethroughout the entire fluidized-bed Gra-nulation Process using Microwave Reso-nance Technology, Pharm. Ind. 71 (2009),1403-1408.
[5] V. Lourenço, T. Herdling, G. Reich, J. Me-nezesa, D. Lochmann, Combining micro-wave resonance technology to multi-variate data analysis as a novel PAT toolto improve process understanding in fluidbed granulation, Eur. J. Pharm. Biopharm.78 (2011) 513–521.
[6] Guidance for industry PAT – A frameworkfor innovative pharmaceutical develop-ment, manufacturing and quality assu-rance. Sept. 2004 http://www.fda.gov/downloads/Drugs/.../Guidances/ucm070305.pdf
[7] ICH, Guideline Q8R2: PharmaceuticalDevelopment, Technical Report, 2009.
ICH, Guideline Q9: Quality Risk Manage-ment, Technical Report, 2006.ICH, Guideline Q10: PharmaceuticalQuality System, Technical Report, 2008.http://www.ich.org/products/guidelines/quality/article/quality-guidelines.html
[8] R. Knöchel, W. Taute, C. Döscher, Strayfield ring resonator and a novel throughguide resonator for precise microwavemoisture and density measurements,Meas. Sci. Technol. 18 (2007) 1061–1068.
[9] C. Buschmüller, W. Wiedey, C. Döscher, J.Dressler, J. Breitkreutz, In-line monitoringof granule moisture in fluidized-bed dryersusing microwave resonance technology,Eur. J. Pharm. Biopharm. 69 (2008) 380–387.
[10] C. Buschmüller, In-line monitoring ofgranule moisture in fluidized bed granu-lators using microwave resonance tech-nology as novel PAT tool, Diss. (2009),Düsseldorf, Heinrich-Heine-Universität
[11] R. Knöchel, R. Jahns, W. Taute, C. Döscher,A Resonator-based moisture meter forhigh moisture levels, KonferenzbandAQUAMETRY 2010 Weimar, Deutschland
ecv
Bestellung:Tel. +49 (0)8191-97000 358, Fax +49 (0)8191-97000 293, eMail: [email protected], Leseproben und Inhaltsverzeichnisse – www.ecv.de
ECV · Editio Cantor Verlag
Zielgruppen• Pharmazeutische Industrie• Zulieferindustrie• Fachhochschulen /
Universitäten• Beratungsunternehmen• Behörden
Führungskräfte, Unternehmensberater und Wissen-schaftler aus Europa und den USA leisteten einenBeitrag zu diesem zweiten Werk über eine Thema-tik, die bereits dazu führte, dass Tausende von Be-schäftigten ihre Arbeit anders betrachten undausüben. Während im ersten Buch, „OperationalExcellence in the Pharmaceutical Industry“, dasAugenmerk auf den Kernpunkten von OPEX lag,geht es im vorliegenden Werk um Aspekte der Füh-rung und Organisation bei großangelegten OPEX-Programmen.
The Pathway to Operational Excellence in the Pharmaceutical IndustryTh. Friedli, P. K. Basu, T. Gronauer, J. Werani
ISBN 978-3-87193-400-1• € 96,00• 1. Auflage 2010• 17 x 24 cm, 365 Seiten, diverse Abbildungen, gebunden
Möglichkeiten zur Überwindung der inneren Trägheit auf dem Weg zur Operational ExcellenceEine neue Marktrealität erfordert zukunftsgerich-tete Operationsstrategien im Pharmabereich – dieEcksteine dafür liefert Operational Excellence. DasEngagement der Menschen stellt dabei nach wievor die größte Herausforderung dar. Wie die Auto-ren dieses Buchs herausstellen, ist es die Fähig-keit, alle Mitarbeiter dazu zu bringen, ihr Denkenauf beständige Optimierung und Veränderung aus-zurichten, mit der Unternehmen ihre innere Schwer-fälligkeit überwinden können und wodurch sich dieGewinner von den Verlierern unterscheiden.
umgekehrt. So ist die Linearkombina-tion unbrauchbar, bei der Maisstärkewird der Koeffizient sogar negativ mit-0,0536, während die Koeffizientendes Quotientenverfahrens eine guteKorrelation mit 0,9862 für Maisstärkeund 0,9747 für Kartoffelstärke wieder-geben.
Messung in der Wirbel-schicht
Bei der Entwicklung des neuen Zwei-Frequenz-Prototyps wurde die Bau-form darauf hin ausgerichtet, dasser in dem kleinvolumigen Wirbel-schichtgerät GPCG1 der Firma Glatt(Binzen, Deutschland) eingesetztwerden kann.
Wie in Abb. 6 zu sehen, wird derschmale Sensorkopf durch einenStandardflansch in den Expansions-behälter eingebracht und befestigt.Im Inneren des Behältnisses schließtder Sensorkopf bündigmit der Behälterober-fläche ab. Somit kön-nen Beeinträchtigun-gen des Wirbelschicht-betts ausgeschlossenwerden. Nachdem dasProdukt eingefüllt wur-de, befindet sich derSensor im Ruhezustandnoch oberhalb des ge-schütteten Materials.Bei Betrieb strömt undwirbelt das Material an
der Sensoroberfläche vorbei unddurchdringt so das Resonanzfeld. Bei-spielhaft ist in Abb. 7 der Messverlaufeiner Trocknung von zuvor befeuch-teter Maisstärke dargestellt.Die durch LOD/IR ermittelten Wertezeigen eine Abnahme der Feuchteausgehend von über 20 % bis auf 4,4 %rel. Feuchte. Die unterschiedlichenVerläufe der Mikrowellenwerte sinddeutlich im Diagramm erkennbar.Während die Mikrowellenwerte beihohen Feuchten größer 10 % entspre-chend der LOD/IR-Werte abnehmen,zeigen sie im niedrigen Feuchte-bereich kaum Abnahme, obwohl dieLOD/IR-Werte weiter sinken.
Bei dem Sensor älteren Typs (s.Abb. 3) trat diese Differenz bei nied-rigen Feuchten nicht auf. Gerade beiFeuchten kleiner als 6 % stimmtendie Messwerte besonders gut mitden Referenzmethoden überein.Möglicherweise sind auf der Sensor-oberfläche adhärierende Pulver- oderGranulatpartikel dafür verantwort-lich. Für den neuen Sensor, der off-line präzise und richtige Messwerteauch bei niedrigen Feuchten liefert,müsste die Oberflächenbeschaffen-heit optimiert werden, um ähnlichgute Messwerte zu erlangen.
Zukünftig soll durch weitere Mes-sungen die Auswertung so weiterent-wickelt werden, dass die Feuchte inder Wirbelschicht materialunabhän-gig über weite Feuchtebereiche ein-deutig bestimmt werden kann. Letzt-lich soll ein System entstehen, mitdem zum Beispiel eine Trocknungdurch direkte kontinuierliche Feuch-teanzeige von Beginn an stetig über-
Fokus: Herstellung fester Arzneiformen
TechnoPharm 3, Nr. 6, 317–321 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)320 Kollar et al. . Mikrowellenresonanz-Sensoren
Tabelle 1
Korrelationskoeffizient nach Pearson, Mikrowellenwertegegen LOD/IR-Messung.
MLF(Ψ) –LOD/IR
MHF(Ψ) –LOD/IR
Linear-komb. –LOD/IR
QuotientMHF/MLF –LOD/IR
MaisstärkeKorrelations-koeffizient0-15 % rel. F.
0,9947 0,9933 0,9961 0,8752
Korrelations-koeffizient15-40 % rel. F.
-0,5664 0,4639 -0,0536 0,9862
KartoffelstärkeKorrelations-koeffizient0-15 % rel. F.
0,9886 0,9860 0,9889 0,9074
Korrelations-koeffizient15-40 % rel. F.
0,6050 0,9187 0,8108 0,9747
Abb. 6: Glatt GPCG1 mit montiertem Prototyp-Sensor. (Quelle: B. Kollar) Abb. 7: Trocknung von Maisstärke im Glatt GPCG1, LOD/IR n=1.

Einleitung
Aktive biologische Arbeitsstoffe wie beispielsweiseBakterien, Viren oder Zellkulturen werden vor ihrerEntsorgung aus dem biologischen oder medizinischenLaboratorium in einem Autoklaven thermisch inakti-viert. Dieses erfordert eine irreversible Zerstörung derInfektions- und Vermehrungsfähigkeit der Arbeitsstof-fe, einschließlich deren Ruhestadien. Der in einem Au-toklaven ablaufende Sterilisationsprozess basiert aufAbtötungskinetiken unter Einhaltung definierter Pro-zessbedingungen. Um biologische Arbeitsstoffe sicherabzutöten ist neben der Qualität des verwendeten Satt-
dampfes, ebenfalls dessen vollständige Durchdringungder Autoklavenkammer, sowie des darin enthaltenenAutoklavierguts erforderlich. In der Kammer verblei-bende Luftinseln verringern den Sterilisationserfolg.Aus diesem Grund muss vor einer Einleitung der Ste-rilisationsphase eine Entfernung der Luft aus der Kam-mer und dem Sterilisationsgut erfolgen. Der Erfolg derEntfernung der Luft wird im Wesentlichen von der Artder Beladung und dem gewählten Autoklavierverfah-ren bestimmt. Autoklaven werden mittels verschiede-ner Prozesstypen betrieben, welche nach ihrem Verfah-ren der Luftentfernung benannt sind. Einfache Verfah-ren wie das Strömungs- oder Gravitationsverfahrennutzen die Verdrängung der Luft durch den strömen-den Dampf. Technisch aufwändigere Verfahren evaku-ieren vor der eigentlichen Sterilisation die Kammerüber in die Abluftstrecke des Autoklaven integrierteVakuumpumpen. Die auf diesem Wege aus der Auto-klavenkammer austretende Abluft wird normalerweisein die Umgebung abgegeben. Um den Austrag von Mi-kroorganismen in Form von Bioaerosolen zu verhin-dern schreibt die Gentechnik-Sicherheitsverordnung(GenTSV) vor, dieses in Abhängigkeit der Risikogruppe
Abb. 1: Abluftfiltersystem eines Laborautoklaven (Quelle alle: Institute ofBioprocess Engineering and Pharmaceutical Technology, TechnischeHochschule Mittelhessen – University of Applied Science)
Abluftfiltration unter der LupeRisiken bei der Sterilfiltration der Autoklavenabluft
Carsten Grumbach, Peter Czermak . Institute of Bioprocess Engineering and Pharmaceutical Technology,Technische Hochschule Mittelhessen – University of Applied Sciences, GiessenHeike Schulte-Lünzum . Staufenberg
Korrespondenz: Carsten Grumbach, Institute of Bioprocess Engineering and Pharmaceutical Technology, Technische HochschuleMittelhessen – University of Applied Science, Wiesenstr. 14, 35390 Giessen
ZusammenfassungFür die thermische Inaktivierung biologischer oder mikrobiell kontaminierter Arbeitsstoffemit sensibilisierender oder toxischer Wirkung, sind hohe Sicherheitsanforderungen an dieAutoklavieranlage gestellt. Um eine effiziente Sterilisation des Produktes zu erreichen,muss eine vollständige Evakuierung der Autoklavenkammer und des darin befindlichenSterilisierguts erfolgen. Die dabei aus der Autoklavenkammer entfernte Abluft mussmittels geeigneter Verfahren inaktiviert werden. Die Abgasleitungen von Kleinanlagenwerden aus diesem Grund mit Sterilfiltern ausgestattet. Die Sterilfiltration kann unterbestimmten Bedingungen mit Risiken behaftet sein. Um den Stand der Technik beimEinsatz sterilisierbarer Sterilfilter zu gewährleisten, sollten diese daher regelmäßig bzgl.ihrer Integrität geprüft und gewechselt werden [1]. Im folgenden Artikel werden dieRisiken der Sterilfiltration am Praxisbeispiel dargestellt und mögliche Verbesserungswegeaufgezeigt.
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 322–326 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)322 Grumbach et al. . Abluftfiltration unter der Lupe
Autoren
Carsten Grumbach
Nach seiner Ausbildung zum Chemielaborantenbei der Celanese Chemicals Europe GmbH,studierte Carsten Grumbach Chemieingenieurwe-sen an der Hochschule Niederrhein. Nach seinerDiplomarbeit am Lehrstuhl für Bioverfahrenstech-nik der RWTH Aachen, erfolgte der Wechsel zumInstitut für Bioverfahrenstechnik und Pharmazeu-tische Technologie der Technischen HochschuleMittelhessen. Herr Carsten Grumbach arbeitet dortals wissenschaftlicher Angestellter auf dem Gebietder Sterilfiltration mit dem Fokus auf der Ent-wicklung von Testsystemen zur Integritätsprüfungvon Sterilfiltern. Neben seiner Berufsarbeit absol-viert Herr Grumbach derzeit sein Masterstudiumder Biotechnologie/Biopharmazeutische Tech-nologie an der Technischen Hochschule Mittel-hessen.
Heike Schulte-Lünzum
Heike Schulte-Lünzum (M.Sc.) studierte an derFachhochschule Gießen-Friedberg Umwelt- undHygienetechnik und ist seit 2001 beim Regie-rungspräsidium Gießen tätig. Ihr Masterstudiumder Biotechnologie / Biopharmazeutischen Tech-nologie an der Technischen Hochschule Mittel-hessen absolvierte sie von 2010 bis 2012. Im Rah-men ihrer Masterthesis beschäftigte sie sich mitder Sterilfiltration von Autoklavenabluft undFilterintegritätstests.
(Die in diesem Artikel dargestellte Meinung vertrittHeike Schulte-Lünzum als Privatperson).
Peter Czermak
Peter Czermak studierte Chemieingenieurwesenund promovierte in Bioverfahrenstechnik. Er istgeschäftsführender Direktor am Institut für Bio-verfahrenstechnik und Pharmazeutische Tech-nologie der Technischen Hochschule Mittelhessen.Außerdem ist er außerordentlicher Professor amDepartment of Chemical Engineering der KansasState University, USA, und Professor h.c. amFachbereich – Biologie und Chemie – der Justus-Liebig Universität, Giessen. Peter Czermak hatmehr als 25 Jahre Erfahrung in Bioverfahrenstech-nik und Bioseparation. Seit 15 Jahren arbeitet er mitseiner Arbeitsgruppe im Bereich der Prozessent-wicklung mit dem Fokus auf tierische Zellkulturen.

Einleitung
Aktive biologische Arbeitsstoffe wie beispielsweiseBakterien, Viren oder Zellkulturen werden vor ihrerEntsorgung aus dem biologischen oder medizinischenLaboratorium in einem Autoklaven thermisch inakti-viert. Dieses erfordert eine irreversible Zerstörung derInfektions- und Vermehrungsfähigkeit der Arbeitsstof-fe, einschließlich deren Ruhestadien. Der in einem Au-toklaven ablaufende Sterilisationsprozess basiert aufAbtötungskinetiken unter Einhaltung definierter Pro-zessbedingungen. Um biologische Arbeitsstoffe sicherabzutöten ist neben der Qualität des verwendeten Satt-
dampfes, ebenfalls dessen vollständige Durchdringungder Autoklavenkammer, sowie des darin enthaltenenAutoklavierguts erforderlich. In der Kammer verblei-bende Luftinseln verringern den Sterilisationserfolg.Aus diesem Grund muss vor einer Einleitung der Ste-rilisationsphase eine Entfernung der Luft aus der Kam-mer und dem Sterilisationsgut erfolgen. Der Erfolg derEntfernung der Luft wird im Wesentlichen von der Artder Beladung und dem gewählten Autoklavierverfah-ren bestimmt. Autoklaven werden mittels verschiede-ner Prozesstypen betrieben, welche nach ihrem Verfah-ren der Luftentfernung benannt sind. Einfache Verfah-ren wie das Strömungs- oder Gravitationsverfahrennutzen die Verdrängung der Luft durch den strömen-den Dampf. Technisch aufwändigere Verfahren evaku-ieren vor der eigentlichen Sterilisation die Kammerüber in die Abluftstrecke des Autoklaven integrierteVakuumpumpen. Die auf diesem Wege aus der Auto-klavenkammer austretende Abluft wird normalerweisein die Umgebung abgegeben. Um den Austrag von Mi-kroorganismen in Form von Bioaerosolen zu verhin-dern schreibt die Gentechnik-Sicherheitsverordnung(GenTSV) vor, dieses in Abhängigkeit der Risikogruppe
Abb. 1: Abluftfiltersystem eines Laborautoklaven (Quelle alle: Institute ofBioprocess Engineering and Pharmaceutical Technology, TechnischeHochschule Mittelhessen – University of Applied Science)
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-06\Anzeigensatz-keine-Druck-PDFs\pharmaserv-TP-2013-06_105x303.indd Zuletzt gesichert: 12.11.13 (11:43:01 Uhr)
Pharmaserv GmbH & Co. KGEmil-von-Behring-Straße 7635041 MarburgTelefon: (0 64 21) 39 -14
www.pharmaserv.de
Qualität überzeugt immer.Unser bestes Messinstrument dafür: Ihre Zufriedenheit.
Das Dienstleistungsportfolio von Pharmaserv bietet Unter-
nehmen u. a. in den Bereichen Kalibrierung / Qualifizierung,
Reinraum-Messtechnik, Sterilisationstechnik, GMP-Wartung
und Pharmalogistik eine Vielzahl innovativer Lösungen:
Damit werden Abläufe optimiert und gleichzeitig die hohen
Anforderungen der hygienesensiblen Prozessindustrie erfüllt.
Sprechen Sie mit uns – gemeinsam finden wir die
richtige Lösung für Sie.
Abluftfiltration unter der LupeRisiken bei der Sterilfiltration der Autoklavenabluft
Carsten Grumbach, Peter Czermak . Institute of Bioprocess Engineering and Pharmaceutical Technology,Technische Hochschule Mittelhessen – University of Applied Sciences, GiessenHeike Schulte-Lünzum . Staufenberg
Korrespondenz: Carsten Grumbach, Institute of Bioprocess Engineering and Pharmaceutical Technology, Technische HochschuleMittelhessen – University of Applied Science, Wiesenstr. 14, 35390 Giessen
ZusammenfassungFür die thermische Inaktivierung biologischer oder mikrobiell kontaminierter Arbeitsstoffemit sensibilisierender oder toxischer Wirkung, sind hohe Sicherheitsanforderungen an dieAutoklavieranlage gestellt. Um eine effiziente Sterilisation des Produktes zu erreichen,muss eine vollständige Evakuierung der Autoklavenkammer und des darin befindlichenSterilisierguts erfolgen. Die dabei aus der Autoklavenkammer entfernte Abluft mussmittels geeigneter Verfahren inaktiviert werden. Die Abgasleitungen von Kleinanlagenwerden aus diesem Grund mit Sterilfiltern ausgestattet. Die Sterilfiltration kann unterbestimmten Bedingungen mit Risiken behaftet sein. Um den Stand der Technik beimEinsatz sterilisierbarer Sterilfilter zu gewährleisten, sollten diese daher regelmäßig bzgl.ihrer Integrität geprüft und gewechselt werden [1]. Im folgenden Artikel werden dieRisiken der Sterilfiltration am Praxisbeispiel dargestellt und mögliche Verbesserungswegeaufgezeigt.
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 322–326 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)322 Grumbach et al. . Abluftfiltration unter der Lupe
Autoren
Carsten Grumbach
Nach seiner Ausbildung zum Chemielaborantenbei der Celanese Chemicals Europe GmbH,studierte Carsten Grumbach Chemieingenieurwe-sen an der Hochschule Niederrhein. Nach seinerDiplomarbeit am Lehrstuhl für Bioverfahrenstech-nik der RWTH Aachen, erfolgte der Wechsel zumInstitut für Bioverfahrenstechnik und Pharmazeu-tische Technologie der Technischen HochschuleMittelhessen. Herr Carsten Grumbach arbeitet dortals wissenschaftlicher Angestellter auf dem Gebietder Sterilfiltration mit dem Fokus auf der Ent-wicklung von Testsystemen zur Integritätsprüfungvon Sterilfiltern. Neben seiner Berufsarbeit absol-viert Herr Grumbach derzeit sein Masterstudiumder Biotechnologie/Biopharmazeutische Tech-nologie an der Technischen Hochschule Mittel-hessen.
Heike Schulte-Lünzum
Heike Schulte-Lünzum (M.Sc.) studierte an derFachhochschule Gießen-Friedberg Umwelt- undHygienetechnik und ist seit 2001 beim Regie-rungspräsidium Gießen tätig. Ihr Masterstudiumder Biotechnologie / Biopharmazeutischen Tech-nologie an der Technischen Hochschule Mittel-hessen absolvierte sie von 2010 bis 2012. Im Rah-men ihrer Masterthesis beschäftigte sie sich mitder Sterilfiltration von Autoklavenabluft undFilterintegritätstests.
(Die in diesem Artikel dargestellte Meinung vertrittHeike Schulte-Lünzum als Privatperson).
Peter Czermak
Peter Czermak studierte Chemieingenieurwesenund promovierte in Bioverfahrenstechnik. Er istgeschäftsführender Direktor am Institut für Bio-verfahrenstechnik und Pharmazeutische Tech-nologie der Technischen Hochschule Mittelhessen.Außerdem ist er außerordentlicher Professor amDepartment of Chemical Engineering der KansasState University, USA, und Professor h.c. amFachbereich – Biologie und Chemie – der Justus-Liebig Universität, Giessen. Peter Czermak hatmehr als 25 Jahre Erfahrung in Bioverfahrenstech-nik und Bioseparation. Seit 15 Jahren arbeitet er mitseiner Arbeitsgruppe im Bereich der Prozessent-wicklung mit dem Fokus auf tierische Zellkulturen.
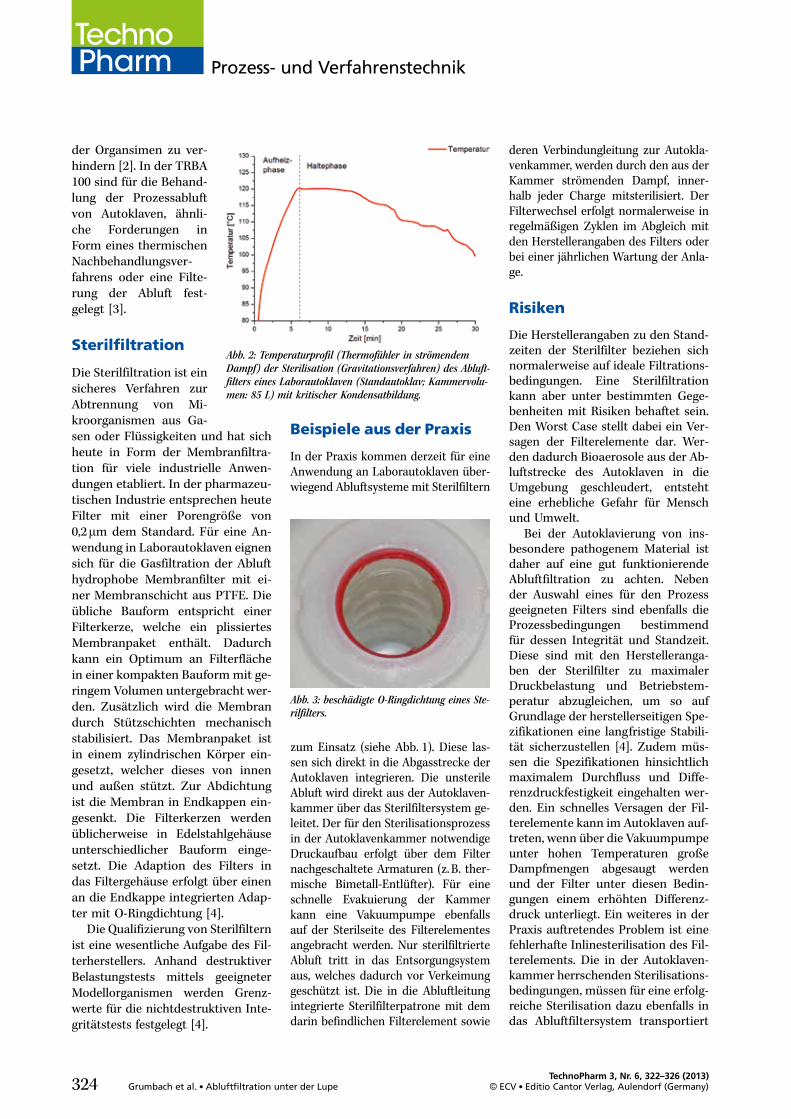
werden. Im Filtergehäuse anfallendesKondensat muss in die Autoklaven-kammer abgeführt werden, um einenKondensatstau zu vermeiden. Dieseswird an Autoklaven technisch bisherüber eine gemeinsame Leitung vonDampf und Kondensat im Gegen-strom realisiert. Fehlende Isolatio-nen, lange Zulaufleitungen und einnicht ablaufendes Kondensat, sindnicht selten Ursache für Kondensats-tau im Filtergehäuse und dem folg-lichen Einbrechen der Sterilisations-bedingungen (siehe Abb. 2).
Eine nicht erfolgte Sterilisation desAbluftfiltersystems birgt eine Gefahrvon Rückkontaminationenen am Auto-klaviergut, durch zurücklaufendes un-steriles Kondensat. Zudem wird durcheinen Kondensatstau die effektive Fil-terfläche des Sterilfilters gemindert. Er-höhte Differenzdrücke können dieFolge sein, was zu einer zusätzlichenBelastung des Filters führt. Im Filter-gehäuse kondensierender Dampf odervom Dampfstrom mitgerissene Was-sertröpfchen, können zu einem Verblo-cken der Membran führen.
Steht der Filter über einen längerenZeitraum weiter im Kondensat (z.B.nach einer Sterilisation oder währendeines Transports der Anlage), könnenebenfalls Verblockungseffekte undTemperatureinbrüche auftreten.
Ebenfalls wird die Sterilisation desFilters in vielen Anlagen nicht mittelseiner Temperaturmessung überwacht,wodurch steuerungstechnisch keineausreichende Durchströmung des Fil-
ters gegeben ist, um die Sterilisations-temperatur im Filter aufrecht zu erhal-ten. Werden an bestehenden AnlagenAbluftfiltersysteme nachgerüstet odermodifiziert, können Undichtigkeitenim System zurückbleiben, welche einenAustritt von pathogenen Keimen zulas-sen. Ein weiteres Risiko birgt die zurAdaption an den Filtersitz notwendige
O-Ringdichtung. Wird diese beim Ein-bau oder Wechseln des Filters beschä-digt oder dichtet diese nicht richtig ab,ist eine Sterilfiltration bis zum näch-sten Filterwechsel nicht mehr gegeben(siehe Abb. 3).
In der Praxis kann dieses 100 Zyklenund mehr bedeuten. Derartige Pro-bleme können häufig dann auftreten,
Abb. 5: Faltwerk eines intakten Filters (links), Faltwerk nach Beanspruchung mittels Pressluft(Mitte), Faltwerk mit Durchbruch nach hoher Druckdifferenz (rechts).
Abb. 4: Sterilfilter.
der Organsimen zu ver-hindern [2]. In der TRBA100 sind für die Behand-lung der Prozessabluftvon Autoklaven, ähnli-che Forderungen inForm eines thermischenNachbehandlungsver-fahrens oder eine Filte-rung der Abluft fest-gelegt [3].
Sterilfiltration
Die Sterilfiltration ist einsicheres Verfahren zurAbtrennung von Mi-kroorganismen aus Ga-sen oder Flüssigkeiten und hat sichheute in Form der Membranfiltra-tion für viele industrielle Anwen-dungen etabliert. In der pharmazeu-tischen Industrie entsprechen heuteFilter mit einer Porengröße von0,2μm dem Standard. Für eine An-wendung in Laborautoklaven eignensich für die Gasfiltration der Ablufthydrophobe Membranfilter mit ei-ner Membranschicht aus PTFE. Dieübliche Bauform entspricht einerFilterkerze, welche ein plissiertesMembranpaket enthält. Dadurchkann ein Optimum an Filterflächein einer kompakten Bauformmit ge-ringem Volumen untergebracht wer-den. Zusätzlich wird die Membrandurch Stützschichten mechanischstabilisiert. Das Membranpaket istin einem zylindrischen Körper ein-gesetzt, welcher dieses von innenund außen stützt. Zur Abdichtungist die Membran in Endkappen ein-gesenkt. Die Filterkerzen werdenüblicherweise in Edelstahlgehäuseunterschiedlicher Bauform einge-setzt. Die Adaption des Filters indas Filtergehäuse erfolgt über einenan die Endkappe integrierten Adap-ter mit O-Ringdichtung [4].
Die Qualifizierung von Sterilfilternist eine wesentliche Aufgabe des Fil-terherstellers. Anhand destruktiverBelastungstests mittels geeigneterModellorganismen werden Grenz-werte für die nichtdestruktiven Inte-gritätstests festgelegt [4].
Beispiele aus der Praxis
In der Praxis kommen derzeit für eineAnwendung an Laborautoklaven über-wiegend Abluftsysteme mit Sterilfiltern
zum Einsatz (siehe Abb. 1). Diese las-sen sich direkt in die Abgasstrecke derAutoklaven integrieren. Die unsterileAbluft wird direkt aus der Autoklaven-kammer über das Sterilfiltersystem ge-leitet. Der für den Sterilisationsprozessin der Autoklavenkammer notwendigeDruckaufbau erfolgt über dem Filternachgeschaltete Armaturen (z.B. ther-mische Bimetall-Entlüfter). Für eineschnelle Evakuierung der Kammerkann eine Vakuumpumpe ebenfallsauf der Sterilseite des Filterelementesangebracht werden. Nur sterilfiltrierteAbluft tritt in das Entsorgungsystemaus, welches dadurch vor Verkeimunggeschützt ist. Die in die Abluftleitungintegrierte Sterilfilterpatrone mit demdarin befindlichen Filterelement sowie
deren Verbindungleitung zur Autokla-venkammer, werden durch den aus derKammer strömenden Dampf, inner-halb jeder Charge mitsterilisiert. DerFilterwechsel erfolgt normalerweise inregelmäßigen Zyklen im Abgleich mitden Herstellerangaben des Filters oderbei einer jährlichen Wartung der Anla-ge.
Risiken
Die Herstellerangaben zu den Stand-zeiten der Sterilfilter beziehen sichnormalerweise auf ideale Filtrations-bedingungen. Eine Sterilfiltrationkann aber unter bestimmten Gege-benheiten mit Risiken behaftet sein.Den Worst Case stellt dabei ein Ver-sagen der Filterelemente dar. Wer-den dadurch Bioaerosole aus der Ab-luftstrecke des Autoklaven in dieUmgebung geschleudert, entstehteine erhebliche Gefahr für Menschund Umwelt.
Bei der Autoklavierung von ins-besondere pathogenem Material istdaher auf eine gut funktionierendeAbluftfiltration zu achten. Nebender Auswahl eines für den Prozessgeeigneten Filters sind ebenfalls dieProzessbedingungen bestimmendfür dessen Integrität und Standzeit.Diese sind mit den Herstelleranga-ben der Sterilfilter zu maximalerDruckbelastung und Betriebstem-peratur abzugleichen, um so aufGrundlage der herstellerseitigen Spe-zifikationen eine langfristige Stabili-tät sicherzustellen [4]. Zudem müs-sen die Spezifikationen hinsichtlichmaximalem Durchfluss und Diffe-renzdruckfestigkeit eingehalten wer-den. Ein schnelles Versagen der Fil-terelemente kann im Autoklaven auf-treten, wenn über die Vakuumpumpeunter hohen Temperaturen großeDampfmengen abgesaugt werdenund der Filter unter diesen Bedin-gungen einem erhöhten Differenz-druck unterliegt. Ein weiteres in derPraxis auftretendes Problem ist einefehlerhafte Inlinesterilisation des Fil-terelements. Die in der Autoklaven-kammer herrschenden Sterilisations-bedingungen, müssen für eine erfolg-reiche Sterilisation dazu ebenfalls indas Abluftfiltersystem transportiert
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 322–326 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)324 Grumbach et al. . Abluftfiltration unter der Lupe
Abb. 2: Temperaturprofil (Thermofühler in strömendemDampf) der Sterilisation (Gravitationsverfahren) des Abluft-filters eines Laborautoklaven (Standautoklav; Kammervolu-men: 85 L) mit kritischer Kondensatbildung.
Abb. 3: beschädigte O-Ringdichtung eines Ste-rilfilters.

werden. Im Filtergehäuse anfallendesKondensat muss in die Autoklaven-kammer abgeführt werden, um einenKondensatstau zu vermeiden. Dieseswird an Autoklaven technisch bisherüber eine gemeinsame Leitung vonDampf und Kondensat im Gegen-strom realisiert. Fehlende Isolatio-nen, lange Zulaufleitungen und einnicht ablaufendes Kondensat, sindnicht selten Ursache für Kondensats-tau im Filtergehäuse und dem folg-lichen Einbrechen der Sterilisations-bedingungen (siehe Abb. 2).
Eine nicht erfolgte Sterilisation desAbluftfiltersystems birgt eine Gefahrvon Rückkontaminationenen am Auto-klaviergut, durch zurücklaufendes un-steriles Kondensat. Zudem wird durcheinen Kondensatstau die effektive Fil-terfläche des Sterilfilters gemindert. Er-höhte Differenzdrücke können dieFolge sein, was zu einer zusätzlichenBelastung des Filters führt. Im Filter-gehäuse kondensierender Dampf odervom Dampfstrom mitgerissene Was-sertröpfchen, können zu einem Verblo-cken der Membran führen.
Steht der Filter über einen längerenZeitraum weiter im Kondensat (z.B.nach einer Sterilisation oder währendeines Transports der Anlage), könnenebenfalls Verblockungseffekte undTemperatureinbrüche auftreten.
Ebenfalls wird die Sterilisation desFilters in vielen Anlagen nicht mittelseiner Temperaturmessung überwacht,wodurch steuerungstechnisch keineausreichende Durchströmung des Fil-
ters gegeben ist, um die Sterilisations-temperatur im Filter aufrecht zu erhal-ten. Werden an bestehenden AnlagenAbluftfiltersysteme nachgerüstet odermodifiziert, können Undichtigkeitenim System zurückbleiben, welche einenAustritt von pathogenen Keimen zulas-sen. Ein weiteres Risiko birgt die zurAdaption an den Filtersitz notwendige
O-Ringdichtung. Wird diese beim Ein-bau oder Wechseln des Filters beschä-digt oder dichtet diese nicht richtig ab,ist eine Sterilfiltration bis zum näch-sten Filterwechsel nicht mehr gegeben(siehe Abb. 3).
In der Praxis kann dieses 100 Zyklenund mehr bedeuten. Derartige Pro-bleme können häufig dann auftreten,
Abb. 5: Faltwerk eines intakten Filters (links), Faltwerk nach Beanspruchung mittels Pressluft(Mitte), Faltwerk mit Durchbruch nach hoher Druckdifferenz (rechts).
Abb. 4: Sterilfilter.
der Organsimen zu ver-hindern [2]. In der TRBA100 sind für die Behand-lung der Prozessabluftvon Autoklaven, ähnli-che Forderungen inForm eines thermischenNachbehandlungsver-fahrens oder eine Filte-rung der Abluft fest-gelegt [3].
Sterilfiltration
Die Sterilfiltration ist einsicheres Verfahren zurAbtrennung von Mi-kroorganismen aus Ga-sen oder Flüssigkeiten und hat sichheute in Form der Membranfiltra-tion für viele industrielle Anwen-dungen etabliert. In der pharmazeu-tischen Industrie entsprechen heuteFilter mit einer Porengröße von0,2μm dem Standard. Für eine An-wendung in Laborautoklaven eignensich für die Gasfiltration der Ablufthydrophobe Membranfilter mit ei-ner Membranschicht aus PTFE. Dieübliche Bauform entspricht einerFilterkerze, welche ein plissiertesMembranpaket enthält. Dadurchkann ein Optimum an Filterflächein einer kompakten Bauformmit ge-ringem Volumen untergebracht wer-den. Zusätzlich wird die Membrandurch Stützschichten mechanischstabilisiert. Das Membranpaket istin einem zylindrischen Körper ein-gesetzt, welcher dieses von innenund außen stützt. Zur Abdichtungist die Membran in Endkappen ein-gesenkt. Die Filterkerzen werdenüblicherweise in Edelstahlgehäuseunterschiedlicher Bauform einge-setzt. Die Adaption des Filters indas Filtergehäuse erfolgt über einenan die Endkappe integrierten Adap-ter mit O-Ringdichtung [4].
Die Qualifizierung von Sterilfilternist eine wesentliche Aufgabe des Fil-terherstellers. Anhand destruktiverBelastungstests mittels geeigneterModellorganismen werden Grenz-werte für die nichtdestruktiven Inte-gritätstests festgelegt [4].
Beispiele aus der Praxis
In der Praxis kommen derzeit für eineAnwendung an Laborautoklaven über-wiegend Abluftsysteme mit Sterilfiltern
zum Einsatz (siehe Abb. 1). Diese las-sen sich direkt in die Abgasstrecke derAutoklaven integrieren. Die unsterileAbluft wird direkt aus der Autoklaven-kammer über das Sterilfiltersystem ge-leitet. Der für den Sterilisationsprozessin der Autoklavenkammer notwendigeDruckaufbau erfolgt über dem Filternachgeschaltete Armaturen (z.B. ther-mische Bimetall-Entlüfter). Für eineschnelle Evakuierung der Kammerkann eine Vakuumpumpe ebenfallsauf der Sterilseite des Filterelementesangebracht werden. Nur sterilfiltrierteAbluft tritt in das Entsorgungsystemaus, welches dadurch vor Verkeimunggeschützt ist. Die in die Abluftleitungintegrierte Sterilfilterpatrone mit demdarin befindlichen Filterelement sowie
deren Verbindungleitung zur Autokla-venkammer, werden durch den aus derKammer strömenden Dampf, inner-halb jeder Charge mitsterilisiert. DerFilterwechsel erfolgt normalerweise inregelmäßigen Zyklen im Abgleich mitden Herstellerangaben des Filters oderbei einer jährlichen Wartung der Anla-ge.
Risiken
Die Herstellerangaben zu den Stand-zeiten der Sterilfilter beziehen sichnormalerweise auf ideale Filtrations-bedingungen. Eine Sterilfiltrationkann aber unter bestimmten Gege-benheiten mit Risiken behaftet sein.Den Worst Case stellt dabei ein Ver-sagen der Filterelemente dar. Wer-den dadurch Bioaerosole aus der Ab-luftstrecke des Autoklaven in dieUmgebung geschleudert, entstehteine erhebliche Gefahr für Menschund Umwelt.
Bei der Autoklavierung von ins-besondere pathogenem Material istdaher auf eine gut funktionierendeAbluftfiltration zu achten. Nebender Auswahl eines für den Prozessgeeigneten Filters sind ebenfalls dieProzessbedingungen bestimmendfür dessen Integrität und Standzeit.Diese sind mit den Herstelleranga-ben der Sterilfilter zu maximalerDruckbelastung und Betriebstem-peratur abzugleichen, um so aufGrundlage der herstellerseitigen Spe-zifikationen eine langfristige Stabili-tät sicherzustellen [4]. Zudem müs-sen die Spezifikationen hinsichtlichmaximalem Durchfluss und Diffe-renzdruckfestigkeit eingehalten wer-den. Ein schnelles Versagen der Fil-terelemente kann im Autoklaven auf-treten, wenn über die Vakuumpumpeunter hohen Temperaturen großeDampfmengen abgesaugt werdenund der Filter unter diesen Bedin-gungen einem erhöhten Differenz-druck unterliegt. Ein weiteres in derPraxis auftretendes Problem ist einefehlerhafte Inlinesterilisation des Fil-terelements. Die in der Autoklaven-kammer herrschenden Sterilisations-bedingungen, müssen für eine erfolg-reiche Sterilisation dazu ebenfalls indas Abluftfiltersystem transportiert
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 322–326 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)324 Grumbach et al. . Abluftfiltration unter der Lupe
Abb. 2: Temperaturprofil (Thermofühler in strömendemDampf) der Sterilisation (Gravitationsverfahren) des Abluft-filters eines Laborautoklaven (Standautoklav; Kammervolu-men: 85 L) mit kritischer Kondensatbildung.
Abb. 3: beschädigte O-Ringdichtung eines Ste-rilfilters.
STERIL
Autoklaven für die MikrobiologieAutoklaven für die Mikrobiologie
EINFACH
GUT
STERILISIEREN
EINFACH
GUT
STERILISIERENEUROPE
HMCHMCEUROPE
Sterilisationstechnik
www.hmc-europe.com
HMC-Europe GmbH Sterilisationstechnik
Kellerstr. 1 84577 Tüssling
Telefon: +49 8633 505 20 -0 Fax +49 8633 505 20 -99
Beste QualitätHöchster Komfort
Bezahlbar
Kammervolumen von 16 - 150 Liter

Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-06\Anzeigensatz-keine-Druck-PDFs\gerresheimer-TP-2013-06_216x303.indd Zuletzt gesichert: 28.10.13 (09:11:40 Uhr)
www.gerresheimer.com
Our broad Duma® Twist-Offproduct range | Child-resistant & senior-friendly closure
| Duma® OneLiner® closure
| Round & square containers
| Camera inspection systems & automated handling equipment
wenn die verwendeten Filter und Filter-gehäuse in ihren Spezifikationen nichtübereinstimmen. Die Verwendung vonGehäuse und Filter unterschiedlicherHersteller sind oft eine Ursache.
Einbauempfehlungfür Neuanlagen
Für eine effektive Sterilfiltration musssichergestellt sein, dass die Membrannicht beschädigt ist und das Filtersys-tem keine Leckagen aufweist. Ein geeig-netes Verfahren zur Prüfung hydropho-ber Filterelemente bildet der Wasser-intrusionstest [5]. Hier wird das hydro-phobe Filterelement (siehe Abb. 4) mitWasser geflutet und ein Testdruck, wel-
cher unterhalb des Penetrationsdrucksdes Filters liegt, auf das Messsystemgebracht. Der an der Filtermembrandurch Evaporation entstehende Was-serdampf passiert die Filtermembran.Eine Messung des Druckabfalls liefertdie Wasserintrusionsrate (WIR), welchesich mit der Bakterienrückhalterate desFilters korrelieren lässt [6, 7].
Einem Durchbruch oder Versagender Filterelemente (siehe Abb. 5) kannauf diese Weise frühzeitig erkannt undvorgebeugt werden.
Undichtigkeiten im System und ander O-Ringdichtung des Filters könnenso sicher festgestellt werden, wennnach der Installation ein Test durch-geführt wird.
Für einen Intrusionstest muss das Fil-tergehäuse mit Wasser gefüllt werden.Aus diesem Grund, muss vor dessenDurchführung sichergestellt sein, dassvon dem Filterelement keine Biogefähr-dung ausgehen kann. Nach dem Testkann das verwendete Testwasser auf ein-
fachem Wege entsorgt werden. Eine Ste-rilisation des Filterelements wird an die-ser Stelle zwingend erforderlich.
Die Forderung nach einem Integri-tätstest der Filterelemente kann daherwichtige Impulse liefern, auch die Ste-rilisation der Filterelemente zukünftigsicherzustellen und zu überwachen.Zusätzlich würde durch einen Inline-test eine automatisierte Dokumenta-tion von Filterüberwachung und Filter-wechsel ermöglicht.
Blick in die Zukunft:Behördenauflagen zurAbluftinaktivierung
Für die pharmazeutische Produktionist ein Integritätstest vor und nachder Filtration obligatorisch [4]. Aktu-elle Entwicklungen deuten darauf hin,dass dieses zukünftig auch für die Ab-luftfiltration in Laborautoklaven demStand der Technik entsprechen wird.Mit Erscheinen des Beschlusses Nr. 3/2009 des Ausschusses für biologischeArbeitsstoffe (ABAS), Stellungnahmezum Thema „Einbauempfehlung fürNeuanlagen, Nachrüstung oder Ergän-zung, zur Wahl der Abluftbehandlungvon Autoklaven“ [1] werden erstmaligkonkrete Anforderungen an die Abluft-nachbehandlung gestellt. In Abhängig-keit der erforderlichen Schutz- bzw. Si-cherheitsstufe, sowie ab Kammervolu-mina größer 54 L, wird eine Abluftfil-tration über zwei in Reihe geschalteteSterilfilter gefordert, die nach dem Ein-bau sowie in regelmäßigen Intervallenzu testen sind. Als geeignete Methodewird derWasserintrusionstest genannt.Neben dem ABAS-Beschluss enthältauch die zum Zeitpunkt der Erstellungdieser Ausarbeitung (Stand Januar2012) im Gründruck vorliegende VDI-Richtlinie VDI 6300 Blatt 1, „Gentech-nische Arbeiten in geschlossenen Sys-temen – Leitfaden zum sicheren Be-trieb gentechnischer Anlagen“ die For-derung, Abluftfilter aus Autoklaven aufihre Funktionsfähigkeit zu testen [8].
Fazit
Die Sterilfiltration hat sich für den Ein-satz in Laborautoklaven als geeigneteMethode zur Abluftreinigung etabliert.
Dies liegt im Besonderen daran, dassdie Sterilfiltration ein vom Filterher-steller qualifiziertes Verfahren darstelltmit für Kleinanlagen geringen Investi-tionskosten.
Jedoch ist die Sterilfiltration nur inVerbindung mit einem Integritätstestein wirklich sicheres Verfahren. Nebender einfachen Detektion von Filterver-sagen bis über die Feststellung von Le-ckagen im System, bedingt eine zu-künftige Forderung nach einer Integri-tätsprüfung der Sterilfilter ein sauberesProzessdesign der Abluftfiltration.
Danksagung
Wir danken dem Hessischen Ministe-rium für Wissenschaft und Kunst fürdie finanzielle Förderung unseres Ent-wicklungsprojekts: „Entwicklung einesmodularen, universell einsetzbarenWasserintrusionstests (WIT) für Klein-anlagen. Zudem danken wir unseremProjektpartner biomedis LaborserviceGmbH für die fruchtbare und vertrau-ensvolle Zusammenarbeit.
Fachliteratur[1] Einbauempfehlung für Neuanlagen,
Nachrüstung oder Ergänzung, zur Wahlder Abluftbehandlung von Autoklaven,Beschluss 3/2009 des ABAS, ELATEC.
[2] Verordnung über die Sicherheitsstufenund sicherheitsmaßnahmen bei gentech-nischen Arbeiten in gentechnischen An-lagen (Gentechniksicherheitsverordnung)in der Fassung der Bekanntgabe vom14. März 1995 (BGBI.I S. 297), zuletzt ge-ändert durch Artikel 4 der Verordnungvom 18. Dezember 2008 (BGBI.I S. 2768).
[3] Ausschuss für biologische Arbeitsstoffe(ABAS): Technische Regeln für biologi-sche Arbeitsstoffe 100, Schutzmaßnah-men für gezielte und nicht gezielte Tä-tigkeiten mit biologischen Arbeitsstoffenin Laboratorien (2006).
[4] U. Brendel-Thimmel, R. Jaenchen, F.Schlamp: Chemie Ingenieur Technik 2006,78, No. II.
[5] C. Grumbach, P. Grace, P. Czermak, T.Pillich, C. Rühl, K. Fey (2011): Einbau-empfehlung für Neuanlagen, SichereÜberwachung der Abluftbehandlung vonAutoklaven mittels Wasserintrusionstestmit hoher Temperaturstabilität; GIT La-borfachzeitschrift 55. 8. 540-542 Aug.
[6] P. Czermak, G. Catapano: PDA Pharma-ceutical Science an Technology, 57 (2003)4, S. 277-286.
[7] P. Czermak, G. Catapano: European Journalof Parenteral Sciences (2000), 5 : 3, 59-63.
[8] VDI-Richtlinie, VDI 6300 Blatt 1 (Gründruck)
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 322–326 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)326 Grumbach et al. . Abluftfiltration unter der Lupe
Dieses Projekt (HA-Projekt-Nr.: 307/11-52)wird im Rahmen von Hessen ModellProjekteaus Mitteln der LOEWE – Landes-Offensive zurEntwicklung Wissenschaftlich-ökonomischerExzellenz, Förderlinie 3: KMU-Verbundvor-haben gefördert.

Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-06\Anzeigensatz-keine-Druck-PDFs\gerresheimer-TP-2013-06_216x303.indd Zuletzt gesichert: 28.10.13 (09:11:40 Uhr)
www.gerresheimer.com
Our broad Duma® Twist-Offproduct range | Child-resistant & senior-friendly closure
| Duma® OneLiner® closure
| Round & square containers
| Camera inspection systems & automated handling equipment
wenn die verwendeten Filter und Filter-gehäuse in ihren Spezifikationen nichtübereinstimmen. Die Verwendung vonGehäuse und Filter unterschiedlicherHersteller sind oft eine Ursache.
Einbauempfehlungfür Neuanlagen
Für eine effektive Sterilfiltration musssichergestellt sein, dass die Membrannicht beschädigt ist und das Filtersys-tem keine Leckagen aufweist. Ein geeig-netes Verfahren zur Prüfung hydropho-ber Filterelemente bildet der Wasser-intrusionstest [5]. Hier wird das hydro-phobe Filterelement (siehe Abb. 4) mitWasser geflutet und ein Testdruck, wel-
cher unterhalb des Penetrationsdrucksdes Filters liegt, auf das Messsystemgebracht. Der an der Filtermembrandurch Evaporation entstehende Was-serdampf passiert die Filtermembran.Eine Messung des Druckabfalls liefertdie Wasserintrusionsrate (WIR), welchesich mit der Bakterienrückhalterate desFilters korrelieren lässt [6, 7].
Einem Durchbruch oder Versagender Filterelemente (siehe Abb. 5) kannauf diese Weise frühzeitig erkannt undvorgebeugt werden.
Undichtigkeiten im System und ander O-Ringdichtung des Filters könnenso sicher festgestellt werden, wennnach der Installation ein Test durch-geführt wird.
Für einen Intrusionstest muss das Fil-tergehäuse mit Wasser gefüllt werden.Aus diesem Grund, muss vor dessenDurchführung sichergestellt sein, dassvon dem Filterelement keine Biogefähr-dung ausgehen kann. Nach dem Testkann das verwendete Testwasser auf ein-
fachem Wege entsorgt werden. Eine Ste-rilisation des Filterelements wird an die-ser Stelle zwingend erforderlich.
Die Forderung nach einem Integri-tätstest der Filterelemente kann daherwichtige Impulse liefern, auch die Ste-rilisation der Filterelemente zukünftigsicherzustellen und zu überwachen.Zusätzlich würde durch einen Inline-test eine automatisierte Dokumenta-tion von Filterüberwachung und Filter-wechsel ermöglicht.
Blick in die Zukunft:Behördenauflagen zurAbluftinaktivierung
Für die pharmazeutische Produktionist ein Integritätstest vor und nachder Filtration obligatorisch [4]. Aktu-elle Entwicklungen deuten darauf hin,dass dieses zukünftig auch für die Ab-luftfiltration in Laborautoklaven demStand der Technik entsprechen wird.Mit Erscheinen des Beschlusses Nr. 3/2009 des Ausschusses für biologischeArbeitsstoffe (ABAS), Stellungnahmezum Thema „Einbauempfehlung fürNeuanlagen, Nachrüstung oder Ergän-zung, zur Wahl der Abluftbehandlungvon Autoklaven“ [1] werden erstmaligkonkrete Anforderungen an die Abluft-nachbehandlung gestellt. In Abhängig-keit der erforderlichen Schutz- bzw. Si-cherheitsstufe, sowie ab Kammervolu-mina größer 54 L, wird eine Abluftfil-tration über zwei in Reihe geschalteteSterilfilter gefordert, die nach dem Ein-bau sowie in regelmäßigen Intervallenzu testen sind. Als geeignete Methodewird derWasserintrusionstest genannt.Neben dem ABAS-Beschluss enthältauch die zum Zeitpunkt der Erstellungdieser Ausarbeitung (Stand Januar2012) im Gründruck vorliegende VDI-Richtlinie VDI 6300 Blatt 1, „Gentech-nische Arbeiten in geschlossenen Sys-temen – Leitfaden zum sicheren Be-trieb gentechnischer Anlagen“ die For-derung, Abluftfilter aus Autoklaven aufihre Funktionsfähigkeit zu testen [8].
Fazit
Die Sterilfiltration hat sich für den Ein-satz in Laborautoklaven als geeigneteMethode zur Abluftreinigung etabliert.
Dies liegt im Besonderen daran, dassdie Sterilfiltration ein vom Filterher-steller qualifiziertes Verfahren darstelltmit für Kleinanlagen geringen Investi-tionskosten.
Jedoch ist die Sterilfiltration nur inVerbindung mit einem Integritätstestein wirklich sicheres Verfahren. Nebender einfachen Detektion von Filterver-sagen bis über die Feststellung von Le-ckagen im System, bedingt eine zu-künftige Forderung nach einer Integri-tätsprüfung der Sterilfilter ein sauberesProzessdesign der Abluftfiltration.
Danksagung
Wir danken dem Hessischen Ministe-rium für Wissenschaft und Kunst fürdie finanzielle Förderung unseres Ent-wicklungsprojekts: „Entwicklung einesmodularen, universell einsetzbarenWasserintrusionstests (WIT) für Klein-anlagen. Zudem danken wir unseremProjektpartner biomedis LaborserviceGmbH für die fruchtbare und vertrau-ensvolle Zusammenarbeit.
Fachliteratur[1] Einbauempfehlung für Neuanlagen,
Nachrüstung oder Ergänzung, zur Wahlder Abluftbehandlung von Autoklaven,Beschluss 3/2009 des ABAS, ELATEC.
[2] Verordnung über die Sicherheitsstufenund sicherheitsmaßnahmen bei gentech-nischen Arbeiten in gentechnischen An-lagen (Gentechniksicherheitsverordnung)in der Fassung der Bekanntgabe vom14. März 1995 (BGBI.I S. 297), zuletzt ge-ändert durch Artikel 4 der Verordnungvom 18. Dezember 2008 (BGBI.I S. 2768).
[3] Ausschuss für biologische Arbeitsstoffe(ABAS): Technische Regeln für biologi-sche Arbeitsstoffe 100, Schutzmaßnah-men für gezielte und nicht gezielte Tä-tigkeiten mit biologischen Arbeitsstoffenin Laboratorien (2006).
[4] U. Brendel-Thimmel, R. Jaenchen, F.Schlamp: Chemie Ingenieur Technik 2006,78, No. II.
[5] C. Grumbach, P. Grace, P. Czermak, T.Pillich, C. Rühl, K. Fey (2011): Einbau-empfehlung für Neuanlagen, SichereÜberwachung der Abluftbehandlung vonAutoklaven mittels Wasserintrusionstestmit hoher Temperaturstabilität; GIT La-borfachzeitschrift 55. 8. 540-542 Aug.
[6] P. Czermak, G. Catapano: PDA Pharma-ceutical Science an Technology, 57 (2003)4, S. 277-286.
[7] P. Czermak, G. Catapano: European Journalof Parenteral Sciences (2000), 5 : 3, 59-63.
[8] VDI-Richtlinie, VDI 6300 Blatt 1 (Gründruck)
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 322–326 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)326 Grumbach et al. . Abluftfiltration unter der Lupe
Dieses Projekt (HA-Projekt-Nr.: 307/11-52)wird im Rahmen von Hessen ModellProjekteaus Mitteln der LOEWE – Landes-Offensive zurEntwicklung Wissenschaftlich-ökonomischerExzellenz, Förderlinie 3: KMU-Verbundvor-haben gefördert.

1.4 AMWHV (Arzneimittel- undWirkstoffherstellungs-verordnung) [5]:Im Abschnitt 2 (Allgemeine Anforde-rungen) und § 5 (Betriebsräume undAusrüstungen) werden ebenfalls An-forderungen festgeschrieben, dassu. a. Betriebsräume und Ausrüstun-gen auf ihre Eignung überprüft wer-den müssen (Qualifizierung), dassdiese ausreichend beleuchtet sindund geeignete klimatische Verhält-nisse aufweisen und dass diesegründlich zu reinigen und instandzu halten sind.
1.5 DIN EN ISO 14644 [6]:Die DIN EN ISO 14644 beschreibt inihren einzelnen Teilen verschie-denste Anforderungen an Reinräumeund zugehörige Reinraumbereiche.Angefangen mit der Klassifizierungder Luftreinheit anhand der Partikel-konzentration und deren fortlaufen-den Übereinstimmung (Teil 1 und 2)bis hin zu SD-Modulen (Teil 7) oderKlassifizierung der chemischenOberflächenreinheit (Teil 10 – Ent-wurf)
1.6 VDI Richtlinie, VDI 2083 [7]:Die VDI 2083 beschreibt in ihren ein-zelnen Blättern verschiedenste An-forderungen, die seitens der Rein-raumtechnik gestellt werden. Diesekönnen teils recht spezifisch sein(Blatt 16.1 – Barrieresysteme, Blatt17 – Reinheitstauglichkeit von Werk-stoffen oder Blatt 18 – Biokontami-nationskontrolle)
2. Hygienezonen
Bei der Definition der Hygienezonensind nicht nur die Anforderungenaus den bereits erwähnten Regula-rien zugrunde zu legen, sondern viel-mehr eine Kombination aus Pro-zessanforderungen, Produkt-anforderungen und Regularien. Da-bei ist der eigentliche Herstellungs-prozess von besonderer Wichtigkeit.Je nach Art und Weise der Herstel-lung resultieren daraus auch unter-schiedliche Reinheitsanforderun-gen:
. Herstellung von sterilen Produk-ten,
. Herstellung von nicht sterilenProdukten,
. Herstellung mit partikelabgeben-den Prozessschritten,
. Herstellung von Radiopharmaka,
. etc.Durch Kenntnis der Prozessanforde-rungen, Produktanforderungen undRegularien kann die notwendigeRaumklasse ermittelt werden. Da-raus kann man dann das entspre-chende Raumkonzept ableiten, was
die Anforderungen an die Ausfüh-rung der Räume, an die raumluft-technische Anlage und an die Moni-toringprogramme und -system be-schreibt.
3. Schalenmodell
Das Schalenmodel ist eine Form derkonzeptionellen Vorüberlegung umRäume höherer Reinheitsklassen zuschützen. Dabei folgt man demGrundsatz, dass der Reinheitsgradder Räume von außen nach innenzunimmt.
Wie in Abb. 1 (steriler Herstel-lungsprozess) ersichtlich, ist dieReinheitsklasse A/B die Reinheits-klasse, die von den übrigen Rein-heitsklassen wie „Schalen“ umgebenist. Diese Anordnung kann auch aufdie nicht sterilen Herstellungspro-zesse übertragen werden.
Außer dem Schalenmodell, undder daraus resultierenden Anord-nung der Räume zueinander, mussman bei der Planung auch die not-
wendigen Druckstufen beachten, so-wie die Anforderungen an Personal-und Materialschleusen berücksichti-gen.
4. Druckstufenkonzept
Durch Einrichtung von unterschied-lichen Druckstufen wird gewollterund ungewollter Luftaustausch zwi-schen verschiedenen Räumen/Syste-men gezielt gesteuert.
Im Allgemeinen haben Räume mithöherer Reinraumklassifizierung einhöheres Druckniveau als die umge-benden Räume (Vermeiden von Ein-dringen kontaminierter Luft – Pro-duktschutz).
In seltenen Spezialfällen (gesund-heitsgefährdende Stoffe) könnenRäume (z.B. Isolatoren) mit höhererReinraumklassifizierung auch einniedrigeres Druckniveau als die um-gebenden Räume haben (Vermeidendes Austretens gesundheitsgefähr-dender Stoffe – Personenschutz).
Die Druckdifferenz vom Reinraumzu den umgebenden Räumen sollteetwa 10 – 15 Pascal betragen [1].
5. Schleusenkonzept
Gemäß Definition des EG-Leitfadeneiner Guten Herstellungspraxis fürArzneimittel – Teil 1 [1], ist eineSchleuse ein geschlossener Raummit zwei oder mehr Türen, der sichzwischen zwei oder mehreren Räu-men, z.B. verschiedener Reinheits-klassen, befindet und dem Zweckdient, den Luftstrom zwischen denRäumen unter Kontrolle zu halten,wenn diese betreten werden müssen.Eine Schleuse kann entweder für Per-sonen oder für Waren vorgesehenund entsprechend benutzt werden(Abb. 2).
Schleusen sind Umwandlungs-zonen für Personal und Material,die zwischen Bereichen mit unter-schiedlichen Raumklassen angeord-net sind und müssen jeweils die An-forderungen der höheren Raum-klasse hinsichtlich Lüftung, Ausstat-tung und Hygieneanforderungen er-füllen.
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 329Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Support Bereich
unkontrollierter Bereich
RRKL
DRRKL
CRRKL
A/B
Abb. 1 Schalenmodell steriler Herstellungs-prozess (Quelle: G. Heuwes).
Ausstattung, Gestaltung undQualifizierung von RäumenRäume, Luft, Technik
Guido Heuwes . Ingenieurbüro Heuwes, Vallendar
Korrespondenz: Guido Heuwes, Ingenieurbüro Heuwes, Oskar-Hasenclever-Straße 8, 56179 Vallendar;e-Mail: [email protected]
ZusammenfassungWenn man sich mit der Thematik der Ausstattung und Gestaltung von Räumen beschäf-tigt, muss man sich zum einen mit den zugrundeliegenden rechtlichen Anforderungenauseinandersetzen, die an die Umgebung und Ausstattung von Räumen gestellt werden.Zum anderen sind aber auch konzeptionelle Vorüberlegungen notwendig, die dann zumeigentlichen Raum, dessen Gestaltung und Beschaffenheit führen. Dazu stehen ver-schiedene Instrumente zur Verfügung:. Definition der Hygienezonen: abgeleitet aus den Regularien, den Prozessanforderungen
und den Produktanforderungen,. Schalenmodell: Beschreibung der Anordnung der Räume zueinander,. Druckstufenkonzept: Beschreibung von unterschiedlichen Druckniveau in Räumen
unterschiedlicher Reinheitsklassen,. Schleusenkonzept: Festlegung der Zugänge zu den einzelnen Räumen unterschiedlicher
Reinheitsklassen und. Material- und Personalfluss: Beschreibung von Material- und Personalbewegungen.Der Prozess der Planung, Realisierung und Inbetriebnahme wird begleitet durch die Qua-lifizierung. Diese erbringt in Form der Risikoanalyse (RA), Design Qualilfication (DQ),Installation Qualification (IQ), Operational Qualilfication (OQ) und Performance Qualifi-cation (PQ) den dokumentierten Nachweis, dass die errichteten Räume für den bestim-mungsgemäßen Gebrauch geeignet sind.
1. Rechtliche Anforde-rungen/Regelwerke
Es gibt eine Vielzahl von Regularien,die bei der Planung und Realisierungvon Räumen zu berücksichtigensind. Da wir es in der pharmazeuti-schen Industrie mit vielfältigen Pro-dukten unterschiedlicher Wirkungs-spektren und Wirkstoffen sowie ver-schiedenster Darreichungsformen zutun haben, seien hier nur einigeexemplarisch aufgeführt.
1.1 EG-Leitfaden einer GutenHerstellungspraxis fürArzneimittel – Teil 1 [1]:Im Kapitel 3 (Räumlichkeit und Aus-rüstung), Kapitel 5 (Produktion) und
im Anhang 15 (Qualifizierung undValidierung) wird u. a. eine einfacheReinigung und einfache Wartung ge-fordert. Darüber hinaus sollen dieProduktionsbereiche ausreichendbelüftet sein und Kreuzkontamina-tion soll verhindert werden. Des Wei-teren ist für eine ausreichende Belüf-tung zu sorgen und auf eine GMP-konforme Ausführung zu achten.Die Anordnung von Abflüssen istfestgeschrieben.
1.2 EG-Leitfaden einer GutenHerstellungspraxis fürWirkstoffe – Teil 2 [2]:Im Kapitel 4 (Gebäude und Anlagen)und Kapitel 12 (Validierung) wirdu. a. eine angemessene Belüftung
inkl. Kontrolle der Mikroorganismengefordert, sowie ein Schutz vorKreuzkontamination. Installierte Ab-flüsse sollen drucklos ausgeführtwerden. Es besteht eine generelleForderung an die Qualifizierungund Validierung.
1.3 21 CFR Part 210 und211 [3, 4]:Wenn man sich mit diesen Regelwer-ken der U.S. Food and Drug Adminis-tration beschäftigt, findet man einehohe inhaltliche Übereinstimmungzu den beiden Erstgenannten. Auchhier haben wir die Anforderung nachangemessener Belüftung, Kontrolleauf Mikroorganismen, Verhinderungvon Kreuzkontamination, etc.
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)328 Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen

1.4 AMWHV (Arzneimittel- undWirkstoffherstellungs-verordnung) [5]:Im Abschnitt 2 (Allgemeine Anforde-rungen) und § 5 (Betriebsräume undAusrüstungen) werden ebenfalls An-forderungen festgeschrieben, dassu. a. Betriebsräume und Ausrüstun-gen auf ihre Eignung überprüft wer-den müssen (Qualifizierung), dassdiese ausreichend beleuchtet sindund geeignete klimatische Verhält-nisse aufweisen und dass diesegründlich zu reinigen und instandzu halten sind.
1.5 DIN EN ISO 14644 [6]:Die DIN EN ISO 14644 beschreibt inihren einzelnen Teilen verschie-denste Anforderungen an Reinräumeund zugehörige Reinraumbereiche.Angefangen mit der Klassifizierungder Luftreinheit anhand der Partikel-konzentration und deren fortlaufen-den Übereinstimmung (Teil 1 und 2)bis hin zu SD-Modulen (Teil 7) oderKlassifizierung der chemischenOberflächenreinheit (Teil 10 – Ent-wurf)
1.6 VDI Richtlinie, VDI 2083 [7]:Die VDI 2083 beschreibt in ihren ein-zelnen Blättern verschiedenste An-forderungen, die seitens der Rein-raumtechnik gestellt werden. Diesekönnen teils recht spezifisch sein(Blatt 16.1 – Barrieresysteme, Blatt17 – Reinheitstauglichkeit von Werk-stoffen oder Blatt 18 – Biokontami-nationskontrolle)
2. Hygienezonen
Bei der Definition der Hygienezonensind nicht nur die Anforderungenaus den bereits erwähnten Regula-rien zugrunde zu legen, sondern viel-mehr eine Kombination aus Pro-zessanforderungen, Produkt-anforderungen und Regularien. Da-bei ist der eigentliche Herstellungs-prozess von besonderer Wichtigkeit.Je nach Art und Weise der Herstel-lung resultieren daraus auch unter-schiedliche Reinheitsanforderun-gen:
. Herstellung von sterilen Produk-ten,
. Herstellung von nicht sterilenProdukten,
. Herstellung mit partikelabgeben-den Prozessschritten,
. Herstellung von Radiopharmaka,
. etc.Durch Kenntnis der Prozessanforde-rungen, Produktanforderungen undRegularien kann die notwendigeRaumklasse ermittelt werden. Da-raus kann man dann das entspre-chende Raumkonzept ableiten, was
die Anforderungen an die Ausfüh-rung der Räume, an die raumluft-technische Anlage und an die Moni-toringprogramme und -system be-schreibt.
3. Schalenmodell
Das Schalenmodel ist eine Form derkonzeptionellen Vorüberlegung umRäume höherer Reinheitsklassen zuschützen. Dabei folgt man demGrundsatz, dass der Reinheitsgradder Räume von außen nach innenzunimmt.
Wie in Abb. 1 (steriler Herstel-lungsprozess) ersichtlich, ist dieReinheitsklasse A/B die Reinheits-klasse, die von den übrigen Rein-heitsklassen wie „Schalen“ umgebenist. Diese Anordnung kann auch aufdie nicht sterilen Herstellungspro-zesse übertragen werden.
Außer dem Schalenmodell, undder daraus resultierenden Anord-nung der Räume zueinander, mussman bei der Planung auch die not-
wendigen Druckstufen beachten, so-wie die Anforderungen an Personal-und Materialschleusen berücksichti-gen.
4. Druckstufenkonzept
Durch Einrichtung von unterschied-lichen Druckstufen wird gewollterund ungewollter Luftaustausch zwi-schen verschiedenen Räumen/Syste-men gezielt gesteuert.
Im Allgemeinen haben Räume mithöherer Reinraumklassifizierung einhöheres Druckniveau als die umge-benden Räume (Vermeiden von Ein-dringen kontaminierter Luft – Pro-duktschutz).
In seltenen Spezialfällen (gesund-heitsgefährdende Stoffe) könnenRäume (z.B. Isolatoren) mit höhererReinraumklassifizierung auch einniedrigeres Druckniveau als die um-gebenden Räume haben (Vermeidendes Austretens gesundheitsgefähr-dender Stoffe – Personenschutz).
Die Druckdifferenz vom Reinraumzu den umgebenden Räumen sollteetwa 10 – 15 Pascal betragen [1].
5. Schleusenkonzept
Gemäß Definition des EG-Leitfadeneiner Guten Herstellungspraxis fürArzneimittel – Teil 1 [1], ist eineSchleuse ein geschlossener Raummit zwei oder mehr Türen, der sichzwischen zwei oder mehreren Räu-men, z.B. verschiedener Reinheits-klassen, befindet und dem Zweckdient, den Luftstrom zwischen denRäumen unter Kontrolle zu halten,wenn diese betreten werden müssen.Eine Schleuse kann entweder für Per-sonen oder für Waren vorgesehenund entsprechend benutzt werden(Abb. 2).
Schleusen sind Umwandlungs-zonen für Personal und Material,die zwischen Bereichen mit unter-schiedlichen Raumklassen angeord-net sind und müssen jeweils die An-forderungen der höheren Raum-klasse hinsichtlich Lüftung, Ausstat-tung und Hygieneanforderungen er-füllen.
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 329Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Support Bereich
unkontrollierter Bereich
RRKL
DRRKL
CRRKL
A/B
Abb. 1 Schalenmodell steriler Herstellungs-prozess (Quelle: G. Heuwes).
Ausstattung, Gestaltung undQualifizierung von RäumenRäume, Luft, Technik
Guido Heuwes . Ingenieurbüro Heuwes, Vallendar
Korrespondenz: Guido Heuwes, Ingenieurbüro Heuwes, Oskar-Hasenclever-Straße 8, 56179 Vallendar;e-Mail: [email protected]
ZusammenfassungWenn man sich mit der Thematik der Ausstattung und Gestaltung von Räumen beschäf-tigt, muss man sich zum einen mit den zugrundeliegenden rechtlichen Anforderungenauseinandersetzen, die an die Umgebung und Ausstattung von Räumen gestellt werden.Zum anderen sind aber auch konzeptionelle Vorüberlegungen notwendig, die dann zumeigentlichen Raum, dessen Gestaltung und Beschaffenheit führen. Dazu stehen ver-schiedene Instrumente zur Verfügung:. Definition der Hygienezonen: abgeleitet aus den Regularien, den Prozessanforderungen
und den Produktanforderungen,. Schalenmodell: Beschreibung der Anordnung der Räume zueinander,. Druckstufenkonzept: Beschreibung von unterschiedlichen Druckniveau in Räumen
unterschiedlicher Reinheitsklassen,. Schleusenkonzept: Festlegung der Zugänge zu den einzelnen Räumen unterschiedlicher
Reinheitsklassen und. Material- und Personalfluss: Beschreibung von Material- und Personalbewegungen.Der Prozess der Planung, Realisierung und Inbetriebnahme wird begleitet durch die Qua-lifizierung. Diese erbringt in Form der Risikoanalyse (RA), Design Qualilfication (DQ),Installation Qualification (IQ), Operational Qualilfication (OQ) und Performance Qualifi-cation (PQ) den dokumentierten Nachweis, dass die errichteten Räume für den bestim-mungsgemäßen Gebrauch geeignet sind.
1. Rechtliche Anforde-rungen/Regelwerke
Es gibt eine Vielzahl von Regularien,die bei der Planung und Realisierungvon Räumen zu berücksichtigensind. Da wir es in der pharmazeuti-schen Industrie mit vielfältigen Pro-dukten unterschiedlicher Wirkungs-spektren und Wirkstoffen sowie ver-schiedenster Darreichungsformen zutun haben, seien hier nur einigeexemplarisch aufgeführt.
1.1 EG-Leitfaden einer GutenHerstellungspraxis fürArzneimittel – Teil 1 [1]:Im Kapitel 3 (Räumlichkeit und Aus-rüstung), Kapitel 5 (Produktion) und
im Anhang 15 (Qualifizierung undValidierung) wird u. a. eine einfacheReinigung und einfache Wartung ge-fordert. Darüber hinaus sollen dieProduktionsbereiche ausreichendbelüftet sein und Kreuzkontamina-tion soll verhindert werden. Des Wei-teren ist für eine ausreichende Belüf-tung zu sorgen und auf eine GMP-konforme Ausführung zu achten.Die Anordnung von Abflüssen istfestgeschrieben.
1.2 EG-Leitfaden einer GutenHerstellungspraxis fürWirkstoffe – Teil 2 [2]:Im Kapitel 4 (Gebäude und Anlagen)und Kapitel 12 (Validierung) wirdu. a. eine angemessene Belüftung
inkl. Kontrolle der Mikroorganismengefordert, sowie ein Schutz vorKreuzkontamination. Installierte Ab-flüsse sollen drucklos ausgeführtwerden. Es besteht eine generelleForderung an die Qualifizierungund Validierung.
1.3 21 CFR Part 210 und211 [3, 4]:Wenn man sich mit diesen Regelwer-ken der U.S. Food and Drug Adminis-tration beschäftigt, findet man einehohe inhaltliche Übereinstimmungzu den beiden Erstgenannten. Auchhier haben wir die Anforderung nachangemessener Belüftung, Kontrolleauf Mikroorganismen, Verhinderungvon Kreuzkontamination, etc.
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)328 Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen

. Auswahl geeigneter Materialien,deren Oberflächen glatt und mög-lichst fugenlos ist.
. Keine Bodenabläufe Klasse A/B,ansonsten atmosphärische Tren-nung.
. Besonderheiten bei Förderbändernzum Austragen aus dem Raumsind zu beachten.
8. Anforderungen anOberflächen inReinräumen
Wenn von Oberflächen im Reinraumgesprochen wird, betrifft dies sowohldie Umschließungsflächen, die demReinraum zugewandt sind, als auchggf. produktberührte Flächen.
Die Oberflächen sollten daher fol-gende Eigenschaften besitzen:. nicht partikelabgebend,. glatt, porenfrei, abriebfest, leichtreinigbar,
. beständig gegen Produkt, Rei-nigungs- und Desinfektionsmittel,
. beständig gegen Alterung undKorrosion und
. antistatisch und geerdet (Vermei-dung von Partikelablagerungdurch elektrostatische Aufladungder Oberfläche)
9. Gestaltungshinweisefür Reinraumoberflächen
Für alle Reinraumoberflächen gilt,dass nachMöglichkeit keine horizon-talen Flächen und keine unzugäng-lichen Ecken und Nischen vorhandensind.
Die Ausrüstungsaufstellung sollteso erfolgen, dass eine Reinigung ein-fach und zuverlässig möglich ist z.B.durch:. gerundete Übergänge zu Wänden,Decken und Böden,
. Bodenfreiheit unter den Anlagenund
. Zugänglichkeit von möglichst allenSeiten.
Eine Partikelansammlung kann nurdann vermieden werden, wenn einegeeignete Ausführung und Anord-nung von Oberflächen realisiertwird.
10. Anforderungen anReinraum-Wand- undDeckensystem sowieBodenbeläge
10.1 Allgemein:. Schallschutz, Wärmedämmung,mechanische und statische Belas-tung, Druck-/Dichtheitsanforde-rungen, etc.
. Sicherheitsaspekte: z.B. Brand-schutz, Blendfreiheit, Rutschfrei-heit, Fenster zur Sichtverbindungnach außen, Fluchtwege (Türen,die nur von innen zu öffnen sindfür Notfälle, etc.)
. Oberflächen reinraumgerecht(glatt, dicht, beständig, abwasch-bar, antistatisch, etc.)
. Beständigkeit gegen Produkt undProzessmedien
. Beständigkeit gegen Chemikalien(Reinigungs-, Desinfektionsmittel,etc.)
. Lufteinlässe und Luftauslässe so-wie andere Einbauten (z.B.Leuchten) spaltfrei und flächen-bündig eingebaut
. Türen und Zargen flächenbündig,ohne Türschwellen, Aufhängungmit abriebarmer Mechanik, ohneBodenreibung (absenkbare Dicht-leiste)
. Durchführungen von Rohrleitun-gen, Kabelkanälen und Ausrüstun-gen luft- und partikeldicht (z.B.Rohrdurchführungen, Rosetten,etc.)
. Fugen elastisch abgedichtet mitz.B. Silikon
. Reinraumgerechte Boden-, De-cken-, Wand- und Ausrüstungs-anschlüsse (z.B. gerundet, mitHohlkehle, etc.)
. Montage-, wartungs- und rei-nigungsfreundlich, leicht anpass-und abdichtbar
10.2 Wandsysteme (spezifisch):. Rammschutz im Bereich vonStapler-, Hubwagenverkehr
10.3 Deckensysteme(spezifisch):. Bei abgehängten KassettendeckenVermeidung des Eindringens von
Partikeln in Reinraum aus De-ckenservicebereich durch spalt-freien Einbau von Deckenpaneelen(Kassetten). z.B. Ausspritzen derFugen mit Silikon
. Begehbare Decken vorteilhaft fürWartungsarbeiten an Deckenele-menten, Luftführung, Rohr- undElektroleitungen, Armaturen, etc.sowie für zusätzliche Installatio-nen
10.4 Bodenbeläge (spezifisch):. Belastbarkeit gegen mechanischeEinflüsse (z.B. Stapler-, Hubwa-genverkehr)
. Integrierte leitfähige Schicht fürEx-Bereiche und ggf. FTS-Fahr-zeugverkehr (Fahrerlose Trans-portsysteme)
. Nach Möglichkeit fugenlose Aus-führung. Bei gefliesten Böden aufflächenbündige Fugen achten
. Bei Gitterrosten keine scharfenKanten (Beschädigung von Schu-hen). Gitterroste nach Möglichkeitganz vermeiden (schlecht zu rei-nigen)
. Bodengefälle im Bereich von Ab-flüssen vorsehen (ca. 2 %)
. Abflüsse so ausführen, dass keineVerunreinigungen in den Raumzurückgelangen (Rückstauklap-pen, Geruchsverschlüsse, Spezial-design – keine direkte Verbindungmit Abwasserkanalsystem)
11. Raumbuch
Alle Anforderungen, die in den zuvorgenannten Abschnitten aufgeführtwurden, müssen natürlich sinnvol-lerweise in einem Dokument zusam-mengefasst beschrieben werden.
Dies sollte im sogenannten Raum-buch geschehen. Mit dem Raumbuchwird jeder Raum im Detail beschrie-ben, sowohl hinsichtlich der Raum-ausstattung (z.B. Wände, Deckenund Böden sowie Raumhöhe und-fläche) als auch hinsichtlich derRaumparameter (z.B. Temperatur,Luftfeuchtigkeit und Luftwechsel-zahlen). In der Praxis hat sich ge-zeigt, dass das Raumbuch aus GMP-Sicht zweigeteilt aufgebaut sein soll-
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 331Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Schleusen haben 2 Zonen (Tren-nung durch Reinraumbank oderKennzeich-nung am Bo-den). Die Zoneder „unreine-ren“ Seite, inder die Um-wandlung vor-bereitet wird(z.B. Ablegender Straßen-leidung, Rei-nigen der Ver-packungen)und die Zoneder „reineren“Seite, in derdie Umwandlung vollendet wird (An-legen der Reinraumkleidung).
Der Wechsel von Personen zwi-schen unterschiedlichen Hygienezo-nen (Reinraumklassen) ist nur überPersonalschleusen möglich. Für denzu betretenden Raum ist die jeweilsfür die entsprechende Hygieneklassevorgesehene Bekleidung anzulegenund Reinigungs-/Desinfektionsmaß-nahmen sind durchzuführen.
Material wird über getrennte Ma-terialschleusen eingeschleust. Gege-benenfalls sind Reinigungs- und Des-infektionsschritte an Material undVerpackung durchzuführen.
Drücke in den Schleusen sind nor-malerweise so angelegt, dass immerein Druckgefälle vom reineren zumunreineren Bereich vorherrscht.
6. Material- / Personalfluss
Bei der Entwicklung des Material-/Personalflusskonzeptes sollte aufeine logische Anordnung der Räumegemäß notwendiger Herstellungs-schritte geachtet werden. Dabei istauch der Mengenfluss an Rohstoffen,Verpackungs- und Hilfsmitteln zu be-rücksichtigen, sowie die Art undWeise der Produktförderung (Gravi-metrische Förderung des Produktes(z.B. Solida), Produktförderung mit-tels Pumpen oder Vakuum (z.B. Li-quida)). Dies gilt auch für die herzu-stellende Mengen (z.B. Infusions-lösungen in Drei-Schichtbetrieb oder
feste Arzneiformen für klinische For-schung) und deren Realisierung in
einer Produk-tion in Ein-oder Mehrebe-nenkonzept.
Des Wei-teren ist da-rauf zu ach-ten, dass Kon-taminationen(z.B. Kreuz-kontaminatio-nen) und Ver-wechslungenvermiedenwerden.
Aus diesemGrund müssen die Verarbeitungs-schritte in geeigneter Umgebung(Reinräume, Hygienezonen) durch-geführt werden, inklusive definier-tem Wechsel zwischen verschiede-nen Bereichen (Schleusen). Dabeisind notwendige Maßnahmen beimWechseln von Hygienezonen (z.B.Umpalettieren von Holz- auf Rein-raumpaletten) umzusetzen.
Generell sollte die Personenzahl,die sich in kritischen Bereichen (offe-nes Produkt) aufhält, minimiert wer-den. Bei der Herstellung sollten mög-lichst geschlossene Systeme verwen-det werden, insbesondere bei hoch-potenten oder kritischen Arzneistof-fen (z.B. Cytostatika, Penicilline).
Die Planung sollte, wie bereitsbeim Schalenmodell beschrieben,von innen nach außen mit dem ge-planten Prozess als führende Größeerfolgen.
Für den Materialfluss gilt außer-dem, dass die Möglichkeit der Abrei-nigung von „Niedriger Hygienezone“nach „Höherer Hygienezone“ gege-ben ist. Dabei ist besonders hervor-zuheben, dass eine Trennung vonverschmutztem und sauberemMate-rial, insbesondere von Ausrüstungen,vor und nach Reinigungsschrittenrealisiert wird.
Ebenso sind kurze und gerichtetePersonalwege zur Vermeidung vonBelastungen von reinen Bereichen(Vermeiden von Personalwegendurch Prozessräume) einzuplanen.
Darüber hinaus sind Besucher imPersonalfluss zu berücksichtigen(z.B. über geeignete Besucherschleu-sen oder separat angeordnete Besu-chergänge).
7. Generelle Design-Anforderungen anReinräume
Im Nachfolgenden sind einige gene-rellen Design-Anforderungen auf-gelistet, die sich z.T. aus den zugrun-deliegenden Regularien ableiten las-sen, zum anderen aber auch aus derPraxis resultieren:. Reinräume müssen so beschaffensein, dass die festgelegte Rein-raumklasse eingehalten wird.
. Zugang von Personal und Einbrin-gung von Material sollte über ge-trennte Schleusensysteme erfolgen.
. Angrenzende Schleusen undDurchreichen müssen den glei-chen Anforderungen wie derReinraum entsprechen.
. Türspalte und andere Raumöff-nungen sind so klein wie möglichzu halten, zur Minimierung vonLeckluftströmen.
. Medienzuführung und -abführungsind so auszuführen, dass keinezusätzliche Partikelbelastung ent-steht (möglichst vertikale Ver-legung mit Rosetten an denDurchführungen).
. Einbauten in Decken und Wände(Türen, Fenster, Leuchten, etc.)sind möglichst flächenbündig,spaltfrei und partikeldicht aus-zuführen.
. Fugenmaterial sollte hochelas-tisch, nicht klebend, beständig undbakterizid/fungizid sein, zur Ver-meidung von Bakterien-/Pilz-wachstum.
. Es sollte nur notwendige Ausrüs-tungen installieren sein.
. Kabel sollten in geschlossenenKanälen verlegt werden (Vermei-dung von Partikelablagerung).
. Bodenbeläge sollten glatt, wider-standsfähig, mit geringem Abrieb,elektrisch leitend/geerdet sein.Wandübergänge sollten gerundetausgeführt werden.
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)330 Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Umwandlungszone (Schleuse)
Personal
Ausrüstung
RohstoffeRRKL
C
RRKLD
Abb. 2 Schleusenkonzept (Quelle: G. Heuwes).

. Auswahl geeigneter Materialien,deren Oberflächen glatt und mög-lichst fugenlos ist.
. Keine Bodenabläufe Klasse A/B,ansonsten atmosphärische Tren-nung.
. Besonderheiten bei Förderbändernzum Austragen aus dem Raumsind zu beachten.
8. Anforderungen anOberflächen inReinräumen
Wenn von Oberflächen im Reinraumgesprochen wird, betrifft dies sowohldie Umschließungsflächen, die demReinraum zugewandt sind, als auchggf. produktberührte Flächen.
Die Oberflächen sollten daher fol-gende Eigenschaften besitzen:. nicht partikelabgebend,. glatt, porenfrei, abriebfest, leichtreinigbar,
. beständig gegen Produkt, Rei-nigungs- und Desinfektionsmittel,
. beständig gegen Alterung undKorrosion und
. antistatisch und geerdet (Vermei-dung von Partikelablagerungdurch elektrostatische Aufladungder Oberfläche)
9. Gestaltungshinweisefür Reinraumoberflächen
Für alle Reinraumoberflächen gilt,dass nachMöglichkeit keine horizon-talen Flächen und keine unzugäng-lichen Ecken und Nischen vorhandensind.
Die Ausrüstungsaufstellung sollteso erfolgen, dass eine Reinigung ein-fach und zuverlässig möglich ist z.B.durch:. gerundete Übergänge zu Wänden,Decken und Böden,
. Bodenfreiheit unter den Anlagenund
. Zugänglichkeit von möglichst allenSeiten.
Eine Partikelansammlung kann nurdann vermieden werden, wenn einegeeignete Ausführung und Anord-nung von Oberflächen realisiertwird.
10. Anforderungen anReinraum-Wand- undDeckensystem sowieBodenbeläge
10.1 Allgemein:. Schallschutz, Wärmedämmung,mechanische und statische Belas-tung, Druck-/Dichtheitsanforde-rungen, etc.
. Sicherheitsaspekte: z.B. Brand-schutz, Blendfreiheit, Rutschfrei-heit, Fenster zur Sichtverbindungnach außen, Fluchtwege (Türen,die nur von innen zu öffnen sindfür Notfälle, etc.)
. Oberflächen reinraumgerecht(glatt, dicht, beständig, abwasch-bar, antistatisch, etc.)
. Beständigkeit gegen Produkt undProzessmedien
. Beständigkeit gegen Chemikalien(Reinigungs-, Desinfektionsmittel,etc.)
. Lufteinlässe und Luftauslässe so-wie andere Einbauten (z.B.Leuchten) spaltfrei und flächen-bündig eingebaut
. Türen und Zargen flächenbündig,ohne Türschwellen, Aufhängungmit abriebarmer Mechanik, ohneBodenreibung (absenkbare Dicht-leiste)
. Durchführungen von Rohrleitun-gen, Kabelkanälen und Ausrüstun-gen luft- und partikeldicht (z.B.Rohrdurchführungen, Rosetten,etc.)
. Fugen elastisch abgedichtet mitz.B. Silikon
. Reinraumgerechte Boden-, De-cken-, Wand- und Ausrüstungs-anschlüsse (z.B. gerundet, mitHohlkehle, etc.)
. Montage-, wartungs- und rei-nigungsfreundlich, leicht anpass-und abdichtbar
10.2 Wandsysteme (spezifisch):. Rammschutz im Bereich vonStapler-, Hubwagenverkehr
10.3 Deckensysteme(spezifisch):. Bei abgehängten KassettendeckenVermeidung des Eindringens von
Partikeln in Reinraum aus De-ckenservicebereich durch spalt-freien Einbau von Deckenpaneelen(Kassetten). z.B. Ausspritzen derFugen mit Silikon
. Begehbare Decken vorteilhaft fürWartungsarbeiten an Deckenele-menten, Luftführung, Rohr- undElektroleitungen, Armaturen, etc.sowie für zusätzliche Installatio-nen
10.4 Bodenbeläge (spezifisch):. Belastbarkeit gegen mechanischeEinflüsse (z.B. Stapler-, Hubwa-genverkehr)
. Integrierte leitfähige Schicht fürEx-Bereiche und ggf. FTS-Fahr-zeugverkehr (Fahrerlose Trans-portsysteme)
. Nach Möglichkeit fugenlose Aus-führung. Bei gefliesten Böden aufflächenbündige Fugen achten
. Bei Gitterrosten keine scharfenKanten (Beschädigung von Schu-hen). Gitterroste nach Möglichkeitganz vermeiden (schlecht zu rei-nigen)
. Bodengefälle im Bereich von Ab-flüssen vorsehen (ca. 2 %)
. Abflüsse so ausführen, dass keineVerunreinigungen in den Raumzurückgelangen (Rückstauklap-pen, Geruchsverschlüsse, Spezial-design – keine direkte Verbindungmit Abwasserkanalsystem)
11. Raumbuch
Alle Anforderungen, die in den zuvorgenannten Abschnitten aufgeführtwurden, müssen natürlich sinnvol-lerweise in einem Dokument zusam-mengefasst beschrieben werden.
Dies sollte im sogenannten Raum-buch geschehen. Mit dem Raumbuchwird jeder Raum im Detail beschrie-ben, sowohl hinsichtlich der Raum-ausstattung (z.B. Wände, Deckenund Böden sowie Raumhöhe und-fläche) als auch hinsichtlich derRaumparameter (z.B. Temperatur,Luftfeuchtigkeit und Luftwechsel-zahlen). In der Praxis hat sich ge-zeigt, dass das Raumbuch aus GMP-Sicht zweigeteilt aufgebaut sein soll-
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 331Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Schleusen haben 2 Zonen (Tren-nung durch Reinraumbank oderKennzeich-nung am Bo-den). Die Zoneder „unreine-ren“ Seite, inder die Um-wandlung vor-bereitet wird(z.B. Ablegender Straßen-leidung, Rei-nigen der Ver-packungen)und die Zoneder „reineren“Seite, in derdie Umwandlung vollendet wird (An-legen der Reinraumkleidung).
Der Wechsel von Personen zwi-schen unterschiedlichen Hygienezo-nen (Reinraumklassen) ist nur überPersonalschleusen möglich. Für denzu betretenden Raum ist die jeweilsfür die entsprechende Hygieneklassevorgesehene Bekleidung anzulegenund Reinigungs-/Desinfektionsmaß-nahmen sind durchzuführen.
Material wird über getrennte Ma-terialschleusen eingeschleust. Gege-benenfalls sind Reinigungs- und Des-infektionsschritte an Material undVerpackung durchzuführen.
Drücke in den Schleusen sind nor-malerweise so angelegt, dass immerein Druckgefälle vom reineren zumunreineren Bereich vorherrscht.
6. Material- / Personalfluss
Bei der Entwicklung des Material-/Personalflusskonzeptes sollte aufeine logische Anordnung der Räumegemäß notwendiger Herstellungs-schritte geachtet werden. Dabei istauch der Mengenfluss an Rohstoffen,Verpackungs- und Hilfsmitteln zu be-rücksichtigen, sowie die Art undWeise der Produktförderung (Gravi-metrische Förderung des Produktes(z.B. Solida), Produktförderung mit-tels Pumpen oder Vakuum (z.B. Li-quida)). Dies gilt auch für die herzu-stellende Mengen (z.B. Infusions-lösungen in Drei-Schichtbetrieb oder
feste Arzneiformen für klinische For-schung) und deren Realisierung in
einer Produk-tion in Ein-oder Mehrebe-nenkonzept.
Des Wei-teren ist da-rauf zu ach-ten, dass Kon-taminationen(z.B. Kreuz-kontaminatio-nen) und Ver-wechslungenvermiedenwerden.
Aus diesemGrund müssen die Verarbeitungs-schritte in geeigneter Umgebung(Reinräume, Hygienezonen) durch-geführt werden, inklusive definier-tem Wechsel zwischen verschiede-nen Bereichen (Schleusen). Dabeisind notwendige Maßnahmen beimWechseln von Hygienezonen (z.B.Umpalettieren von Holz- auf Rein-raumpaletten) umzusetzen.
Generell sollte die Personenzahl,die sich in kritischen Bereichen (offe-nes Produkt) aufhält, minimiert wer-den. Bei der Herstellung sollten mög-lichst geschlossene Systeme verwen-det werden, insbesondere bei hoch-potenten oder kritischen Arzneistof-fen (z.B. Cytostatika, Penicilline).
Die Planung sollte, wie bereitsbeim Schalenmodell beschrieben,von innen nach außen mit dem ge-planten Prozess als führende Größeerfolgen.
Für den Materialfluss gilt außer-dem, dass die Möglichkeit der Abrei-nigung von „Niedriger Hygienezone“nach „Höherer Hygienezone“ gege-ben ist. Dabei ist besonders hervor-zuheben, dass eine Trennung vonverschmutztem und sauberemMate-rial, insbesondere von Ausrüstungen,vor und nach Reinigungsschrittenrealisiert wird.
Ebenso sind kurze und gerichtetePersonalwege zur Vermeidung vonBelastungen von reinen Bereichen(Vermeiden von Personalwegendurch Prozessräume) einzuplanen.
Darüber hinaus sind Besucher imPersonalfluss zu berücksichtigen(z.B. über geeignete Besucherschleu-sen oder separat angeordnete Besu-chergänge).
7. Generelle Design-Anforderungen anReinräume
Im Nachfolgenden sind einige gene-rellen Design-Anforderungen auf-gelistet, die sich z.T. aus den zugrun-deliegenden Regularien ableiten las-sen, zum anderen aber auch aus derPraxis resultieren:. Reinräume müssen so beschaffensein, dass die festgelegte Rein-raumklasse eingehalten wird.
. Zugang von Personal und Einbrin-gung von Material sollte über ge-trennte Schleusensysteme erfolgen.
. Angrenzende Schleusen undDurchreichen müssen den glei-chen Anforderungen wie derReinraum entsprechen.
. Türspalte und andere Raumöff-nungen sind so klein wie möglichzu halten, zur Minimierung vonLeckluftströmen.
. Medienzuführung und -abführungsind so auszuführen, dass keinezusätzliche Partikelbelastung ent-steht (möglichst vertikale Ver-legung mit Rosetten an denDurchführungen).
. Einbauten in Decken und Wände(Türen, Fenster, Leuchten, etc.)sind möglichst flächenbündig,spaltfrei und partikeldicht aus-zuführen.
. Fugenmaterial sollte hochelas-tisch, nicht klebend, beständig undbakterizid/fungizid sein, zur Ver-meidung von Bakterien-/Pilz-wachstum.
. Es sollte nur notwendige Ausrüs-tungen installieren sein.
. Kabel sollten in geschlossenenKanälen verlegt werden (Vermei-dung von Partikelablagerung).
. Bodenbeläge sollten glatt, wider-standsfähig, mit geringem Abrieb,elektrisch leitend/geerdet sein.Wandübergänge sollten gerundetausgeführt werden.
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)330 Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Umwandlungszone (Schleuse)
Personal
Ausrüstung
RohstoffeRRKL
C
RRKLD
Abb. 2 Schleusenkonzept (Quelle: G. Heuwes).

men der Qualifizierung der Lüftungs-anlage durchgeführt werden. Diesmuss jedoch, wie bereits erläutert,über eine Definition der SchnittstelleRaum/Lüftungsanlage festgelegtwerden. Die Luftqualität wird dannim Betriebszustand „at rest“ und „inoperation“ überprüft.
13. Fazit
Abschließend kann man sagen, dassdie Ausstattung und Gestaltung vonRäumen maßgeblich von den Prozess-und Produktanforderungen abhängigist sowie von den zugrundeliegendenregulatorischen Anforderungen.
Sind diese bekannt, so ist man inder Lage über entsprechende Pla-nungstools die Ausstattung und dieGestaltung der Räume genau zu spe-zifizieren und diese Spezifikationenim Raumbuch festzuschreiben.
Mit Hilfe einer Risikoanalyse kön-nen bereits während der Planungs-phase die GMP-Risiken betrachtetwerden, Maßnahmen definiert wer-den und die festgelegten Prüfungenim Rahmen der Qualifizierung (DQ,IQ, OQ, PQ) beschrieben werden.
Fachliteratur[1] European Commission: EudraLex, The
Rules Governing Medicinal Products inthe European Union EU, Guidelines toGood Manufacturing Practice MedicinalProducts for Human and Veterinary Use
[2] European Commission: EudraLex, TheRules Governing Medicinal Products inthe European Union EU, Guidelines toGood Manufacturing Practice MedicinalProducts for Human and Veterinary Use
[3] CFR – Code of Federal Regulations Title21, Part 210 Current Good ManufacturingPractice in Manufacturing, Processing,Packaging or Holding of Drugs, General
[4] CFR – Code of Federal Regulations Title21, Part 211 Current Good ManufacturingPractices for Finished Pharmaceuticals
[5] AMWHV (Arzneimittel- und Wirkstoff-herstellungsverordnung), Verordnungüber die Anwendung der Guten Herstel-lungspraxis bei der Herstellung von Arz-neimitteln und Wirkstoffen und über dieAnwendung der Guten fachlichen Praxisbei der Herstellung von Produktenmenschlicher Herkunft
[6] DIN EN ISO 14644 Cleanrooms and asso-ciated controlled environments Part 1:Klassifizierung der Luftreinheit anhandder Partikelkonzentration, Part 2: Fest-legungen zur Prüfung und Überwachungzum Nachweis der fortlaufenden Über-einstimmung mit ISO 14644-1, Part 7: SD-Module (Reinlufthauben, Handschuhbo-xen, Isolatoren und Minienvironments),Part 10: Klassifizierung der chemischenOberflächenreinheit, Beuth Verlag GmbH
[7] VDI Richtlinie, VDI 2083, Reinraumtech-nik, Blatt 16.1: Barrieresysteme (Isolatoren,Mini-Environments, Reinraummodule) –Wirksamkeit und Zertifizierung, Blatt 17:Reinheitstauglichkeit von Werkstoffen,Blatt 18: Biokontaminationskontrolle, Ver-ein Deutscher Ingenieure e.V. Düsseldorf
[8] ZLG (Zentralstelle der Länder für Ge-sundheitsschutz bei Arzneimitteln undMedizinprodukten), Aide memoire07121105, Inspektion von Qualifizierungund Validierung in pharmazeutischerHerstellung und Qualitätskontrolle
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-01\Anzeigensatz-keine-Druck-PDFs\harter-tp-2013-01-216x102.indd Zuletzt gesichert: 10.01.13 (07:51:46 Uhr)
HARTER Oberfl ächen- und Umwelttechnik GmbH Telefon +49 (0) 83 83 / 92 23 - 0 www.harter-med.de
Harter trocknet alles. Sicher.Ihr Produkt hat beste Behandlung verdient. Und intensivste Trocknung.AIRGENEX®med von Harter trocknet alle Produkte nach Sterilisation oder Reinigung sowie Materialien und pharmazeutische Erzeugnisse in allen Stadien der Produktion – energiesparend im geschlossenen System, ohne unerwünschte Produkterhitzung, in kurzer Zeit und mit höchster Qualität.
AIRGENEX®med Trocknungssysteme für Pharma und Medizintechnik
„WIR LÖSEN IHRE PROBLEME IN LUFT AUF!“
Erleben Sie
Trocknungstechnik
LIVEim HARTER
Technikum.
Harter_Anz_Pharma_216x102_Wir_loesen_RZ.indd 1 21.12.12 12:29
te. Zum einen sollte ein Raumbuchexistieren, das als Instrument für denProzessplaner/TGA-Planer und Lo-gistiker dient, damit diese auf glei-cher Datenbasis planen können (Be-schreibung von GMP- und nichtGMP-Anforderungen) und zum an-deren ein Raumbuch, das nur dieGMP-Anforderungen beinhaltet.
Dadurch werden in einem Raum-buch die GMP-Anforderungen ver-ankert. Dieses Raumbuch kanndann im weiteren Verlauf der Qua-lifizierung als Testdokument ge-nutzt werden und später als Anlagein die Qualifizierung eingebundenwerden.
Die Voraussetzungen, damit einRaumbuch erstellt werden kann ist,dass das Lastenheft vorliegt, das Ge-neral Layout erstellt wurde, die Rein-raumklassifizierung vorgenommenwurde und dass der Material- undPersonalfluss feststeht.
Je nach gewählter Form desRaumbuchs sollte dies folgendenUmfang abdecken:. Heizung, Klima, Lüftung, Sanitär(HKLS)
. Elektrotechnik (ELT)
. Prozessmedien
. Pharmainnenausbau
. ArchitekturMan sollte sich beim Raumbuch je-doch darüber im Klaren sein, dassdies ein lebendes Dokument ist, d. h.dass das Raumbuch nach Änderun-gen in der Planung angepasst werdenmuss. Nur so kann sichergestellt wer-den, dass der im Detail beschriebeneRaum auch mit dem „as built“ – Sta-tus übereinstimmt und somit dieQualifizierung, die auf den Datendes Raumbuches basiert, erfolgreichabgeschlossen werden kann.
12. Qualifizierung
Wenn man sich mit der Qualifizie-rung der Räume beschäftigt, wirdman schnell feststellen, dass mansich Gedanken darüber machenmuss, wo die Schnittstelle zwischenQualifizierung der Lüftungsanlageund Qualifizierung der Räume gelegtwerden muss.
Die Qualifizierung (Abb. 3) derRäume beinhaltet die Ausstattungund die Gestaltung der Räume mitall ihren Anforderungen gemäßRaumbuch. Die Qualifizierung derLüftungsanlage hingegen beinhaltetdie Versorgung der Räume mit vor-konditionierter Luft entsprechenderQualität.
Genau an dieser Stelle sollte mansauber definieren, was an welcherStelle überprüft wird. Hier kannman auch keine allgemeingültigeVorgehensweise festlegen, da diesesich aus den Projektanforderungenund aus den grundlegenden Fest-legungen des jeweiligen Herstel-lungsbetriebes ergeben.
Aus diesem Grund sollte man zumProjektstart bereits in der Risikoana-lyse (RA) genau beschreiben, wasUmfang der Qualifizierung derRäume ist und was zum Umfangder Qualifizierung der Lüftungs-anlage zählt.
Räume werden qualifiziert, weilProzess und Produkt durch dieRaumbedingungen beeinflusst wer-den können. Dazu zählen Partikel-kontamination von Produkten, Mi-krobiologische Belastung von Pro-dukten durch Hygienestandard desRaumes sowie Temperatur (ther-mischer Abbau), Luftfeuchte (Hydro-lysen) und Licht (Abbaureaktion).
Die Design Qualification (DQ)sollte bei der Qualifizierung von Räu-men mindestens folgendes beinhal-ten:
. zu verwendete Materialien für De-cken, Böden, Wände und Fenster
. Aufbau der Decken, Böden, Wände
. Anforderungen an Verarbeitung
. allgemeine zu liefernde Dokumen-tation seitens der Lieferanten und
. Anforderung an Temperatur undFeuchte, Beleuchtung, Schleusen-funktion
Bei der Installation Qualification (IQ)erfolgt die Prüfung dann vor Ort bzw.gegen die Lieferantendokumentati-on. Dabei werden für die Materialiender Decken, Böden und Wände dieentsprechenden Materialzertifikate/Werkstoffzeugnisse der Lieferantenbenötigt. Aufbau, Beschaffenheitund Verarbeitung der Decken, Böden
und Wände wer-den einer visuel-len Prüfung vorOrt unterzogenebenso wie dieReinigbarkeit. Un-ter Zuhilfenahmevon genehmigtenZeichnungen wer-den die Bezeich-nung und Maßeder Räume über-prüft.
Die Prüfungdes Vorhanden-seins der spezifi-zierten Kom-
ponenten (z.B.: Beleuchtung, Boden-ablauf und Lüftungsauslässe) erfolgtformal und systematisch gegen dasRaumbuch, indem jede durch-geführte Prüfung im Raumbuch ab-gezeichnet wird.
Die Prüfungen im Rahmen derOperational Qualification (OQ) fol-gen den Festlegungen der Risikoana-lyse (RA). Hier sollten die Schleusen-funktionen hinsichtlich ihrer korrek-ten Verriegelung überprüft werden.Des Weiteren sollten Messungenvon Temperatur und Luftfeuchtigkeitdurchgeführt werden. Darüber hi-naus ist, an festgelegten Arbeitsplät-zen der Mitarbeiter, die Beleuch-tungsintensität zu prüfen.
Was die Durchführung einer Per-formance Qualification (PQ) betrifft,so kann diese vollumfänglich im Rah-
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)332 Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Abb. 3 Lebenszyklus der Qualifizierung (Quelle: ZLG)[8].

men der Qualifizierung der Lüftungs-anlage durchgeführt werden. Diesmuss jedoch, wie bereits erläutert,über eine Definition der SchnittstelleRaum/Lüftungsanlage festgelegtwerden. Die Luftqualität wird dannim Betriebszustand „at rest“ und „inoperation“ überprüft.
13. Fazit
Abschließend kann man sagen, dassdie Ausstattung und Gestaltung vonRäumen maßgeblich von den Prozess-und Produktanforderungen abhängigist sowie von den zugrundeliegendenregulatorischen Anforderungen.
Sind diese bekannt, so ist man inder Lage über entsprechende Pla-nungstools die Ausstattung und dieGestaltung der Räume genau zu spe-zifizieren und diese Spezifikationenim Raumbuch festzuschreiben.
Mit Hilfe einer Risikoanalyse kön-nen bereits während der Planungs-phase die GMP-Risiken betrachtetwerden, Maßnahmen definiert wer-den und die festgelegten Prüfungenim Rahmen der Qualifizierung (DQ,IQ, OQ, PQ) beschrieben werden.
Fachliteratur[1] European Commission: EudraLex, The
Rules Governing Medicinal Products inthe European Union EU, Guidelines toGood Manufacturing Practice MedicinalProducts for Human and Veterinary Use
[2] European Commission: EudraLex, TheRules Governing Medicinal Products inthe European Union EU, Guidelines toGood Manufacturing Practice MedicinalProducts for Human and Veterinary Use
[3] CFR – Code of Federal Regulations Title21, Part 210 Current Good ManufacturingPractice in Manufacturing, Processing,Packaging or Holding of Drugs, General
[4] CFR – Code of Federal Regulations Title21, Part 211 Current Good ManufacturingPractices for Finished Pharmaceuticals
[5] AMWHV (Arzneimittel- und Wirkstoff-herstellungsverordnung), Verordnungüber die Anwendung der Guten Herstel-lungspraxis bei der Herstellung von Arz-neimitteln und Wirkstoffen und über dieAnwendung der Guten fachlichen Praxisbei der Herstellung von Produktenmenschlicher Herkunft
[6] DIN EN ISO 14644 Cleanrooms and asso-ciated controlled environments Part 1:Klassifizierung der Luftreinheit anhandder Partikelkonzentration, Part 2: Fest-legungen zur Prüfung und Überwachungzum Nachweis der fortlaufenden Über-einstimmung mit ISO 14644-1, Part 7: SD-Module (Reinlufthauben, Handschuhbo-xen, Isolatoren und Minienvironments),Part 10: Klassifizierung der chemischenOberflächenreinheit, Beuth Verlag GmbH
[7] VDI Richtlinie, VDI 2083, Reinraumtech-nik, Blatt 16.1: Barrieresysteme (Isolatoren,Mini-Environments, Reinraummodule) –Wirksamkeit und Zertifizierung, Blatt 17:Reinheitstauglichkeit von Werkstoffen,Blatt 18: Biokontaminationskontrolle, Ver-ein Deutscher Ingenieure e.V. Düsseldorf
[8] ZLG (Zentralstelle der Länder für Ge-sundheitsschutz bei Arzneimitteln undMedizinprodukten), Aide memoire07121105, Inspektion von Qualifizierungund Validierung in pharmazeutischerHerstellung und Qualitätskontrolle
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-01\Anzeigensatz-keine-Druck-PDFs\harter-tp-2013-01-216x102.indd Zuletzt gesichert: 10.01.13 (07:51:46 Uhr)
HARTER Oberfl ächen- und Umwelttechnik GmbH Telefon +49 (0) 83 83 / 92 23 - 0 www.harter-med.de
Harter trocknet alles. Sicher.Ihr Produkt hat beste Behandlung verdient. Und intensivste Trocknung.AIRGENEX®med von Harter trocknet alle Produkte nach Sterilisation oder Reinigung sowie Materialien und pharmazeutische Erzeugnisse in allen Stadien der Produktion – energiesparend im geschlossenen System, ohne unerwünschte Produkterhitzung, in kurzer Zeit und mit höchster Qualität.
AIRGENEX®med Trocknungssysteme für Pharma und Medizintechnik
„WIR LÖSEN IHRE PROBLEME IN LUFT AUF!“
Erleben Sie
Trocknungstechnik
LIVEim HARTER
Technikum.
Harter_Anz_Pharma_216x102_Wir_loesen_RZ.indd 1 21.12.12 12:29
te. Zum einen sollte ein Raumbuchexistieren, das als Instrument für denProzessplaner/TGA-Planer und Lo-gistiker dient, damit diese auf glei-cher Datenbasis planen können (Be-schreibung von GMP- und nichtGMP-Anforderungen) und zum an-deren ein Raumbuch, das nur dieGMP-Anforderungen beinhaltet.
Dadurch werden in einem Raum-buch die GMP-Anforderungen ver-ankert. Dieses Raumbuch kanndann im weiteren Verlauf der Qua-lifizierung als Testdokument ge-nutzt werden und später als Anlagein die Qualifizierung eingebundenwerden.
Die Voraussetzungen, damit einRaumbuch erstellt werden kann ist,dass das Lastenheft vorliegt, das Ge-neral Layout erstellt wurde, die Rein-raumklassifizierung vorgenommenwurde und dass der Material- undPersonalfluss feststeht.
Je nach gewählter Form desRaumbuchs sollte dies folgendenUmfang abdecken:. Heizung, Klima, Lüftung, Sanitär(HKLS)
. Elektrotechnik (ELT)
. Prozessmedien
. Pharmainnenausbau
. ArchitekturMan sollte sich beim Raumbuch je-doch darüber im Klaren sein, dassdies ein lebendes Dokument ist, d. h.dass das Raumbuch nach Änderun-gen in der Planung angepasst werdenmuss. Nur so kann sichergestellt wer-den, dass der im Detail beschriebeneRaum auch mit dem „as built“ – Sta-tus übereinstimmt und somit dieQualifizierung, die auf den Datendes Raumbuches basiert, erfolgreichabgeschlossen werden kann.
12. Qualifizierung
Wenn man sich mit der Qualifizie-rung der Räume beschäftigt, wirdman schnell feststellen, dass mansich Gedanken darüber machenmuss, wo die Schnittstelle zwischenQualifizierung der Lüftungsanlageund Qualifizierung der Räume gelegtwerden muss.
Die Qualifizierung (Abb. 3) derRäume beinhaltet die Ausstattungund die Gestaltung der Räume mitall ihren Anforderungen gemäßRaumbuch. Die Qualifizierung derLüftungsanlage hingegen beinhaltetdie Versorgung der Räume mit vor-konditionierter Luft entsprechenderQualität.
Genau an dieser Stelle sollte mansauber definieren, was an welcherStelle überprüft wird. Hier kannman auch keine allgemeingültigeVorgehensweise festlegen, da diesesich aus den Projektanforderungenund aus den grundlegenden Fest-legungen des jeweiligen Herstel-lungsbetriebes ergeben.
Aus diesem Grund sollte man zumProjektstart bereits in der Risikoana-lyse (RA) genau beschreiben, wasUmfang der Qualifizierung derRäume ist und was zum Umfangder Qualifizierung der Lüftungs-anlage zählt.
Räume werden qualifiziert, weilProzess und Produkt durch dieRaumbedingungen beeinflusst wer-den können. Dazu zählen Partikel-kontamination von Produkten, Mi-krobiologische Belastung von Pro-dukten durch Hygienestandard desRaumes sowie Temperatur (ther-mischer Abbau), Luftfeuchte (Hydro-lysen) und Licht (Abbaureaktion).
Die Design Qualification (DQ)sollte bei der Qualifizierung von Räu-men mindestens folgendes beinhal-ten:
. zu verwendete Materialien für De-cken, Böden, Wände und Fenster
. Aufbau der Decken, Böden, Wände
. Anforderungen an Verarbeitung
. allgemeine zu liefernde Dokumen-tation seitens der Lieferanten und
. Anforderung an Temperatur undFeuchte, Beleuchtung, Schleusen-funktion
Bei der Installation Qualification (IQ)erfolgt die Prüfung dann vor Ort bzw.gegen die Lieferantendokumentati-on. Dabei werden für die Materialiender Decken, Böden und Wände dieentsprechenden Materialzertifikate/Werkstoffzeugnisse der Lieferantenbenötigt. Aufbau, Beschaffenheitund Verarbeitung der Decken, Böden
und Wände wer-den einer visuel-len Prüfung vorOrt unterzogenebenso wie dieReinigbarkeit. Un-ter Zuhilfenahmevon genehmigtenZeichnungen wer-den die Bezeich-nung und Maßeder Räume über-prüft.
Die Prüfungdes Vorhanden-seins der spezifi-zierten Kom-
ponenten (z.B.: Beleuchtung, Boden-ablauf und Lüftungsauslässe) erfolgtformal und systematisch gegen dasRaumbuch, indem jede durch-geführte Prüfung im Raumbuch ab-gezeichnet wird.
Die Prüfungen im Rahmen derOperational Qualification (OQ) fol-gen den Festlegungen der Risikoana-lyse (RA). Hier sollten die Schleusen-funktionen hinsichtlich ihrer korrek-ten Verriegelung überprüft werden.Des Weiteren sollten Messungenvon Temperatur und Luftfeuchtigkeitdurchgeführt werden. Darüber hi-naus ist, an festgelegten Arbeitsplät-zen der Mitarbeiter, die Beleuch-tungsintensität zu prüfen.
Was die Durchführung einer Per-formance Qualification (PQ) betrifft,so kann diese vollumfänglich im Rah-
Prozess- und Verfahrenstechnik
TechnoPharm 3, Nr. 6, 328–333 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)332 Heuwes . Ausstattung, Gestaltung und Qualifizierung von Räumen
Abb. 3 Lebenszyklus der Qualifizierung (Quelle: ZLG)[8].

Die nachgewiesen sicherste und ambesten akzeptierte Methode ist hierdie Leitfähigkeitsmessung. Entspre-chende Messzellen, die die hohenAnforderungen an Zuverlässigkeitund Prozesstauglichkeit erfüllen, ste-hen am Markt zur Verfügung.
Leitfähigkeit als Maß
Grundsätzlich ist die Leitfähigkeitdes Messmediums von der Anzahl,
der Ladungszahl und der Beweglich-keit der Ionen abhängig. Ein Leitfä-higkeitssensor (Abb. 2). erfasst dieSumme aller in der Lösung befind-lichen Ionen und liefert damitschnell und sicher Aussagen zur ak-tuellen Wasserqualität. Die Mes-sung in Reinstwasser erfolgt mitLeitfähigkeitsensoren, die nachdem Zwei-Elektroden-Verfahren ar-beiten. Bei dieser Anwendung sinddie Elektroden konzentrisch ange-ordnet, wobei die äußere die innereElektrode abschirmt. Da die elektro-lytische Leitfähigkeit einer Flüssig-keit stark temperaturabhängig ist,wird der Messwert normalerweiseauf die international anerkannte Re-ferenztemperatur von 25 °C bezo-gen (temperaturkompensiert). Dasspezielle Bewertungsverfahren „Wa-ter Conductivity <645>“ (nach USP)zur Leitfähigkeitsmessung in
Reinstwasserbildet eineAusnahme:Hier sind ent-sprechendeMessumformer(Abb. 1) er-hältlich, beidenen dieTemperatur-kompensationausgeschaltetwerden kann.
Zwei-Elektroden-Sensor –koaxial
Ein Zwei-Elektroden-Sensor nutztzwei leitfähige Messelektroden, diefür Messungen in Reinstwasser ausEdelstahl oder Titan bestehen und ineiner bestimmten Geometrie angeord-
Abb. 1: dTRANS CR 02 Messumformer fürkonduktive Leitfähigkeitssensoren (Quelle alleBilder: JUMO).
Abb. 2: tecLine CR kon-duktiver Leitfähigkeits-sensor für Pharmawasser.
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-02\Anzeigensatz-keine-Druck-PDFs\schuelke-tp-2013-02-216x149.indd Zuletzt gesichert: 14.03.13 (01:07:14 Uhr)
Schülke & Mayr GmbH | Customer Care | Tel. +49 40 521 00- 666 | www.schuelke.com
perform® sterile wipes IPA, mit 70 % v/v IPA getränkte Tücher zur Desinfektion von Flächen. • Softpackmit20Tüchern•Farbstoff-undparfümfrei•Partikelarm,geeignet fürReinraumklasseA/B/ISOKlasse5• Biozidrichtlinien-konformeWirkstoffe•SterilesProduktentsprechendAnnexIderEUGMP-Guideline• NachEuro-NormengeprüfteDesinfektionswirksamkeit
perform® sterile perform® advanced perform® classic perform® selectDesinfektionsmittelsicherverwenden.VorGebrauchstetsKennzeichnungundProduktinformationlesen.
Volle Performance für den Reinraum.
NEU: perform ® sterile wipes IPA
Rufen Sie uns an und erfahren Sie mehr über unser umfangreiches Produktsortiment für Ihre Produktionshygiene.
Inline Leitfähigkeitsmessungfür pharmazeutische ProzesseChristina Hoffmann . JUMO GmbH & Co. KG, Fulda
Korrespondenz: Christina Hoffmann, JUMO GmbH & Co. KG, Moritz-Juchheim-Straße 1, 36039 Fulda,e-mail: [email protected]
ZusammenfassungDie pharmazeutische Industrie ist eine Branche in der hochsensible Produkte mit ebensohochsensiblen Prozessen hergestellt werden. Kleinste Prozessschwankungen könnenfolgenschwere Auswirkungen haben, und damit sind nicht die Produktionskostengemeint. Mit Hilfe geeigneter Mess- und Regeltechnik werden Prozesse wesentlichsicherer, das Prozessmonitoring verbessert und die Qualitätsstandards erhöht, wie sichhier am Beispiel Reinstwasser zeigen lässt.
Einleitung
In den meisten pharmazeutischenProzessen werden die ParameterTemperatur, Druck, pH-Wert, Leitfä-higkeit, Füllstand, Feuchte, Sauer-stoff und Durchfluss erfasst, über-wacht und/oder geregelt. Dies isteine Voraussetzung zur Standardisie-rung, Validierung und Optimierungder Prozesse, wobei die eingesetzteMesstechnik dabei sehr unterschied-lich sein kann. Messgeräte innerhalbder pharmazeutischen Industriemüssen unterschiedlichsten Anfor-derungen in Bezug auf Material, Kon-struktion und insbesondere Prozess-stabilität gerecht werden. Beispiele:Beim Sterilisieren von Produktensind Messgeräte wie Temperaturfüh-ler zu verwenden, die für den Einsatzin wasserdampfhaltiger und unterDruck stehendender Atmosphäre ge-mäß Schutzart IP 69 konstruiert sind.Oder bei der Verarbeitung pulverför-miger Materialien: Die dort einge-setzten Messgeräte wie Druckmess-umformer, Temperaturfühler usw.müssen speziell für den Einsatz inBereichen geeignet sein, in denen ex-plosive Staubwolken vorkommenkönnen. Das setzt entsprechendeKonstruktionsmerkmale und dieZertifizierung gemäß den ATEX-Richtlinien für Zone 21 (gelegentli-
che explosive Atmosphäre durch ent-flammbare Staubwolken) voraus.
Wasserreinheitnach Norm
Einer der wichtigsten Prozesse beiallen pharmazeutischen Produktio-nen ist jedoch die Wasseraufberei-tung (Abb. 3). Je nach Anwendunggibt es hier unterschiedliche Anforde-rungen und Aufbereitungsmethoden.Von der Aufbereitung über einen Io-nenaustauscher für entmineralisierteWässer, über dieUmkehrosmosemit Elektrodenio-nisation fürReinstwasser biszur Herstellungvon WFI (Waterfor Injection) mit-tels Destillation.Dennoch werdendiese Prozesseüberwiegend mitder gleichenMesstechnik aus-gestattet, da diezu messendenGrößen unter an-derem in Regel-werken vor-geschrieben sind.
Die Qualität von Reinstwasser –highly purified water(hpw), purifiedwater(pw), ultra pure water (upw),water for injection (wfi) usw. – istin Normen und Empfehlungen be-schrieben, zum Beispiel von ASTMInternational (American Society forTesting and Materials), in der Phar-macopoea Europaea (EP), der UnitedStates Pharmacopeia (USP) und inDIN- oder ISO-Normen (Tab. 1).Sämtliche der genannten Normen er-fordern eine zuverlässige Qualitäts-kontrolle des hergestellten Wassers.
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 334–336 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)334 Hoffmann . Leitfähigkeitsmessung
Tabelle 1
Reinheit unterschiedlicher Wässernach Europaea (EP) und United StatesPharmacopeia (USP)
PW HPW WFI
LeitfähigkeitμS/cm (20 ºC)
4,3 (EP)1,3 (USP)
1,1 (EP) 1,1 (EP,USP)
TOC (mg/l) 0,5 0,5 0,5
Keimzahl(KBE)
100/1 ml 10/100 ml 10/100 ml
Bakterien-Endotoxin(EU)
---- < 0,25/ml < 0,25/ml

Die nachgewiesen sicherste und ambesten akzeptierte Methode ist hierdie Leitfähigkeitsmessung. Entspre-chende Messzellen, die die hohenAnforderungen an Zuverlässigkeitund Prozesstauglichkeit erfüllen, ste-hen am Markt zur Verfügung.
Leitfähigkeit als Maß
Grundsätzlich ist die Leitfähigkeitdes Messmediums von der Anzahl,
der Ladungszahl und der Beweglich-keit der Ionen abhängig. Ein Leitfä-higkeitssensor (Abb. 2). erfasst dieSumme aller in der Lösung befind-lichen Ionen und liefert damitschnell und sicher Aussagen zur ak-tuellen Wasserqualität. Die Mes-sung in Reinstwasser erfolgt mitLeitfähigkeitsensoren, die nachdem Zwei-Elektroden-Verfahren ar-beiten. Bei dieser Anwendung sinddie Elektroden konzentrisch ange-ordnet, wobei die äußere die innereElektrode abschirmt. Da die elektro-lytische Leitfähigkeit einer Flüssig-keit stark temperaturabhängig ist,wird der Messwert normalerweiseauf die international anerkannte Re-ferenztemperatur von 25 °C bezo-gen (temperaturkompensiert). Dasspezielle Bewertungsverfahren „Wa-ter Conductivity <645>“ (nach USP)zur Leitfähigkeitsmessung in
Reinstwasserbildet eineAusnahme:Hier sind ent-sprechendeMessumformer(Abb. 1) er-hältlich, beidenen dieTemperatur-kompensationausgeschaltetwerden kann.
Zwei-Elektroden-Sensor –koaxial
Ein Zwei-Elektroden-Sensor nutztzwei leitfähige Messelektroden, diefür Messungen in Reinstwasser ausEdelstahl oder Titan bestehen und ineiner bestimmten Geometrie angeord-
Abb. 1: dTRANS CR 02 Messumformer fürkonduktive Leitfähigkeitssensoren (Quelle alleBilder: JUMO).
Abb. 2: tecLine CR kon-duktiver Leitfähigkeits-sensor für Pharmawasser.
Abgelegt auf: F:\GK\ECV\Satz\TechnoPharm\TP_2013-02\Anzeigensatz-keine-Druck-PDFs\schuelke-tp-2013-02-216x149.indd Zuletzt gesichert: 14.03.13 (01:07:14 Uhr)
Schülke & Mayr GmbH | Customer Care | Tel. +49 40 521 00- 666 | www.schuelke.com
perform® sterile wipes IPA, mit 70 % v/v IPA getränkte Tücher zur Desinfektion von Flächen. • Softpackmit20Tüchern•Farbstoff-undparfümfrei•Partikelarm,geeignet fürReinraumklasseA/B/ISOKlasse5• Biozidrichtlinien-konformeWirkstoffe•SterilesProduktentsprechendAnnexIderEUGMP-Guideline• NachEuro-NormengeprüfteDesinfektionswirksamkeit
perform® sterile perform® advanced perform® classic perform® selectDesinfektionsmittelsicherverwenden.VorGebrauchstetsKennzeichnungundProduktinformationlesen.
Volle Performance für den Reinraum.
NEU: perform ® sterile wipes IPA
Rufen Sie uns an und erfahren Sie mehr über unser umfangreiches Produktsortiment für Ihre Produktionshygiene.
Inline Leitfähigkeitsmessungfür pharmazeutische ProzesseChristina Hoffmann . JUMO GmbH & Co. KG, Fulda
Korrespondenz: Christina Hoffmann, JUMO GmbH & Co. KG, Moritz-Juchheim-Straße 1, 36039 Fulda,e-mail: [email protected]
ZusammenfassungDie pharmazeutische Industrie ist eine Branche in der hochsensible Produkte mit ebensohochsensiblen Prozessen hergestellt werden. Kleinste Prozessschwankungen könnenfolgenschwere Auswirkungen haben, und damit sind nicht die Produktionskostengemeint. Mit Hilfe geeigneter Mess- und Regeltechnik werden Prozesse wesentlichsicherer, das Prozessmonitoring verbessert und die Qualitätsstandards erhöht, wie sichhier am Beispiel Reinstwasser zeigen lässt.
Einleitung
In den meisten pharmazeutischenProzessen werden die ParameterTemperatur, Druck, pH-Wert, Leitfä-higkeit, Füllstand, Feuchte, Sauer-stoff und Durchfluss erfasst, über-wacht und/oder geregelt. Dies isteine Voraussetzung zur Standardisie-rung, Validierung und Optimierungder Prozesse, wobei die eingesetzteMesstechnik dabei sehr unterschied-lich sein kann. Messgeräte innerhalbder pharmazeutischen Industriemüssen unterschiedlichsten Anfor-derungen in Bezug auf Material, Kon-struktion und insbesondere Prozess-stabilität gerecht werden. Beispiele:Beim Sterilisieren von Produktensind Messgeräte wie Temperaturfüh-ler zu verwenden, die für den Einsatzin wasserdampfhaltiger und unterDruck stehendender Atmosphäre ge-mäß Schutzart IP 69 konstruiert sind.Oder bei der Verarbeitung pulverför-miger Materialien: Die dort einge-setzten Messgeräte wie Druckmess-umformer, Temperaturfühler usw.müssen speziell für den Einsatz inBereichen geeignet sein, in denen ex-plosive Staubwolken vorkommenkönnen. Das setzt entsprechendeKonstruktionsmerkmale und dieZertifizierung gemäß den ATEX-Richtlinien für Zone 21 (gelegentli-
che explosive Atmosphäre durch ent-flammbare Staubwolken) voraus.
Wasserreinheitnach Norm
Einer der wichtigsten Prozesse beiallen pharmazeutischen Produktio-nen ist jedoch die Wasseraufberei-tung (Abb. 3). Je nach Anwendunggibt es hier unterschiedliche Anforde-rungen und Aufbereitungsmethoden.Von der Aufbereitung über einen Io-nenaustauscher für entmineralisierteWässer, über dieUmkehrosmosemit Elektrodenio-nisation fürReinstwasser biszur Herstellungvon WFI (Waterfor Injection) mit-tels Destillation.Dennoch werdendiese Prozesseüberwiegend mitder gleichenMesstechnik aus-gestattet, da diezu messendenGrößen unter an-derem in Regel-werken vor-geschrieben sind.
Die Qualität von Reinstwasser –highly purified water(hpw), purifiedwater(pw), ultra pure water (upw),water for injection (wfi) usw. – istin Normen und Empfehlungen be-schrieben, zum Beispiel von ASTMInternational (American Society forTesting and Materials), in der Phar-macopoea Europaea (EP), der UnitedStates Pharmacopeia (USP) und inDIN- oder ISO-Normen (Tab. 1).Sämtliche der genannten Normen er-fordern eine zuverlässige Qualitäts-kontrolle des hergestellten Wassers.
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 334–336 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)334 Hoffmann . Leitfähigkeitsmessung
Tabelle 1
Reinheit unterschiedlicher Wässernach Europaea (EP) und United StatesPharmacopeia (USP)
PW HPW WFI
LeitfähigkeitμS/cm (20 ºC)
4,3 (EP)1,3 (USP)
1,1 (EP) 1,1 (EP,USP)
TOC (mg/l) 0,5 0,5 0,5
Keimzahl(KBE)
100/1 ml 10/100 ml 10/100 ml
Bakterien-Endotoxin(EU)
---- < 0,25/ml < 0,25/ml

Basiswissen KalibriermanagementDanica Schwarzkopf . WIKA Alexander Wiegand SE & Co. KG, Klingenberg
Korrespondenz: Danica Schwarzkopf, WIKA Alexander WiegandSE & Co. KG, Alexander-Wiegand-Straße30, 63911 Klingenberg
ZusammenfassungJedes Messgerät altert aufgrund von mechanischen, chemischen oder thermischen Belas-tungen und liefert deshalb im Laufe der Zeit sich verändernde Messwerte. Dies kannzwar nicht verhindert, aber durch Kalibrierung rechtzeitig erkannt werden. Bei der Kali-brierung wird die Anzeige des Messgeräts mit dem Messergebnis eines anderen Mess-mittels verglichen. Dessen korrekte und genaue Funktion ist bekannt und seinerseitsdirekt oder indirekt mit einem Referenzgerät in Übereinstimmung gebracht. Warum eineKalibrierung immer wichtiger wird, soll folgender Artikel erläutern.
Einführung
Definition der BegrifflichkeitenKalibrieren in der Messtechnik be-deutet, die Messabweichungen amvollständigen Messgerät festzustel-len. Beim Kalibrieren erfolgt keintechnischer Eingriff am Messgerätwie z.B. Nullpunktabgleich, Spanneund Linearität einstellen. Bei anzei-genden Messgeräten wird durch dasKalibrieren die Messabweichung zwi-schen Anzeige und dem als richtiggeltenden Wert der Messgröße fest-gestellt. Bei Maßverkörperungen,z.B. Massen, wird die Messabwei-chung zwischen der Aufschrift unddem richtigen Wert bestimmt. BeiMessketten stellt man die Mess-abweichung zwischen dem Wert desAusgangssignales und demWert, dendieses Signal bei idealem Übertra-gungsverhalten und vorgegebenemEingangswert haben müsste, fest.Justieren bedeutet, ein Messgerät soeinzustellen oder abzugleichen, dassdie Messabweichungen möglichstklein werden oder dass die Beträgeder Messabweichungen die Fehler-grenzen nicht überschreiten. Das Jus-tieren erfordert also einen Eingriff, derdas Messgerät oder die Maßverkörpe-
rung meistens bleibend verändert, z. B.den Zeiger neu zu positionieren oderein neues Zifferblatt aufbringen.
Das Eichen eines Messgerätes um-fasst die von der zuständigen Eichbe-hörde nach den Eichvorschriften vor-zunehmenden Prüfungen und dieStempelung. Durch die Prüfung wirdfestgestellt, ob das vorgelegte Mess-gerät den Eichvorschriften ent-spricht, d. h. ob es den an seine Be-schaffenheit und seine messtech-nischen Eigenschaften zu stellendenAnforderungen genügt, insbesondereob die Messabweichungen die Feh-lergrenzen nicht überschreiten.Durch die Stempelung wird beurkun-det, dass das Messgerät zum Zeit-punkt der Prüfung diesen Anforde-rungen genügt hat und dass auf-grund seiner Beschaffenheit zu er-warten ist, dass es bei einer Hand-habung entsprechend den Regelnder Technik innerhalb der Nacheich-frist in dem angegebenen Toleranz-bereich bleibt. Welche Messgeräteder Eichpflicht unterliegen und wel-che davon befreit sind, ist gesetzlichgeregelt. Das Eichen soll nur in die-sem Sinne verwendet werden undnicht – wie vielfach üblich – für Jus-tieren und Kalibrieren.
GültigkeitsdauerAnders als bei einer Eichung, dienach gesetzlich vorgegebener Zeit-spanne ihre Gültigkeit verliert, folgtdie Gültigkeitsdauer einer Kalibrie-rung eher praktischen Vorgaben wieHerstellerangaben, Forderungen ei-ner Qualitätssicherungsnorm oderfirmeninternen und kundenspezi-fischen Regelungen. Eine wichtigeRolle spielt hierbei die Risikobetrach-tung des Nutzers. Kalibriert werdenmuss auch dann, wenn das Mess-gerät bei der Herstellung von Pro-dukten eingesetzt wird, die staatli-cher Überwachung unterliegen, wiez.B. Medikamenten oder Nahrungs-mitteln.
BedeutungIm Rahmen einer Befragung von 100Führungskräften internationaler Un-ternehmen ermittelte das Marktfor-schungsunternehmen Nielsen Re-search Company im Jahr 2008, dassproduzierende Unternehmen auf-grund fehlerhafter Kalibrierungendurchschnittlich über 1,7 MillionenDollar pro Jahr verlieren, Firmenmit mehr als einer Milliarde DollarUmsatz sogar 4 Millionen Dollar. ImZusammenhang mit notwendigen
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 337Schwarzkopf . Basiswissen Kalibriermanagement
net sind. Den Abstand der Sensorflä-chen zueinander (l) im Verhältnis zurwirksamen Messfläche (A) nennt manZellenkonstante K, mit der Einheit [1/cm]. Bei Reinstwasser muss diese Zel-lenkonstante K = 0,01 betragen. RealeSensoren sind meist koaxial auf-gebaut, d. h. die beiden Messelektro-den sind konzentrisch angeordnet.Außerdem ist in diesen Sensorenmeist ein Fühler zur Erfassung derTemperatur des Mediums integriert.Ein externer elektronischer Mess-umformer beaufschlagt den Zwei-Elektroden-Sensor mit einer Wechsel-spannung. Entsprechend dem elektri-schen Widerstand der Messlösung(Reinheitsgrad) stellt sich ein Wech-selstrom ein, der durch den Mess-umformer unter Berücksichtigungder Zellenkonstante und eventuellder Temperatur des Messmediums inden Wert für die Leitfähigkeit derMesslösung umgerechnet wird.
Eine komplette Messkette fürReinstwasser-Leitfähigkeitsmessun-gen besteht aus:. Reinstwasser-Messumfomer/Regler
. Reinstwasser-Leitfähigkeitssensormit exakt vermessener Zellenkon-stante
. Temperaturfühler (meist im Leit-fähigkeitssensor integriert)
. AnschlussleitungDie mediumberührenden Teile derGeräte sind aus Edelstahl 14435(316L) in elektropolierter Qualität ge-fertigt. Für sie können Abnahmeprüf-zeugnisse nach DIN EN 10204 3.1 so-wie Rauigkeitszertifikate (< 0,8μmgemäß Ph. Eur. bzw. ASTM-Interna-tional) zur Verfügung gestellt wer-den. Die verwendeten Dichtungenund Kunststoffe entsprechen den An-forderungen der FDA und sind phy-siologisch unbedenklich. Die Instal-lation konduktiver Leitfähigkeitssen-soren sollte am Besten in Fließrich-tung erfolgen, so dass der Sensor di-rekt angeströmt wird. Hierbei sollteder Sensor so eingebaut werden, dasssich im Kopfraum keine Luftblasenbilden können.
Fazit
Die Reinstwasserherstellung zähltzu den wichtigsten Prozessen beider Herstellung von Arzneimittelnund Pharmazeutika. Um die Quali-tät des Reinstwassers sicher zu stel-len, muss die Leitfähigkeit kontinu-ierlich erfasst und überwacht wer-
den. So wird eine gleich bleibendeQualität und hohe Prozesssicher-heit gewährleistet. Koaxial konstru-ierte Zwei-Elektroden-Sensoren bie-ten hier zusammen mit einem ex-ternen Messumformer beste Vo-raussetzungen für zuverlässigeMesswerte.
Die online Messung der Leitfähig-keit ist das sicherste und zuverläs-sigste Messverfahren zur Über-wachung der chem./phy. Qualitätvon Reinstwasser in der pharm. In-dustrie.
Fachliteratur[1] K. Marquardt, Rein- und Reinstwasser-
aufbereitung, Expert Verlag,Renningen Malmsheim 1994
[2] P. Henderson, Z. Phys. Chemie 59, 118 –127 (1907)
[3] H. Galster, VGB Kraftwerkstechnik 59, 885– 889 (1979)
[4] VDI 2083 Blatt 13.1 Reinraumtechnik –Qualität, Erzeugung und Verteilung vonReinstwasser – Grundlagen
[5] VDI 2083 Blatt 13.2 Reinraumtechnik -Qualität, Erzeugung und Verteilung vonReinstwasser – Mikroelektronik und an-dere technische Anwendungen
[6] GMP Praxisbuch Reinstwasser – Planung,Realisierung, Qualifizierung von Reinst-wasseranlagen 2011
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 334–336 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)336 Hoffmann . Leitfähigkeitsmessung
Abb. 3: Prozess der Reinstwasserherstellung.

Basiswissen KalibriermanagementDanica Schwarzkopf . WIKA Alexander Wiegand SE & Co. KG, Klingenberg
Korrespondenz: Danica Schwarzkopf, WIKA Alexander WiegandSE & Co. KG, Alexander-Wiegand-Straße30, 63911 Klingenberg
ZusammenfassungJedes Messgerät altert aufgrund von mechanischen, chemischen oder thermischen Belas-tungen und liefert deshalb im Laufe der Zeit sich verändernde Messwerte. Dies kannzwar nicht verhindert, aber durch Kalibrierung rechtzeitig erkannt werden. Bei der Kali-brierung wird die Anzeige des Messgeräts mit dem Messergebnis eines anderen Mess-mittels verglichen. Dessen korrekte und genaue Funktion ist bekannt und seinerseitsdirekt oder indirekt mit einem Referenzgerät in Übereinstimmung gebracht. Warum eineKalibrierung immer wichtiger wird, soll folgender Artikel erläutern.
Einführung
Definition der BegrifflichkeitenKalibrieren in der Messtechnik be-deutet, die Messabweichungen amvollständigen Messgerät festzustel-len. Beim Kalibrieren erfolgt keintechnischer Eingriff am Messgerätwie z.B. Nullpunktabgleich, Spanneund Linearität einstellen. Bei anzei-genden Messgeräten wird durch dasKalibrieren die Messabweichung zwi-schen Anzeige und dem als richtiggeltenden Wert der Messgröße fest-gestellt. Bei Maßverkörperungen,z.B. Massen, wird die Messabwei-chung zwischen der Aufschrift unddem richtigen Wert bestimmt. BeiMessketten stellt man die Mess-abweichung zwischen dem Wert desAusgangssignales und demWert, dendieses Signal bei idealem Übertra-gungsverhalten und vorgegebenemEingangswert haben müsste, fest.Justieren bedeutet, ein Messgerät soeinzustellen oder abzugleichen, dassdie Messabweichungen möglichstklein werden oder dass die Beträgeder Messabweichungen die Fehler-grenzen nicht überschreiten. Das Jus-tieren erfordert also einen Eingriff, derdas Messgerät oder die Maßverkörpe-
rung meistens bleibend verändert, z. B.den Zeiger neu zu positionieren oderein neues Zifferblatt aufbringen.
Das Eichen eines Messgerätes um-fasst die von der zuständigen Eichbe-hörde nach den Eichvorschriften vor-zunehmenden Prüfungen und dieStempelung. Durch die Prüfung wirdfestgestellt, ob das vorgelegte Mess-gerät den Eichvorschriften ent-spricht, d. h. ob es den an seine Be-schaffenheit und seine messtech-nischen Eigenschaften zu stellendenAnforderungen genügt, insbesondereob die Messabweichungen die Feh-lergrenzen nicht überschreiten.Durch die Stempelung wird beurkun-det, dass das Messgerät zum Zeit-punkt der Prüfung diesen Anforde-rungen genügt hat und dass auf-grund seiner Beschaffenheit zu er-warten ist, dass es bei einer Hand-habung entsprechend den Regelnder Technik innerhalb der Nacheich-frist in dem angegebenen Toleranz-bereich bleibt. Welche Messgeräteder Eichpflicht unterliegen und wel-che davon befreit sind, ist gesetzlichgeregelt. Das Eichen soll nur in die-sem Sinne verwendet werden undnicht – wie vielfach üblich – für Jus-tieren und Kalibrieren.
GültigkeitsdauerAnders als bei einer Eichung, dienach gesetzlich vorgegebener Zeit-spanne ihre Gültigkeit verliert, folgtdie Gültigkeitsdauer einer Kalibrie-rung eher praktischen Vorgaben wieHerstellerangaben, Forderungen ei-ner Qualitätssicherungsnorm oderfirmeninternen und kundenspezi-fischen Regelungen. Eine wichtigeRolle spielt hierbei die Risikobetrach-tung des Nutzers. Kalibriert werdenmuss auch dann, wenn das Mess-gerät bei der Herstellung von Pro-dukten eingesetzt wird, die staatli-cher Überwachung unterliegen, wiez.B. Medikamenten oder Nahrungs-mitteln.
BedeutungIm Rahmen einer Befragung von 100Führungskräften internationaler Un-ternehmen ermittelte das Marktfor-schungsunternehmen Nielsen Re-search Company im Jahr 2008, dassproduzierende Unternehmen auf-grund fehlerhafter Kalibrierungendurchschnittlich über 1,7 MillionenDollar pro Jahr verlieren, Firmenmit mehr als einer Milliarde DollarUmsatz sogar 4 Millionen Dollar. ImZusammenhang mit notwendigen
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 337Schwarzkopf . Basiswissen Kalibriermanagement
net sind. Den Abstand der Sensorflä-chen zueinander (l) im Verhältnis zurwirksamen Messfläche (A) nennt manZellenkonstante K, mit der Einheit [1/cm]. Bei Reinstwasser muss diese Zel-lenkonstante K = 0,01 betragen. RealeSensoren sind meist koaxial auf-gebaut, d. h. die beiden Messelektro-den sind konzentrisch angeordnet.Außerdem ist in diesen Sensorenmeist ein Fühler zur Erfassung derTemperatur des Mediums integriert.Ein externer elektronischer Mess-umformer beaufschlagt den Zwei-Elektroden-Sensor mit einer Wechsel-spannung. Entsprechend dem elektri-schen Widerstand der Messlösung(Reinheitsgrad) stellt sich ein Wech-selstrom ein, der durch den Mess-umformer unter Berücksichtigungder Zellenkonstante und eventuellder Temperatur des Messmediums inden Wert für die Leitfähigkeit derMesslösung umgerechnet wird.
Eine komplette Messkette fürReinstwasser-Leitfähigkeitsmessun-gen besteht aus:. Reinstwasser-Messumfomer/Regler
. Reinstwasser-Leitfähigkeitssensormit exakt vermessener Zellenkon-stante
. Temperaturfühler (meist im Leit-fähigkeitssensor integriert)
. AnschlussleitungDie mediumberührenden Teile derGeräte sind aus Edelstahl 14435(316L) in elektropolierter Qualität ge-fertigt. Für sie können Abnahmeprüf-zeugnisse nach DIN EN 10204 3.1 so-wie Rauigkeitszertifikate (< 0,8μmgemäß Ph. Eur. bzw. ASTM-Interna-tional) zur Verfügung gestellt wer-den. Die verwendeten Dichtungenund Kunststoffe entsprechen den An-forderungen der FDA und sind phy-siologisch unbedenklich. Die Instal-lation konduktiver Leitfähigkeitssen-soren sollte am Besten in Fließrich-tung erfolgen, so dass der Sensor di-rekt angeströmt wird. Hierbei sollteder Sensor so eingebaut werden, dasssich im Kopfraum keine Luftblasenbilden können.
Fazit
Die Reinstwasserherstellung zähltzu den wichtigsten Prozessen beider Herstellung von Arzneimittelnund Pharmazeutika. Um die Quali-tät des Reinstwassers sicher zu stel-len, muss die Leitfähigkeit kontinu-ierlich erfasst und überwacht wer-
den. So wird eine gleich bleibendeQualität und hohe Prozesssicher-heit gewährleistet. Koaxial konstru-ierte Zwei-Elektroden-Sensoren bie-ten hier zusammen mit einem ex-ternen Messumformer beste Vo-raussetzungen für zuverlässigeMesswerte.
Die online Messung der Leitfähig-keit ist das sicherste und zuverläs-sigste Messverfahren zur Über-wachung der chem./phy. Qualitätvon Reinstwasser in der pharm. In-dustrie.
Fachliteratur[1] K. Marquardt, Rein- und Reinstwasser-
aufbereitung, Expert Verlag,Renningen Malmsheim 1994
[2] P. Henderson, Z. Phys. Chemie 59, 118 –127 (1907)
[3] H. Galster, VGB Kraftwerkstechnik 59, 885– 889 (1979)
[4] VDI 2083 Blatt 13.1 Reinraumtechnik –Qualität, Erzeugung und Verteilung vonReinstwasser – Grundlagen
[5] VDI 2083 Blatt 13.2 Reinraumtechnik -Qualität, Erzeugung und Verteilung vonReinstwasser – Mikroelektronik und an-dere technische Anwendungen
[6] GMP Praxisbuch Reinstwasser – Planung,Realisierung, Qualifizierung von Reinst-wasseranlagen 2011
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 334–336 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)336 Hoffmann . Leitfähigkeitsmessung
Abb. 3: Prozess der Reinstwasserherstellung.

InnerbetrieblicheKalibrierung
Die Ausgestaltung eines inner-betrieblichen Kalibriersystems bleibtdem einzelnen Unternehmen über-lassen, jedoch müssen alle Mess-und Prüfmittel mit eigenen Bezugs-normalen, die wiederum auf das na-tionale Normal rückführbar seinmüssen, kalibriert werden. Als Nach-weise werden innerbetriebliche Kali-brierscheine ausgestellt.
FachgerechteKalibrierung
Für die fachgerechte Aus-führung von Kalibrierun-gen gelten verschiedeneNormen, Bestimmungenund Richtlinien. Damit einMessinstrument überhauptkalibriert werden kann,muss es gewisse Grund-voraussetzungen erfüllen.Auch die physikalischenBedingungen, unter denendie Kalibrierung durch-geführt wird, müssen be-kannt sein und berücksich-tigt werden. Unter diesenVoraussetzungen ist esmöglich, einen Kalibrier-ablauf zu wählen, der dengestellten Anforderungengerecht wird.
Normen und Regelwerke
ImWesentlichen greifen Vorschriftenfür die Kalibrierung von Messinstru-menten dann, wenn ein Unterneh-men Produkte herstellt, deren Pro-duktion gesetzlichen Bestimmungenunterliegt.
Normen zur QualitätssicherungGroße Bedeutung haben Normenund Richtlinien zur Qualitätssiche-rung wie etwa die ISO-9000-Nor-menreihe, die in allen Industriena-tionen immer häufiger angewendetwird. In Kapitel 7.6 „Lenkung vonÜberwachungs- und Messmitteln“der Norm ISO 9001:2008 „Qualitäts-
managementsysteme – Anforderun-gen“ wird gefordert, dass alle Prüf-mittel kalibriert werden müssen, diedirekten oder indirekten Einfluss aufdie Qualität der Produkte haben.Dazu zählen z.B. Prüfmittel, die alsReferenzen in Messräumen oder un-mittelbar im Produktionsprozesseingesetzt werden. Die ISO-9000-Normen schreiben keine Gültig-keitsdauer der Kalibrierung vorwohl aber, dass alle Prüfmittel er-fasst und danach unterschieden
werden müssen, ob sie regelmäßigzu kalibrieren sind oder nicht. Esmüssen Prüfpläne erstellt werden,in denen Umfang, Häufigkeit, Me-thode und Annahmekriterien fest-gelegt werden. Anhand von Plaket-ten auf den Messinstrumenten odergeeigneten Listen muss ersichtlichsein, wann jedes Prüfmittel erneutkalibriert werden muss. Unabding-bar ist eine Neukalibrierung, wennein Messinstrument während derHandhabung, Instandhaltung undLagerung verstellt oder beschädigtwird.
Gesetzliche BestimmungenNormen und Richtlinien zur Quali-tätssicherungmüssen nur von Unter-nehmen eingehalten werden, die zer-
tifiziert sein wollen. Ganz anders istdas, wenn z.B. Medikamente, Kosme-tika oder Lebensmittel hergestelltwerden. Hier greifen oft gesetzlicheBestimmungen, deren Einhaltungvon staatlichen Stellen überwachtwird. Aufgrund der internationalenHandelsbeziehungen sind weltweitdie Regelwerke der amerikanischenFood and Drug Administration(FDA) von Bedeutung. So verlangtder Code of Federal Regulation (CFR)die Kalibrierung von Geräten, Appa-
raten, Messgeräten undAufzeichnungsgeräten ingeeigneten Intervallen inÜbereinstimmung mit ei-nem schriftlich erstelltenProgramm, das nach einerRisikobetrachtung erstelltwird. Die europäische Ge-setze fordern ein vergleich-bares Vorgehen.
Kalibrierabläufe
In der Regel wird beimEinsatz eines Messgerätsdessen Messbereich nurzum Teil benutzt. Die Ka-librierpunkte sind dann soauszuwählen, dass sie imArbeitsbereich des Prüf-mittels liegen. Bei der Ka-librierung muss mindes-tens der obere und der un-
tere Prozesspunkt berücksichtigtwerden. Um die Linearität der An-zeige zu überprüfen, wird mindes-tens an einem weiteren Kontroll-punkt gemessen, der zwischen die-sen Extrempunkten liegt. Für denVergleich der Messwerte von Mess-gerät und Bezugs- oder Gebrauchs-normal kann die Messgröße in vie-len Fällen entweder nach der An-zeige des Prüflings oder nach derAnzeige des Normals eingestelltwerden. In vielen Fällen ist die An-zahl der Messpunkte zur Kalibrie-rung eines Messgeräts nicht in einerNorm festgelegt. Dann ist eine engeAbstimmung zwischen dem Kali-brierlabor und dem Anwender erfor-derlich, um geeignete Prüfpunkte zudefinieren.
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 339Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 2: Kalibrierabläufe Druck.
Optimierungen von Herstellprozes-sen gewinnt die Kalibrierung zuneh-mend an Bedeutung. Eine Erhöhungder Messgenauigkeit kann zu Roh-stoffeinsparungen und wenigerSchadstoffemissionen führen, etwawenn bei einer chemischen Reaktiondie exakt richtige Menge an Sauer-stoff zugeführt wird. Die Kalibrierungvon Messgeräten kann sicherheits-relevant sein. Liefern Druck- oderTemperatursensoren in der che-mischen Industrie nicht die korrek-ten Werte, besteht aufgrund falscherSteuerung chemischer Prozesse un-ter Umständen sogar Explosions-gefahr. Die Bedeutung der Kalibrie-rung zeigt sich auch an alltäglichenBeispielen, etwa an Zählern für denGas- oder Wasserverbrauch im Haus-halt und an Benzinuhren an Zapfsäu-len.
Rückführung undKalibrierhierarchie
Um Messergebnisse vergleichen zukönnen, müssen sie sich über eineKette von Vergleichsmessungen aufein nationales oder internationalesNormal „rückführen“ lassen. Die An-zeige des eingesetzten Messgerätsoder eine Maßverkörperung wirddazu in einer oder mehreren Stufenmit diesem Normal verglichen. Aufjeder dieser Stufen erfolgt eine Kali-brierung mit einem Normal, das zu-vor mit einem höherwertigen Nor-mal kalibriert wurde. Entsprechendder Rangfolge der Normale – von Ge-brauchs- oder Werksnormalen überBezugsnormale bis hin zum nationa-len Normal – gibt es eine Kalibrier-hierarchie der durchführenden Stel-len. Diese reicht vom innerbetriebli-chen Kalibrierlabor über akkredi-tierte Kalibrierlaboratorien bis hinzum nationalen metrologischen In-stitut (Abb. 1).
Rückführung in der PraxisDer Deutsche Kalibrierdienst (DKD)nennt als wesentliche Elemente derRückführung:. Die Vergleichskette darf nicht un-terbrochen sein.
. Bei jedem Schritt in der Kalibrier-kette muss die Messunsicherheitbekannt sein, sodass eine Gesamt-messunsicherheit berechnet wer-den kann. In der Regel sollte einvorrangiges Messgerät eine drei-bis viermal so hohe Messgenau-igkeit besitzen wie das Gerät, dasmit seiner Hilfe kalibriert wird.
. Jeder Schritt der Kalibrierungs-kette muss dokumentiert werden,ebenso das Ergebnis.
. Alle Stellen, die einen Schritt indieser Kette durchführen, müssenetwa durch eine Akkreditierungihre Kompetenz nachweisen.
. Kalibrierungen müssen, je nachgeforderter Messgenauigkeit undtechnischen Anforderungen, inangemessenen Zeiträumen wie-derholt werden.
AkkreditierteKalibrierlaboratorien
Akkreditierte Kalibrierlaboratorienübernehmen oft als externe Dienst-leister die Kalibrierung für Unterneh-men, die nicht selbst über die not-wendige Ausrüstung verfügen. Siekönnen aber auch selbst Teil einesUnternehmenssein und in die-sem alle Mess-geräte kalibrie-ren. Dazu ver-fügen sie über ei-gene Werks- oderGebrauchsnor-male, die in ange-messenen Zeit-abständen mit-hilfe der Bezugs-normale des zu-ständigen metro-logischen Staats-instituts oder an-derer akkreditier-ter Laboratorien mit kleinstmögli-cher Messunsicherheit kalibriertwerden.
Koordination durch die EADamit überall ein gleiches Niveau beiKalibrierungen herrscht, müssen diedurchführenden Laboratorien nach
international anerkannten Regeln(DIN EN ISO IEC 17025) akkreditiertwerden. Innerhalb der EU wird diesvon der Europäischen Kooperationfür Akkreditierung (European co-ope-ration for Accreditation, kurz EA)koordiniert. In den Mitgliedsländernkönnen Kalibrierlaboratorien beiden für sie zuständigen staatlichenStellen ein Zertifikat erlangen, dasbelegt, dass sie nach diesen Vor-schriften arbeiten. In Deutschlandist dafür die Deutsche Akkreditierungs-stelle (DAkkS) zuständig, die dieseAufgabe mit Wirkung vom 17.12.2009vom DKD übernommen hat. AlsFachgremium der PTB und Zusam-menschluss von Kalibrierlaborato-rien in Industrieunternehmen, Prüf-institutionen und technischen Be-hörden betreibt der DKD heute aus-schließlich fachliche Basisarbeit wiedie Entwicklung von Normen undRichtlinien. Die DAkkS führt alle fünfJahre eine vollständige Begutach-tung jedes akkreditierten Kalibrier-laboratoriums und alle 18 MonateÜberwachungsbesuche durch, umdie Erfüllung der hohen Ansprüchean die Messprozesse sicherzustellen.Neben der beschriebenen Prozessüb-
erwachung derLaboratoriengibt es auchExpertenkreisezur Sicherstel-lung der tech-nischen Ent-wicklung so-wie des Know-how- Trans-fers. Weil alleeuropäischenStellen, die Ka-librierlabora-torien akkre-ditieren, inder EA zu-
sammenarbeiten, wurden die Ar-beitsweisen der Laboratorien anei-nander angepasst. Von einem be-liebigen Kalibrierlaboratoriumausgestellte Kalibrierscheine wer-den deshalb auch in allen andereneuropäischen Ländern anerkannt.
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)338 Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 1: Kalibrierhierachie Deutschland(Quelle aller Bilder: Wika Alexander Wiegand SE& Co. KG).
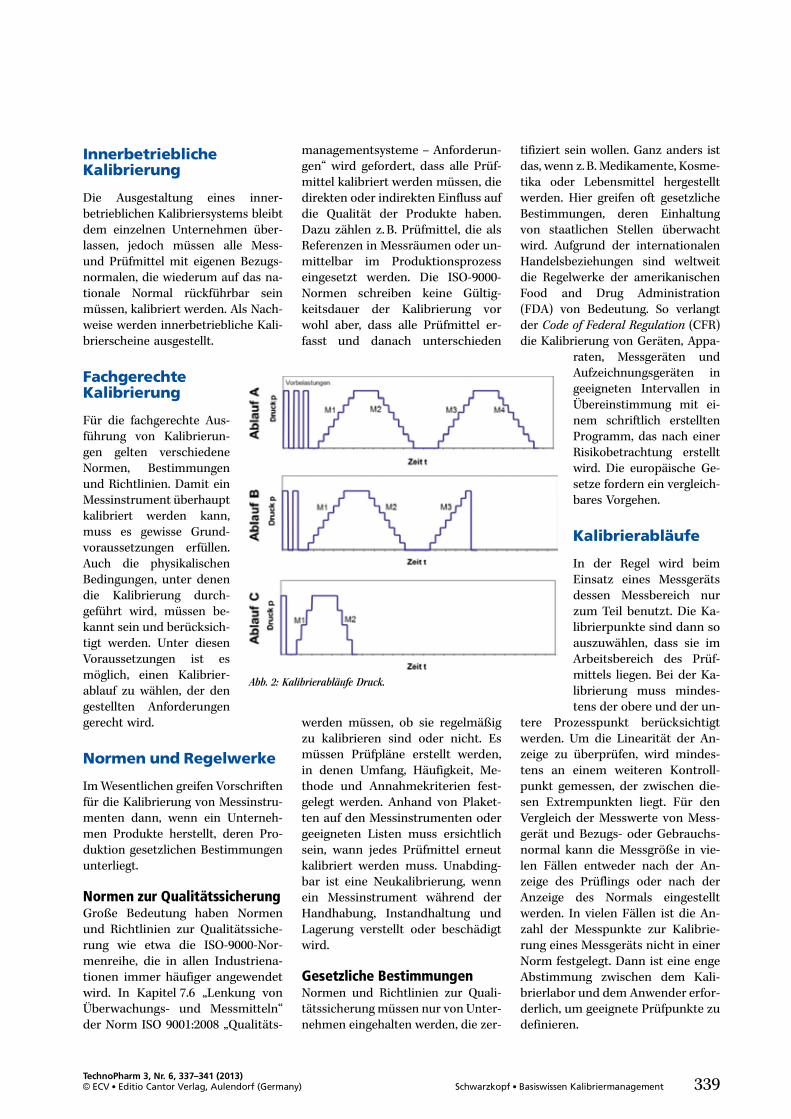
InnerbetrieblicheKalibrierung
Die Ausgestaltung eines inner-betrieblichen Kalibriersystems bleibtdem einzelnen Unternehmen über-lassen, jedoch müssen alle Mess-und Prüfmittel mit eigenen Bezugs-normalen, die wiederum auf das na-tionale Normal rückführbar seinmüssen, kalibriert werden. Als Nach-weise werden innerbetriebliche Kali-brierscheine ausgestellt.
FachgerechteKalibrierung
Für die fachgerechte Aus-führung von Kalibrierun-gen gelten verschiedeneNormen, Bestimmungenund Richtlinien. Damit einMessinstrument überhauptkalibriert werden kann,muss es gewisse Grund-voraussetzungen erfüllen.Auch die physikalischenBedingungen, unter denendie Kalibrierung durch-geführt wird, müssen be-kannt sein und berücksich-tigt werden. Unter diesenVoraussetzungen ist esmöglich, einen Kalibrier-ablauf zu wählen, der dengestellten Anforderungengerecht wird.
Normen und Regelwerke
ImWesentlichen greifen Vorschriftenfür die Kalibrierung von Messinstru-menten dann, wenn ein Unterneh-men Produkte herstellt, deren Pro-duktion gesetzlichen Bestimmungenunterliegt.
Normen zur QualitätssicherungGroße Bedeutung haben Normenund Richtlinien zur Qualitätssiche-rung wie etwa die ISO-9000-Nor-menreihe, die in allen Industriena-tionen immer häufiger angewendetwird. In Kapitel 7.6 „Lenkung vonÜberwachungs- und Messmitteln“der Norm ISO 9001:2008 „Qualitäts-
managementsysteme – Anforderun-gen“ wird gefordert, dass alle Prüf-mittel kalibriert werden müssen, diedirekten oder indirekten Einfluss aufdie Qualität der Produkte haben.Dazu zählen z.B. Prüfmittel, die alsReferenzen in Messräumen oder un-mittelbar im Produktionsprozesseingesetzt werden. Die ISO-9000-Normen schreiben keine Gültig-keitsdauer der Kalibrierung vorwohl aber, dass alle Prüfmittel er-fasst und danach unterschieden
werden müssen, ob sie regelmäßigzu kalibrieren sind oder nicht. Esmüssen Prüfpläne erstellt werden,in denen Umfang, Häufigkeit, Me-thode und Annahmekriterien fest-gelegt werden. Anhand von Plaket-ten auf den Messinstrumenten odergeeigneten Listen muss ersichtlichsein, wann jedes Prüfmittel erneutkalibriert werden muss. Unabding-bar ist eine Neukalibrierung, wennein Messinstrument während derHandhabung, Instandhaltung undLagerung verstellt oder beschädigtwird.
Gesetzliche BestimmungenNormen und Richtlinien zur Quali-tätssicherungmüssen nur von Unter-nehmen eingehalten werden, die zer-
tifiziert sein wollen. Ganz anders istdas, wenn z.B. Medikamente, Kosme-tika oder Lebensmittel hergestelltwerden. Hier greifen oft gesetzlicheBestimmungen, deren Einhaltungvon staatlichen Stellen überwachtwird. Aufgrund der internationalenHandelsbeziehungen sind weltweitdie Regelwerke der amerikanischenFood and Drug Administration(FDA) von Bedeutung. So verlangtder Code of Federal Regulation (CFR)die Kalibrierung von Geräten, Appa-
raten, Messgeräten undAufzeichnungsgeräten ingeeigneten Intervallen inÜbereinstimmung mit ei-nem schriftlich erstelltenProgramm, das nach einerRisikobetrachtung erstelltwird. Die europäische Ge-setze fordern ein vergleich-bares Vorgehen.
Kalibrierabläufe
In der Regel wird beimEinsatz eines Messgerätsdessen Messbereich nurzum Teil benutzt. Die Ka-librierpunkte sind dann soauszuwählen, dass sie imArbeitsbereich des Prüf-mittels liegen. Bei der Ka-librierung muss mindes-tens der obere und der un-
tere Prozesspunkt berücksichtigtwerden. Um die Linearität der An-zeige zu überprüfen, wird mindes-tens an einem weiteren Kontroll-punkt gemessen, der zwischen die-sen Extrempunkten liegt. Für denVergleich der Messwerte von Mess-gerät und Bezugs- oder Gebrauchs-normal kann die Messgröße in vie-len Fällen entweder nach der An-zeige des Prüflings oder nach derAnzeige des Normals eingestelltwerden. In vielen Fällen ist die An-zahl der Messpunkte zur Kalibrie-rung eines Messgeräts nicht in einerNorm festgelegt. Dann ist eine engeAbstimmung zwischen dem Kali-brierlabor und dem Anwender erfor-derlich, um geeignete Prüfpunkte zudefinieren.
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 339Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 2: Kalibrierabläufe Druck.
Optimierungen von Herstellprozes-sen gewinnt die Kalibrierung zuneh-mend an Bedeutung. Eine Erhöhungder Messgenauigkeit kann zu Roh-stoffeinsparungen und wenigerSchadstoffemissionen führen, etwawenn bei einer chemischen Reaktiondie exakt richtige Menge an Sauer-stoff zugeführt wird. Die Kalibrierungvon Messgeräten kann sicherheits-relevant sein. Liefern Druck- oderTemperatursensoren in der che-mischen Industrie nicht die korrek-ten Werte, besteht aufgrund falscherSteuerung chemischer Prozesse un-ter Umständen sogar Explosions-gefahr. Die Bedeutung der Kalibrie-rung zeigt sich auch an alltäglichenBeispielen, etwa an Zählern für denGas- oder Wasserverbrauch im Haus-halt und an Benzinuhren an Zapfsäu-len.
Rückführung undKalibrierhierarchie
Um Messergebnisse vergleichen zukönnen, müssen sie sich über eineKette von Vergleichsmessungen aufein nationales oder internationalesNormal „rückführen“ lassen. Die An-zeige des eingesetzten Messgerätsoder eine Maßverkörperung wirddazu in einer oder mehreren Stufenmit diesem Normal verglichen. Aufjeder dieser Stufen erfolgt eine Kali-brierung mit einem Normal, das zu-vor mit einem höherwertigen Nor-mal kalibriert wurde. Entsprechendder Rangfolge der Normale – von Ge-brauchs- oder Werksnormalen überBezugsnormale bis hin zum nationa-len Normal – gibt es eine Kalibrier-hierarchie der durchführenden Stel-len. Diese reicht vom innerbetriebli-chen Kalibrierlabor über akkredi-tierte Kalibrierlaboratorien bis hinzum nationalen metrologischen In-stitut (Abb. 1).
Rückführung in der PraxisDer Deutsche Kalibrierdienst (DKD)nennt als wesentliche Elemente derRückführung:. Die Vergleichskette darf nicht un-terbrochen sein.
. Bei jedem Schritt in der Kalibrier-kette muss die Messunsicherheitbekannt sein, sodass eine Gesamt-messunsicherheit berechnet wer-den kann. In der Regel sollte einvorrangiges Messgerät eine drei-bis viermal so hohe Messgenau-igkeit besitzen wie das Gerät, dasmit seiner Hilfe kalibriert wird.
. Jeder Schritt der Kalibrierungs-kette muss dokumentiert werden,ebenso das Ergebnis.
. Alle Stellen, die einen Schritt indieser Kette durchführen, müssenetwa durch eine Akkreditierungihre Kompetenz nachweisen.
. Kalibrierungen müssen, je nachgeforderter Messgenauigkeit undtechnischen Anforderungen, inangemessenen Zeiträumen wie-derholt werden.
AkkreditierteKalibrierlaboratorien
Akkreditierte Kalibrierlaboratorienübernehmen oft als externe Dienst-leister die Kalibrierung für Unterneh-men, die nicht selbst über die not-wendige Ausrüstung verfügen. Siekönnen aber auch selbst Teil einesUnternehmenssein und in die-sem alle Mess-geräte kalibrie-ren. Dazu ver-fügen sie über ei-gene Werks- oderGebrauchsnor-male, die in ange-messenen Zeit-abständen mit-hilfe der Bezugs-normale des zu-ständigen metro-logischen Staats-instituts oder an-derer akkreditier-ter Laboratorien mit kleinstmögli-cher Messunsicherheit kalibriertwerden.
Koordination durch die EADamit überall ein gleiches Niveau beiKalibrierungen herrscht, müssen diedurchführenden Laboratorien nach
international anerkannten Regeln(DIN EN ISO IEC 17025) akkreditiertwerden. Innerhalb der EU wird diesvon der Europäischen Kooperationfür Akkreditierung (European co-ope-ration for Accreditation, kurz EA)koordiniert. In den Mitgliedsländernkönnen Kalibrierlaboratorien beiden für sie zuständigen staatlichenStellen ein Zertifikat erlangen, dasbelegt, dass sie nach diesen Vor-schriften arbeiten. In Deutschlandist dafür die Deutsche Akkreditierungs-stelle (DAkkS) zuständig, die dieseAufgabe mit Wirkung vom 17.12.2009vom DKD übernommen hat. AlsFachgremium der PTB und Zusam-menschluss von Kalibrierlaborato-rien in Industrieunternehmen, Prüf-institutionen und technischen Be-hörden betreibt der DKD heute aus-schließlich fachliche Basisarbeit wiedie Entwicklung von Normen undRichtlinien. Die DAkkS führt alle fünfJahre eine vollständige Begutach-tung jedes akkreditierten Kalibrier-laboratoriums und alle 18 MonateÜberwachungsbesuche durch, umdie Erfüllung der hohen Ansprüchean die Messprozesse sicherzustellen.Neben der beschriebenen Prozessüb-
erwachung derLaboratoriengibt es auchExpertenkreisezur Sicherstel-lung der tech-nischen Ent-wicklung so-wie des Know-how- Trans-fers. Weil alleeuropäischenStellen, die Ka-librierlabora-torien akkre-ditieren, inder EA zu-
sammenarbeiten, wurden die Ar-beitsweisen der Laboratorien anei-nander angepasst. Von einem be-liebigen Kalibrierlaboratoriumausgestellte Kalibrierscheine wer-den deshalb auch in allen andereneuropäischen Ländern anerkannt.
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)338 Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 1: Kalibrierhierachie Deutschland(Quelle aller Bilder: Wika Alexander Wiegand SE& Co. KG).

den können, setzen vieleDAkkS Laboratorien ins-besondere zur Kalibrie-rung von Thermoelemen-ten oberhalb 500 °C Pla-tinrhodium-Platin Ther-moelemente (Typ S oderR nach DIN IEC 584-1)als Normalthermometerein. Die Vergleichsmes-sungen finden hier meistim Rohrofen mit eineman die Thermoelementeangepassten Ausgleichs-block statt. Vor Ort setztman sogenannte Block-kalibratoren ein.Bei der Durchführung ei-ner solchen Messreihe istdarauf zu achten, dass:. genügend Zeit vorhan-den ist, dass der Prüf-ling die Temperatur desReferenzgeräts (Abb. 4)annehmen kann
. die Umgebung eineräumlich und zeitlichhomogene Tempera-turverteilung liefert,sodass die Temperaturam Referenzgerät dengleichen Wert hat wieam Prüfling
. eine ausreichende Eintauchtiefevon mindestens dem Zehnfachendes Fühlerrohrdurchmessers gege-ben ist, damit eine Wärmeablei-tung in die Umgebung aus-geschlossen werden kann.
Empfehlung zur Wahl der Prüfpunk-te:. Kalibrierbereich = Einsatzbereichbeim Kunden ≠ möglicher Mess-bereich des Thermometers
. Anzahl der Prüfpunkte ist demKunden überlassen
. Empfehlung: Referenzthermo-meter 6 Prüfpunkte, SonstigeThermometer 3 Prüfpunkte
. Werte der Prüfpunkte werdenmeist linear über den Kalibrier-bereich verteilt
. Eine bestimmte Reihenfolge derTemperaturen muss beim Kali-brieren nicht eingehaltenwerden
Dokumentation
Der Kalibrierschein gibt die Ergeb-nisse der Kalibrierung wieder unddokumentiert die Rückführbarkeitder Kalibrierung auf nationale Nor-male zur Darstellung der physika-lischen Einheiten in Übereinstim-mung mit dem Internationalen Ein-heitensystem (SI). Kalibrierscheine(Abb. 5), die von einem Labor derDeutschen Akkrediterungsstelle,DAkkS, erstellt werden, bestehenaus einem Deckblatt, auf dem all-gemeine Angaben zum Kalibrier-gegenstand, zum Auftraggeber, zumausführenden DKD/ DAkkS Laborsowie Aussagen über die internatio-nale Anerkennung von DKD/ DAkkSKalibrierscheinen im Rahmen der EA– Mitgliedsländer (EA – EuropeanAssociation for Accreditation) ge-macht werden, die sich auf multilate-
rale Abkommen stützen.Auf den Folgeseiten wirddie Kalibrierung doku-mentiert. Dazu gehörteine kurze Beschreibungdes Kalibriergegenstan-des, des angewandten Ka-librierverfahrens mit An-gabe der geltenden Richt-linien, der Messbedingun-gen wie z.B. verwendeteNormale und Messein-richtungen sowie der fürdie Kalibrierung relevan-ten Umgebungsbedingun-gen. Zu einer vollständi-gen Angabe der Mess-ergebnisse gehören derWert der Messgröße, derangezeigte Wert, dieMessabweichung und dieGesamtmessunsicherheit.Ergänzend können Kon-formitätsaussagen getrof-fen werden, z.B. dass dieMessabweichung inner-halb einer bestimmtenToleranzklasse bleibt.Nach der Kalibrierungwird der Kalibriergegen-stand mit einer Kalibrier-marke versehen, derenKennzeichen auf allen
Seiten des Kalibrierscheins angege-ben wird. Eine ausführliche Anlei-tung zum Erstellen eines Kalibrier-scheins wird in der DKD/ DAkkSSchrift Nr. 5 gegeben.
Fazit
Die Bedeutung der Kalibrierung wirdimmer größer. Es steigen die Anfor-derungen der Anwender an dieMesstechnik der Referenzgeräte.Die damit verbundene Weiterent-wicklung erfolgt häufig auf Heraus-forderungen, die auf den ersten Blickgar nicht mit einer Kalibrierung inVerbindung gebracht werden. DerAnwender soll deshalb mit kom-petenten Partnern zusammenarbei-ten, welche Ihm bei der Wahl desrichtigen Referenzgerätes und derkorrekten Kalibriermethode hilfreichzur Seite stehen.
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 341Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 5: Deckblatt DAkkS Kalibrierschein.
Kalibrierung vonDruckmessgerätenDie Kalibrierung von Druckmess-geräten ist sehr detailliert geregelt.In der Schrift DKD-R 6-1 des Deut-schen Kalibrierdienstes werden dreiKalibrierabläufe für unterschiedlicheGenauigkeitsklassen beschrieben.Der in Abb. 2 mit A bezeichnete Ka-librierablauffür die Genau-igkeitsklasse< 0,1 schreibtdrei Belastun-gen bis zumMessbereichs-endwert vor,die vor den ei-gentlichenMessreihen zuerfolgen ha-ben. Der maximale Druck wird min-destens 30 Sekunden lang gehaltenund dann wieder vollständig abge-baut. Im Verlauf der ersten Messreihewerden neun gleichmäßig über denMessbereich verteilte Messpunktedurch allmähliche Druckerhöhung„von unten nach oben“ angefahren.Der Nullpunkt zählt als erster Mess-punkt, wird aber bei der statistischenAuswertung nur mitberücksichtigt,wenn er frei ist, d. h., wenn die Bewe-gung des Zeigers nicht durch einenAnschlagsstift eingeschränkt ist. DerDruck muss so langsam erhöht wer-den, dass er den anvisierten Mess-punkt nicht übersteigt, weil dies auf-grund der Hysterese zu einer Verfäl-schung der Ergebnisse führen könn-te. Gegebenenfalls muss der Druckwieder stark verringert werden, so-dass der Messpunkt auf jeden Fallvon unten erreicht wird. Der er-reichte Wert wird nach mindestens30 Sekunden Haltezeit abgelesen.Diese Haltezeit ist notwendig, weilder am Prüfling angezeigte Wertnicht unmittelbar angenommen,sondern erst nach einer gewissen Re-laxationszeit erreicht wird. Bei Fe-dermanometern sollte vor dem Able-sen leicht am Gehäuse geklopft wer-den, um Einflüsse der Reibung aufdas Zeigerwerk zu minimieren. Aucham letzten Prüfpunkt wird der End-
wert nach 30 Sekunden abgelesenund dokumentiert. Nach zwei Minu-ten bei konstantem Druck wird die-ser Wert ein weiteres Mal abgelesen.Bei Federmanometern sollte der Ma-ximaldruck sogar fünf Minuten ge-halten werden. Bei piezoelektrischenSensoren können die Haltezeitenhingegen verringert werden. Vom
Maximaldruckausgehendwerden in derzweiten Mess-reihe diesel-ben Mess-punkte „vonoben nach un-ten“ angefah-ren. Der Druckwird dazu sogesenkt, dass
der jeweils angepeilte Wert nicht un-terschritten wird. Zum Schluss die-ses ersten Messzyklus wird der Null-punkt gemessen. Das Gerät verbleibtdazu zwei Minuten im drucklosenZustand. Kalibrierablauf A sieht vor,dass der beschriebene Zyklus der stu-fenweisen Druckerhöhung und -ver-ringerung einmal wiederholt wird.Falls beiDruckmessge-räten mit gro-ßem Mess-bereich oderoffenliegender,frontbündigerMembran Ein-spanneffektezu beobachtensind, kann eindritter Mess-zyklus durch-geführt wer-den, um eineAbhängigkeitdes Nullsignals vom Anzugsdrehmo-ment nachzuweisen – ein Effekt, derhäufig bei kostengünstigen elektri-schen Sensoren auftritt. Kalibrier-ablauf B, der für Druckmessgeräteder Genauigkeitsklassen 0,1 bis 0,6eingesetzt wird, verlangt etwas weni-ger Aufwand: Es gibt nur zwei Vor-belastungen bis zum Maximalwert,und die dritte Messreihe endet be-
reits kurz nachdem der Druck denMaximalwert erreicht hat. Von dortwird sofort bis zum Nullwert abge-lassen. Kalibrierablauf C kann beiDruckmessgeräten der Genauigkeits-klasse > 0,6 angewendet werden. Erverlangt nur eine Vorbelastung biszum Maximalwert und nur eineMessreihe, die einschließlich desNullwerts und des Maximalwertsaus fünf Messpunkten besteht undbei der der Druck stufenweise erhöhtund anschließend wieder abgesenktwird (Abb. 3).
Kalibrierung vonTemperaturmessgerätenDie Erzeugung einer genau definier-ten Temperatur ist weit aufwendigerals die Erzeugung eines bestimmtenDrucks. Deshalb werden bei der Ka-librierung von Temperaturmessgerä-ten in der Regel keine so umfangrei-chen Messreihen durchgeführt. Invielen Fällen werden Thermometernur an einem einzigen Fixpunkt ka-libriert, wie z.B. dem Tripelpunkt desWassers. Für das Justieren des Tem-peraturmessgeräts wird seine meistsehr gut bekannte Kennlinie so nach
oben oder untenverschoben,dass das Gerätam Fixpunktden richtigenWert zeigt.Messreihen sinddann praktika-bel, wenn einTemperatur-messgerät nichtan einem Fix-punkt, sonderndurch Vergleichmit einem hö-herwertigen
Messgerät kalibriert werden soll.Dies ist in Tauchbädern oder Öfenmöglich. Hat sich ein thermischesGleichgewicht zwischen den Ther-mometern und der Badflüssigkeiteingestellt, so vergleicht man die An-zeige der Prüflinge mit der desNormalthermometers. Obwohl diemeisten Normal-Widerstandsther-mometer bis 660 °C verwendet wer-
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)340 Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 3: Referenzgerät CPC8000.
Abb. 4: Temperatur-BlockkalibratorCTD9100-165.

den können, setzen vieleDAkkS Laboratorien ins-besondere zur Kalibrie-rung von Thermoelemen-ten oberhalb 500 °C Pla-tinrhodium-Platin Ther-moelemente (Typ S oderR nach DIN IEC 584-1)als Normalthermometerein. Die Vergleichsmes-sungen finden hier meistim Rohrofen mit eineman die Thermoelementeangepassten Ausgleichs-block statt. Vor Ort setztman sogenannte Block-kalibratoren ein.Bei der Durchführung ei-ner solchen Messreihe istdarauf zu achten, dass:. genügend Zeit vorhan-den ist, dass der Prüf-ling die Temperatur desReferenzgeräts (Abb. 4)annehmen kann
. die Umgebung eineräumlich und zeitlichhomogene Tempera-turverteilung liefert,sodass die Temperaturam Referenzgerät dengleichen Wert hat wieam Prüfling
. eine ausreichende Eintauchtiefevon mindestens dem Zehnfachendes Fühlerrohrdurchmessers gege-ben ist, damit eine Wärmeablei-tung in die Umgebung aus-geschlossen werden kann.
Empfehlung zur Wahl der Prüfpunk-te:. Kalibrierbereich = Einsatzbereichbeim Kunden ≠ möglicher Mess-bereich des Thermometers
. Anzahl der Prüfpunkte ist demKunden überlassen
. Empfehlung: Referenzthermo-meter 6 Prüfpunkte, SonstigeThermometer 3 Prüfpunkte
. Werte der Prüfpunkte werdenmeist linear über den Kalibrier-bereich verteilt
. Eine bestimmte Reihenfolge derTemperaturen muss beim Kali-brieren nicht eingehaltenwerden
Dokumentation
Der Kalibrierschein gibt die Ergeb-nisse der Kalibrierung wieder unddokumentiert die Rückführbarkeitder Kalibrierung auf nationale Nor-male zur Darstellung der physika-lischen Einheiten in Übereinstim-mung mit dem Internationalen Ein-heitensystem (SI). Kalibrierscheine(Abb. 5), die von einem Labor derDeutschen Akkrediterungsstelle,DAkkS, erstellt werden, bestehenaus einem Deckblatt, auf dem all-gemeine Angaben zum Kalibrier-gegenstand, zum Auftraggeber, zumausführenden DKD/ DAkkS Laborsowie Aussagen über die internatio-nale Anerkennung von DKD/ DAkkSKalibrierscheinen im Rahmen der EA– Mitgliedsländer (EA – EuropeanAssociation for Accreditation) ge-macht werden, die sich auf multilate-
rale Abkommen stützen.Auf den Folgeseiten wirddie Kalibrierung doku-mentiert. Dazu gehörteine kurze Beschreibungdes Kalibriergegenstan-des, des angewandten Ka-librierverfahrens mit An-gabe der geltenden Richt-linien, der Messbedingun-gen wie z.B. verwendeteNormale und Messein-richtungen sowie der fürdie Kalibrierung relevan-ten Umgebungsbedingun-gen. Zu einer vollständi-gen Angabe der Mess-ergebnisse gehören derWert der Messgröße, derangezeigte Wert, dieMessabweichung und dieGesamtmessunsicherheit.Ergänzend können Kon-formitätsaussagen getrof-fen werden, z.B. dass dieMessabweichung inner-halb einer bestimmtenToleranzklasse bleibt.Nach der Kalibrierungwird der Kalibriergegen-stand mit einer Kalibrier-marke versehen, derenKennzeichen auf allen
Seiten des Kalibrierscheins angege-ben wird. Eine ausführliche Anlei-tung zum Erstellen eines Kalibrier-scheins wird in der DKD/ DAkkSSchrift Nr. 5 gegeben.
Fazit
Die Bedeutung der Kalibrierung wirdimmer größer. Es steigen die Anfor-derungen der Anwender an dieMesstechnik der Referenzgeräte.Die damit verbundene Weiterent-wicklung erfolgt häufig auf Heraus-forderungen, die auf den ersten Blickgar nicht mit einer Kalibrierung inVerbindung gebracht werden. DerAnwender soll deshalb mit kom-petenten Partnern zusammenarbei-ten, welche Ihm bei der Wahl desrichtigen Referenzgerätes und derkorrekten Kalibriermethode hilfreichzur Seite stehen.
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 341Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 5: Deckblatt DAkkS Kalibrierschein.
Kalibrierung vonDruckmessgerätenDie Kalibrierung von Druckmess-geräten ist sehr detailliert geregelt.In der Schrift DKD-R 6-1 des Deut-schen Kalibrierdienstes werden dreiKalibrierabläufe für unterschiedlicheGenauigkeitsklassen beschrieben.Der in Abb. 2 mit A bezeichnete Ka-librierablauffür die Genau-igkeitsklasse< 0,1 schreibtdrei Belastun-gen bis zumMessbereichs-endwert vor,die vor den ei-gentlichenMessreihen zuerfolgen ha-ben. Der maximale Druck wird min-destens 30 Sekunden lang gehaltenund dann wieder vollständig abge-baut. Im Verlauf der ersten Messreihewerden neun gleichmäßig über denMessbereich verteilte Messpunktedurch allmähliche Druckerhöhung„von unten nach oben“ angefahren.Der Nullpunkt zählt als erster Mess-punkt, wird aber bei der statistischenAuswertung nur mitberücksichtigt,wenn er frei ist, d. h., wenn die Bewe-gung des Zeigers nicht durch einenAnschlagsstift eingeschränkt ist. DerDruck muss so langsam erhöht wer-den, dass er den anvisierten Mess-punkt nicht übersteigt, weil dies auf-grund der Hysterese zu einer Verfäl-schung der Ergebnisse führen könn-te. Gegebenenfalls muss der Druckwieder stark verringert werden, so-dass der Messpunkt auf jeden Fallvon unten erreicht wird. Der er-reichte Wert wird nach mindestens30 Sekunden Haltezeit abgelesen.Diese Haltezeit ist notwendig, weilder am Prüfling angezeigte Wertnicht unmittelbar angenommen,sondern erst nach einer gewissen Re-laxationszeit erreicht wird. Bei Fe-dermanometern sollte vor dem Able-sen leicht am Gehäuse geklopft wer-den, um Einflüsse der Reibung aufdas Zeigerwerk zu minimieren. Aucham letzten Prüfpunkt wird der End-
wert nach 30 Sekunden abgelesenund dokumentiert. Nach zwei Minu-ten bei konstantem Druck wird die-ser Wert ein weiteres Mal abgelesen.Bei Federmanometern sollte der Ma-ximaldruck sogar fünf Minuten ge-halten werden. Bei piezoelektrischenSensoren können die Haltezeitenhingegen verringert werden. Vom
Maximaldruckausgehendwerden in derzweiten Mess-reihe diesel-ben Mess-punkte „vonoben nach un-ten“ angefah-ren. Der Druckwird dazu sogesenkt, dass
der jeweils angepeilte Wert nicht un-terschritten wird. Zum Schluss die-ses ersten Messzyklus wird der Null-punkt gemessen. Das Gerät verbleibtdazu zwei Minuten im drucklosenZustand. Kalibrierablauf A sieht vor,dass der beschriebene Zyklus der stu-fenweisen Druckerhöhung und -ver-ringerung einmal wiederholt wird.Falls beiDruckmessge-räten mit gro-ßem Mess-bereich oderoffenliegender,frontbündigerMembran Ein-spanneffektezu beobachtensind, kann eindritter Mess-zyklus durch-geführt wer-den, um eineAbhängigkeitdes Nullsignals vom Anzugsdrehmo-ment nachzuweisen – ein Effekt, derhäufig bei kostengünstigen elektri-schen Sensoren auftritt. Kalibrier-ablauf B, der für Druckmessgeräteder Genauigkeitsklassen 0,1 bis 0,6eingesetzt wird, verlangt etwas weni-ger Aufwand: Es gibt nur zwei Vor-belastungen bis zum Maximalwert,und die dritte Messreihe endet be-
reits kurz nachdem der Druck denMaximalwert erreicht hat. Von dortwird sofort bis zum Nullwert abge-lassen. Kalibrierablauf C kann beiDruckmessgeräten der Genauigkeits-klasse > 0,6 angewendet werden. Erverlangt nur eine Vorbelastung biszum Maximalwert und nur eineMessreihe, die einschließlich desNullwerts und des Maximalwertsaus fünf Messpunkten besteht undbei der der Druck stufenweise erhöhtund anschließend wieder abgesenktwird (Abb. 3).
Kalibrierung vonTemperaturmessgerätenDie Erzeugung einer genau definier-ten Temperatur ist weit aufwendigerals die Erzeugung eines bestimmtenDrucks. Deshalb werden bei der Ka-librierung von Temperaturmessgerä-ten in der Regel keine so umfangrei-chen Messreihen durchgeführt. Invielen Fällen werden Thermometernur an einem einzigen Fixpunkt ka-libriert, wie z.B. dem Tripelpunkt desWassers. Für das Justieren des Tem-peraturmessgeräts wird seine meistsehr gut bekannte Kennlinie so nach
oben oder untenverschoben,dass das Gerätam Fixpunktden richtigenWert zeigt.Messreihen sinddann praktika-bel, wenn einTemperatur-messgerät nichtan einem Fix-punkt, sonderndurch Vergleichmit einem hö-herwertigen
Messgerät kalibriert werden soll.Dies ist in Tauchbädern oder Öfenmöglich. Hat sich ein thermischesGleichgewicht zwischen den Ther-mometern und der Badflüssigkeiteingestellt, so vergleicht man die An-zeige der Prüflinge mit der desNormalthermometers. Obwohl diemeisten Normal-Widerstandsther-mometer bis 660 °C verwendet wer-
Messen/Steuern/Regeln
TechnoPharm 3, Nr. 6, 337–341 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)340 Schwarzkopf . Basiswissen Kalibriermanagement
Abb. 3: Referenzgerät CPC8000.
Abb. 4: Temperatur-BlockkalibratorCTD9100-165.

management & Projektmanagementfür Pharma-Ingenieure. Die zusammen-gestellten Informationen dienen als Hil-festellung und können keine individu-elle Auseinandersetzung mit der jewei-ligen Situation ersetzen. Auch könnendie Angaben nicht als Best-Practice-Lö-sung verstanden werden, die es in einerstark vom Einzelfall abhängigen Weltdes Rechts in der Regel nicht gibt. Ziel-setzung der Beiträge ist es vielmehr, diegeneigten Leserinnen und Leser inso-weit zu sensibilisieren. Sie sollten sichin die Lage versetzen können, in derjeweiligen Situation die Tragweite undBedeutung notwendiger Schritte zu er-kennen und gegebenenfalls auch haus-intern dementsprechend zu kommuni-zieren.
Vertragscharakter
In einem ersten Schritt ist zunächst derRechtscharakter des zwischen den Par-teien geschlossenen Vertrages fest-
zustellen, da hiermit der Einstieg indie jeweilige Rechtsmaterie erfolgt. Vo-ranstehend wurde bereits begrifflichzwischen „Lieferant“ und „Subunter-nehmer“ unterschieden. An diese Diffe-renzierung wird im Folgenden ange-knüpft, da die weitergehende Bewer-tung der sich stellenden Risiken undMöglichkeiten hiervon maßgeblich ab-hängt: Mit Blick auf die vorgenanntenGegebenheiten Käufer/Lieferant bzw.Besteller/Subunternehmer muss stetszwischen dem Vorliegen eines Kaufver-trages (§ 433 BGB) oder eines Werkver-trages (§ 631 BGB) unterschieden wer-den. Ein Kaufvertrag ist regelmäßig einVertrag zwischen zwei Parteien, bei de-nen sich der Verkäufer verpflichtet,dem Käufer den Besitz an einem Pro-dukt – einer Sache – zu verschaffen unddas Eigentum daran zu übertragen.Hierbei ist entscheidend, dass es demKäufer nicht darauf ankommt, dass derVerkäufer das Produkt auch tatsächlichhergestellt hat. Dieses Kriterium ist
vielmehr bedeutsam für die Unterschei-dung zumWerkvertrag. ImWerkvertraggeht es dem Besteller gegenüber demWerkunternehmer auch darum, dassder Werkunternehmer das Produkt –seine Leistung – tatsächlich selbst her-stellt oder zumindest durch andere her-stellen lässt. Im Ergebnis übernimmtalso der Werkunternehmer die Verant-wortung für das Produkt auch herstel-lerseitig. In bestimmten Konstellatio-nen kann trotz einer werkvertraglichenGrundcharakteristik dennoch das Kauf-recht zur Anwendung kommen. Diesgilt für den besonderen Fall des soge-nannten „Werklieferungsvertrages“(§ 651 BGB), bei dem der Unternehmerverpflichtet ist, bewegliche Sachen zuerzeugen oder herzustellen. Die Unter-scheidung zwischen Kauf- und Werk-vertrag ist bei der Problemabwicklungim Rahmen der Vertragserfüllung maß-geblich. In der Regel wird bei individu-ell angefertigten Produkten, wie auchbei Planungs- und sonstigen geis-
ProjektmanagementRechtssicherheit für Pharma-Ingenieure: Teil 1
Daniel Wuhrmann und Philipp Reusch . Reusch Rechtsanwälte, Saarbrücken
Korrespondenz: Daniel Wuhrmann, Reusch Rechtsanwälte, Nell-Breuning-Allee 10, 66115 Saarbrücken;e-mail: [email protected]
ZusammenfassungDie Aufgaben von technischen Mitarbeitern im Rahmen der Durchführung von Projekten impharmazeutischen Bereich sind vielschichtig. Neben einer gesteigerten Sensibilität für auf-kommende Probleme in der Projektrealisierung und einer schnellen Reaktion auf akute Ent-wicklungen wird eine konsequente Umsetzung geregelter Vorgaben verlangt. Im Bereichdes Projektmanagements zählen zu den Aufgaben mitunter die Nachverfolgung von Ter-minen, Kosten und Qualitäten. Der nachfolgende Beitrag soll in zwei Teilen grundlegendejuristische Zusammenhänge erläutern, um so dem Leser eine Hilfestellung bei derBewertung von Problemen mit juristischem Hintergrund und deren Handling zu geben.
Normative Hintergründe
Bei der Gestaltung und Durchführungvon Projekten in der pharmazeuti-schen Industrie sind vielfach Inge-nieure an deren Umsetzung beteiligt.Dies liegt naturgemäß daran, dass dieumfangreichen Vorgaben des europä-ischen und deutschen Arzneimittel-rechts, insbesondere die für die Um-setzung der EU-GMP-Leitfäden not-wendigen Inhalte, ohne detaillierteKenntnisse der Grundsätze und Leit-linien der guten Herstellungspraxisnicht sinnvoll umzusetzen sind. Somitzählen aber auch die Steuerung vonLieferanten bei der Durchführung ei-nes Green-Field- Projektes sowie derUmgang mit Planern und Subunter-nehmern zu den Aufgaben der verant-wortlichen Personen. Unabhängig vonden internationalen und den deut-schen arzneimittelrechtlichen Vor-gaben werden die Geschäfte zwischenAuftraggeber und den verschiedenenAuftragnehmern hauptsächlich durchdas deutsche Zivilrecht geregelt. InBetracht kommen hierbei insbeson-
dere die Regelungen des BürgerlichenGesetzbuches (BGB) und teilweise diedes Handelsgesetzbuches (HGB). Aus-ländische Rechtsordnungen sowie dasUN-Kaufrecht sollen bei der folgendenDarstellung außer Betracht bleiben.
MöglicheProblemstellungen
In der Regel obliegt die Vertrags-gestaltung nicht den beteiligten Pro-jektingenieuren, sondern dem Ein-kauf. Die Beauftragung des Lieferan-ten erfolgt durch den Einkauf, nachentsprechender Abstimmung mitdem Bedarfsträger in der Produktionund Technik. Die Ingenieure tragenallerdings die alleinige Verantwor-tung für die spätere Realisierungdes Projektes. Dementsprechendmüssen sich Projektingenieure auchmit rechtlichen Themen der Projekt-umsetzung befassen. Regelmäßig las-sen insbesondere folgende Themenakuten Handlungsbedarf entstehen:. der Lieferant liefert zu spät odergar nicht
. der Lieferant liefert eine zu geringeMenge der bestellten Produkte
. der Lieferant liefert fehlerhafteProdukte
. der Subunternehmer wird das Ge-werk voraussichtlich nicht odernicht rechtzeitig beenden
. der Subunternehmer wird das Ge-werk voraussichtlich mangelhaftbeenden
. der Subunternehmer hat das Ge-werk nicht, nicht rechtzeitig odermangelhaft beendet
. der Subunternehmer besteht aufAbnahme und Vergütung, obwohldas Werk erhebliche Mängel hat.
Solche Situationen tauchen in Projek-ten regelmäßig auf, sind aber auch beiprofessioneller und vorbildlicher Pla-nung nicht auszuschließen und müssenmeist unter Zeitdruck gelöst werden.Hierbei handelt es sich offensichtlichum rechtlich relevante Vorgänge, denenman nur mit dem notwendigen recht-lichen Rüstzeug sinnvoll und rechts-sicher begegnen kann. Dieser und auchder nachfolgende Beitrag beschäftigensich daher mit der Thematik Vertrags-
Maschinen- und Anlagenbau
TechnoPharm 3, Nr. 6, 342–345 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)342 Wuhrmann und Reusch . Projektmanagement

management & Projektmanagementfür Pharma-Ingenieure. Die zusammen-gestellten Informationen dienen als Hil-festellung und können keine individu-elle Auseinandersetzung mit der jewei-ligen Situation ersetzen. Auch könnendie Angaben nicht als Best-Practice-Lö-sung verstanden werden, die es in einerstark vom Einzelfall abhängigen Weltdes Rechts in der Regel nicht gibt. Ziel-setzung der Beiträge ist es vielmehr, diegeneigten Leserinnen und Leser inso-weit zu sensibilisieren. Sie sollten sichin die Lage versetzen können, in derjeweiligen Situation die Tragweite undBedeutung notwendiger Schritte zu er-kennen und gegebenenfalls auch haus-intern dementsprechend zu kommuni-zieren.
Vertragscharakter
In einem ersten Schritt ist zunächst derRechtscharakter des zwischen den Par-teien geschlossenen Vertrages fest-
zustellen, da hiermit der Einstieg indie jeweilige Rechtsmaterie erfolgt. Vo-ranstehend wurde bereits begrifflichzwischen „Lieferant“ und „Subunter-nehmer“ unterschieden. An diese Diffe-renzierung wird im Folgenden ange-knüpft, da die weitergehende Bewer-tung der sich stellenden Risiken undMöglichkeiten hiervon maßgeblich ab-hängt: Mit Blick auf die vorgenanntenGegebenheiten Käufer/Lieferant bzw.Besteller/Subunternehmer muss stetszwischen dem Vorliegen eines Kaufver-trages (§ 433 BGB) oder eines Werkver-trages (§ 631 BGB) unterschieden wer-den. Ein Kaufvertrag ist regelmäßig einVertrag zwischen zwei Parteien, bei de-nen sich der Verkäufer verpflichtet,dem Käufer den Besitz an einem Pro-dukt – einer Sache – zu verschaffen unddas Eigentum daran zu übertragen.Hierbei ist entscheidend, dass es demKäufer nicht darauf ankommt, dass derVerkäufer das Produkt auch tatsächlichhergestellt hat. Dieses Kriterium ist
vielmehr bedeutsam für die Unterschei-dung zumWerkvertrag. ImWerkvertraggeht es dem Besteller gegenüber demWerkunternehmer auch darum, dassder Werkunternehmer das Produkt –seine Leistung – tatsächlich selbst her-stellt oder zumindest durch andere her-stellen lässt. Im Ergebnis übernimmtalso der Werkunternehmer die Verant-wortung für das Produkt auch herstel-lerseitig. In bestimmten Konstellatio-nen kann trotz einer werkvertraglichenGrundcharakteristik dennoch das Kauf-recht zur Anwendung kommen. Diesgilt für den besonderen Fall des soge-nannten „Werklieferungsvertrages“(§ 651 BGB), bei dem der Unternehmerverpflichtet ist, bewegliche Sachen zuerzeugen oder herzustellen. Die Unter-scheidung zwischen Kauf- und Werk-vertrag ist bei der Problemabwicklungim Rahmen der Vertragserfüllung maß-geblich. In der Regel wird bei individu-ell angefertigten Produkten, wie auchbei Planungs- und sonstigen geis-
Ihr Partner für die Planung undschlüsselfertige Realisierungvon Produktionsanlagen
Ob Neubau, Erweiterung, maßgeschneiderte Prozesssysteme oder Prozessoptimierung – unsere Projektteams erarbeiten die besten Lösungen für Sie. Lassen Sie sich überzeugen, z.B. durch einen Blick in die nahe Zukunft Ihrer Anlage mittels modernster Simulationstechnik.
Von Ihrer Idee zum erfolgreichen Projekt – alle Leistungen aus einer Hand.
Beratung Planung Realisierung Qualifizierung Validierung TFM
M+W Process Industries GmbHA Company of the M+W Groupwww.pi.mwgroup.net
ProjektmanagementRechtssicherheit für Pharma-Ingenieure: Teil 1
Daniel Wuhrmann und Philipp Reusch . Reusch Rechtsanwälte, Saarbrücken
Korrespondenz: Daniel Wuhrmann, Reusch Rechtsanwälte, Nell-Breuning-Allee 10, 66115 Saarbrücken;e-mail: [email protected]
ZusammenfassungDie Aufgaben von technischen Mitarbeitern im Rahmen der Durchführung von Projekten impharmazeutischen Bereich sind vielschichtig. Neben einer gesteigerten Sensibilität für auf-kommende Probleme in der Projektrealisierung und einer schnellen Reaktion auf akute Ent-wicklungen wird eine konsequente Umsetzung geregelter Vorgaben verlangt. Im Bereichdes Projektmanagements zählen zu den Aufgaben mitunter die Nachverfolgung von Ter-minen, Kosten und Qualitäten. Der nachfolgende Beitrag soll in zwei Teilen grundlegendejuristische Zusammenhänge erläutern, um so dem Leser eine Hilfestellung bei derBewertung von Problemen mit juristischem Hintergrund und deren Handling zu geben.
Normative Hintergründe
Bei der Gestaltung und Durchführungvon Projekten in der pharmazeuti-schen Industrie sind vielfach Inge-nieure an deren Umsetzung beteiligt.Dies liegt naturgemäß daran, dass dieumfangreichen Vorgaben des europä-ischen und deutschen Arzneimittel-rechts, insbesondere die für die Um-setzung der EU-GMP-Leitfäden not-wendigen Inhalte, ohne detaillierteKenntnisse der Grundsätze und Leit-linien der guten Herstellungspraxisnicht sinnvoll umzusetzen sind. Somitzählen aber auch die Steuerung vonLieferanten bei der Durchführung ei-nes Green-Field- Projektes sowie derUmgang mit Planern und Subunter-nehmern zu den Aufgaben der verant-wortlichen Personen. Unabhängig vonden internationalen und den deut-schen arzneimittelrechtlichen Vor-gaben werden die Geschäfte zwischenAuftraggeber und den verschiedenenAuftragnehmern hauptsächlich durchdas deutsche Zivilrecht geregelt. InBetracht kommen hierbei insbeson-
dere die Regelungen des BürgerlichenGesetzbuches (BGB) und teilweise diedes Handelsgesetzbuches (HGB). Aus-ländische Rechtsordnungen sowie dasUN-Kaufrecht sollen bei der folgendenDarstellung außer Betracht bleiben.
MöglicheProblemstellungen
In der Regel obliegt die Vertrags-gestaltung nicht den beteiligten Pro-jektingenieuren, sondern dem Ein-kauf. Die Beauftragung des Lieferan-ten erfolgt durch den Einkauf, nachentsprechender Abstimmung mitdem Bedarfsträger in der Produktionund Technik. Die Ingenieure tragenallerdings die alleinige Verantwor-tung für die spätere Realisierungdes Projektes. Dementsprechendmüssen sich Projektingenieure auchmit rechtlichen Themen der Projekt-umsetzung befassen. Regelmäßig las-sen insbesondere folgende Themenakuten Handlungsbedarf entstehen:. der Lieferant liefert zu spät odergar nicht
. der Lieferant liefert eine zu geringeMenge der bestellten Produkte
. der Lieferant liefert fehlerhafteProdukte
. der Subunternehmer wird das Ge-werk voraussichtlich nicht odernicht rechtzeitig beenden
. der Subunternehmer wird das Ge-werk voraussichtlich mangelhaftbeenden
. der Subunternehmer hat das Ge-werk nicht, nicht rechtzeitig odermangelhaft beendet
. der Subunternehmer besteht aufAbnahme und Vergütung, obwohldas Werk erhebliche Mängel hat.
Solche Situationen tauchen in Projek-ten regelmäßig auf, sind aber auch beiprofessioneller und vorbildlicher Pla-nung nicht auszuschließen und müssenmeist unter Zeitdruck gelöst werden.Hierbei handelt es sich offensichtlichum rechtlich relevante Vorgänge, denenman nur mit dem notwendigen recht-lichen Rüstzeug sinnvoll und rechts-sicher begegnen kann. Dieser und auchder nachfolgende Beitrag beschäftigensich daher mit der Thematik Vertrags-
Maschinen- und Anlagenbau
TechnoPharm 3, Nr. 6, 342–345 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)342 Wuhrmann und Reusch . Projektmanagement

Abbedingung dieser Käuferpflichtennicht möglich ist, insbesondere nichtim Rahmen von Allgemeinen Ge-schäfts-/Einkaufsbedingungen odersonstigen Musterformulierungen, soempfiehlt es sich jedoch, den Pflichten-kreis des Käufers möglichst klein undsimpel zu halten. Dabei sind die gesetz-lichen Vorgaben einzuhalten und diejeweiligen Abläufe möglichst konkretdarzustellen. Ansonsten könnten dieseVereinbarungen letztlich juristisch re-lativ wertlos sein – was oftmals dasManko vieler sogenannter „Qualitäts-sicherungsvereinbarungen“ und ähnli-cher Vertragswerke ist. Die verantwort-lichen Personen sollten daher prüfen,
a) ob und welche Vereinbarungenzur Wareneingangskontrolle und zurRüge bestehen und
b) welche Maßnahmen diesbezüg-lich im jeweiligen Fall ergriffen wurden.
Ist die Ware mangelhaft, stehtdem Käufer vorrangig das Recht zu,Nacherfüllung (Nachbesserung oderNachlieferung) zu verlangen (§§ 437Nr. 1, 439 Abs. 1 BGB). Ohne vor-herige Vereinbarung mit dem Liefe-ranten darf er dies nicht eigenmäch-tig vornehmen – es sind allerdingsdurchaus Situationen denkbar, in de-nen die sogenannte Selbstvornahmefür beide Seiten im Hinblick auf Kos-ten und Dringlichkeit vorteilhaft istund sollte, sofern nicht schon vorabvereinbart, mit dem Lieferanten dis-kutiert werden. Die Kosten der Nach-erfüllung (Transport-, Arbeits- undMaterialkosten etc.) hat der Verkäu-fer zu tragen (§ 439 Abs. 2 BGB). ImFalle von bereits verbauter Warekann sich im BZB-Bereich unterdem Gesichtspunkt des Schadens-ersatzes auch ein weitergehenderAnspruch auf Ersatz der nunmehraufgewandten Aus- und Einbaukos-ten ergeben. Um den eventuell fol-genden Regress zu erleichtern, solltedies im Einzelfall geprüft und detail-liert dokumentiert werden. Als wei-tergehende Mängelansprüche stehendem Käufer zudem zu:– die Minderung des Kaufpreisesoder
– der Rücktritt vom Vertrag– und/oder Schadensersatz wegenNichterfüllung oder Ersatz vergeb-licher Aufwendungen.
Zwingende Voraussetzung hierfür istjedoch, dass entweder eine angemes-sen gesetzte Frist zur Nacherfüllungfruchtlos verstrichen ist, ein zweiterNachbesserungsversuch gescheitertoder der Verkäufer die Nacherfüllungendgültig verweigert hat. Auch hier gilt,dass das Nacherfüllungsverlangen unddie Fristsetzung deutlich und konkretformuliert sowie in nachweisbarerForm übermittelt werden. In speziellenFällen (z.B. just-in-time) kann es sein,dass eine Fristsetzung entbehrlich ist.
Darüber hinaus sind auch Schädendenkbar, die aus der Lieferung mangel-hafter Ware resultieren und durch eine(hypothetische) Nacherfüllung nichtwieder entfallen. Auch hier sind Scha-densersatzansprüche gegen den Liefe-ranten denkbar, wenn er seine Pflicht-verletzung nicht entschuldigen kann.Insbesondere für den Fall von nachträg-lich auftretenden Mängeln ist es wis-senswert, dass die gesetzliche Gewähr-leistungsfrist 24 Monate ab Ablieferungbeträgt. Mittels vertraglicher Verein-barungen kann sie jedoch sowohl ver-kürzt als auch verlängert werden. Da-her sollten im Fall eines nachträglichauftretenden Mangels die zugrunde lie-genden Vereinbarungen hiernachgründlich untersucht werden. Zudemsollte die Regelung des § 320 BGB nichtaußer Acht gelassen werden. Vorbehalt-lich abweichender Vereinbarungensteht es dem Gläubiger der nicht gehö-rig – also mangelhaft – erbrachten Leis-tung grundsätzlich zu, seine Leistung –in unserem Fall die Kaufpreiszahlung –so lange zurückzuhalten, bis der andereTeil pflichtgemäß erfüllt hat.
Fazit 1. Teil
Wie aus den voranstehenden Erläute-rungen erkennbar ist, reichen oft ein-zelne Maßnahmen und ein vom Um-fang her überschaubares Qualitätscon-trolling, um die Ansprüche des eigenenUnternehmens zu wahren und entste-hende bzw. drohende Schäden mög-lichst gering zu halten oder gar zu ver-meiden. Im zweiten Teil der Veröffent-lichungsreihe werden weitere Beson-derheiten des Vertragsmanagements,insbesondere auch im Hinblick aufWerkverträge erläutert.
TechnoPharm 3, Nr. 6, 342–345 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 345Wuhrmann und Reusch . Projektmanagement
tigen Leistung eher ein Werkvertrag an-zunehmen zu sein als ein Kaufvertrag.Dieser ist besser geeignet für die Fälle,in denen Ware quasi „von der Stange“bestellt wird.
Kaufverträge
Beschäftigt man sich in einem erstenSchritt mit den kaufvertraglichenPflichten des Lieferanten, so lässt sichfesthalten, dass dieser die gekaufteWare. zur rechten Zeit. am richtigen Ort. in mangelfreiem Zustandzur Verfügung stellen muss. Aus dendrei genannten Pflichtenbündeln ent-springen überwiegend auch die Pflicht-verletzungen des Lieferanten. Alltäg-liche Probleme entstehen meist daraus,dass der Lieferant gar nicht oder nichtrechtzeitig leistet und/oder aber derInhalt seiner Leistung nicht den ver-traglichen Absprachen entspricht.
Lieferverzug desLieferanten im Rahmeneines Kaufvertrages
Regelmäßig wird ein Lieferant ein Lie-ferdatum beachten müssen, welchesder Käufer vertraglich mit ihm verein-bart hat, etwa „Lieferung erfolgt am21. Dezember“, „ ...Ende der KW 41“,„…14 Tage nach Zugang dieser Bestel-lung“, etc. Es ist demnach ganz eindeu-tig festzustellen, wann der Lieferantmit seiner Leistung zu spät ist. Sobaldein kalendarisch zu bestimmendes Da-tum für die Erbringung der geschulde-ten Leistung (also der Lieferung dergekauften Ware) vereinbart wurde, istauch die häufig angeführte Mahnungnicht mehr erforderlich – dies wird oft-mals übersehen. Der Lieferant befindetsich ab dem vereinbarten aber erfolglosverstrichenen Zeitpunkt in Verzug mitseiner Leistung (§ 286 Abs. 2 Nr. 1BGB). Er hat damit eine Pflicht ausdem zugrunde liegenden Vertrag ver-letzt und macht sich dementsprechendschadensersatzpflichtig. In der Regelexistiert für den Lieferanten auchkaum eine Möglichkeit, dieser Scha-densersatzhaftung durch eine ausrei-chende Entschuldigung (sog. „Exkulpa-
tion“) zu entgehen. Eine oftmals vonLieferanten aufgeführte, aber ebensohäufig folgenlose Entschuldigung ist,dass sie für die Verspätung ihres Vor-lieferanten nicht verantwortlich ge-macht werden könnten. Lediglich inFällen höherer Gewalt, also einemvon beiden Parteien nicht vorher zusehenden Ereignis, das insbesonderevon keiner der Parteien beeinflusstwerden kann (z.B. Kriege, Naturkata-strophen etc.), ist ein Wegfall der Haf-tung sicher. Sobald sich also der Liefe-rant im Verzug befindet und dies nichtentschuldigen kann, haftet er für sämt-liche daraus entstehenden Schäden(§§ 280 I, II, 286 BGB). Der Käufer istso zu stellen, wie er bei rechtzeitigerLieferung gestanden hätte. DieserSchadensersatzanspruch ist der Höhenach nicht begrenzt und korreliert we-der mit dem Wert, noch mit dem Preisdes Vertragsgegenstandes. Theoretischist demnach ein Schadensersatz-anspruch für einen Umsatzausfall desKäufers realisierbar, der mit einemniedrigpreisigen C-Teil gekoppelt ist.Allerdings ist der Käufer dazu angehal-ten, den Schaden möglichst gering zuhalten, wenn und soweit ihm dies mitangemessenen Bemühungen möglichist. Jedoch darf nicht außer Acht gelas-sen werden, dass der geschädigte Käu-fer die Beweislast für den Schaden unddessen Umfang trägt. Aufgabe der be-teiligten Personen im Unternehmendes Käufers ist es daher insbesondere,a) die Fristen für die Lieferung der
einzelnen Lieferanten eindeutigfestzulegen und zu überwachen und
b) bei Fristüberschreitungen alle da-raus entstehenden Schäden und dieAbläufe exakt zu dokumentieren.
Darüber hinaus können weiterge-hende Rechte in Anspruch genom-men werden, so z.B. der Rücktrittvom Vertrag oder der Schadensersatzstatt der (ganzen) Leistung. Um diesedurchsetzen zu können, muss derLieferant im Regelfall unter Fristset-zung erneut (aber erfolglos) zur ord-nungsgemäßen Erbringung seinerLeistung aufgefordert werden. Hier-bei sollten Soll- und Istzustand mög-lichst genau beschrieben und dieFrist genau beziffert werden. Auchhier empfiehlt sich aus Nachweis-erwägungen dies nicht nur rein fern-
mündlich (sondern per Fax – Über-mittlungsbericht sichern – oder perEinschreiben) zu tun.
Lieferung mangelhafterWare im Rahmen einesKaufvertrages
Zur Erfüllung des Kaufvertrages mussder Verkäufer die Ware in mangel-freiem Zustand liefern. Weicht dieWare von der vereinbarten Beschaffen-heit ab, oder ist sie nicht für die nachdem Kaufvertrag vorausgesetzte Ver-wendung geeignet oder entspricht sienicht der üblichen Beschaffenheit, soist sie mangelhaft (§ 434 BGB). Glei-ches gilt auch, wenn eine falscheMenge geliefert wird oder aber dieWare mit Rechten Dritter belastet ist,z.B. mit einem Pfandrecht (sogenann-ter Rechtsmangel nach § 435 BGB). Istdie Ware bei Gefahrübergang mangel-haft, so stehen dem Käufer die Behelfedes Gewährleistungsrechts zur Ver-fügung. Wann der Gefahrübergang er-folgt, hängt von Vereinbarungen zwi-schen den Parteien ab – im Grundfallist es die Übergabe der Ware vom Ver-käufer an den Käufer. Im B2B-Bereichist es zur Wahrung dieser Rechts-behelfe allerdings grundsätzlich zwin-gend notwendig, dass der Käufer dieWare nach Eingang unverzüglich aufihre Mangelfreiheit hin untersuchtund sich hierbei zeigende Mängel (sog.„offene Mängel“) dem Verkäufer ohneVerzögerung anzeigt. Versteckte Män-gel, also solche, die sich erst später zei-gen, müssen unverzüglich nach ihrerEntdeckung gemeldet werden. Fallseine solche Rüge unterbleibt, so giltdie Ware als genehmigt und Mängel-ansprüche sind ausgeschlossen. DerUmfang der Prüfung richtet sichgrundsätzlich nach dem „was tunlichist“ – also nach Branchenüblichkeit.Hierbei ist eine aus objektiver Sichtvorgenommene Interessenabwägungvorzunehmen: Kosten, Zeit und Inten-sität müssen in Relation zu Art, Mengeund Einsatzbereich der Ware stehen.Oftmals existieren jedoch vertraglicheAbreden zwischen Käufer und Liefe-rant bezüglich Umfang und Inhalt derPrüfung sowie Ausmaß der Rügepflich-ten. Auch wenn zwar eine vollständige
Maschinen- und Anlagenbau
TechnoPharm 3, Nr. 6, 342–345 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)344 Wuhrmann und Reusch . Projektmanagement

Abbedingung dieser Käuferpflichtennicht möglich ist, insbesondere nichtim Rahmen von Allgemeinen Ge-schäfts-/Einkaufsbedingungen odersonstigen Musterformulierungen, soempfiehlt es sich jedoch, den Pflichten-kreis des Käufers möglichst klein undsimpel zu halten. Dabei sind die gesetz-lichen Vorgaben einzuhalten und diejeweiligen Abläufe möglichst konkretdarzustellen. Ansonsten könnten dieseVereinbarungen letztlich juristisch re-lativ wertlos sein – was oftmals dasManko vieler sogenannter „Qualitäts-sicherungsvereinbarungen“ und ähnli-cher Vertragswerke ist. Die verantwort-lichen Personen sollten daher prüfen,
a) ob und welche Vereinbarungenzur Wareneingangskontrolle und zurRüge bestehen und
b) welche Maßnahmen diesbezüg-lich im jeweiligen Fall ergriffen wurden.
Ist die Ware mangelhaft, stehtdem Käufer vorrangig das Recht zu,Nacherfüllung (Nachbesserung oderNachlieferung) zu verlangen (§§ 437Nr. 1, 439 Abs. 1 BGB). Ohne vor-herige Vereinbarung mit dem Liefe-ranten darf er dies nicht eigenmäch-tig vornehmen – es sind allerdingsdurchaus Situationen denkbar, in de-nen die sogenannte Selbstvornahmefür beide Seiten im Hinblick auf Kos-ten und Dringlichkeit vorteilhaft istund sollte, sofern nicht schon vorabvereinbart, mit dem Lieferanten dis-kutiert werden. Die Kosten der Nach-erfüllung (Transport-, Arbeits- undMaterialkosten etc.) hat der Verkäu-fer zu tragen (§ 439 Abs. 2 BGB). ImFalle von bereits verbauter Warekann sich im BZB-Bereich unterdem Gesichtspunkt des Schadens-ersatzes auch ein weitergehenderAnspruch auf Ersatz der nunmehraufgewandten Aus- und Einbaukos-ten ergeben. Um den eventuell fol-genden Regress zu erleichtern, solltedies im Einzelfall geprüft und detail-liert dokumentiert werden. Als wei-tergehende Mängelansprüche stehendem Käufer zudem zu:– die Minderung des Kaufpreisesoder
– der Rücktritt vom Vertrag– und/oder Schadensersatz wegenNichterfüllung oder Ersatz vergeb-licher Aufwendungen.
Zwingende Voraussetzung hierfür istjedoch, dass entweder eine angemes-sen gesetzte Frist zur Nacherfüllungfruchtlos verstrichen ist, ein zweiterNachbesserungsversuch gescheitertoder der Verkäufer die Nacherfüllungendgültig verweigert hat. Auch hier gilt,dass das Nacherfüllungsverlangen unddie Fristsetzung deutlich und konkretformuliert sowie in nachweisbarerForm übermittelt werden. In speziellenFällen (z.B. just-in-time) kann es sein,dass eine Fristsetzung entbehrlich ist.
Darüber hinaus sind auch Schädendenkbar, die aus der Lieferung mangel-hafter Ware resultieren und durch eine(hypothetische) Nacherfüllung nichtwieder entfallen. Auch hier sind Scha-densersatzansprüche gegen den Liefe-ranten denkbar, wenn er seine Pflicht-verletzung nicht entschuldigen kann.Insbesondere für den Fall von nachträg-lich auftretenden Mängeln ist es wis-senswert, dass die gesetzliche Gewähr-leistungsfrist 24 Monate ab Ablieferungbeträgt. Mittels vertraglicher Verein-barungen kann sie jedoch sowohl ver-kürzt als auch verlängert werden. Da-her sollten im Fall eines nachträglichauftretenden Mangels die zugrunde lie-genden Vereinbarungen hiernachgründlich untersucht werden. Zudemsollte die Regelung des § 320 BGB nichtaußer Acht gelassen werden. Vorbehalt-lich abweichender Vereinbarungensteht es dem Gläubiger der nicht gehö-rig – also mangelhaft – erbrachten Leis-tung grundsätzlich zu, seine Leistung –in unserem Fall die Kaufpreiszahlung –so lange zurückzuhalten, bis der andereTeil pflichtgemäß erfüllt hat.
Fazit 1. Teil
Wie aus den voranstehenden Erläute-rungen erkennbar ist, reichen oft ein-zelne Maßnahmen und ein vom Um-fang her überschaubares Qualitätscon-trolling, um die Ansprüche des eigenenUnternehmens zu wahren und entste-hende bzw. drohende Schäden mög-lichst gering zu halten oder gar zu ver-meiden. Im zweiten Teil der Veröffent-lichungsreihe werden weitere Beson-derheiten des Vertragsmanagements,insbesondere auch im Hinblick aufWerkverträge erläutert.
TechnoPharm 3, Nr. 6, 342–345 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 345Wuhrmann und Reusch . Projektmanagement
Neu:Pharma-DispenserVPHD
ViscoTec Pumpen- u. Dosiertechnik GmbHAmperstraße 13 | 84513 Töging a.InnTelefon: + 49 (0) 8631/ 9274 - 0 mail@ viscotec.de | www.viscotec.de
Perfekt dosiert!
Innenraum totraumoptimiert
Design nach EHEDG-Richtlinien
FDA-konforme Materialien
werkzeuglose, schnelle und einfach
Demontage/Montage
Rotorstrang dichtungslos, demontierbar
autoklavierbar
prozesssichere, hochpräzise Abfüllung
für alle Pharmazeutika, kosmetische
Produkte und Lebensmittel besonders
geeignet
für abrasive, hochgefüllte oder
schersensitive Medien
VPHD-Dispenser.indd 1 31.10.2013 10:07:30

ware besitzen heute in der Regel vieleunterschiedliche Funktionen, so zumBeispiel der Signalwandler ACT20X,der per FDT/DTM-Software konfigu-riert wird. Erst nach der Installationin der Anlage wird die finale Funk-tion des Signalwandlers für die spezi-fische Applikation festgelegt – erwird zur Instanz.
Um diese bis zum Ende der Anla-gennutzungszeit zu erhalten, müssenProduktinnovationen mit gleichenInstanz-Eigenschaften wie bei älte-ren Produktversionen entwickeltwerden. Dabei berücksichtigt mannicht nur die Kompatibilität, dasheißt die Austauschbarkeit, von Ge-räten, sondern ebenso die Interope-rabilität: die Aufrechterhaltung derFunktion ohne Einflussnahme aufweitere Komponenten. Damit jedesProdukt eindeutig identifizierbar ist,wird es über die Versionskennungunverwechselbare beschrieben.Durch Berücksichtigung von Versio-nierung, Kompatibilität und Inter-operabilität sind die wesentlichenAnforderungen an ein effizientes Li-fe-Cycle-Management bereits bei derProduktentwicklung erfüllt.
Umsetzung mit FDTTechnologie in derVersion 2
Die FDT Technologie ist eine nachIEC 62453 und ISA103 standardi-sierte Schnittstellenspezifikation
für Gerätesoftware, die einen offe-nen Datenaustausch zwischen Feld-geräte und Engineering-System er-möglicht. Zwei Komponenten wer-den für den Einsatz der FDT Tech-nologie benötigt – der DTM (DeviceType Manager oder auch „Geräte-treiber“) und die FDT Rahmen-anwendung. Beides sind Software-komponenten, die ihre Funktionennur gemeinsam auf einer Bedien-station ausführen können. Speziellfür die Anforderungen an das Life-Cycle-Management wurden erwei-terte Funktionen und Eigenschaftenhierfür in der neusten FDT Spezifi-kation 2.0 [FDT1] beschrieben. WirdLife-Cycle-Management auf einFDT-basiertes Automatisierungs-system angewendet, ist über die
Nutzungszeit eine konsistente Kon-figuration der eingesetzten Hard-ware und Software sicherzustellen.Hierbei bestehen zahlreiche Abhän-gigkeiten (Abb. 2): von der einge-setzten Hardware und implemen-tierten Firmware über die Konfigu-rationssoftware auf dem PC (FDT-Rahmenanwendung und Device-Ty-pe-Manager) bis zu dem installier-ten Betriebssystem. ErfolgreichesLife-Cycle-Management bedeutetan dieser Stelle, den Anlagenbetriebauch dann sicherzustellen, wenneinzelne Systembestandteile oderihre Konfiguration geändert werden.
Das technische Life-Cycle-Ma-nagement Konzept der FDT Spezifi-kation in der Version 2.0 [FDT1] de-finiert sowohl Richtlinien zur Identi-
Abb 1: Übersicht Life-Cycle-Management (Quelle alle: M. Schade)
Jahre vergehen,Funktionen bestehenMarkus Schade . Weidmüller Interface GmbH & Co. KG, Detmold
Korrespondenz: Markus Schade, Weidmüller Interface GmbH & Co. KG, Klingenbergstr. 16, 32758 Detmold;e-mail: [email protected]
Einleitung
Die Verfügbarkeit von Produktenüber ihre gesamte Nutzungszeit ge-winnt in der Automatisierungstech-nik immer mehr an Gewicht. DieFDT Group und Ihre Mitgliedsfirmenhandhaben dies zukunftsgerechtdurch den effizienten Einsatz von Li-fe-Cycle-Management-Richtlinien.
Life-Cycle-Managementfür Produkte und Systemein der Automation
Der reine Lebenszyklus eines Pro-duktes reicht von seiner Marktein-führung bis zur Abkündigung. Ein-mal eingesetzt, überdauern Produkteihren eigenen Lebenszyklus jedochzum Teil noch Jahrzehnte, bis sieschließlich entsorgt werden. Wiewird in dieser Zeit sichergestellt, dassbei dem Austausch eines Produktesweiterhin die Verfügbarkeit des Ge-samtsystems gegeben ist? Wie blei-ben eingerichtete Funktionen erhal-ten? Die FDT Group Mitgliedsfirmenbegegnen dieser Herausforderungdurch die Integration von Life-Cy-cle-Management-Richtlinien als fes-tem Bestandteil in der Produktent-wicklung.
Funktionen über dieNutzungszeit aufrecht-erhalten
Steigende Funktionalität, fortschrei-tende Entwicklung der Elektronik so-wie die hohen Innovationsraten beiHard- und Software verkürzen kon-tinuierlich den Lebenszyklus einzelnerProdukte. Daher kann die Nutzungs-zeit von Produkten in einer Automati-sierungsanlage heute je nach Brancheweitaus länger sein als der eigentlicheLebenszyklus der eingesetzten Kom-ponente. Eine Produktionsstrecke imAutomobilbereich wird in der Regelüber den Zeitraum der Fertigung einesneuen Modells genutzt, also um die 7bis 8 Jahre. Die Laufzeit einer verfah-renstechnischen Anlage in der Che-mieindustrie beträgt typischerweise15 Jahre, bei Kraftwerken können essogar 50 Jahre und mehr sein. Ver-gleichbar mit passiven Komponentenwie beispielsweise Weidmüllers erstekunststoffisolierte Anreihklemme von1948, die SAK, die heute noch im Sor-timent ist und damit bei Wartungs-arbeiten oder Anlagenerweiterung ge-nutzt werden kann, so ist der An-spruch heute auch bei Produkten ausdem Bereich Elektronik und Software:Auch hier ist die Intention, stets Kom-patibilität sicherzustellen.
Life-Cycle-Managementin der Anwendung
Der Fachverband AUTOMATION in-nerhalb des Zentralverbandes Elek-trotechnik und Elektronikindustrie
hat das Thema „Life-Cycle-Manage-ment“ in dem Leitfaden „Life-Cycle-Management für Produkte und Sys-tem der Automation“ [ZVEI] be-schrieben. Ziel ist eine einheitlicheBeschreibung von Definitionen undBegriffen, die in diesem Zusammen-hang angewendet werden. Als Resul-tat definiert Life-Cycle-ManagementMethoden und Aktivitäten zum Pla-nen, Realisieren und Betreuen vonProdukten für den Lebenszyklus (Li-fe-Cycle) von Typen und die Lebens-zeit von Instanzen“ (Abb. 1). Elektro-nikprodukte mit integrierter Soft-
Automation
TechnoPharm 3, Nr. 6, 346–348 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)346 Schade . FDT Standards
Autor
Markus Schade
Markus Schade ist bei Weidmüller als GlobalIndustry Manager Energy tätig. Seine Karriere be-gann er bei den Firmen Siemens und ABB. HerrSchade hat langjährige Erfahrungen im Enginee-ring, Projekt- und Produktmanagement, Design-in,Inbetriebnahmen sowie Fehleranalysen von Auto-matisierungsanlagen. Nach seiner elektrotech-nischen Ausbildung im Kraftwerksbereich studierteMarkus Schade an der Hochschule Lippe und er-warb dort den akademischen Grad des Diplom-Ingenieurs Elektrotechnik.
Key Words. Life-Cycle-Management. FDT2. Life Cycle Policy

ware besitzen heute in der Regel vieleunterschiedliche Funktionen, so zumBeispiel der Signalwandler ACT20X,der per FDT/DTM-Software konfigu-riert wird. Erst nach der Installationin der Anlage wird die finale Funk-tion des Signalwandlers für die spezi-fische Applikation festgelegt – erwird zur Instanz.
Um diese bis zum Ende der Anla-gennutzungszeit zu erhalten, müssenProduktinnovationen mit gleichenInstanz-Eigenschaften wie bei älte-ren Produktversionen entwickeltwerden. Dabei berücksichtigt mannicht nur die Kompatibilität, dasheißt die Austauschbarkeit, von Ge-räten, sondern ebenso die Interope-rabilität: die Aufrechterhaltung derFunktion ohne Einflussnahme aufweitere Komponenten. Damit jedesProdukt eindeutig identifizierbar ist,wird es über die Versionskennungunverwechselbare beschrieben.Durch Berücksichtigung von Versio-nierung, Kompatibilität und Inter-operabilität sind die wesentlichenAnforderungen an ein effizientes Li-fe-Cycle-Management bereits bei derProduktentwicklung erfüllt.
Umsetzung mit FDTTechnologie in derVersion 2
Die FDT Technologie ist eine nachIEC 62453 und ISA103 standardi-sierte Schnittstellenspezifikation
für Gerätesoftware, die einen offe-nen Datenaustausch zwischen Feld-geräte und Engineering-System er-möglicht. Zwei Komponenten wer-den für den Einsatz der FDT Tech-nologie benötigt – der DTM (DeviceType Manager oder auch „Geräte-treiber“) und die FDT Rahmen-anwendung. Beides sind Software-komponenten, die ihre Funktionennur gemeinsam auf einer Bedien-station ausführen können. Speziellfür die Anforderungen an das Life-Cycle-Management wurden erwei-terte Funktionen und Eigenschaftenhierfür in der neusten FDT Spezifi-kation 2.0 [FDT1] beschrieben. WirdLife-Cycle-Management auf einFDT-basiertes Automatisierungs-system angewendet, ist über die
Nutzungszeit eine konsistente Kon-figuration der eingesetzten Hard-ware und Software sicherzustellen.Hierbei bestehen zahlreiche Abhän-gigkeiten (Abb. 2): von der einge-setzten Hardware und implemen-tierten Firmware über die Konfigu-rationssoftware auf dem PC (FDT-Rahmenanwendung und Device-Ty-pe-Manager) bis zu dem installier-ten Betriebssystem. ErfolgreichesLife-Cycle-Management bedeutetan dieser Stelle, den Anlagenbetriebauch dann sicherzustellen, wenneinzelne Systembestandteile oderihre Konfiguration geändert werden.
Das technische Life-Cycle-Ma-nagement Konzept der FDT Spezifi-kation in der Version 2.0 [FDT1] de-finiert sowohl Richtlinien zur Identi-
Abb 1: Übersicht Life-Cycle-Management (Quelle alle: M. Schade)
IT-ValidierungOhne Umwege zum Ziel.
pragmatisch · verständlich · transparent

Cloud ComputingClaus Bauer . CerDat GmbH, Mannheim
Korrespondenz: Claus Bauer, CerDat GmbH, Carl-Reuther-Str. 3, 68305 Mannheim:e-mail: [email protected]
ZusammenfassungDieser Artikel soll einen Überblick der unterschiedlichen Modelle des Cloud Computingvermitteln. Er bespricht die Faktoren, die bei der Migration von der klassischen IT zuCloud basierten Architekturen eine Rolle spielen sowie die Potentiale des Cloud Com-puting für die pharmazeutische Industrie. Eines der zentralen Themen beim Cloud Com-puting ist der Datenschutz. Im Folgenden soll insbesondere auf diesen wichtigen Aspekt– Datenschutz in Verbindung mit Cloud Computing – eingegangen und Lösungsmöglich-keiten aufgezeigt werden.
Paradigmenwechselin der IT
Cloud Computing ermöglicht Unter-nehmen IT-Leistungen per Internetzu beziehen, mit der Folge, dass in-terne IT-Kapazitäten wie Server, Da-tenspeicher und Software nicht mehrin vollem Umfang im Unternehmenvorgehalten werden müssen. Die da-bei entstehenden Ergebnisse, wieKostenreduktion durch Skalierungs-effekte, ermöglichen vollkommenneue Gestaltungsmöglichkeiten fürdie Fachabteilungen.
Die Mehrzahl der Unternehmenin der Pharmabranche betreiben
heute noch ihre eigenen Rechen-zentren, doch das Thema Cloudrückt immer mehr in den Fokus.Die Anforderungen an die Branche,ihr Geschäftsmodell kontinuierlichweiterzuentwickeln, ist eine Seite,die andere besteht in den zuneh-menden Herausforderungen im Be-reich R&D, vor allem was beispiels-weise die Gensequenzierung be-trifft. Beim Cloud Computing liegendie Vorteile vor allem in der welt-weiten Verfügbarkeit der Daten inEchtzeit und der Redundanz, daauch durch einen Serverausfall dieDaten theoretisch nicht verlorengehen.
Beschleunigungsfaktorenfür Cloud Computing
Das Zusammenwirken der unter-schiedlichsten Faktoren hat die heu-tige Entwicklung erst ermöglicht. DieInternationalisierung der Geschäfts-modelle sowie Änderungen im Nut-zerverhalten haben einen wesentli-chen Anteil an dieser Entwicklung(Abb. 1).
Begriffsbestimmung
Was versteht man unter Cloud Com-puting? Das Bundesamt für Sicher-heit in der Informationstechnik hat
IT
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 349Bauer . Cloud Computing
Autor
Claus Bauer
Claus Bauer studierte an der FH Aalen Wirt-schaftsingenieurwesen. Nach mehrjähriger Tätig-keit bei der Siemens AG im Bereich weltweiter Just-In-Time Beschaffung von Halbleiter Produktions-komponenten wechselte er als Kfm. Leiter für dasVertriebsgebiet Bayern zur Klöckner Möller GmbH.1995 gründete er ein IT-Unternehmen im BereichNetzwerktechnik. Nach dem Verkauf des Unter-nehmens 2010 gründete er die CerDat GmbH mitdem Schwerpunkt Cloud Computing und Daten-schutz.
fikation von Software- und Hard-warekomponenten (= Kennungen)als auch Richtlinien zur Sicherstel-lung der Abwärtskompatibilität zwi-schen unterschiedlichen Ausgabe-ständen (= Versionen) dieser Kom-ponente. Hierzu wurden speziell zweiAttributseigenschaften definiert, diefür die Bestandteile des Betriebssys-tems auf der Bedienstation, der FDTRahmenanwendung, der Device TypeManager sowie der Gerätefirmwareinklusive Gerätedatensatz zur Ver-fügung stehen:. Das Identifikationsattribut stelltInformationen zur Identität derauf der FDT Technologie basie-renden Komponente zur Ver-fügung, unter anderem der Pro-duktname mit dem zugehörigenAusgabestand. Hierdurch kanneine eindeutige Zuordnung bei-spielsweise zwischen DTM undGeräte-Firmware erfolgen sowiezwischen neuen und älteren Aus-gabeständen unterschieden wer-den.
. Das Kompatibilitätsattribut er-laubt die Bewertung der Kompati-bilität von Bestandteilen vor undnach deren Installation sowiewährend des Betriebs.
Die Attribute und ihre Verbindungzu den jeweiligen Komponenten imFDT basierten System ermöglichenbeispielsweise dem Device Type
Manager gezielt Informationenüber den angeschlossenen Geräte-typ, die Firmware-Version, das ge-nutzte Kommunikationsprotokollund das unterstützte Betriebssys-tem einschließlich deren Kompati-bilität untereinander abzufragen.
Damit dieses Life-Cycle-Manage-ment Konzept effizient genutzt wer-den kann, tragen die Hersteller derFeldgeräte mit ihrem Softwareanteil(Firmware und Device Type Mana-ger), die Hersteller der Engineering-Systeme (FDT Rahmenanwendung)und die FDT Group als Inhaber derFDT Spezifikation ihre Verantwor-tung, alle Attributselemente zu un-terstützen und kompatibel zu pfle-gen. Hierzu unterstützen wir die Li-fe-Cycle-Management Schnittstellender FDT-Spezifikation 2.0.
Fazit
Life-Cycle-Management ist bei Her-stellerfirmen ein entscheidender Be-standteil der Produktentwicklung.Durch den effizienten Einsatz vonLife-Cycle-Management-Richtlinienkann eine Interoperabilität undKompatibilität über die Nutzungs-dauer der Produkte erreicht werden.
Grundlage hierfür ist eine ein-deutige Identifikation von Kom-ponenten sowie deren Prüfung zurLaufzeit in einer Automation.
Hierzu werden Standards und ver-fügbare Technologien genutzt wiebeispielsweise die neue FDT 2 Spe-zifikation. Diese bietet spezifischeErweiterungen in der Schnittstel-len- und Attributsbeschreibung, ins-besondere für Life-Cycle-Manage-ment. Zusammen mit der FDTRichtlinie „Life-Cycle-Policy“[FDT2], die den praktischen EinsatzFDT basierter Komponenten überden Lebenszyklus einer Anlage be-schreibt, kann bereits während derEntwicklung von Gerätefirmwareund Device Type Manager (DTM)eine Life-Cycle-Management Strate-gie umgesetzt werden.
Über Field-Device-Tool/Device-Type-Manager(FDT/DTM)
FDT/DTM ist ein herstellerüber-greifendes Konzept zur Flexibilisie-rung komplexer Anlagenprozesse.Konkret spezifiziert und standar-disiert die FDT/DTM-Technologiedie Anbindung kommunikations-fähiger Geräte unterschiedlicherHersteller durch ein übergelagertesGeräte-Management-Programm.Das FDT-basierte Geräte-Manage-ment-System (FDT-Frame) ver-waltet die einzelnen Gerätetreiberder Automatisierungsgeräte, dievom Hersteller als DTM (DeviceType Manager) bereitgestellt wer-den. FDT/DTM ist für eine Viel-zahl von in der Industrie einge-setzten Feldbussen spezifiziert,etwa PROFIBUS und PROFINETIO sowie HART und IO-Link. Da-durch erlaubt die Technologie ei-nen Gerätezugriff vom Werkstatt-platz über die bestehenden Netz-werk-Typologien.
Fachliteratur[FDT1] FDT 2.0 Technical Specification,FDT Group, Order No: 0001-0008-000[FDT2] FDT Life Cycle Policy Version 2,FDT Group, Order No: 0001-0011-000[ZVEI] ZVEI Leitfaden „Life-Cycle-Manage-ment für Produkte und System der Automati-on“,ISBN: 978-3-939265-00-9
Automation
TechnoPharm 3, Nr. 6, 346–348 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)348 Schade . FDT Standards
Abb 2: Übersicht der Abhängigkeiten von Komponenten

Cloud ComputingClaus Bauer . CerDat GmbH, Mannheim
Korrespondenz: Claus Bauer, CerDat GmbH, Carl-Reuther-Str. 3, 68305 Mannheim:e-mail: [email protected]
ZusammenfassungDieser Artikel soll einen Überblick der unterschiedlichen Modelle des Cloud Computingvermitteln. Er bespricht die Faktoren, die bei der Migration von der klassischen IT zuCloud basierten Architekturen eine Rolle spielen sowie die Potentiale des Cloud Com-puting für die pharmazeutische Industrie. Eines der zentralen Themen beim Cloud Com-puting ist der Datenschutz. Im Folgenden soll insbesondere auf diesen wichtigen Aspekt– Datenschutz in Verbindung mit Cloud Computing – eingegangen und Lösungsmöglich-keiten aufgezeigt werden.
Paradigmenwechselin der IT
Cloud Computing ermöglicht Unter-nehmen IT-Leistungen per Internetzu beziehen, mit der Folge, dass in-terne IT-Kapazitäten wie Server, Da-tenspeicher und Software nicht mehrin vollem Umfang im Unternehmenvorgehalten werden müssen. Die da-bei entstehenden Ergebnisse, wieKostenreduktion durch Skalierungs-effekte, ermöglichen vollkommenneue Gestaltungsmöglichkeiten fürdie Fachabteilungen.
Die Mehrzahl der Unternehmenin der Pharmabranche betreiben
heute noch ihre eigenen Rechen-zentren, doch das Thema Cloudrückt immer mehr in den Fokus.Die Anforderungen an die Branche,ihr Geschäftsmodell kontinuierlichweiterzuentwickeln, ist eine Seite,die andere besteht in den zuneh-menden Herausforderungen im Be-reich R&D, vor allem was beispiels-weise die Gensequenzierung be-trifft. Beim Cloud Computing liegendie Vorteile vor allem in der welt-weiten Verfügbarkeit der Daten inEchtzeit und der Redundanz, daauch durch einen Serverausfall dieDaten theoretisch nicht verlorengehen.
Beschleunigungsfaktorenfür Cloud Computing
Das Zusammenwirken der unter-schiedlichsten Faktoren hat die heu-tige Entwicklung erst ermöglicht. DieInternationalisierung der Geschäfts-modelle sowie Änderungen im Nut-zerverhalten haben einen wesentli-chen Anteil an dieser Entwicklung(Abb. 1).
Begriffsbestimmung
Was versteht man unter Cloud Com-puting? Das Bundesamt für Sicher-heit in der Informationstechnik hat
IT
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 349Bauer . Cloud Computing
Autor
Claus Bauer
Claus Bauer studierte an der FH Aalen Wirt-schaftsingenieurwesen. Nach mehrjähriger Tätig-keit bei der Siemens AG im Bereich weltweiter Just-In-Time Beschaffung von Halbleiter Produktions-komponenten wechselte er als Kfm. Leiter für dasVertriebsgebiet Bayern zur Klöckner Möller GmbH.1995 gründete er ein IT-Unternehmen im BereichNetzwerktechnik. Nach dem Verkauf des Unter-nehmens 2010 gründete er die CerDat GmbH mitdem Schwerpunkt Cloud Computing und Daten-schutz.
fikation von Software- und Hard-warekomponenten (= Kennungen)als auch Richtlinien zur Sicherstel-lung der Abwärtskompatibilität zwi-schen unterschiedlichen Ausgabe-ständen (= Versionen) dieser Kom-ponente. Hierzu wurden speziell zweiAttributseigenschaften definiert, diefür die Bestandteile des Betriebssys-tems auf der Bedienstation, der FDTRahmenanwendung, der Device TypeManager sowie der Gerätefirmwareinklusive Gerätedatensatz zur Ver-fügung stehen:. Das Identifikationsattribut stelltInformationen zur Identität derauf der FDT Technologie basie-renden Komponente zur Ver-fügung, unter anderem der Pro-duktname mit dem zugehörigenAusgabestand. Hierdurch kanneine eindeutige Zuordnung bei-spielsweise zwischen DTM undGeräte-Firmware erfolgen sowiezwischen neuen und älteren Aus-gabeständen unterschieden wer-den.
. Das Kompatibilitätsattribut er-laubt die Bewertung der Kompati-bilität von Bestandteilen vor undnach deren Installation sowiewährend des Betriebs.
Die Attribute und ihre Verbindungzu den jeweiligen Komponenten imFDT basierten System ermöglichenbeispielsweise dem Device Type
Manager gezielt Informationenüber den angeschlossenen Geräte-typ, die Firmware-Version, das ge-nutzte Kommunikationsprotokollund das unterstützte Betriebssys-tem einschließlich deren Kompati-bilität untereinander abzufragen.
Damit dieses Life-Cycle-Manage-ment Konzept effizient genutzt wer-den kann, tragen die Hersteller derFeldgeräte mit ihrem Softwareanteil(Firmware und Device Type Mana-ger), die Hersteller der Engineering-Systeme (FDT Rahmenanwendung)und die FDT Group als Inhaber derFDT Spezifikation ihre Verantwor-tung, alle Attributselemente zu un-terstützen und kompatibel zu pfle-gen. Hierzu unterstützen wir die Li-fe-Cycle-Management Schnittstellender FDT-Spezifikation 2.0.
Fazit
Life-Cycle-Management ist bei Her-stellerfirmen ein entscheidender Be-standteil der Produktentwicklung.Durch den effizienten Einsatz vonLife-Cycle-Management-Richtlinienkann eine Interoperabilität undKompatibilität über die Nutzungs-dauer der Produkte erreicht werden.
Grundlage hierfür ist eine ein-deutige Identifikation von Kom-ponenten sowie deren Prüfung zurLaufzeit in einer Automation.
Hierzu werden Standards und ver-fügbare Technologien genutzt wiebeispielsweise die neue FDT 2 Spe-zifikation. Diese bietet spezifischeErweiterungen in der Schnittstel-len- und Attributsbeschreibung, ins-besondere für Life-Cycle-Manage-ment. Zusammen mit der FDTRichtlinie „Life-Cycle-Policy“[FDT2], die den praktischen EinsatzFDT basierter Komponenten überden Lebenszyklus einer Anlage be-schreibt, kann bereits während derEntwicklung von Gerätefirmwareund Device Type Manager (DTM)eine Life-Cycle-Management Strate-gie umgesetzt werden.
Über Field-Device-Tool/Device-Type-Manager(FDT/DTM)
FDT/DTM ist ein herstellerüber-greifendes Konzept zur Flexibilisie-rung komplexer Anlagenprozesse.Konkret spezifiziert und standar-disiert die FDT/DTM-Technologiedie Anbindung kommunikations-fähiger Geräte unterschiedlicherHersteller durch ein übergelagertesGeräte-Management-Programm.Das FDT-basierte Geräte-Manage-ment-System (FDT-Frame) ver-waltet die einzelnen Gerätetreiberder Automatisierungsgeräte, dievom Hersteller als DTM (DeviceType Manager) bereitgestellt wer-den. FDT/DTM ist für eine Viel-zahl von in der Industrie einge-setzten Feldbussen spezifiziert,etwa PROFIBUS und PROFINETIO sowie HART und IO-Link. Da-durch erlaubt die Technologie ei-nen Gerätezugriff vom Werkstatt-platz über die bestehenden Netz-werk-Typologien.
Fachliteratur[FDT1] FDT 2.0 Technical Specification,FDT Group, Order No: 0001-0008-000[FDT2] FDT Life Cycle Policy Version 2,FDT Group, Order No: 0001-0011-000[ZVEI] ZVEI Leitfaden „Life-Cycle-Manage-ment für Produkte und System der Automati-on“,ISBN: 978-3-939265-00-9
Automation
TechnoPharm 3, Nr. 6, 346–348 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)348 Schade . FDT Standards
Abb 2: Übersicht der Abhängigkeiten von Komponenten

Cloud-TypenNeben den Leistungen der Cloud-An-bieter, die in den Service-Ebenen auf-gezeigt werden, gibt es zwei Grund-formen des Cloud Computing(Abb. 2).
In der Praxis gibt es eine ganzeReihe von Derivaten der beidenGrundformen Public und PrivateCloud.
Horizontal (Application) CloudDie Horizontal Cloud leitet sich vonder Private Cloud ab und stellt aufder SaaS-Ebene branchenunspezfi-zische Anwendungen bereit. Manch-mal wird sie auch als „CRM-Cloud“oder „Communication-Cloud“ be-zeichnet.
Vertical (Application) CloudIhre Ausprägung entspricht der Ho-rizontal Cloud, richtet sich aber anUnternehmen mit gleichen oder zu-mindest ähnlichen Geschäftsprozes-sen und Applikationsanforderungen.Sie wird deswegen auch als Commu-nity-Cloud bezeichnet.
Alle Cloud-Typen lassen sichdurch eine Sourcing Option weiterdifferenzieren.. Insourced: die Cloud wird vomKunden selbst betrieben.
. Managed: die Cloud wird von ei-nem externen Dienstleister nachKundenvorgaben betrieben.
. Outsourced: die Cloud wird voneinem Provider betrieben (z.B. Pu-blic Cloud).
Zum Vergleich der Cloud-Typen sieheTab. 1.
Business Potenzial für diePharma Branche
Einer der wichtigsten Gründe für dieEinführung von Cloud Computing istder Aspekt der Flexibilisierung vonIT-Ressourcen. Auch auf die wach-senden Anforderungen, wie die, dasGeschäftsmodell kontinuierlich wei-terzuentwickeln sowie im R&D Be-reich immer umfangreicher wer-dende Speicherung der Sequenzie-rungsdaten, bietet Cloud Computingdie Antwort. Ein weltweites „Log-in“
der Mitarbeiter und ein direkter Zu-griff auf Daten und Kommunikati-onskanäle beschleunigt und verein-facht betriebliche Abläufe. Darüberhinaus kann beim Einsatz von Appli-kationen in der Cloud auf die Be-schaffung, Implementierung undWartung derselben verzichtet wer-den. Trotz der Versprechen derCloud-Anbieter gibt es für Phar-maunternehmen wichtige Hin-derungsgründe für die Migration indie Cloud. Der Aufwand bei der Über-führung der bestehenden IT-Infra-struktur in eine Cloud basierte Archi-tektur kann einen beträchtlichenUmfang annehmen. Ein weiterer Hin-derungsgrund ist die Sicherheit derDaten, die ein ganz besonderes An-liegen der Branche ist. Angaben überdie Krankengeschichten der Patien-ten und Forschungsergebnisse sindbesonders sensitive Daten, die nichtnur nach deutschem Datenschutz-recht einer sicheren Handhabung be-dürfen.
Marktsituation fürCloud Computing
Die Marktentwicklung für CloudComputing Produkte und Serviceshat sich in jüngster Vergangenheitenorm beschleunigt. Es ist davonauszugehen, dass allein auf demdeutschen Markt heute mehr als300 Anbieter aktiv sind. In den näch-sten Jahren wird eine starke Konsoli-dierung stattfinden. Umso wichtigerist es, strenge Kriterien für die Aus-wahl des geeigneten Cloud-Anbietersanzulegen.
Hier eine unvollständige Auswahlführender Cloud-Anbieter:
IBM, Google, Salesforce, Amazon,Microsoft, Siemens, Telekom, SAP,EMC, HP, Oracle, Rackspace etc.
Die führenden Cloud-Anbieterversuchen möglichst viele Cloud-Ty-pen und Service-Ebenen abzude-cken, wobei es auch viele attraktivekleinere Anbieter gibt wie z.B. Joy-ent, Cloudworks etc., die unter Um-ständen sogar den Anforderungenbesser gerecht werden als große An-bieter.
Traditionelle IT-Strukturen und CloudComputing
Neben den bekannten ökonomischenVorteilen hat die Nutzung von CloudComputing auch Auswirkungen aufklassische IT-Strukturen. Eine Ände-rung der Sicherheitsanforderungenkann sich kurzfristig ergeben, z.B.durch Hackerattacken. Cloud Com-puting Architekturen ermöglichenkurzfristige Kapazitätsanpassungenzur Neutralisierung der Sicherheits-bedrohung (z.B. bei DDoS-Attacken).Die Einführung neuer Release Ständezur Erhöhung der Sicherheitsfunktio-nen einer Anwendung erfolgt durchden Cloud-Provider, ohne dass in-terne Ressourcen gebunden werden.Die Anwender arbeiten stets mit demaktuellen Sicherheitsstandard.
Bei IT-Rollouts sind Cloud Com-puting Architekturen gegenüberklassischen IT-Systemen schnellerverfügbar. Neben vollständigen Si-cherheitsfunktionen bieten sie aucheine zeitlich beschränkte Nutzungder Ressourcen.
Erfolgsfaktoren für dieEinführung von CloudComputing
Die folgenden Faktoren stellen ausSicht der CIOs Eckpunkte dar, diezufriedenstellend geregelt sein müs-sen.
InformationssicherheitDas Thema Gewährleistung der Da-tensicherheit ist eine der am wichtigs-ten eingestuften Anforderungen andas Cloud Computing. Neben der Ab-sicherung gegen physikalische und lo-gische Fehler müssen auch gesetzli-che Vorgaben beachtet werden, z.B.was den Transfer von personenbezo-genen Daten in Drittländer (Länderaußerhalb des Europäischen Wirt-schaftsraums) betrifft. Da die Datenin der Cloud nicht mehr vollständigunter eigener Kontrolle stehen, kannes in diesem Spannungsfeld bei IT-Verantwortlichen zu Bedenken kom-men (Wo liegen die Daten?Wo stehen
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 351Bauer . Cloud Computing
folgenden Begriff für das Cloud Com-puting festgelegt: [1]
„Cloud Computing bezeichnet dasdynamische an den Bedarf ange-passte Arbeiten, Nutzen und Abrech-nen von IT-Dienstleistungen über einNetz. Angebot und Nutzung dieserDienstleistungen erfolgt dabei aus-schließlich über definierte tech-nische Schnittstellen und Protokolle.Die Spannbreite der im Rahmen vonCloud Computing angebotenenDienstleistungen umfasst das kom-plette Spektrum der Informations-technik und beinhaltet unter ande-rem Infrastruktur (z.B. Rechenleis-tung, Speicherplatz), Plattformenund Software.“
Service-EbenenEs lassen sich folgende Service-Ebe-nen des Cloud Computing unter-scheiden:
Infrastructure as a Service (IaaS):Hierunter versteht man die Bereit-stellung von skalierbaren IT-Res-sourcen über das Internet. Tech-nisch wird IaaS durch die Bereitstel-lung von virtualisierten Servern undNetzwerkfunktionalität realisiert.Ein hoher Grad der Standardisie-rung und Automatisierung sowieder Einsatz von System-Manage-ment-Software charakterisierendiese Serviceebene.
Platform as a Service (PaaS): Un-ter dieser Form wird eine integrierteLaufzeitumgebung verstanden, die esz.B. Systemarchitekten sowie An-wendungsentwicklern ermöglicht,
Anwendungskomponentenplattformübergreifend zuentwickeln.
Software as a Service(SaaS): Darunter kann einGeschäftsmodell verstan-den werden, mit dem Soft-ware als Service bereit-gestellt wird. Die Softwarewird vom Cloud-Anbietergestellt und muss nichtvom Anwender käuflich er-worben werden.
IT
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)350 Bauer . Cloud Computing
CloudComputing
Weiterentwicklungdes Webs zurPlattformz.B. Facebook
Standards, Skalierbarkeit,Big Data
Globalisierung der Märkte,
Kostensituation der IT
Nachfrage u. AngebotInternet-Technologien,Mobiler Zugriff,globale Verfügbarkeit,Bandbreite (LTE etc.)
Technische Entwicklungen
Nutzerverhalten
Industrialisierung der IT
Abb. 1: Beschleunigungsfaktoren (Quelle alleBilder: CerDat GmbH).
Tabelle 1
Vergleich der Cloud-Typen.
HybridCloud
VerticalCloud
VirtualCloud
HorizontalCloud
PublicCloud
PrivateCloud
Abb. 2: Cloud-Typen

Cloud-TypenNeben den Leistungen der Cloud-An-bieter, die in den Service-Ebenen auf-gezeigt werden, gibt es zwei Grund-formen des Cloud Computing(Abb. 2).
In der Praxis gibt es eine ganzeReihe von Derivaten der beidenGrundformen Public und PrivateCloud.
Horizontal (Application) CloudDie Horizontal Cloud leitet sich vonder Private Cloud ab und stellt aufder SaaS-Ebene branchenunspezfi-zische Anwendungen bereit. Manch-mal wird sie auch als „CRM-Cloud“oder „Communication-Cloud“ be-zeichnet.
Vertical (Application) CloudIhre Ausprägung entspricht der Ho-rizontal Cloud, richtet sich aber anUnternehmen mit gleichen oder zu-mindest ähnlichen Geschäftsprozes-sen und Applikationsanforderungen.Sie wird deswegen auch als Commu-nity-Cloud bezeichnet.
Alle Cloud-Typen lassen sichdurch eine Sourcing Option weiterdifferenzieren.. Insourced: die Cloud wird vomKunden selbst betrieben.
. Managed: die Cloud wird von ei-nem externen Dienstleister nachKundenvorgaben betrieben.
. Outsourced: die Cloud wird voneinem Provider betrieben (z.B. Pu-blic Cloud).
Zum Vergleich der Cloud-Typen sieheTab. 1.
Business Potenzial für diePharma Branche
Einer der wichtigsten Gründe für dieEinführung von Cloud Computing istder Aspekt der Flexibilisierung vonIT-Ressourcen. Auch auf die wach-senden Anforderungen, wie die, dasGeschäftsmodell kontinuierlich wei-terzuentwickeln sowie im R&D Be-reich immer umfangreicher wer-dende Speicherung der Sequenzie-rungsdaten, bietet Cloud Computingdie Antwort. Ein weltweites „Log-in“
der Mitarbeiter und ein direkter Zu-griff auf Daten und Kommunikati-onskanäle beschleunigt und verein-facht betriebliche Abläufe. Darüberhinaus kann beim Einsatz von Appli-kationen in der Cloud auf die Be-schaffung, Implementierung undWartung derselben verzichtet wer-den. Trotz der Versprechen derCloud-Anbieter gibt es für Phar-maunternehmen wichtige Hin-derungsgründe für die Migration indie Cloud. Der Aufwand bei der Über-führung der bestehenden IT-Infra-struktur in eine Cloud basierte Archi-tektur kann einen beträchtlichenUmfang annehmen. Ein weiterer Hin-derungsgrund ist die Sicherheit derDaten, die ein ganz besonderes An-liegen der Branche ist. Angaben überdie Krankengeschichten der Patien-ten und Forschungsergebnisse sindbesonders sensitive Daten, die nichtnur nach deutschem Datenschutz-recht einer sicheren Handhabung be-dürfen.
Marktsituation fürCloud Computing
Die Marktentwicklung für CloudComputing Produkte und Serviceshat sich in jüngster Vergangenheitenorm beschleunigt. Es ist davonauszugehen, dass allein auf demdeutschen Markt heute mehr als300 Anbieter aktiv sind. In den näch-sten Jahren wird eine starke Konsoli-dierung stattfinden. Umso wichtigerist es, strenge Kriterien für die Aus-wahl des geeigneten Cloud-Anbietersanzulegen.
Hier eine unvollständige Auswahlführender Cloud-Anbieter:
IBM, Google, Salesforce, Amazon,Microsoft, Siemens, Telekom, SAP,EMC, HP, Oracle, Rackspace etc.
Die führenden Cloud-Anbieterversuchen möglichst viele Cloud-Ty-pen und Service-Ebenen abzude-cken, wobei es auch viele attraktivekleinere Anbieter gibt wie z.B. Joy-ent, Cloudworks etc., die unter Um-ständen sogar den Anforderungenbesser gerecht werden als große An-bieter.
Traditionelle IT-Strukturen und CloudComputing
Neben den bekannten ökonomischenVorteilen hat die Nutzung von CloudComputing auch Auswirkungen aufklassische IT-Strukturen. Eine Ände-rung der Sicherheitsanforderungenkann sich kurzfristig ergeben, z.B.durch Hackerattacken. Cloud Com-puting Architekturen ermöglichenkurzfristige Kapazitätsanpassungenzur Neutralisierung der Sicherheits-bedrohung (z.B. bei DDoS-Attacken).Die Einführung neuer Release Ständezur Erhöhung der Sicherheitsfunktio-nen einer Anwendung erfolgt durchden Cloud-Provider, ohne dass in-terne Ressourcen gebunden werden.Die Anwender arbeiten stets mit demaktuellen Sicherheitsstandard.
Bei IT-Rollouts sind Cloud Com-puting Architekturen gegenüberklassischen IT-Systemen schnellerverfügbar. Neben vollständigen Si-cherheitsfunktionen bieten sie aucheine zeitlich beschränkte Nutzungder Ressourcen.
Erfolgsfaktoren für dieEinführung von CloudComputing
Die folgenden Faktoren stellen ausSicht der CIOs Eckpunkte dar, diezufriedenstellend geregelt sein müs-sen.
InformationssicherheitDas Thema Gewährleistung der Da-tensicherheit ist eine der am wichtigs-ten eingestuften Anforderungen andas Cloud Computing. Neben der Ab-sicherung gegen physikalische und lo-gische Fehler müssen auch gesetzli-che Vorgaben beachtet werden, z.B.was den Transfer von personenbezo-genen Daten in Drittländer (Länderaußerhalb des Europäischen Wirt-schaftsraums) betrifft. Da die Datenin der Cloud nicht mehr vollständigunter eigener Kontrolle stehen, kannes in diesem Spannungsfeld bei IT-Verantwortlichen zu Bedenken kom-men (Wo liegen die Daten?Wo stehen
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 351Bauer . Cloud Computing
folgenden Begriff für das Cloud Com-puting festgelegt: [1]
„Cloud Computing bezeichnet dasdynamische an den Bedarf ange-passte Arbeiten, Nutzen und Abrech-nen von IT-Dienstleistungen über einNetz. Angebot und Nutzung dieserDienstleistungen erfolgt dabei aus-schließlich über definierte tech-nische Schnittstellen und Protokolle.Die Spannbreite der im Rahmen vonCloud Computing angebotenenDienstleistungen umfasst das kom-plette Spektrum der Informations-technik und beinhaltet unter ande-rem Infrastruktur (z.B. Rechenleis-tung, Speicherplatz), Plattformenund Software.“
Service-EbenenEs lassen sich folgende Service-Ebe-nen des Cloud Computing unter-scheiden:
Infrastructure as a Service (IaaS):Hierunter versteht man die Bereit-stellung von skalierbaren IT-Res-sourcen über das Internet. Tech-nisch wird IaaS durch die Bereitstel-lung von virtualisierten Servern undNetzwerkfunktionalität realisiert.Ein hoher Grad der Standardisie-rung und Automatisierung sowieder Einsatz von System-Manage-ment-Software charakterisierendiese Serviceebene.
Platform as a Service (PaaS): Un-ter dieser Form wird eine integrierteLaufzeitumgebung verstanden, die esz.B. Systemarchitekten sowie An-wendungsentwicklern ermöglicht,
Anwendungskomponentenplattformübergreifend zuentwickeln.
Software as a Service(SaaS): Darunter kann einGeschäftsmodell verstan-den werden, mit dem Soft-ware als Service bereit-gestellt wird. Die Softwarewird vom Cloud-Anbietergestellt und muss nichtvom Anwender käuflich er-worben werden.
IT
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)350 Bauer . Cloud Computing
CloudComputing
Weiterentwicklungdes Webs zurPlattformz.B. Facebook
Standards, Skalierbarkeit,Big Data
Globalisierung der Märkte,
Kostensituation der IT
Nachfrage u. AngebotInternet-Technologien,Mobiler Zugriff,globale Verfügbarkeit,Bandbreite (LTE etc.)
Technische Entwicklungen
Nutzerverhalten
Industrialisierung der IT
Abb. 1: Beschleunigungsfaktoren (Quelle alleBilder: CerDat GmbH).
Tabelle 1
Vergleich der Cloud-Typen.
HybridCloud
VerticalCloud
VirtualCloud
HorizontalCloud
PublicCloud
PrivateCloud
Abb. 2: Cloud-Typen

chen Vertragstyps (was unmittel-bare Konsequenzen für nachfol-genden Punkt hat)
. Gewährleistung und Haftung
. Source-Governance und Audit-Rechte
. Vergütungsmodell
. Regelung für eine Vertragsbeendi-gung
Ein wichtiger Punkt gerade im Hin-blick auf den gesetzlichen Daten-schutz ist die Frage, wo die Nutzer-daten gespeichert werden. Die wenigs-ten Cloud-Anbieter speichern die Da-ten ausschließlich in Deutschland.Größere Provider überlassen den Kun-den oft die Entscheidung hinsichtlichdes Speicherortes. Bei kleinerenCloud-Anbietern kann es vorkommen,dass Subunternehmen eingesetzt wer-den und dadurch der Standort für dieDatenspeicherung unklar oder zumin-dest nicht frei wählbar ist.
DatenschutzDer Nutzer als verantwortliche Stelleim Sinne des Bundesdatenschutz-gesetzes muss den Datenschutz nachdeutschem Recht für personenbezo-gene Daten in der Cloud sicherstel-len. Sämtliche gesetzlichen Daten-schutzvorschriften sind grundsätz-lich auch auf die Cloud anzuwenden.An dieser Stelle sollen drei wesentli-che Aspekte aufgegriffen werden:
Das deutsche Bundesdaten-schutzgesetz regelt den Umgangmit personenbezogenen Daten. Soll-ten besonders schützenswerte per-sonenbezogene Daten (im Sinne des§ 3 Abs. 9 BDSG, z.B. Gesundheits-daten) verarbeitet werden, müssenbesondere Maßnahmen getroffenwerden. Andere Daten wie Finanz-daten oder R&D- Daten, etc. müssennatürlich auch geschützt werden, fal-len jedoch nicht unter das Bundes-datenschutzgesetz.
Wenn personenbezogene Datenan einen Cloud Provider übermitteltwerden, darf dies nur dann erfolgen,wenn die Betroffenen eingewilligt ha-ben oder ein gesetzlicher Erlaubnis-tatbestand vorliegt, auch wenn essich bei dem Cloud-Provider um einkonzerneigenes Tochterunterneh-
men handelt. Im Bundesdaten-schutzgesetz gibt es das Konstruktder Auftragsdatenverarbeitung, wasbedeutet, dass der Cloud Providerdie Daten im Auftrag des Nutzersverarbeitet kann (quasi als Hilfsfunk-tion für den Nutzer). Er ist damit imrechtlichen Sinn kein Dritter, somitentfällt die Notwendigkeit einer Er-laubnis der Betroffenen. Dies gilt al-lerdings nur, wenn der Cloud-Pro-vider die Daten innerhalb Deutsch-lands, der EU oder des EWRs ver-arbeitet (gilt auch für Subunterneh-men). Bei einem Datentransfer inDrittstaaten kommt hinzu, dass dietechnisch organisatorischen Maß-nahmen zum Schutz der personen-bezogenen Daten des Cloud-Pro-viders (auch seiner Subunterneh-men) vom Nutzer regelmäßig kon-trolliert werden müssen. Dabei istes ausreichend, wenn regelmäßigentsprechende Prüfberichte zur Ver-fügung gestellt werden.
Preismodelle für CloudLeistungen
Die Preismodelle der Cloud-Anbietersind meist klarer als die Lizenzmo-delle für den Kauf von Software.Trotzdem können sie sich für denBezug von Cloud-Leistungen starkunterscheiden. Viele Modelle sindzeitbasiert, andere werden anhanddes Datenaufkommens abgerechnetetc. Wichtig ist, dass die Konditionenfür den Bezug von Cloud-Leistungenumstellbar sind. Anhand von zweiausgewählten Anbietern für PublicClouds erhalten Sie einen ersten Ein-druck der Preisgrößenordnung fürInfrastructure as a Service – Leistun-gen (Stand: 06/2012) (Tab. 2 und 3).
Fazit
Cloud Computing bietet den Phar-maunternehmen die Möglichkeit ei-nes grundsätzlichen Richtungswech-sels beim Einsatz von IT. Investitionenin interne IT-Ressourcen können zu-rückgefahren werden zugunsten einerSteigerung der Wettbewerbsfähigkeitim eigentlichen Kerngeschäft.
Trotz mancher Risiken sehe ichfür die Pharmaindustrie vier wesent-liche Vorteile des Cloud Computings:. Niedrigere Kosten für die ITEin Kostenvorteil des Cloud Compu-tings gegenüber dem Eigenerwerblässt sich allerdings nur dann realisie-ren, wenn die Nutzung der Cloud-Ser-vices dynamisch erfolgt. Die Zu- undAbschaltung von IT-Kapazitäten mussautomatisiert erfolgen, ohne dasshohe Transaktionskosten für das IT-Ressourcenmanagement entstehen.. Anpassung von IT-Ressourcen anbetriebliche Notwendigkeiten
Werden z.B. für R&D mehr IT-Res-sourcen benötigt, können sie alsCloud-Service kurzfristig aktiviertwerden. Werden sie nicht mehr benö-tigt, werden sie abgestellt. Die Kapa-zitäten der IT bleiben flexibel und ori-entieren sich am aktuellen Bedarf. DerLeerstand von IT-Ressourcen solltedamit der Vergangenheit angehören.. Verbesserung der internen undexternen Zusammenarbeit
Durch die beim Cloud Computingeingesetzten Techniken wie z.B. Vir-tualisierung ist es möglich, die fürProjekte notwendigen IT-Ressourceninnerhalb kürzester Zeit mit allenfachlichen und sicherheitstech-nischen Features zur Verfügung zustellen. Projekte werden dadurchschneller implementiert und die„Time to Market“ wird kürzer.. SicherheitDer Ausfall von IT-Ressourcen kannein ernstes Problem für Pharmaunter-nehmen darstellen. Die redundanteAuslegung klassischer IT-Infrastruk-tur ist kostenintensiv und unflexibel.Die schnelle Wiederverfügbarkeit imDesasterfall sowie laufend aktuali-sierte Sicherheitsfeatures und vertrag-liche Regelungen im Bereich der Si-cherheit und bei der Einhaltung ge-setzlicher Datenschutzvorschriftenmachen Cloud Computing für phar-mazeutische Unternehmen attraktiv.
Fachliteratur[1] Eckpunktpapier Sicherheitsempfehlun-
gen für Cloud Computer Anbieter, BSI,S. 15/16
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 353Bauer . Cloud Computing
die Server? Wer hat Zugriff?). Infor-mationssicherheit und Cloud Compu-ting erfordern gegenüber der klassi-schen IT eine vollkommene Neube-wertung der Sicherheitsrisiken. DieAusgangssituation von Cloud basier-ten Lösungen besteht im Grunde da-rin, dass sich mehrere Nutzer die phy-sikalischen Systemressourcen teilen.Dies könnte dazu führen, dass durchFehlfunktionen unbeabsichtigte man-datenübergreifende Zugriffe stattfin-den oder dass Sicherheitslücken dieDaten in der Cloud gefährden. DieProvider müssen ein angemessenesSicherheitsniveau herstellen und sichan laufend ändernden Gegebenheitenim Unternehmen anpassen.
Die Sicherstellung der Informati-onssicherheit erfolgt durch Risiko-analysen und -bewertung. Hier spieltdie ISO/IEC 27001 mit der Einfüh-rung eines Informations SicherheitsManagement System (ISMS) einezentrale Rolle. Zertifizierungen inder Informationssicherheit bieten inder Regel Cloud-Provider, die grö-ßere Unternehmen betreuen. Wich-tig ist in jedem Fall, dass die Ver-tragspartner Notfallpläne verein-baren, damit entsprechende Vorkeh-rungen bei Sicherheitsvorfällen ge-troffen werden.
VerfügbarkeitDie Einführung von Service LevelAgreements (SLA) ist eine essentielleVoraussetzung für die Einführung vonCloud Services. Die Verfügbarkeit
muss sich ebenso wiedie Performance an denBelangen der Geschäfts-prozesse im Unterneh-men orientieren, welchebisher durch klassischeIT-Leistungen bedientwurden.
Service IntegrationIn der Regel werden Ge-schäftsprozesse im Un-ternehmen durch Appli-kationen abgedeckt.Übernehmen Cloud Ser-vices Teil- oder Gesamt-prozesse stellt sich die
Frage der Integrationsfähigkeit zumeinen mit klassischen IT-Systemen,zum anderen der Kopplung vonCloud Systemen miteinander. DieAbschätzung der damit verbundenenRisiken und die Prozessintegrationmüssen im Vorfeld geklärt werden.
AbhängigkeitDie Nutzer-Schnittstelle hat für dieIaaS- und PaaS-Ebene eine besondereBedeutung. Sie dient dazu die Pro-visionierung und Steuerung derCloud-Anwendung durchzuführen.Bei nicht standardisierten Schnittstel-len wird es für den Nutzer schwierig,den Anbieter zu wechseln. Anbieter-spezifische APIs, die notwendig sind,um einen größtmöglichen Automati-sierungsgrad zur erreichen, erzeugenaber eine Abhängigkeit des Nutzersvom jeweiligen Cloud-Anbieter. Beider Planung von Cloud Projekten istes deshalb sinnvoll, darauf zu achtenwie eine Rückführbarkeit der Cloud-Services (technisch, öko-nomisch und rechtlich)sichergestellt werdenkann.
TransparenzDie Einführung von Mo-nitoring- und ReportingTools bringt Klarheit indie Kostenermittlungfür die bezogenen Ser-vices, bzw. deren Nut-zung durch die unter-schiedlichen Unterneh-
mensbereiche. Darüber hinaus bele-gen diese Tools die Einhaltung dervereinbarten SLAs sowie der Cloud-Service Performance. Sie stellen da-durch eine Quelle zur Analyse künf-tiger Einsparpotentiale dar.
Vertragliche AspekteVertragliche Regelungen bilden dieGrundlage und definieren den Um-fang der Leistungserbringung wieauch die gegenseitigen Ansprücheder Vertragspartner. Werden auf derSeite der Anbieter Subunternehmenzur Leistungserbringung eingebun-den, müssen auch für die Subunter-nehmen datenschutzrechtliche Ver-einbarungen getroffen werden. Alleindie Anzahl der beteiligten Vertrags-partner kann die Komplexität und da-mit den Aufwand für den Kundendrastisch beeinflussen. Die Beschrei-bung der vertraglichen Leistungenund die Folgen für deren Nichterfül-lung werden in den Service LevelAgreements niedergelegt. Je nach derArt der Cloud Nutzung müssen auchdie länderspezifischen Nutzungs-rechte der Software geregelt werden.Dies gilt sowohl für Software, die derCloud-Anbieter seinen Kunden bereit-stellt (hier muss in der Regel kein ur-heberrechtliches Nutzungsrecht fürden Anwender eingeräumt werden),wie auch für Software, die durch denKunden beigestellt wird.
Weitere Aspekte einer vertragli-chen Regelung für Cloud Computing:. Datenschutz. Geltendes Recht. Leistungsbeschreibung und damitauch eine Festlegung des gesetzli-
IT
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)352 Bauer . Cloud Computing
Tabelle 3
Telekom Preise.
Telekom HostingServicesDeployments
Nutzung in e
pro Stunde
1 PU/1 GB RAM/Speicher 160 GB
0,13
8 PU/15 GB RAM/Speicher 1690 GB
1,39
Tabelle 2
Amazon Preise.
Amazon EC2Standard onDemandInstances
Linux/UnixNutzungin $ proStunde
WindowsNutzungin $ proStunde
Small(Standard)
0,085 0,115
Medium 0,170 0,230
Large 0,340 0,460
Extra Large 0,680 0,920

chen Vertragstyps (was unmittel-bare Konsequenzen für nachfol-genden Punkt hat)
. Gewährleistung und Haftung
. Source-Governance und Audit-Rechte
. Vergütungsmodell
. Regelung für eine Vertragsbeendi-gung
Ein wichtiger Punkt gerade im Hin-blick auf den gesetzlichen Daten-schutz ist die Frage, wo die Nutzer-daten gespeichert werden. Die wenigs-ten Cloud-Anbieter speichern die Da-ten ausschließlich in Deutschland.Größere Provider überlassen den Kun-den oft die Entscheidung hinsichtlichdes Speicherortes. Bei kleinerenCloud-Anbietern kann es vorkommen,dass Subunternehmen eingesetzt wer-den und dadurch der Standort für dieDatenspeicherung unklar oder zumin-dest nicht frei wählbar ist.
DatenschutzDer Nutzer als verantwortliche Stelleim Sinne des Bundesdatenschutz-gesetzes muss den Datenschutz nachdeutschem Recht für personenbezo-gene Daten in der Cloud sicherstel-len. Sämtliche gesetzlichen Daten-schutzvorschriften sind grundsätz-lich auch auf die Cloud anzuwenden.An dieser Stelle sollen drei wesentli-che Aspekte aufgegriffen werden:
Das deutsche Bundesdaten-schutzgesetz regelt den Umgangmit personenbezogenen Daten. Soll-ten besonders schützenswerte per-sonenbezogene Daten (im Sinne des§ 3 Abs. 9 BDSG, z.B. Gesundheits-daten) verarbeitet werden, müssenbesondere Maßnahmen getroffenwerden. Andere Daten wie Finanz-daten oder R&D- Daten, etc. müssennatürlich auch geschützt werden, fal-len jedoch nicht unter das Bundes-datenschutzgesetz.
Wenn personenbezogene Datenan einen Cloud Provider übermitteltwerden, darf dies nur dann erfolgen,wenn die Betroffenen eingewilligt ha-ben oder ein gesetzlicher Erlaubnis-tatbestand vorliegt, auch wenn essich bei dem Cloud-Provider um einkonzerneigenes Tochterunterneh-
men handelt. Im Bundesdaten-schutzgesetz gibt es das Konstruktder Auftragsdatenverarbeitung, wasbedeutet, dass der Cloud Providerdie Daten im Auftrag des Nutzersverarbeitet kann (quasi als Hilfsfunk-tion für den Nutzer). Er ist damit imrechtlichen Sinn kein Dritter, somitentfällt die Notwendigkeit einer Er-laubnis der Betroffenen. Dies gilt al-lerdings nur, wenn der Cloud-Pro-vider die Daten innerhalb Deutsch-lands, der EU oder des EWRs ver-arbeitet (gilt auch für Subunterneh-men). Bei einem Datentransfer inDrittstaaten kommt hinzu, dass dietechnisch organisatorischen Maß-nahmen zum Schutz der personen-bezogenen Daten des Cloud-Pro-viders (auch seiner Subunterneh-men) vom Nutzer regelmäßig kon-trolliert werden müssen. Dabei istes ausreichend, wenn regelmäßigentsprechende Prüfberichte zur Ver-fügung gestellt werden.
Preismodelle für CloudLeistungen
Die Preismodelle der Cloud-Anbietersind meist klarer als die Lizenzmo-delle für den Kauf von Software.Trotzdem können sie sich für denBezug von Cloud-Leistungen starkunterscheiden. Viele Modelle sindzeitbasiert, andere werden anhanddes Datenaufkommens abgerechnetetc. Wichtig ist, dass die Konditionenfür den Bezug von Cloud-Leistungenumstellbar sind. Anhand von zweiausgewählten Anbietern für PublicClouds erhalten Sie einen ersten Ein-druck der Preisgrößenordnung fürInfrastructure as a Service – Leistun-gen (Stand: 06/2012) (Tab. 2 und 3).
Fazit
Cloud Computing bietet den Phar-maunternehmen die Möglichkeit ei-nes grundsätzlichen Richtungswech-sels beim Einsatz von IT. Investitionenin interne IT-Ressourcen können zu-rückgefahren werden zugunsten einerSteigerung der Wettbewerbsfähigkeitim eigentlichen Kerngeschäft.
Trotz mancher Risiken sehe ichfür die Pharmaindustrie vier wesent-liche Vorteile des Cloud Computings:. Niedrigere Kosten für die ITEin Kostenvorteil des Cloud Compu-tings gegenüber dem Eigenerwerblässt sich allerdings nur dann realisie-ren, wenn die Nutzung der Cloud-Ser-vices dynamisch erfolgt. Die Zu- undAbschaltung von IT-Kapazitäten mussautomatisiert erfolgen, ohne dasshohe Transaktionskosten für das IT-Ressourcenmanagement entstehen.. Anpassung von IT-Ressourcen anbetriebliche Notwendigkeiten
Werden z.B. für R&D mehr IT-Res-sourcen benötigt, können sie alsCloud-Service kurzfristig aktiviertwerden. Werden sie nicht mehr benö-tigt, werden sie abgestellt. Die Kapa-zitäten der IT bleiben flexibel und ori-entieren sich am aktuellen Bedarf. DerLeerstand von IT-Ressourcen solltedamit der Vergangenheit angehören.. Verbesserung der internen undexternen Zusammenarbeit
Durch die beim Cloud Computingeingesetzten Techniken wie z.B. Vir-tualisierung ist es möglich, die fürProjekte notwendigen IT-Ressourceninnerhalb kürzester Zeit mit allenfachlichen und sicherheitstech-nischen Features zur Verfügung zustellen. Projekte werden dadurchschneller implementiert und die„Time to Market“ wird kürzer.. SicherheitDer Ausfall von IT-Ressourcen kannein ernstes Problem für Pharmaunter-nehmen darstellen. Die redundanteAuslegung klassischer IT-Infrastruk-tur ist kostenintensiv und unflexibel.Die schnelle Wiederverfügbarkeit imDesasterfall sowie laufend aktuali-sierte Sicherheitsfeatures und vertrag-liche Regelungen im Bereich der Si-cherheit und bei der Einhaltung ge-setzlicher Datenschutzvorschriftenmachen Cloud Computing für phar-mazeutische Unternehmen attraktiv.
Fachliteratur[1] Eckpunktpapier Sicherheitsempfehlun-
gen für Cloud Computer Anbieter, BSI,S. 15/16
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 353Bauer . Cloud Computing
die Server? Wer hat Zugriff?). Infor-mationssicherheit und Cloud Compu-ting erfordern gegenüber der klassi-schen IT eine vollkommene Neube-wertung der Sicherheitsrisiken. DieAusgangssituation von Cloud basier-ten Lösungen besteht im Grunde da-rin, dass sich mehrere Nutzer die phy-sikalischen Systemressourcen teilen.Dies könnte dazu führen, dass durchFehlfunktionen unbeabsichtigte man-datenübergreifende Zugriffe stattfin-den oder dass Sicherheitslücken dieDaten in der Cloud gefährden. DieProvider müssen ein angemessenesSicherheitsniveau herstellen und sichan laufend ändernden Gegebenheitenim Unternehmen anpassen.
Die Sicherstellung der Informati-onssicherheit erfolgt durch Risiko-analysen und -bewertung. Hier spieltdie ISO/IEC 27001 mit der Einfüh-rung eines Informations SicherheitsManagement System (ISMS) einezentrale Rolle. Zertifizierungen inder Informationssicherheit bieten inder Regel Cloud-Provider, die grö-ßere Unternehmen betreuen. Wich-tig ist in jedem Fall, dass die Ver-tragspartner Notfallpläne verein-baren, damit entsprechende Vorkeh-rungen bei Sicherheitsvorfällen ge-troffen werden.
VerfügbarkeitDie Einführung von Service LevelAgreements (SLA) ist eine essentielleVoraussetzung für die Einführung vonCloud Services. Die Verfügbarkeit
muss sich ebenso wiedie Performance an denBelangen der Geschäfts-prozesse im Unterneh-men orientieren, welchebisher durch klassischeIT-Leistungen bedientwurden.
Service IntegrationIn der Regel werden Ge-schäftsprozesse im Un-ternehmen durch Appli-kationen abgedeckt.Übernehmen Cloud Ser-vices Teil- oder Gesamt-prozesse stellt sich die
Frage der Integrationsfähigkeit zumeinen mit klassischen IT-Systemen,zum anderen der Kopplung vonCloud Systemen miteinander. DieAbschätzung der damit verbundenenRisiken und die Prozessintegrationmüssen im Vorfeld geklärt werden.
AbhängigkeitDie Nutzer-Schnittstelle hat für dieIaaS- und PaaS-Ebene eine besondereBedeutung. Sie dient dazu die Pro-visionierung und Steuerung derCloud-Anwendung durchzuführen.Bei nicht standardisierten Schnittstel-len wird es für den Nutzer schwierig,den Anbieter zu wechseln. Anbieter-spezifische APIs, die notwendig sind,um einen größtmöglichen Automati-sierungsgrad zur erreichen, erzeugenaber eine Abhängigkeit des Nutzersvom jeweiligen Cloud-Anbieter. Beider Planung von Cloud Projekten istes deshalb sinnvoll, darauf zu achtenwie eine Rückführbarkeit der Cloud-Services (technisch, öko-nomisch und rechtlich)sichergestellt werdenkann.
TransparenzDie Einführung von Mo-nitoring- und ReportingTools bringt Klarheit indie Kostenermittlungfür die bezogenen Ser-vices, bzw. deren Nut-zung durch die unter-schiedlichen Unterneh-
mensbereiche. Darüber hinaus bele-gen diese Tools die Einhaltung dervereinbarten SLAs sowie der Cloud-Service Performance. Sie stellen da-durch eine Quelle zur Analyse künf-tiger Einsparpotentiale dar.
Vertragliche AspekteVertragliche Regelungen bilden dieGrundlage und definieren den Um-fang der Leistungserbringung wieauch die gegenseitigen Ansprücheder Vertragspartner. Werden auf derSeite der Anbieter Subunternehmenzur Leistungserbringung eingebun-den, müssen auch für die Subunter-nehmen datenschutzrechtliche Ver-einbarungen getroffen werden. Alleindie Anzahl der beteiligten Vertrags-partner kann die Komplexität und da-mit den Aufwand für den Kundendrastisch beeinflussen. Die Beschrei-bung der vertraglichen Leistungenund die Folgen für deren Nichterfül-lung werden in den Service LevelAgreements niedergelegt. Je nach derArt der Cloud Nutzung müssen auchdie länderspezifischen Nutzungs-rechte der Software geregelt werden.Dies gilt sowohl für Software, die derCloud-Anbieter seinen Kunden bereit-stellt (hier muss in der Regel kein ur-heberrechtliches Nutzungsrecht fürden Anwender eingeräumt werden),wie auch für Software, die durch denKunden beigestellt wird.
Weitere Aspekte einer vertragli-chen Regelung für Cloud Computing:. Datenschutz. Geltendes Recht. Leistungsbeschreibung und damitauch eine Festlegung des gesetzli-
IT
TechnoPharm 3, Nr. 6, 349–353 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)352 Bauer . Cloud Computing
Tabelle 3
Telekom Preise.
Telekom HostingServicesDeployments
Nutzung in e
pro Stunde
1 PU/1 GB RAM/Speicher 160 GB
0,13
8 PU/15 GB RAM/Speicher 1690 GB
1,39
Tabelle 2
Amazon Preise.
Amazon EC2Standard onDemandInstances
Linux/UnixNutzungin $ proStunde
WindowsNutzungin $ proStunde
Small(Standard)
0,085 0,115
Medium 0,170 0,230
Large 0,340 0,460
Extra Large 0,680 0,920

Produkte
TechnoPharm 3, Nr. 6, 355 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 355Produkte
Feuchte- und Temperatursen-soren zur Innenraum-MontageDie Bedeutung der Energieeffizienzin der Gebäudeausstattung, aberauch die Sicherung eines behagli-chen Raumklimas wächst. Zur Ein-haltung der optimalen Parameterfür Klima-, Gebäude-, und Lüftungs-technik gilt die DIN EN 15251. Um dietechnischen Regeln optimal zu be-dienen, bietet die Galltec-Mela mitder L-Serie neue Feuchte- und Tem-peratursensoren zur Innenraum-Montage. Mit der neuen erweitertenDIN EN 15251 werden europaweitParameter für das Raumklima fest-gesetzt – unter anderen zur Bewer-tung von Energieeffizienz, Raumluft-qualität und Temperatur. Zur Umset-zung dieser technischen Regeln fürRaumluft- und Klimatechnik sindSensoren zur Messung von Tempera-tur und Luftfeuchte notwendig. Mitder L-Serie bietet Galltec-Mela* eineSensorlinie, die Temperatur, Feuch-tigkeit oder beides misst. Die Pro-
duktlinie wurde mit einem anspre-chenden Gehäuse in Weiß und Licht-grau ausgestattet. Zwischen 30 %und 65 % Luftfeuchte und Tempera-turen zwischen 20 °C und 28 °C sindlaut dem neuen nationalen Anhangzur DIN EN 15251 für Wohn- undNichtwohngebäude vorgeschrieben.Die Sensoren der L-Serie bieten zurEinhaltung dieser Kriterien optimaleMess-Eigenschaften. Die Geräte sindals Kanal-, Wand-, Stab oder Raum-sensor verfügbar. Je nach Anforde-rung bieten die Sensoren aktive oderpassive Ausgänge. Für erweiterteEinsatzmöglichkeiten werden dieSensoren zum Schutz vor Vibratio-nen oder erhöhten Anforderungenwie beispielsweise Kondensation inSpezialausführungen angeboten. Dieoptisch überarbeiteten Gehäuse-For-men erlauben nicht nur eine äußersteinfache Montage, sondern fügensich fließend in den Innenraum ein.Das gleiche Design für Kanal, Wandund Innenraum, aber technisch er-
weitert, bietet die D-Se-rie, optional mit Dis-play und in den Indus-trieversionen mit inte-griertem hx-Converter,der bei Bedarf auch ab-geleitete Größen wieTaupunkttemperatur,Wassergehalt, Enthal-pie, absolute Feuchteoder Feuchtkugeltem-peratur berechnet.
PCT Touchscreen-Technikund RDPDie Systec & Solutions* Systeme un-terstützen ab sofort auch einen PCT(Projected Capacitive Touch) Multi-touchscreen bei Verwendung desRDP 8 Protokolls in Kombinationmit einem integrierten Ultra-Thin-Client und einer Microsoft-Umge-bung mit Windows 8 oder WindowsServer 2012 R2.Mit einem Multitouchscreen eröff-nen sich für Software-Anwendungen,die eine präzise Touch-Bedienungmit Gesten erfordern, ganz neueMöglichkeiten. Der energiesparsameUltra-Thin-Client unterstützt RDP 8,welches eine Multitouch-Bedienungper Remotezugriff auf einen geeig-neten Windows basierten Server er-möglicht.Der PCT Multitouchscreen ermög-licht eine Bedienung mit bis zu 10Fingern. Damit können konventio-nelle Keyboards eingespart werden,da virtuelle Keyboards unterschiedli-
cher Sprachen auf dem Display abge-bildet und bedient werden können.Zudem besteht die Frontseite desMultitouchscreen komplett aus Glas,was hinsichtlich der Brillanz, Rei-nigung und Robustheit weitere Vor-teile bietet.Der Multitouchscreen mit Ultra-Thin-Client kann mit wenigen Klicks überdie IP-Adresse mit dem Server ver-bunden werden. Dabei unterstützt
der Ultra-Thin-Client USB-Gerätewie z.B. Scanner, Smartcard-Reader,Audio, etc.. Es ist kein lokales Be-triebssystem und keine Software not-wendig. Zusätzlich ist eine lokale Ad-ministration der Clients nicht erfor-derlich, da sie über ein Softwaretoolzentral aus der Ferne per LAN admi-nistriert werden können. Dies umfasstalle wichtigen Konfigurationen undist an unbegrenzt vielen Clients mög-lich. Die Technologie auf Basis vonUltra-Thin-Clients ist zudem kosten-günstiger als herkömmliche KVM-oder PC-Lösungen. Die integriertenUltra-Thin-Client mit Multitouchs-creen sind in den Größen 21,5 Zollund 24 Zoll erhältlich. Weitere Dis-playgrößen mit einem Ultra-Thin-Client und Multitouchscreen werdenin Kürze folgen.
* Galltec Mess- und Regeltechnik GmbHBoschstraße 471149 Bondorfwww.galltec-mela.de
* Systec & Solutions GmbHEmmy-Noether-Str. 1776131 Karlsruhewww.systec-solutions.com
Der schlanke Stabsensor LP vonGalltec+Mela ist eine von 4 mögli-chen Versionen für verschiedeneEinbausituationen.
WAVE 221 Multitouch mit integriertem Ultra-Thin-Client.
Produkte
TechnoPharm 3, Nr. 6, 354 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)354 Produkte
Mehrkanalpumpe für kleineFördervolumina(5μl – 3 ml/min.)Das neue Pumpensystem von DNEmicrotechnology und 2E* mecha-tronic zur Förderung verschiedens-ter Fluide vereint die Vorteile gän-giger Pumpentypen. Der Fluidikteilist als Disposable ausgeführt undkann sehr einfach komplett aus-getauscht werden. Die Pumpe istmodular aufgebaut, so dass siemit 2 bis 10 Schläuchen parallelausgestattet werden kann. In jedemKanal kann, unabhängig von
Druckschwankungen in Nachbar-kanälen, ein definiertes Volumengefördert werden. Zudem bietet
die Pumpe folgendeweitere Benefits: Peris-taltisches Förderprinzipmit integriertem Flow-Stop, BidirektionaleFörderung möglich, op-timiertes Preis-/Leis-tungsverhältnis, ver-schiedene Schlauch-durchmesser und Mo-tortypen wählbar.
Neues Dichtsystem für JUMO-WiderstandsthermometerSTEAMtemp für den Einsatz inSterilisatorenTemperaturfühler werden in Sterili-satoren häufig extremen klimati-schen Verhältnissen ausgesetzt. EineErwärmung bis zu 137 °C, wech-selnde Druckverhältnisse und die zu-sätzliche Belastung durch den ver-wendeten Wasserdampf stellen be-sondere Anforderungen an die Dich-tigkeit des Fühlers. JUMO* hat fürdas Einsteck-Widerstandsthermo-meter STEAMtemp deshalb ein kom-plett neues Dichtsystem entwickelt.Die neue Abdichtung zwischenSchutzarmatur und Kabel erfolgtüber einen Schrumpfschlauch ausbesonders widerstandsfähigen Mate-rialien, der mit beiden Teilen absolutdicht und zugentlastet verschweißtwird. Durch diesen Aufbau reduziertsich die mechanische Belastung aufden Messeinsatz. Das wirkt sich po-sitiv auf die Genauigkeit, die Lang-zeitstabilität und erheblich auf dieStandzeit aus. Die Konstruktion des
STEAMtemp erlaubt die Verwen-dung in wasserdampfhaltiger, unterDruck stehender Atmosphäre. NebenSterilisatoren finden sich unter ande-rem im Apparatebau, in der Labor-technik oder bei Klimaprüfschrän-ken weitere Einsatzmöglichkeiten.Alle STEAMtemp-Varianten werdenin Schutzart IP69 ausgeführt. Sie sindabsolut wasserdicht sowie druckfestbis 3,5 bar. Das Widerstandsthermo-meter kann bei Prozesstemperaturen
von –50 bis +205 °C ein-gesetzt werden. Je nachAnforderung sind ver-schiedene Schutzarma-turen möglich. Beson-ders schnelle Reaktions-zeiten können dabei übereine Miniatur-Variantemit einer Armatur vonnur zwei MillimeternDurchmesser erreichtwerden. Der Messeinsatzwird mit einem oderzwei Platin-SensorenPt100 Klasse A aufgebautund generell in Vierlei-
terschaltung angeschlossen. Die An-schlussleitung hat eine Standard-länge von 2,5 m, andere Längen sindmöglich. Das Kabel kann über Steck-hülsen oder mehrpolige Stecker andie Elektronik angeschlossen wer-den.
* 2E mechatronic GmbH & Co. KGMaria-Merian-Str. 2973230 Kirchheim unter Teckwww.2e-mechatronic.de
* JUMO GmbH & Co. KGMoritz-Juchheim-Straße 136039 Fuldawww.jumo.net
Modulare Mikroliterpumpe
Die Thermometer der STEAMtemp-Serie sind jetzt nochbesser abgedichtet und damit widerstandsfähiger.

Produkte
TechnoPharm 3, Nr. 6, 355 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany) 355Produkte
Feuchte- und Temperatursen-soren zur Innenraum-MontageDie Bedeutung der Energieeffizienzin der Gebäudeausstattung, aberauch die Sicherung eines behagli-chen Raumklimas wächst. Zur Ein-haltung der optimalen Parameterfür Klima-, Gebäude-, und Lüftungs-technik gilt die DIN EN 15251. Um dietechnischen Regeln optimal zu be-dienen, bietet die Galltec-Mela mitder L-Serie neue Feuchte- und Tem-peratursensoren zur Innenraum-Montage. Mit der neuen erweitertenDIN EN 15251 werden europaweitParameter für das Raumklima fest-gesetzt – unter anderen zur Bewer-tung von Energieeffizienz, Raumluft-qualität und Temperatur. Zur Umset-zung dieser technischen Regeln fürRaumluft- und Klimatechnik sindSensoren zur Messung von Tempera-tur und Luftfeuchte notwendig. Mitder L-Serie bietet Galltec-Mela* eineSensorlinie, die Temperatur, Feuch-tigkeit oder beides misst. Die Pro-
duktlinie wurde mit einem anspre-chenden Gehäuse in Weiß und Licht-grau ausgestattet. Zwischen 30 %und 65 % Luftfeuchte und Tempera-turen zwischen 20 °C und 28 °C sindlaut dem neuen nationalen Anhangzur DIN EN 15251 für Wohn- undNichtwohngebäude vorgeschrieben.Die Sensoren der L-Serie bieten zurEinhaltung dieser Kriterien optimaleMess-Eigenschaften. Die Geräte sindals Kanal-, Wand-, Stab oder Raum-sensor verfügbar. Je nach Anforde-rung bieten die Sensoren aktive oderpassive Ausgänge. Für erweiterteEinsatzmöglichkeiten werden dieSensoren zum Schutz vor Vibratio-nen oder erhöhten Anforderungenwie beispielsweise Kondensation inSpezialausführungen angeboten. Dieoptisch überarbeiteten Gehäuse-For-men erlauben nicht nur eine äußersteinfache Montage, sondern fügensich fließend in den Innenraum ein.Das gleiche Design für Kanal, Wandund Innenraum, aber technisch er-
weitert, bietet die D-Se-rie, optional mit Dis-play und in den Indus-trieversionen mit inte-griertem hx-Converter,der bei Bedarf auch ab-geleitete Größen wieTaupunkttemperatur,Wassergehalt, Enthal-pie, absolute Feuchteoder Feuchtkugeltem-peratur berechnet.
PCT Touchscreen-Technikund RDPDie Systec & Solutions* Systeme un-terstützen ab sofort auch einen PCT(Projected Capacitive Touch) Multi-touchscreen bei Verwendung desRDP 8 Protokolls in Kombinationmit einem integrierten Ultra-Thin-Client und einer Microsoft-Umge-bung mit Windows 8 oder WindowsServer 2012 R2.Mit einem Multitouchscreen eröff-nen sich für Software-Anwendungen,die eine präzise Touch-Bedienungmit Gesten erfordern, ganz neueMöglichkeiten. Der energiesparsameUltra-Thin-Client unterstützt RDP 8,welches eine Multitouch-Bedienungper Remotezugriff auf einen geeig-neten Windows basierten Server er-möglicht.Der PCT Multitouchscreen ermög-licht eine Bedienung mit bis zu 10Fingern. Damit können konventio-nelle Keyboards eingespart werden,da virtuelle Keyboards unterschiedli-
cher Sprachen auf dem Display abge-bildet und bedient werden können.Zudem besteht die Frontseite desMultitouchscreen komplett aus Glas,was hinsichtlich der Brillanz, Rei-nigung und Robustheit weitere Vor-teile bietet.Der Multitouchscreen mit Ultra-Thin-Client kann mit wenigen Klicks überdie IP-Adresse mit dem Server ver-bunden werden. Dabei unterstützt
der Ultra-Thin-Client USB-Gerätewie z.B. Scanner, Smartcard-Reader,Audio, etc.. Es ist kein lokales Be-triebssystem und keine Software not-wendig. Zusätzlich ist eine lokale Ad-ministration der Clients nicht erfor-derlich, da sie über ein Softwaretoolzentral aus der Ferne per LAN admi-nistriert werden können. Dies umfasstalle wichtigen Konfigurationen undist an unbegrenzt vielen Clients mög-lich. Die Technologie auf Basis vonUltra-Thin-Clients ist zudem kosten-günstiger als herkömmliche KVM-oder PC-Lösungen. Die integriertenUltra-Thin-Client mit Multitouchs-creen sind in den Größen 21,5 Zollund 24 Zoll erhältlich. Weitere Dis-playgrößen mit einem Ultra-Thin-Client und Multitouchscreen werdenin Kürze folgen.
* Galltec Mess- und Regeltechnik GmbHBoschstraße 471149 Bondorfwww.galltec-mela.de
* Systec & Solutions GmbHEmmy-Noether-Str. 1776131 Karlsruhewww.systec-solutions.com
Der schlanke Stabsensor LP vonGalltec+Mela ist eine von 4 mögli-chen Versionen für verschiedeneEinbausituationen.
WAVE 221 Multitouch mit integriertem Ultra-Thin-Client.
Produkte
TechnoPharm 3, Nr. 6, 354 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)354 Produkte
Mehrkanalpumpe für kleineFördervolumina(5μl – 3 ml/min.)Das neue Pumpensystem von DNEmicrotechnology und 2E* mecha-tronic zur Förderung verschiedens-ter Fluide vereint die Vorteile gän-giger Pumpentypen. Der Fluidikteilist als Disposable ausgeführt undkann sehr einfach komplett aus-getauscht werden. Die Pumpe istmodular aufgebaut, so dass siemit 2 bis 10 Schläuchen parallelausgestattet werden kann. In jedemKanal kann, unabhängig von
Druckschwankungen in Nachbar-kanälen, ein definiertes Volumengefördert werden. Zudem bietet
die Pumpe folgendeweitere Benefits: Peris-taltisches Förderprinzipmit integriertem Flow-Stop, BidirektionaleFörderung möglich, op-timiertes Preis-/Leis-tungsverhältnis, ver-schiedene Schlauch-durchmesser und Mo-tortypen wählbar.
Neues Dichtsystem für JUMO-WiderstandsthermometerSTEAMtemp für den Einsatz inSterilisatorenTemperaturfühler werden in Sterili-satoren häufig extremen klimati-schen Verhältnissen ausgesetzt. EineErwärmung bis zu 137 °C, wech-selnde Druckverhältnisse und die zu-sätzliche Belastung durch den ver-wendeten Wasserdampf stellen be-sondere Anforderungen an die Dich-tigkeit des Fühlers. JUMO* hat fürdas Einsteck-Widerstandsthermo-meter STEAMtemp deshalb ein kom-plett neues Dichtsystem entwickelt.Die neue Abdichtung zwischenSchutzarmatur und Kabel erfolgtüber einen Schrumpfschlauch ausbesonders widerstandsfähigen Mate-rialien, der mit beiden Teilen absolutdicht und zugentlastet verschweißtwird. Durch diesen Aufbau reduziertsich die mechanische Belastung aufden Messeinsatz. Das wirkt sich po-sitiv auf die Genauigkeit, die Lang-zeitstabilität und erheblich auf dieStandzeit aus. Die Konstruktion des
STEAMtemp erlaubt die Verwen-dung in wasserdampfhaltiger, unterDruck stehender Atmosphäre. NebenSterilisatoren finden sich unter ande-rem im Apparatebau, in der Labor-technik oder bei Klimaprüfschrän-ken weitere Einsatzmöglichkeiten.Alle STEAMtemp-Varianten werdenin Schutzart IP69 ausgeführt. Sie sindabsolut wasserdicht sowie druckfestbis 3,5 bar. Das Widerstandsthermo-meter kann bei Prozesstemperaturen
von –50 bis +205 °C ein-gesetzt werden. Je nachAnforderung sind ver-schiedene Schutzarma-turen möglich. Beson-ders schnelle Reaktions-zeiten können dabei übereine Miniatur-Variantemit einer Armatur vonnur zwei MillimeternDurchmesser erreichtwerden. Der Messeinsatzwird mit einem oderzwei Platin-SensorenPt100 Klasse A aufgebautund generell in Vierlei-
terschaltung angeschlossen. Die An-schlussleitung hat eine Standard-länge von 2,5 m, andere Längen sindmöglich. Das Kabel kann über Steck-hülsen oder mehrpolige Stecker andie Elektronik angeschlossen wer-den.
* 2E mechatronic GmbH & Co. KGMaria-Merian-Str. 2973230 Kirchheim unter Teckwww.2e-mechatronic.de
* JUMO GmbH & Co. KGMoritz-Juchheim-Straße 136039 Fuldawww.jumo.net
Modulare Mikroliterpumpe
Die Thermometer der STEAMtemp-Serie sind jetzt nochbesser abgedichtet und damit widerstandsfähiger.

Verlag / PublisherECV · Editio Cantor Verlag für Medizinund Naturwissenschaften GmbHBaendelstockweg 2088326 Aulendorf (Germany)GF/MD: Claudius Arndt,Andreas GerthEingetragen/Registered:Amtsgericht Ulm, HRB 600174Tel. +49 (0) 7525-9400Fax +49 (0) 7525-940 180www.ecv.de
Redaktion / Editorial officeChefredakteur / Editor-in-Chief:Claudius ArndtTel. +49 (0) 7525-940 159Leitende Redakteurin/Managing Editor:Kerstin Jarosch (V. i. S. d. P.)Tel. +49 (0) 8191-98578 12Fax +49 (0) 8191-98578 19e-mail: [email protected]
Anzeigen / AdvertisementsVerantwortlich/Responsible:Lara LehmannTel. +49 (0) 8191-98578 11Fax +49 (0) 8191-98578 19e-mail: [email protected]
Herstellung / ProductionRombach Druck- und VerlagshausGmbH & Co. KGUnterwerkstr. 5, 79115 FreiburgTel. +49 (0) 761-4500-0Fax +49 (0) 761-4500-2122e-mail: [email protected]
Druck / PrintHOLZMANN DRUCK GmbH & Co. KGGewerbestraße 2, 86825 Bad WörishofenTel.: +49-(0) 8247-993 0Fax:: +49-(0) 8247-993 208e-mail: [email protected]
Bezugsbedingungen (gültig ab Januar 2011)Die Zeitschrift erscheint sechsmal pro Jahr und kannvom Verlag, von der Arbeitsgemeinschaft für Pharma-zeutische Verfahrenstechnik e. V. (APV) oder durch eineBuchhandlung (ISSN 2191-8341) bezogen werden.Preise für das Jahresabonnement als Printausgabe ein-schließlich Online-Zugang (inkl. MwSt., zzgl. Versand):Inland: 72,00 €, APV-Mitglieder und Studenten 54,00 €,zzgl. 14,00 € Versand; Ausland (Europa mit VAT Ident.Nr.): 67,29 €, APV-Mitglieder und Studenten 50,47 €,Versandkosten 16,82 €Ausland (Europa ohne VAT Ident. Nr. und weiteresAusland): 72,00 €, APV-Mitglieder und Studenten 54,00 €,Versandkosten 18,00 €Preis für das Einzelheft: 19,00 € (inkl. MwSt., zzgl.Versand). Das Abonnement ist weiter rechtsverbindlich,wenn es nicht mindestens 3 Monate vor Ende desBerechnungszeitraums gekündigt wird.Konten des Verlages: Commerzbank Friedrichshafen(BLZ 651 400 72) 17 080 80;IBAN: DE21 6514 0072 0170 8080 00;SWIFT-BIC: COBADEFF651.Landesbank Baden-Württemberg(BLZ 600 501 01) 4 508 560;IBAN: DE57 6005 0101 0004 5085 60;SWIFT-BIC: SOLADEST.Deutsche Postbank AG(BLZ 600 100 70) 29 487 703;IBAN: DE08 6001 0070 0029 4877 03;SWIFT-BIC: PBNKDEFF600.
ManuskripteManuskripte sind an die Redaktion zu senden. Es werdennur Beiträge zur Erstveröffentlichung angenommen.Die Autoren versichern, dass sie allein berechtigt sind,über die urheberrechtlichen Nutzungsrechte an ihrenArbeiten einschließlich etwaiger Bilder, Tabellen etc. zuverfügen und dass keine Rechte Dritter verletzt werden.Sie versichern außerdem, dass keine Doppelpublikation(Veröffentlichung auch in anderen Zeitschriften)beabsichtigt ist bzw. erfolgt. Mit der Annahme desManuskriptes zur Veröffentlichung räumt der Autor demVerlag das ausschließliche, zeitlich, räumlich und in-haltlich unbeschränkte Recht zur Veröffentlichung, Ver-vielfältigung, Verbreitung und öffentlichen Wiedergabe inallen Sprachen und Ländern ein, einschließlich desRechts zur Speicherung in und Nutzung durch Daten-banken jeder Art (Online, auch Internet, und Offline)sowie der weiteren Vervielfältigung zu gewerblichenZwecken im Wege des fotomechanischen oder einesanderen Verfahrens.
Urheber- und VerlagsrechteSämtliche in dieser Zeitschrift veröffentlichten Beiträgegenießen urheberrechtlichen Schutz. Kein Teil der Zeit-schrift darf außerhalb der engen Grenzen des Urheber-rechtsgesetzes ohne schriftliche Einwilligung des Ver-lages in irgendeiner Form vervielfältigt, verbreitet odersonst verwertet werden oder in eine von Maschinenverwendbare Sprache übertragen oder übersetzt werden.Insbesondere ist jede Digitalisierung, Speicherung undNutzung in und durch elektronische Datenbanken jederArt untersagt.
HaftungDer Inhalt dieses Heftes wurde sorgfältig erarbeitet.Dennoch übernehmen Autoren, Beirat, Redaktion undVerlag für die Richtigkeit von Angaben sowie für even-tuelle Satz- oder Druckfehler keine Haftung.
WarenzeichenDas Fehlen des Symbols ® nach Namen bedeutet nicht,dass der Name nicht durch Warenzeichen geschützt ist.
SonderdruckeVon Veröffentlichungen erhält der Korrespondenzautor20 Sonderdrucke in Form guter Fotokopierqualität ohneBerechnung. Weitere Sonderdrucke oder Ausführungenin höherwertiger Form oder Aufmachung können gegenÜbernahme der Kosten beim Verlag bezogen werden.
TechnoPharm® ist eine eingetragene Marke
Terms of subscription (valid from January 2011)The journal is published bimonthly beginning in 2012and can be obtained from the Publisher, from the Ar-beitsgemeinschaft Pharmazeutische Verfahrenstechnike. V. (APV) or via bookstores (ISSN 2191-8341).Rates per annum for the print + online subscription(including VAT, plus postage):Germany: 72.00 €, APV members and students 54.00 €,plus postage 14.00 €;Abroad (Europe with VAT ID number): 67.29 €, APV mem-bers and students 50.47 €, plus postage 16,82 € (surface mail);Abroad (Europe without VAT ID number and othercountries): 72.00 €, APV members and students 54.00 €,plus postage 18.00 € (surface mail);Price for single copy: 19.00 € (including VAT, plus postage).A subscription will continue unless cancelled with threemonths’ notice prior to the end of the invoiced period.Publisher’s bank accounts:Commerzbank Friedrichshafen(Bank Code 651 400 72) Account no. 17 080 80;IBAN: DE21 6514 0072 0170 8080 00;SWIFT-BIC: COBADEFF651.Landesbank Baden-Württemberg(Bank Code 600 501 01) Account no. 4 508 560;IBAN: DE57 6005 0101 0004 5085 60;SWIFT-BIC: SOLADEST.Deutsche Postbank AG(Bank Code 600 100 70) Account no. 29 487 703;IBAN: DE08 6001 0070 0029 4877 03;SWIFT-BIC: PBNKDEFF600.
ManuscriptsManuscripts have to be sent to the editorial office. Onlypreviously unpublished papers are eligible for publicati-on. Authors affirm that they are the sole owners of thecopyright to their manuscripts including any figures,tables, etc., if applicable, and that they do not infringe anyrights of third parties. They further affirm that no doublepublication (additional publication in other journals) isintended or will occur. The acceptance of a manuscriptfor publication implies that the author assigns to thePublisher the copyright to the paper whereby the Pu-blisher acquires the exclusive right – unrestricted withregard to time, territory and contents – of publication,reproduction, distribution and public communication inany languages and countries, including the right of sto-rage and use in databases of any kind (online includinginternet and offline) and the right of reproduction forcommercial purposes by photomechanical or other me-ans.
Copyright and the publisher’s rightAll articles published in this journal are protected bycopyright. No part of this journal may be reproduced,distributed or otherwise exploited in any form or trans-ferred into a machine-readable language or translatedbeyond the narrow scope of the Copyright Act withoutthe Publisher's consent in writing. In particular, any kindof digitizing, storage or utilization in and by means ofdatabases is prohibited.
LiabilityThe contents of this issue have been carefully compiled. Ne-vertheless, the authors, editorial advisory board, editorial officeand Publisher do not assume any liability for the correctness ofinformation or for typographical errors or misprints.
TrademarksThe absence of the symbol ® after any name does notimply that this name is not under trademark protection.
ReprintsThe corresponding author receives 20 reprints of a pu-blished article in good photocopy quality free of charge.Additional reprints or reprints in higher quality form ordesign can be obtained from the Publisher against pay-ment.
TechnoPharm® is a registered trademark
TechnoPharm 3, Nr. 6, 356 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)356 Impressum / Masthead
Impressum / Masthead
APVNEWS 06 • 2013
Nachrichten und Mitteilungen
APV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e. V.
MAKING SCIENCE WORK
Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V.Gemeinnütziger wissenschaftlicher VereinInternational Association for Pharmaceutical Technology
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 1

Verlag / PublisherECV · Editio Cantor Verlag für Medizinund Naturwissenschaften GmbHBaendelstockweg 2088326 Aulendorf (Germany)GF/MD: Claudius Arndt,Andreas GerthEingetragen/Registered:Amtsgericht Ulm, HRB 600174Tel. +49 (0) 7525-9400Fax +49 (0) 7525-940 180www.ecv.de
Redaktion / Editorial officeChefredakteur / Editor-in-Chief:Claudius ArndtTel. +49 (0) 7525-940 159Leitende Redakteurin/Managing Editor:Kerstin Jarosch (V. i. S. d. P.)Tel. +49 (0) 8191-98578 12Fax +49 (0) 8191-98578 19e-mail: [email protected]
Anzeigen / AdvertisementsVerantwortlich/Responsible:Lara LehmannTel. +49 (0) 8191-98578 11Fax +49 (0) 8191-98578 19e-mail: [email protected]
Herstellung / ProductionRombach Druck- und VerlagshausGmbH & Co. KGUnterwerkstr. 5, 79115 FreiburgTel. +49 (0) 761-4500-0Fax +49 (0) 761-4500-2122e-mail: [email protected]
Druck / PrintHOLZMANN DRUCK GmbH & Co. KGGewerbestraße 2, 86825 Bad WörishofenTel.: +49-(0) 8247-993 0Fax:: +49-(0) 8247-993 208e-mail: [email protected]
Bezugsbedingungen (gültig ab Januar 2011)Die Zeitschrift erscheint sechsmal pro Jahr und kannvom Verlag, von der Arbeitsgemeinschaft für Pharma-zeutische Verfahrenstechnik e. V. (APV) oder durch eineBuchhandlung (ISSN 2191-8341) bezogen werden.Preise für das Jahresabonnement als Printausgabe ein-schließlich Online-Zugang (inkl. MwSt., zzgl. Versand):Inland: 72,00 €, APV-Mitglieder und Studenten 54,00 €,zzgl. 14,00 € Versand; Ausland (Europa mit VAT Ident.Nr.): 67,29 €, APV-Mitglieder und Studenten 50,47 €,Versandkosten 16,82 €Ausland (Europa ohne VAT Ident. Nr. und weiteresAusland): 72,00 €, APV-Mitglieder und Studenten 54,00 €,Versandkosten 18,00 €Preis für das Einzelheft: 19,00 € (inkl. MwSt., zzgl.Versand). Das Abonnement ist weiter rechtsverbindlich,wenn es nicht mindestens 3 Monate vor Ende desBerechnungszeitraums gekündigt wird.Konten des Verlages: Commerzbank Friedrichshafen(BLZ 651 400 72) 17 080 80;IBAN: DE21 6514 0072 0170 8080 00;SWIFT-BIC: COBADEFF651.Landesbank Baden-Württemberg(BLZ 600 501 01) 4 508 560;IBAN: DE57 6005 0101 0004 5085 60;SWIFT-BIC: SOLADEST.Deutsche Postbank AG(BLZ 600 100 70) 29 487 703;IBAN: DE08 6001 0070 0029 4877 03;SWIFT-BIC: PBNKDEFF600.
ManuskripteManuskripte sind an die Redaktion zu senden. Es werdennur Beiträge zur Erstveröffentlichung angenommen.Die Autoren versichern, dass sie allein berechtigt sind,über die urheberrechtlichen Nutzungsrechte an ihrenArbeiten einschließlich etwaiger Bilder, Tabellen etc. zuverfügen und dass keine Rechte Dritter verletzt werden.Sie versichern außerdem, dass keine Doppelpublikation(Veröffentlichung auch in anderen Zeitschriften)beabsichtigt ist bzw. erfolgt. Mit der Annahme desManuskriptes zur Veröffentlichung räumt der Autor demVerlag das ausschließliche, zeitlich, räumlich und in-haltlich unbeschränkte Recht zur Veröffentlichung, Ver-vielfältigung, Verbreitung und öffentlichen Wiedergabe inallen Sprachen und Ländern ein, einschließlich desRechts zur Speicherung in und Nutzung durch Daten-banken jeder Art (Online, auch Internet, und Offline)sowie der weiteren Vervielfältigung zu gewerblichenZwecken im Wege des fotomechanischen oder einesanderen Verfahrens.
Urheber- und VerlagsrechteSämtliche in dieser Zeitschrift veröffentlichten Beiträgegenießen urheberrechtlichen Schutz. Kein Teil der Zeit-schrift darf außerhalb der engen Grenzen des Urheber-rechtsgesetzes ohne schriftliche Einwilligung des Ver-lages in irgendeiner Form vervielfältigt, verbreitet odersonst verwertet werden oder in eine von Maschinenverwendbare Sprache übertragen oder übersetzt werden.Insbesondere ist jede Digitalisierung, Speicherung undNutzung in und durch elektronische Datenbanken jederArt untersagt.
HaftungDer Inhalt dieses Heftes wurde sorgfältig erarbeitet.Dennoch übernehmen Autoren, Beirat, Redaktion undVerlag für die Richtigkeit von Angaben sowie für even-tuelle Satz- oder Druckfehler keine Haftung.
WarenzeichenDas Fehlen des Symbols ® nach Namen bedeutet nicht,dass der Name nicht durch Warenzeichen geschützt ist.
SonderdruckeVon Veröffentlichungen erhält der Korrespondenzautor20 Sonderdrucke in Form guter Fotokopierqualität ohneBerechnung. Weitere Sonderdrucke oder Ausführungenin höherwertiger Form oder Aufmachung können gegenÜbernahme der Kosten beim Verlag bezogen werden.
TechnoPharm® ist eine eingetragene Marke
Terms of subscription (valid from January 2011)The journal is published bimonthly beginning in 2012and can be obtained from the Publisher, from the Ar-beitsgemeinschaft Pharmazeutische Verfahrenstechnike. V. (APV) or via bookstores (ISSN 2191-8341).Rates per annum for the print + online subscription(including VAT, plus postage):Germany: 72.00 €, APV members and students 54.00 €,plus postage 14.00 €;Abroad (Europe with VAT ID number): 67.29 €, APV mem-bers and students 50.47 €, plus postage 16,82 € (surface mail);Abroad (Europe without VAT ID number and othercountries): 72.00 €, APV members and students 54.00 €,plus postage 18.00 € (surface mail);Price for single copy: 19.00 € (including VAT, plus postage).A subscription will continue unless cancelled with threemonths’ notice prior to the end of the invoiced period.Publisher’s bank accounts:Commerzbank Friedrichshafen(Bank Code 651 400 72) Account no. 17 080 80;IBAN: DE21 6514 0072 0170 8080 00;SWIFT-BIC: COBADEFF651.Landesbank Baden-Württemberg(Bank Code 600 501 01) Account no. 4 508 560;IBAN: DE57 6005 0101 0004 5085 60;SWIFT-BIC: SOLADEST.Deutsche Postbank AG(Bank Code 600 100 70) Account no. 29 487 703;IBAN: DE08 6001 0070 0029 4877 03;SWIFT-BIC: PBNKDEFF600.
ManuscriptsManuscripts have to be sent to the editorial office. Onlypreviously unpublished papers are eligible for publicati-on. Authors affirm that they are the sole owners of thecopyright to their manuscripts including any figures,tables, etc., if applicable, and that they do not infringe anyrights of third parties. They further affirm that no doublepublication (additional publication in other journals) isintended or will occur. The acceptance of a manuscriptfor publication implies that the author assigns to thePublisher the copyright to the paper whereby the Pu-blisher acquires the exclusive right – unrestricted withregard to time, territory and contents – of publication,reproduction, distribution and public communication inany languages and countries, including the right of sto-rage and use in databases of any kind (online includinginternet and offline) and the right of reproduction forcommercial purposes by photomechanical or other me-ans.
Copyright and the publisher’s rightAll articles published in this journal are protected bycopyright. No part of this journal may be reproduced,distributed or otherwise exploited in any form or trans-ferred into a machine-readable language or translatedbeyond the narrow scope of the Copyright Act withoutthe Publisher's consent in writing. In particular, any kindof digitizing, storage or utilization in and by means ofdatabases is prohibited.
LiabilityThe contents of this issue have been carefully compiled. Ne-vertheless, the authors, editorial advisory board, editorial officeand Publisher do not assume any liability for the correctness ofinformation or for typographical errors or misprints.
TrademarksThe absence of the symbol ® after any name does notimply that this name is not under trademark protection.
ReprintsThe corresponding author receives 20 reprints of a pu-blished article in good photocopy quality free of charge.Additional reprints or reprints in higher quality form ordesign can be obtained from the Publisher against pay-ment.
TechnoPharm® is a registered trademark
TechnoPharm 3, Nr. 6, 356 (2013)© ECV . Editio Cantor Verlag, Aulendorf (Germany)356 Impressum / Masthead
Impressum / Masthead
APVNEWS 06 • 2013
Nachrichten und Mitteilungen
APV – Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e. V.
MAKING SCIENCE WORK
Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V.Gemeinnütziger wissenschaftlicher VereinInternational Association for Pharmaceutical Technology
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 1

APV NEWS 5-2013
APV NEWS – Aus der Geschäftsstelle
9th World Meeting on Pharmaceutics,Biopharmaceutics and Pharmaceutical Technology and ResearchPharm®
31 March to 03 April 2014, Lisbon, Portugal
Skin Forum 14th Annual Meeting04 to 05 September 2014, Prague, Czech Republic
TechnoPharm® 201430 September to 02 October 2014, Nuremberg, Germany
Upcoming international events – Save the date!
Die APV vergibt im Jahr 2014 erneut zwei Preise für den wissenschaftlichen Nachwuchs. Der Doktorandenpreis, dessen Preisgeldin Höhe von 5.000 Euro in diesem Jahr das Unternehmen Evonik Industries AG zur Verfügung stellt, wird für eine exzellenteDissertation aus dem Jahr 2012 oder 2013 vergeben. Der Promovierte kann sich selbst um den Preis bewerben. Das Gutach-terkomitee wird von Prof. Dr. Reinhard Neubert geleitet. Der Forschungspreis, dessen Preisgeld in Höhe von 5.000 Euro vonBayer Healthcare zur Verfügung gestellt wird, ehrt einen erfolgreichen Wissenschaftler unter 46 Jahren in der Pharmazie, derin den letzten Jahren herausragende Fortschritte in seinem Fachgebiet erzielt hat. Prof. Dr. H.P. Merkle leitet das Kuratorium.Der Einsendeschluss ist jeweils der 15.12.2013. Die Preisträger werden auf dem 9th PBP World Meeting in Lissabon geehrt.Weitere Details entnehmen Sie bitte der Internetseitewww.apv-mainz.de/english/apv/research-support/apv-awards/
Doktoranden- und Forschungspreis der APV
Lokale Gruppen
Neue Treffpunkte und -zeiten der lokalen Gruppen:
Montag, 20. Januar 2014 Lokale APV-Gruppe Berlin um 19:00 Uhr im Pestana Hotel Berlin Tiergarten, Stülerstrasse 6, 10787 Berlin.Bitte melden Sie sich bis zum 10.01.2014 bei Dr. Andreas Sachse an.
Weitere Informationen zu unseren Lokalen Gruppen finden Sie auf http://www.apv-mainz.de/apv/lokale-gruppen/
APV NEWS – Aus dem Vereinsleben
Liebe APV-Mitglieder, nach der erfolgreichen Gründung mehrerer lokaler Gruppen würden wir das Konzept der lokalenAPV-Gruppen gerne auch in weiteren Regionen etablieren. Bitte sprechen Sie uns an, wenn SieInteresse an einer Teilnahme an einer lokalen Gruppe in Ihrer Region haben oder als Ansprechpartner, unterstützt durch die APV-Geschäftsstelle, für eine neue lokale Gruppe zur Verfügung stehen würden. Wir freuen uns auf Ihre Rückmeldung! Ansprechpartner: Dr. Martin Bornhöft, Email: [email protected], Tel: + 49 6131 9769-35
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 2
APV NEWS – Aus dem Vereinsleben
APV NEWS 6-2013
Renate Lieckfeldt ist am 08. September 2013 für immer von uns gegangen. Fassungslos und bestürzthaben wir die traurige Nachricht entgegennehmen müssen.
Im Jahr 1988 schloss Renate Lieckfeldt ihr Studium der Pharmazie an der Universität Heidelberg ab, imJahr darauf erfolgte ihre Approbation zur Apothekerin. 1993 promovierte sie im Fach PharmazeutischeTechnologie am Institut für Pharmazeutische Technologie und Biopharmazie der Universität Heidelberg.In den folgenden Jahren war sie Regulatory Affairs Manager for Drugs & Medical Devices bei der Procter& Gamble GmbH in Schwalbach und anschließend Technical External Relations Manager Benelux beider Procter & Gamble Nederland B. V. in Rotterdam. Von 2001 bis 2011 lehrte sie als Professorin fürTechnisches Management und Projektmanagement im Fachbereich Physikalische Technik an der Fachhochschule Gelsenkirchen.Im Januar 2011 wurde Renate Lieckfeldt vom Erweiterten Senat der HTWK Leipzig zur neuen Rektorin gewählt. Renate Lieck-feldt war seit 1994 Mitglied der APV und seit 2002 der Fachgruppe Prozessoptimierung. Neben ihrer Arbeit in der Fachgruppehat sie zahlreiche erfolgreiche Kurse und Seminare konzipiert und unter ihrer Leitung durchgeführt.
Wir kannten Renate Lieckfeldt als engagierte und zielorientierte Kollegin, die mit ihrer Anwesenheit in der Fachgruppe aufvieles eine völlig neue Sicht einbrachte und damit eine ungemeine Bereicherung für unsere Fachgruppe war. Sie war ein Menschvoller Kraft und enormer Willensstärke. Renate Lieckfeldt war aber auch ein humorvoller, hilfsbereiter und liebenswerter Mensch,bei dem gegenseitiger Respekt und Kollegialität immer im Vordergrund standen. Alle Fachgruppenmitglieder, die Geschäftsstelleund der Vorstand der APV werden Renate Lieckfeldt ein ehrendes Gedenken bewahren.
Jürgen Werani
Nachruf Frau Prof. Dr. rer. nat. Renate Lieckfeldt
The School of Pharmacy and Pharmaceutical Sciences at Trinity College Dublin recently hosted the 2nd Galenus Workshop on PulmonaryDrug Delivery at the Panoz Institute from September 18 - 20, 2013, supported by the Galenus Foundation (Vienna / Austria). The work-shop was attended by an international group of PhD students undertaking research in Pharmaceutical Sciences and junior faculty mem-bers from 18 different Universities as well as representatives of the pharmaceutical industry from a variety of companies and of theGalenus Foundation. Both practical and theoretical in scope, the workshop covered many aspects of Pulmonary Formulation and DrugDisposition with lectures and practical sessions delivered by highly experienced faculty members and international speakers.
Participants heard about a series of studies focussing on new targets for the treatment of lung diseases, a particularly important topicas a result of new pipeline drug development and novel clinical entities which may have a narrow therapeutic index or be too toxic forsystemic delivery. Improving the efficiency of inhaled aerosol delivery by targeting drug to the appropriate lung regions/sites, may improvethe therapeutic response and minimise potential adverse effects.Other topics covered included imaging techniques in pulmonary drugdelivery; in vitro models of the air-blood barrier; pulmonary delivery of biopharmaceuticals; nanoparticles for aerosol delivery, and ad-vances in aerosoliser device technology. Participants also received practical training in spray drying of respirable particles; in vitro cha-racterisation of aerosol particles, and uptake and transport studies in lung cell cultures. Speaking about the success of this event, DrCarsten Ehrhardt, Associate Professor in Pharmaceutics and Pharmaceutical Technology at The School of Pharmacy and PharmaceuticalSciences, organizer of this workshop, emphasizes “For Tri-nity College and our School this was a huge opportunityto engage not only academics and scientists but also to in-clude valuable input from the commercial world, wheretheoretical ideas can be tested and eventually rolled out,not only to be commercially viable but also to benefit thou-sands if not millions of patients worldwide. The feedbackfrom the participants and speakers was fantastic and themix of lectures and practical training was seen as being ofreal benefit.”For further information please refer to www.galenusprivatstiftung.at/workshop
2nd Galenus Workshop in Dublin (Sept. 18 – 20, 2013)
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 3

APV NEWS 5-2013
APV NEWS – Aus der Geschäftsstelle
9th World Meeting on Pharmaceutics,Biopharmaceutics and Pharmaceutical Technology and ResearchPharm®
31 March to 03 April 2014, Lisbon, Portugal
Skin Forum 14th Annual Meeting04 to 05 September 2014, Prague, Czech Republic
TechnoPharm® 201430 September to 02 October 2014, Nuremberg, Germany
Upcoming international events – Save the date!
Die APV vergibt im Jahr 2014 erneut zwei Preise für den wissenschaftlichen Nachwuchs. Der Doktorandenpreis, dessen Preisgeldin Höhe von 5.000 Euro in diesem Jahr das Unternehmen Evonik Industries AG zur Verfügung stellt, wird für eine exzellenteDissertation aus dem Jahr 2012 oder 2013 vergeben. Der Promovierte kann sich selbst um den Preis bewerben. Das Gutach-terkomitee wird von Prof. Dr. Reinhard Neubert geleitet. Der Forschungspreis, dessen Preisgeld in Höhe von 5.000 Euro vonBayer Healthcare zur Verfügung gestellt wird, ehrt einen erfolgreichen Wissenschaftler unter 46 Jahren in der Pharmazie, derin den letzten Jahren herausragende Fortschritte in seinem Fachgebiet erzielt hat. Prof. Dr. H.P. Merkle leitet das Kuratorium.Der Einsendeschluss ist jeweils der 15.12.2013. Die Preisträger werden auf dem 9th PBP World Meeting in Lissabon geehrt.Weitere Details entnehmen Sie bitte der Internetseitewww.apv-mainz.de/english/apv/research-support/apv-awards/
Doktoranden- und Forschungspreis der APV
Lokale Gruppen
Neue Treffpunkte und -zeiten der lokalen Gruppen:
Montag, 20. Januar 2014 Lokale APV-Gruppe Berlin um 19:00 Uhr im Pestana Hotel Berlin Tiergarten, Stülerstrasse 6, 10787 Berlin.Bitte melden Sie sich bis zum 10.01.2014 bei Dr. Andreas Sachse an.
Weitere Informationen zu unseren Lokalen Gruppen finden Sie auf http://www.apv-mainz.de/apv/lokale-gruppen/
APV NEWS – Aus dem Vereinsleben
Liebe APV-Mitglieder, nach der erfolgreichen Gründung mehrerer lokaler Gruppen würden wir das Konzept der lokalenAPV-Gruppen gerne auch in weiteren Regionen etablieren. Bitte sprechen Sie uns an, wenn SieInteresse an einer Teilnahme an einer lokalen Gruppe in Ihrer Region haben oder als Ansprechpartner, unterstützt durch die APV-Geschäftsstelle, für eine neue lokale Gruppe zur Verfügung stehen würden. Wir freuen uns auf Ihre Rückmeldung! Ansprechpartner: Dr. Martin Bornhöft, Email: [email protected], Tel: + 49 6131 9769-35
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 2
APV NEWS – Aus dem Vereinsleben
APV NEWS 6-2013
Renate Lieckfeldt ist am 08. September 2013 für immer von uns gegangen. Fassungslos und bestürzthaben wir die traurige Nachricht entgegennehmen müssen.
Im Jahr 1988 schloss Renate Lieckfeldt ihr Studium der Pharmazie an der Universität Heidelberg ab, imJahr darauf erfolgte ihre Approbation zur Apothekerin. 1993 promovierte sie im Fach PharmazeutischeTechnologie am Institut für Pharmazeutische Technologie und Biopharmazie der Universität Heidelberg.In den folgenden Jahren war sie Regulatory Affairs Manager for Drugs & Medical Devices bei der Procter& Gamble GmbH in Schwalbach und anschließend Technical External Relations Manager Benelux beider Procter & Gamble Nederland B. V. in Rotterdam. Von 2001 bis 2011 lehrte sie als Professorin fürTechnisches Management und Projektmanagement im Fachbereich Physikalische Technik an der Fachhochschule Gelsenkirchen.Im Januar 2011 wurde Renate Lieckfeldt vom Erweiterten Senat der HTWK Leipzig zur neuen Rektorin gewählt. Renate Lieck-feldt war seit 1994 Mitglied der APV und seit 2002 der Fachgruppe Prozessoptimierung. Neben ihrer Arbeit in der Fachgruppehat sie zahlreiche erfolgreiche Kurse und Seminare konzipiert und unter ihrer Leitung durchgeführt.
Wir kannten Renate Lieckfeldt als engagierte und zielorientierte Kollegin, die mit ihrer Anwesenheit in der Fachgruppe aufvieles eine völlig neue Sicht einbrachte und damit eine ungemeine Bereicherung für unsere Fachgruppe war. Sie war ein Menschvoller Kraft und enormer Willensstärke. Renate Lieckfeldt war aber auch ein humorvoller, hilfsbereiter und liebenswerter Mensch,bei dem gegenseitiger Respekt und Kollegialität immer im Vordergrund standen. Alle Fachgruppenmitglieder, die Geschäftsstelleund der Vorstand der APV werden Renate Lieckfeldt ein ehrendes Gedenken bewahren.
Jürgen Werani
Nachruf Frau Prof. Dr. rer. nat. Renate Lieckfeldt
The School of Pharmacy and Pharmaceutical Sciences at Trinity College Dublin recently hosted the 2nd Galenus Workshop on PulmonaryDrug Delivery at the Panoz Institute from September 18 - 20, 2013, supported by the Galenus Foundation (Vienna / Austria). The work-shop was attended by an international group of PhD students undertaking research in Pharmaceutical Sciences and junior faculty mem-bers from 18 different Universities as well as representatives of the pharmaceutical industry from a variety of companies and of theGalenus Foundation. Both practical and theoretical in scope, the workshop covered many aspects of Pulmonary Formulation and DrugDisposition with lectures and practical sessions delivered by highly experienced faculty members and international speakers.
Participants heard about a series of studies focussing on new targets for the treatment of lung diseases, a particularly important topicas a result of new pipeline drug development and novel clinical entities which may have a narrow therapeutic index or be too toxic forsystemic delivery. Improving the efficiency of inhaled aerosol delivery by targeting drug to the appropriate lung regions/sites, may improvethe therapeutic response and minimise potential adverse effects.Other topics covered included imaging techniques in pulmonary drugdelivery; in vitro models of the air-blood barrier; pulmonary delivery of biopharmaceuticals; nanoparticles for aerosol delivery, and ad-vances in aerosoliser device technology. Participants also received practical training in spray drying of respirable particles; in vitro cha-racterisation of aerosol particles, and uptake and transport studies in lung cell cultures. Speaking about the success of this event, DrCarsten Ehrhardt, Associate Professor in Pharmaceutics and Pharmaceutical Technology at The School of Pharmacy and PharmaceuticalSciences, organizer of this workshop, emphasizes “For Tri-nity College and our School this was a huge opportunityto engage not only academics and scientists but also to in-clude valuable input from the commercial world, wheretheoretical ideas can be tested and eventually rolled out,not only to be commercially viable but also to benefit thou-sands if not millions of patients worldwide. The feedbackfrom the participants and speakers was fantastic and themix of lectures and practical training was seen as being ofreal benefit.”For further information please refer to www.galenusprivatstiftung.at/workshop
2nd Galenus Workshop in Dublin (Sept. 18 – 20, 2013)
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 3

APV NEWS 6-2013
APVnews – Infos aus der Hochschule
Vol 85 Issue 2
European Journal of Pharmaceutics and Biopharmaceutics85 (2013) 162–169Recent advances and further challengesin lyophilizationJulia Christina Kasper, Gerhard Winter, WolfgangFriess While entering a new century, lyophilization in the phar-maceutical field has been subjected to ongoing develop-ment and steady expansion. This review aims to highlightrecent advances but also to discuss further challenges inlyophilization. At first, the expanded range of pharmaceu-tical applications based on lyophilization is summarized.Moreover, novel formulation aspects and novel containersystems are discussed, and the importance of the freezingstep is outlined. Furthermore, the dogma of ‘‘never lyophi-lize above the glass transition temperature’’ is argued, andrecent insights into novel stabilization concepts are provi-ded. Process analytical technology (PAT) and quality bydesign (QbD) are now leading issues, and the design of thelyophilization equipment also might have to be reconside-red in the future.
European Journal of Pharmaceutics and Biopharmaceutics85 (2013) 189–196Characterization of dynamics in com-plex lyophilized formulations: I. Compa-rison of relaxation times measured byisothermal calorimetry with data esti-mated from the width of the glass tran-sition temperature regionNorman Chieng, Masayasu Mizuno, Michael PikalThe purposes of this study are to characterize the relaxati-on dynamics in complex freeze dried formulations and toinvestigate the quantitative relationship between thestructural relaxation time as measured by thermal activitymonitor (TAM) and that estimated from the width of theglass transition temperature (ΔTg). The latter method hasadvantages over TAM because it is simple and quick. Aspart of this objective, we evaluate the accuracy in estima-ting relaxation time data at higher temperatures (50 °Cand 60 °C) from TAM data at lower temperature (40 °C)and glass transition region width (ΔTg) data obtained bydifferential scanning calorimetry. Formulations studiedhere were hydroxyethyl starch (HES)–disaccharide, HES–polyol, and HES–disaccharide–polyol at various ratios. Wealso re-examine, using TAM derived relaxation times, thecorrelation between protein stability (human growth hor-mone, hGH) and relaxation times explored in a previousreport, which employed relaxation time data obtained
from ΔTg. Results show that most of the freeze dried for-mulations exist in single amorphous phase, and structuralrelaxation times were successfully measured for thesesystems. We find a reasonably good correlation betweenTAM measured relaxation times and corresponding dataobtained from estimates based on ΔTg, but theagreement is only qualitative. The comparison plot showedthat TAM data are directly proportional to the 1/3 power ofΔTg data, after correcting for an offset. Nevertheless, thecorrelation between hGH stability and relaxation timeremained qualitatively the same as found with using ΔTgderived relaxation data, and it was found that the modestextrapolation of TAM data to higher temperatures usingΔTg method and TAM data at 40 °C resulted in quantitati-ve agreement with TAM measurements made at 50 °C and60 °C, provided the TAM experiment temperature, is wellbelow the Tg of the sample.
European Journal of Pharmaceutics and Biopharmaceutics85 (2013) 314–326Investigating factors leading to foggingof glass vials in lyophilized drug pro-ductsAhmad M. Abdul-Fattah, Richard Oeschger, HolgerRoehl, Isabelle Bauer Dauphin, Martin Worgull, Georg Kallmeyer, Hanns-Christian Mahler Vial ‘‘Fogging’’ is a phenomenon observed after lyophiliza-tion due to drug product creeping upwards along theinner vial surface. After the freeze-drying process, a hazeof dried powder is visible inside the drug product vial,making it barely acceptable for commercial distributionfrom a cosmetic point of view. Development studies wereperformed to identify the root cause for fogging duringmanufacturing of a lyophilized monoclonal antibody drugproduct. The results of the studies indicate that drug pro-duct creeping occurs during the filling process, leading tovial fogging after lyophilization. Glass quality/inner surfa-ce, glass conversion/vial processing (vial ‘‘history’’) and for-mulation excipients, e.g., surfactants (three different sur-factants were tested), all affect glass fogging to a certaindegree. Results showed that the main factor to controlfogging is primarily the inner vial surface hydrophili-city/hydrophobicity. While Duran vials were not capable ofreliably improving the level of fogging, hydrophobic con-tainers provided reliable means to improve the cosmeticappearance due to reduction in fogging. Varying vial depy-rogenation treatment conditions did not lead to satisfyingresults in removal of the fogging effect. Processing condi-tions of the vial after filling with drug product had a strongimpact on reducing but not eliminating fogging.
What’s hot in European Journal ofPharmaceutics and Biopharmaceutics?
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 4

APV NEWS 6-2013
APVnews – Infos aus der Hochschule
Vol 85 Issue 2
European Journal of Pharmaceutics and Biopharmaceutics85 (2013) 162–169Recent advances and further challengesin lyophilizationJulia Christina Kasper, Gerhard Winter, WolfgangFriess While entering a new century, lyophilization in the phar-maceutical field has been subjected to ongoing develop-ment and steady expansion. This review aims to highlightrecent advances but also to discuss further challenges inlyophilization. At first, the expanded range of pharmaceu-tical applications based on lyophilization is summarized.Moreover, novel formulation aspects and novel containersystems are discussed, and the importance of the freezingstep is outlined. Furthermore, the dogma of ‘‘never lyophi-lize above the glass transition temperature’’ is argued, andrecent insights into novel stabilization concepts are provi-ded. Process analytical technology (PAT) and quality bydesign (QbD) are now leading issues, and the design of thelyophilization equipment also might have to be reconside-red in the future.
European Journal of Pharmaceutics and Biopharmaceutics85 (2013) 189–196Characterization of dynamics in com-plex lyophilized formulations: I. Compa-rison of relaxation times measured byisothermal calorimetry with data esti-mated from the width of the glass tran-sition temperature regionNorman Chieng, Masayasu Mizuno, Michael PikalThe purposes of this study are to characterize the relaxati-on dynamics in complex freeze dried formulations and toinvestigate the quantitative relationship between thestructural relaxation time as measured by thermal activitymonitor (TAM) and that estimated from the width of theglass transition temperature (ΔTg). The latter method hasadvantages over TAM because it is simple and quick. Aspart of this objective, we evaluate the accuracy in estima-ting relaxation time data at higher temperatures (50 °Cand 60 °C) from TAM data at lower temperature (40 °C)and glass transition region width (ΔTg) data obtained bydifferential scanning calorimetry. Formulations studiedhere were hydroxyethyl starch (HES)–disaccharide, HES–polyol, and HES–disaccharide–polyol at various ratios. Wealso re-examine, using TAM derived relaxation times, thecorrelation between protein stability (human growth hor-mone, hGH) and relaxation times explored in a previousreport, which employed relaxation time data obtained
from ΔTg. Results show that most of the freeze dried for-mulations exist in single amorphous phase, and structuralrelaxation times were successfully measured for thesesystems. We find a reasonably good correlation betweenTAM measured relaxation times and corresponding dataobtained from estimates based on ΔTg, but theagreement is only qualitative. The comparison plot showedthat TAM data are directly proportional to the 1/3 power ofΔTg data, after correcting for an offset. Nevertheless, thecorrelation between hGH stability and relaxation timeremained qualitatively the same as found with using ΔTgderived relaxation data, and it was found that the modestextrapolation of TAM data to higher temperatures usingΔTg method and TAM data at 40 °C resulted in quantitati-ve agreement with TAM measurements made at 50 °C and60 °C, provided the TAM experiment temperature, is wellbelow the Tg of the sample.
European Journal of Pharmaceutics and Biopharmaceutics85 (2013) 314–326Investigating factors leading to foggingof glass vials in lyophilized drug pro-ductsAhmad M. Abdul-Fattah, Richard Oeschger, HolgerRoehl, Isabelle Bauer Dauphin, Martin Worgull, Georg Kallmeyer, Hanns-Christian Mahler Vial ‘‘Fogging’’ is a phenomenon observed after lyophiliza-tion due to drug product creeping upwards along theinner vial surface. After the freeze-drying process, a hazeof dried powder is visible inside the drug product vial,making it barely acceptable for commercial distributionfrom a cosmetic point of view. Development studies wereperformed to identify the root cause for fogging duringmanufacturing of a lyophilized monoclonal antibody drugproduct. The results of the studies indicate that drug pro-duct creeping occurs during the filling process, leading tovial fogging after lyophilization. Glass quality/inner surfa-ce, glass conversion/vial processing (vial ‘‘history’’) and for-mulation excipients, e.g., surfactants (three different sur-factants were tested), all affect glass fogging to a certaindegree. Results showed that the main factor to controlfogging is primarily the inner vial surface hydrophili-city/hydrophobicity. While Duran vials were not capable ofreliably improving the level of fogging, hydrophobic con-tainers provided reliable means to improve the cosmeticappearance due to reduction in fogging. Varying vial depy-rogenation treatment conditions did not lead to satisfyingresults in removal of the fogging effect. Processing condi-tions of the vial after filling with drug product had a strongimpact on reducing but not eliminating fogging.
What’s hot in European Journal ofPharmaceutics and Biopharmaceutics?
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 4
APV NEWS – Mitteilungen
APV NEWS 2-2012
Impressum:
Redaktion
Prof. Dr. Jörg Breitkreutz (Präsident)Dr. Martin Bornhöft (Leiter Geschäftsstelle)
Vorstand der APV
Dr. Rainer Alex · Dr. Hermann Allgaier ·Prof. Dr. Jörg Breitkreutz · Dr. HubertusFolttmann · Prof. Dr. Achim Göpferich · Prof. Dr. Heribert Häusler · Dr. Hermann P.Osterwald · Dr. Andreas Rummelt
Arbeitsgemeinschaft für Pharmazeu tischeVerfahrenstechnik e. V. (APV) Kurfürstenstraße 5955118 Mainz (Germany)Telefon +49 6131 9769-0Telefax +49 6131 9769-69e-mail: [email protected]://www.apv-mainz.de
Verlag
ECV · Editio Cantor Verlag für Medizinund Naturwissenschaften GmbHBaendelstockweg 2088326 Aulendorf, Germany
Telefon +49 7525 940-0Telefax +49 7525 940-180
e-mail: [email protected]://www.ecv.de
Alle Rechte bei APV e. V.All rights reservedPrinted in GermanyJede Form des Nachdrucks verboten
Druck
Holzmann Druck GmbH & Co. KGGewerbestr. 286825 Bad Wörishofen, Germany
Satz
Arbeitsgemeinschaft für Pharmazeu tischeVerfahrenstechnik e. V. (APV) Kurfürstenstraße 5955118 Mainz (Germany)
6. Galenus Technologie-Preis vergeben
Zum 6. Mal hat die Galenus Privatstiftung den mit 5.000 €dotierten, renommierten Technologie-Preis vergeben. DieAuszeichnung ging an die österreichische Forscherin Ass.-Prof. Dr. Eva Roblegg vom Institut für PharmazeutischeTechnologie der Karl-Franzens-Universität Graz.Das preisgekrönte innovative Forschungsprojekt von FrauRoblegg „Der Mund als alternative Applikationsroute fürNano-Drug Delivery Systeme“ berücksichtigt die wach-sende Bedeutung von „Biologicals“ und deren Stabilitäts-probleme. Moderne Techniken zeigen, dass Nanokapselnje nach Größe und Ladung die Mukosa durchdringen kön-nen und sich als Resorptionsfähren anbieten.
MEETING REPORT on the “InternationalSymposium on Phospholipids inPharmaceutical Research”, Heidelberg,Germany, 16-17 September 2013
Peter van Hoogevest, Phospholipid Research Center Heidelberg,Im Neuenheimer Feld 582, D-69120 Heidelberg, Germany. Cor-respondence: Dr. Peter van Hoogevest, Phospholipid ResearchCenter Heidelberg, Im Neuenheimer Feld 582, D-69120 Heidel-berg, Germany; E-Mail: [email protected]
This two day event was organized at the University of Heidelbergfor the third time by the Phospholipid Research Center Heidelberg.The symposium was visited by 170 scientists from academia, phar-maceutical industry, regulatory authorities etc. from all over theworld. Sixty posters and 16 lectures by highly reputed phospholi-pid experts were presented. The symposium offered an excellentplatform for providing formation on the basic properties of phos-pholipids and latest pharmaceutical use of phospholipids and fornetworking between the various scientific disciplines. This yearevent started with a session on “Physico-Chemical Aspects ofPhospholipids”, chaired by Dr. Frank Martin (San Francisco, USA).This session reviewed the chemical composition, mixing behaviorand phase transitions of phospholipids, phospholipid dynamics(rotational-, lateral diffusion and intermembrane transfer proper-ties), their physicochemical properties as wetting agent and emul-sifier, and their stability characteristics, which are relevant forpharmaceutical development and production. The following ses-sion on “Rational Selection of Phospholipids as Excipients” chairedby Prof. Dr. Gert Fricker (Heidelberg, DE) provided a systemic re-view of the specific use of phospholipids in relation to the admi-nistration route (oral, parenteral, pulmonary and topical/skin). Thesecond day started with a session on “Latest advances in applica-tion of phospholipids”, chaired by Prof. Dr. Jörg Huwyler (Basle,CH). The use of liposomes in photodynamic therapy of solid can-cers, fusogenic peptides for phospholipids-based drug delivery,thermo-sensitive liposomes and lipid based carriers for endothelialcell specific delivery of siRNA targeting by means of liposomes,were reviewed. The symposium was concluded by the session on“Future Use of Phospholipids” chaired by Prof. Dr. Christel Mül-ler-Goymann (Braunschweig, DE). In a “Young Session” six 10 minpresentations on preselected posters were given. The last seminarof this session claimed, that considering the research and deve-lopment experience obtained with liposomes and the presence ofmany liposome based nanotechnology products on the market,liposomes should be the first choice in selecting nano-carriers fordrug targeting.
The following three posters were awarded:Dr. Martin Hossann, Ludwig-Maximilians-University Munich, Uni-versity Hospital Munich, on: „Thermosensitive liposomes encap-sulating gemcitabine - an in-vivo feasibility study”. Silvia Pantze,Ruprecht-Karls University Heidelberg, Institute for Pharmacy andMolecular Biotechnology (IPMB) on: „Matrix Liposomes – A solidliposomal formulation for oral administration”. Dr. Ronny Rüger,Friedrich-Schiller-University Jena, Institute for Pharmacy, on: “Invivo whole-body imaging using a liposomal quenched near-infra-red dye”.
The next International Symposium on Phospholipids in Pharma-ceutical Research will be held in September 2015 in Heidelberg,Germany.
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 5

Hersteller/Typ Listenpreis mtl. Rate
Audi A1 Sportback „admired“ 1.2 TFSI 63kW/86PS inkl. Xenon, Media Paket, BOSE Sound, PDC hinten, Klimaautomatik, 17" Alu-Felgen etc. 18.886,00 € 199,00 €
Audi A5 Coupé 1.8 TFSI 125kW/170PS multitronic inkl. Navi, S line selection Einparkhilfe plus, Komfortpaket, Komfortklimaautomatik etc. 36.538,00 € 339,00 €
Audi Q5 2.0 TDI quattro S tronic 130kW/177PS inkl. Navi, Xenon, Leder-Milano, Einparkhilfe plus, Komfortklimaautomatik, 18" Alu-Felgen etc. 43.647,00 € 499,00 €
BMW 316i Limousine 100kW/136PS inkl. Klimaautomatik, Radio Professional CD MP3, Nebelscheinwerfer, Armauflage vorn etc. 24.412,00 € 289,00 €
BMW 520d Touring 135kW/184PS inkl. Klimaautomatik, LM-Räder, Sitzheizung, PDC, Innovationspaket, Connected Drive etc. 47.092,00 € 515,00 €
BMW X5 xDrive30d 190kW/258PS inkl. 8-Gang-Automatic, Navigations- paket Connected Drive, 2-Zonen-Klimaautomatik, 18" LM-Felgen etc. 52.689,00 € 749,00 €
Jaguar XJ Luxury 3.0 L V6 Diesel S 202kW/275PS inkl. 8-Gang-Automatik, Xenon-Scheinwerfer mit adaptivem Kurvenlicht, PDC v+h etc. 69.576,00 € 859,00 €
LandRover Range Rover Sport 3.0 TDV6 HSE 190kW/258PS inkl. 8-Stufen- Automatik, Metallic, Klimaautomatik, Xenon-Scheinwerfer mit LED etc. 60.924,00 € 849,00 €
Mazda 3 CenterLine 5-Türer 88kW/120PS inkl. Metallic, Navi, Klimaautomatik, Radio/CD, Cruisematic, Freisrecheinrichtung/Bluetooth etc. 18.000,00 € 189,00 €
Mazda CX-5 CenterLine SKYACTIV-G 121kW/165PS inkl. Navi, PDC, Sitzheizung vorne, Bi-Xenon, Klimaautomatik, Cruisematic etc. 24.890,00 € 279,00 €
MINI Cooper Paceman 90kW/122PS inkl. Leichtmetallräder, Klimaanlage, Radio/CD, Heckspoiler, DSC, Spiegelkappen in Wagenfarbe etc. 20.000,00 € 249,00 €
Volvo V60 D3 Kinetic 100kW/136PS inkl. Einparkhilfe hinten, Sitzheizung, Frontscheibenheizung, Klimaautomatik, Radio/CD, LM-Felgen etc. 29.294,00 € 299,00 €
Volvo XC60 D4 FWD Momentum 133kW/181PS inkl. Metallic, LM-Felgen, Navigationssystem, Sitzheizung vorn, Einparkhilfe vorn u. hinten etc. 35.790,00 € 419,00 €
VW Golf Comfortline BMT 1,2l TSI 63kW/86PS inkl. Climatronic, LM-Felgen, Radio/CD Composition Media, Aussenspiegel elektr./beheizbar etc. 16.617,00 € 179,00 €
VW Touran Life 1,2l TSI 77kW/105PS inkl. LM-Räder „Spokane“, Park Assist mit ParkPilot, Radio/CD 310, Climatronic, Sitzheizung vorne etc. 22.595,00 € 229,00 €
VW Tiguan Life BMT 1,4l TSI 90kW/122PS inkl. Metallic, Vordersitze beheizbar, Climatronic, LM-Räder, Park Assist mit ParkPilot etc. 24.185,00 € 239,00 €
Kfz-Leasing: Vorteile für APV-MitgliederDie APV hat für ihre Mitglieder einen Rahmenvertrag mit einem bekannten Leasing-Unternehmen geschlossen. Als Koopera-tionspartner der APV bietet das Unternehmen Leasing von Neu- und Gebrauchtfahrzeugen zu Sonderkonditionen. Alle Markenund Modelle sind lieferbar. Die nachfolgende Tabelle gibt nur wenige aktuelle Beispiele möglicher Modelle und Marken wieder.NEU: Vorführwagen (VFW) aus dem Leasing-Pool zu attraktiven Konditionen erhältlich.
Alle Preise in Euro zuzüglich gesetzlicher Mehrwertsteuer. Beschaffung durch die Leasing-Gesellschaft. 36 Monate Laufzeit,15.000 km pro Jahr, Angebote freibleibend. Der Nachlass auf den Listenpreis ist in die ermäßigte Rate einkalkuliert.
Anfragen bitte an [email protected], das Leasing-Unternehmen wird sich dann mit Ihnen in Verbindung setzen.
Leasing und Finanzierung zu günstigen Konditionen sind auch für Investitionsgüter wie Walzenpressen,Verpackungsmaschinen, Laboreinrichtungen etc. über die APV möglich. Sprechen Sie uns an.
JETZT NEU: Leasing auch für andere Investitionsgüter
APV NEWS – Leasing-Highlights zu Sonderkonditionen
APV NEWS 06_2013_APVnews TP 13.11.2013 15:21 Seite 6

Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-11\Anzeigensatz-keine-Druck-PDFs\apv-pi-2013-11-216x303.indd Zuletzt gesichert: 12.11.13 (11:03:29 Uhr)
APV Seminare 2014
Über 50 Jahre Erfahrung, Kompetenz und Qualifikation
International Association for Pharmaceutical Technology
Kurs: 6531
27. - 28. Februar 2014, Berlin, Germany
APV basics:Praktikum Tablettieren
Kurs: 6535
18. - 19. März 2014, Wiesbaden, Germany
Der PharmaExperte® mit APV-Diplom:Qualitätssicherung: Basistraining Reinigungsvalidierung
Kurs: 6541
12. - 13. Mai 2014, Wiesbaden, Germany
Der PharmaExperte® mit APV-Diplom:GMP-konforme Chargendokumentation:Herstellungsvorschrift und Batch Record Review
Kurs: 6548
12. - 14. Mai 2014, Darmstadt, Germany
Seminar:Die Stabilitätsprüfung 2014
Kurs: 6550
20. - 21. Mai 2014, Ludwigshafen, Germany
APV basics:Praktikum Pelletieren
Kurs: 6551
20. - 21. Mai 2014, Mannheim, Germany
Workshop:Good Engineering Practice – Anlagenbeschaffung undQualifizierung in der Praxis
Weitere Informationen unter www.apv-mainz.de
MAKING SCIENCE WORK
Arbeitsgemeinschaft für Pharmazeutische Verfahrenstechnik e.V.Gemeinnütziger wissenschaftlicher VereinInternational Association for Pharmaceutical Technology
www.apv-mainz.de
APV Anzeige_November_2013_pi_APV Anzeige pi 11.11.13 16:39 Seite 1

Abgelegt auf: F:\GK\ECV\Satz\Pharmind\PI_2013-06\Anzeigensatz-keine-Druck-PDFs\caesar-pi-2013-06-216x303.indd Zuletzt gesichert: 14.06.13 (12:07:01 Uhr)
Caesar & Loretz GmbH | Herderstr. 31 | D-40721 Hilden | Tel: +49 (0)21 03/49 94 0 | Mail: [email protected] | www.caelo.de
Bleiben Sie fl exibel, schlank und schnell. Wir fertigen Ihre Produkte – von halbfesten, festen und fl üssigen Formen bis hin zu Teemischungen. Je nach Darreichungsform in Chargengrößen von 10 bis 1000 kg. In höchster pharma-zeutischer Qualität und nach GMP-zertifi zierten Prozessen. Alle Vorteile fi nden Sie unter www.caelo.de/_industrie.
Oder besuchen Sie uns auf den Messen: Expopharm und CPhl.
Von der Rohstoffl ieferung bis zum fertigen Produkt mit Caelo.
So gut wie selbst gemacht. Lohnfertigung für die Pharma-, Kosmetik- und Lebensmittelindustrie.





























