Embed Size (px)
Citation preview

W. DIEMAIR und R. BAIEn: Verfahren zur Bestimmung des Phosphors. 7
Einige Beleganalysen (Tab. 1), fiir deren Durchft ihrung Her rn cand. phil. E. H6~LI?CG gedankt sei, sollen die Genauigkeit der beschriebenen ~¢[ethodik unter Beweis sgellen.
Die Arbeit wurde dutch ein VA~'T HoFF-St ipendium erm5glicht, wofiir an dieser Stelle der Dank zum Ausdruck gebracht werden soll.
Literatur.
1 KICYNITSK¥, J. A., J. E. Jo~-so~ u. H. W. CA~AaT: J. Amer. Chem. Soc. 70, 486 (1948). - - ~ LIEB, H., u. W. SCHS~IGE~: Mikrochem.35, 400 (1950); vgI. diese Z. 132~ 188 (1951). - - 3 U~T~ZAVCRER, J. : Chemie-Ing. Techn. 22, 39 (1950): vgl. diese Z. 132, 291 (1951). - - ~ 1VIEy~, F. 1~., u. G. RONOE: Angew. Chem. 52, 637 (1939).
Dr. W. SCI~51~IOER, Graz, Universit~tsplatz 2.
Aus dem Universit~ts-Institut fiir Lebensmittelchemie Frankfurt a.M.
t~ber ein Verfahren zur 3Iikrobestimmung des Phosphors *.
Von W. DIEMAIR und R. BAIER.
(Eingegangen am 19. Februar 1951.)
Fiir Stoffwechse]versuche in raschwiichsigen N/~hrhefen. (Torula utilis) war eine in Reihenuntersuchungen rasch durchfi ihrbare Methode zur quant i ta t iven Bes t immung yon P notwendig, die auch auf geringe ~ e n g e n anspraeh 1. Die bekannte ~ e t h o d e von J. TILLMANS, I-1. I~IFFART und A. Ki3I~N 2, die recht gute Ergebnisse lieferte, war fiir die vorliegenden Zwecke, infolge der geforderten dauernden Kiihlung, zu umst~tndlich. Das Verfahren von N. v. LOt~ENZ ~ und dasjenige yon R. WoY-FINKENER 4 war besonders bei kleinen P-Mengen zu ungenau. Es wurden hier Schwan- kungen bis zu 4 und 5 ~o beobachte~. Eine bessere Genauigkeit war zwar bei der F/~llung der im Kiihlschrank abgekfihlten LSsung m5glich, die Werte waren aber erst nach 24 S~d zu ermitt~ln. Die colorimetrische ~[ethode zeigte Fehler bis zu etwa o/ 3/o, was durch Ablesefehler im Stufen- fotometer und durch das verz5gerte Einhal ten der Erh i tzungsdauer yon einer Stunde bei kons tanter Tempera tur mit bedingt war. DieseMethoden schieden dasher fiir die vorliegenden Zwecke aus, weshalb nach einem ti tr imetrischen Verfahren gesucht wurde, das bei einer groBen Genauig- keit auch zei tsparend sein sollte.
Eine direkte Ti t ra t ion der Phosphors~ure erwies sich nicht als giinstig, da keine reproduzierbaren Werte erhalten werden konnten . Es wurde
* Herrn Prof. Dr. W. GEIL~ANN zu,'n 60. Geburtstag gewidmet.

8 W. DZE~A~g und t~. B n ~ :
nun versueht, die Molybd~ns~ure nach Entfernung des Ammoniums mit Lauge zu titrieren 5, wobei sich aber herausstellte, dal~ das Ammonium in dem Ammoniumphosphormolybdat-Komplex (PA) sehr lest gebunden ist, so dal~, selbst nach einstiindigem Kochen, das gesamte Ammonium nicht in Freiheit gesetzt werden konnte. Diese Beobachtung veranlaI~te dazu, den Phosphor fiber den Ammoniumgehalt yon (PA) nach KJ~L- DA~L in der PAR~As-WAGNER-Apparatur zu ermitteln. Bekanntlich wird das Phosphor-Stickstoff-Verh~ltnis des heil~ gef~llten Komplexes (NI-Ia)sPO a • 12Mo03 mit 1 :3 angegeben. In Wirklichkeit ist es aber recht unterschiedlich, so dal~ empirisehe Umrechnungsfaktoren erforder- lich sind. Um an deren Stelle den theoretisch errechneten Wert ver- wenden zu k6nnen, wodurch die Sicherheit der P-Bestimmung gewinnt, wurden die gfinstigsten F~llungsbedingungen, vor allem in Hinsicht der Dauer der Erhitzung und Art und Menge der verdfinnten S~uren £est- gestellt und versucht, aus diesen Beobachtungen eine neue Mikromethode zur Phosphor-Bestimmung zu entwickeln.
Eigene Versuche. In den in bekann~er Weise heil~ gef~llten und gravimetrisch bestimmten
(PA)-Niederschl~gen wurden der Phosphor- und der StickstoffgehMt er- mittelt, wobei sich die in Tab. 1 festgehaltenen Werte ergaben.
Tabelle 1. N-Gehalt des gravimetrisch bestimmten Ammoniumphosphormolybdats.
Sollwert gravimetrisch Fehler Titrim. bestimmter Fehler rag P bestimmt To N-Gehalt m a l 0,738 To
mg 1 ~ = m g P
1,4090 1,4090 1,4090 1,4090
1,4466 1,4554 1,4603 1,5013
~- 2,67 ~- 3,29
3,72 6,55
1,2200 1,2830 1,0774 1,5718
- - 13,44 - - 8,94 - - 23,54 -~ 11,55
Die (PA)-Xiederschl~ge sind verschieden zusan~mengesetzt und ent- sprechen nur bedingt der Formel (~I-Ia)aP04.12 ~ o 0 a. Au•erdem gehen, bei zwar ungef~hrer ~bereinstimmung der gravimetrischen Werte~ die Werte ffir den Ammoniumgehalt auseinander. Dies scheint yon den F~llungsbedingungen abzuh~ngen und dutch die Art und Menge der verdiinnten S~uren nnd durch die Erhitzungsdauer bedingt zu sein. Aus diesem Grund wurde die Art und Dauer der Erhitzung genauer ver- folgt und dabei.so verfahren, dal~ zur Bildung des Xiederschlags 5 rain lang mit einem elektrischen l~tihrwerk geriihrt und damit das 1 rain lange Umschiitteln und das viertelstiindige Absitzenlassen des Niederschlages ausgeschaltet.
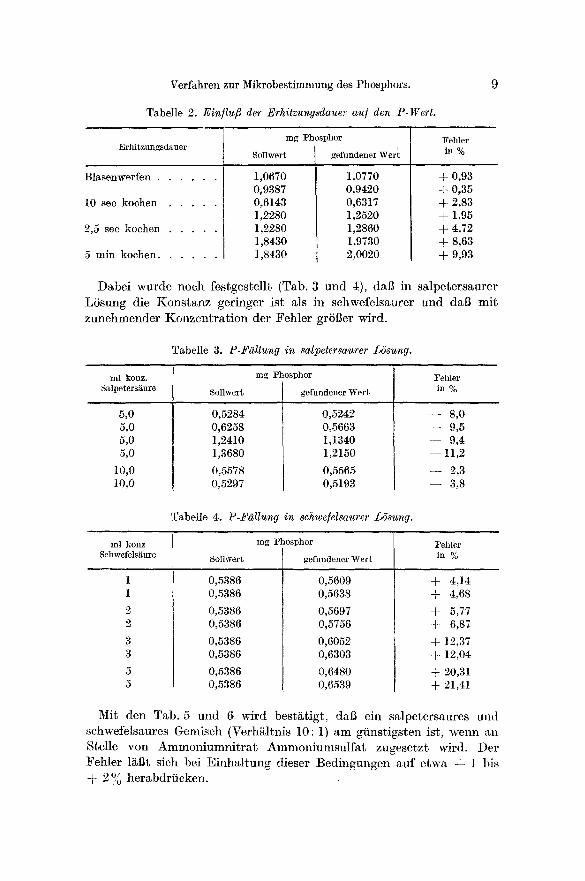
Wie aus der Tab. 2 hervorgeht, nimmt mit zunehmender Erhitzungs- dauer der Ammoniumgehalt des Komplexes zu, am geringsten (~- 0,93 °/o) bleibt der Fehler beim Erhitzen bis zum Blasenwerfen.
Verf~hren zur Mikrobestimmung des Phosphors.
Tabelle 2. Ein/lufi der Erhitzungsdauer au] den P-Wert.
mg Phosphor l%hler Erhi tzungsdauer
Sollwert gefundener Wer~ in %
Blasenwerfen . . . . . .
10 sec kochen . . . . .
2,5 see kochen . . . . .
5 min kochen . . . . . .
1,0670 0,9387 0,6143 1,2280 1,2280 1,8430 1,8430
1,0770 0,9420 0,6317 1,2520 1,2860 1,9730 2,0020
+ 0,93 + 0,35 + 2,83 + 1,95 + 4,72 + 8,63 + 9,93
Dabe i wurde noch festges~ellt (Tab. 3 und 4), d a b in sa lpe te r sau re r LSsung die K o n s t a n z ger inger i s t als in schwefelsaurer und dal~ m i t zunehmender K o n z e n t r a t i o n der Feh l e r grSBer wird.
Tabelle 3. P-F~llung in salpetersaurer L6sung.
ml konz. mg Phosphor Fehler
Salpeters~ture Sollwert gefundener Wer t in %
5,0 5,0 5,0 5,0
10,0 10,0
0,5284 0,6258 1,2410 1,3680
0,5578 0,5297
0,5242 0,5663 1,1340 1,2150
0,5565 0,5193
- - 8,0 - - 9,5 - - 9 , 4
- - 1 1 , 2
- - 2,3 - - 3,8
Tabelle 4. P-F~llunff in schwe]elsaurer Lb'sung.
ml konz mg t 'hosphor Fehler
Schwefels~ure Sollwert gefundener Wert in %
0,5386 0,5386
0,5386 0,5386
0,5386 0,5386
0,5386 0,5386
0,5609 0,5638
0,5697 0,5756
0,6052 0,6303
0,6480 0,6539
+ 4,14 + 4,68
+ 5,77 + 6,87
+ 12,37 + 12,04
+ 20,31 + 21,41
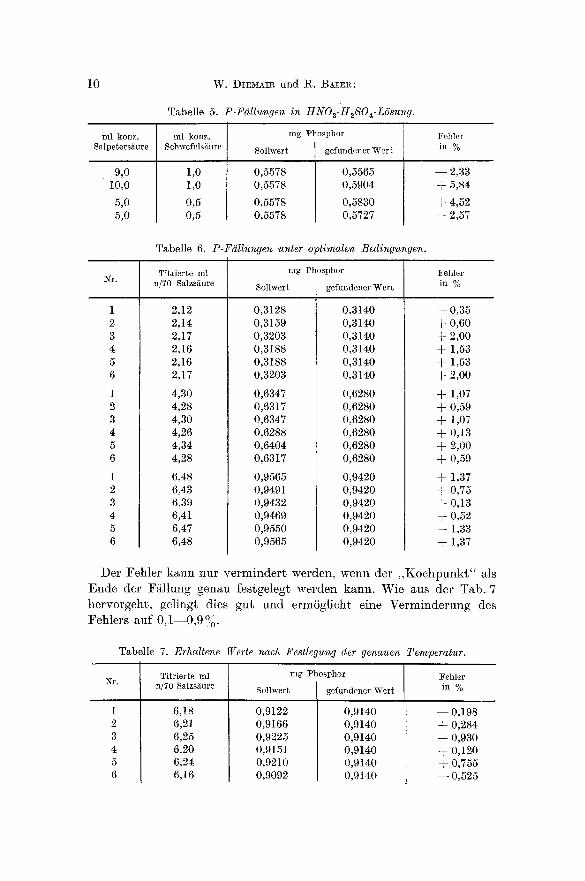
Mit den Tab. 5 und 6 wird bes tg t ig t , daI3 ein sa lpe te r saures und schwefelsaures Gemisch (Verh~ltnis 10: 1) a m gi ins t igs ten ist , wenn an Stelle von A m m o n i u m n i t r a t A m m o n i u m s u l f a t zugese tz t wird. Der Feh le r l~Bt sich bei Ef i lha l tung diesel" Bed ingungen a u f etw~, + 1 bis + 2 °, o herabdr i i cken .
10 W . DIEMAII~ und R. BAIER:
Tabelle 5. P-F(~llungen in HNO3-H~SOa-ZSsung.
ml konz. ml konz. Fehler Salpeters~ure Schwefelsi~ure in %
9,0 10~0
5,0 5,0
1,0 1,0
0,5 0,5
mg Phosphor
Sollwer t ge~ndcner Wert
0,5578 0,5565 0,5578 0,5904
0,5578 0,5830 0,5578 0,5727
- -2 ,33 +5 ,84
+4 ,52 +2 ,57
Tabelle 6. P-F~llungen unter optimalen Bedingungen.
Titrierte ml mg Phosphor Fehler
~r . n/70 8alzsgure Sollwert gefundener Wert in %
1
2 3 4 5 6
1
2 3 4 5 6
1 2 3 4 5 6
2,12 2,14 2,17 2,16 2,16 2,17
4,30 4,28 4,30 4,26 4,34 4,28
6,48 6,43 6,39 6,41 6,47 6,48
0,3128 0,3159 0,3203 0,3188 0,3188 0,3203
0,6347 0,6317 0,6347 0,6288 0,6404 0,6317
0,9565 0,9491 0,9432 0,9469 0,9550 0,9565
0,3140 0,3140 0,3140 0,3140 0,3140 0,3140
0,6280 0,6280 0,6280 0,6280 0,6280 0,6280
0,9420 0,9420 0,9420 0,9420 0,9420 0,9420
- -0 ,35 + 0,60 + 2,0O + 1,53 + 1,53 + 2,00
+ 1,07 + 0,59 + 1,07 + 0,13 + 2,00 + 0,59
+ 1,37 + 0,75 + 0,13 + 0,52 + 1,33 + 1,37
Der Fehle r kann nur v e r m i n d e r t werden, wenn der , , K o c h p u n k t " als Ende der F~tllung genau festgelegt werden kann. Wie aus der Tab. 7 hervorgeht , gelingt dies gut und ermSgl ieht eine Verminderung des
O/ Fehlers auf 0 ,1 - -0 ,9 /o .
Tabelle 7. Erhaltene Werte nach Festlegung der genauen Temperatur.
~r. Titrierte ml n/70 Salzsiiure
6,18 6,21 6,25 6,20 6,24 6,16
mg Phosphor
Sollwert
0,9122 0,9166 0,9225 0,9151 0,9210 0,9092
gefundcncr Weft
0,9140 0,9140 0,9140 0,9140 0,9140 0,9140
Fehler in %
- - 0,198 + 0,284 + 0,930 + 0,120 + 0,755 - - 0,525

Verfahren zur Mikrobestimmung des Phosphors. 11
Ill weiteren Versuchen konnte noch gezeigt werden, dab ffir das In- freiheitsetzen yon Ammoniak aus dem (PA) -Niederschlag nicht die ~[enge, sondern die Konzentration der Lauge mal3geblich ist und ferner, da~ die N-BesLimmung insbesondere b~i den zu ermit~elnden Werten yon etwa 300 #g Stickstoff genaue Zahlen liefert. Schwierigkeiten ergaben sich beim Einstellen der S~ture und Lauge. Mehrmals umkrystallisierte Oxa]- s~ure erwies sich wegen des inkonstanten Wassergehaltes als zu ungenau, da das ,,Ziehen" beim Titrieren (Tashiro-Indicator) st6rt. Beides lief3 sich ausschalten, indem nicht in S~ure, sondern in Wasser destilliert und das NHs unmittelbar (Tashiro-Indicator) titriert wird. Unter der Voraus- setzung einer einwandfreien Kiihlung Lreten keine Verluste an Ntis in der Vorlage auf.
Aus den etwa 2000 Einzelbestimmungen ergab sich folgende Arbeits- vorschrift, die bei Serienbestimmungen nur etwa 15 rain Zeitaufwand ben6tigt.
BenStigte Reagenzien: 34% ige Ammoniumsulfatl6sung, T~shiro-Indicator, 3%ige AmmoniummolybdatlSsung, n/70 Salzsaure, konz. Salpetersgure, 33%ige Natron[auge, ~bsol. Alkohol, 0,25 n Natronlauge.
w~sserfreier Xther, Arbeitsvorschri/t. Die nach dem n~ssen Aufschlul~ erhaltene PO4-LSsung wird
in einen 100 ml Weithals-Erlenmeyerkolben einpipettiert, mit dest. Wasser auf 25 ml ~ufgeftillt und mit 10 ml 34%iger AmmoniumsulfatlSsung, 7,5 ml 3%iger AmmoniummolybdatlSsung und 5 ml konz. S~lpetersgure versetzt. Hierauf wird die L6sung fiber einem Asbestnetz mit einem Bunsenbrenner bis suf 94 ° C erhitzt, der Brenner sofort entfernt und das l~fihrwerk eingeschgltet. Naeh 5 rain langem Rfihren wird der Rfihrer abgestellt und der an ihm haftende Niederschlag in den Kolben gebracht. Der Inhult des Kolbens wird durch einen 3 G 4-Tiegel ~bgessugt, der in dem Kolben verbliebene IN~iedersehlag mit 40--50 ml dest. Wasser in den Tiegel gesptilt. Der so erh~ltene Ammoniumphosphormolybdatniederschlag wird unter Aufwirbeln und Absaugen mit je 20 ml absolutem Alkohol und wasserfreiem *ther getrocknet. Dann wird der Niederschlag im Tiegel mit dreimal 5 ml 0,25 n Natronlauge gel6st und die LSsung in einen Erlenmeyerkolben ~bgesaugt. Mit dieser LSsung wird die Stickstoffbestimmung n~ch PAm~As-WAa~EI~ durchgeffihrt. Der erhaltene Stickstoffweri~ wird mi~ dem Faktor 0,738 multipliziert und ergibt den Phosphorgehalt in der untersuchten LSsung.
Zusammenfassung. Es wird fiber eine Mikromethode zur Phosphor-Bestimmung beriehtet,
deren Prinzip darauf beruht, dal~ man den Ammoniumgehalt des Am- moniumphosphormolybdat-Komplexes nach K~EL~AI4L in der PARKAS- WAs~xI~-Apparatur bestimmt und darius den P-Gehal~ berechnet. 2000 Einzelbestimmungen haben gezeigt, da6 die Methode bei genauer Einhaltung der angegebenen Bedingungen gut reproduzierbare Werte lie fern.

12 B. WUI~ZSCttMITT :
Literatur. 1 D:[EMAIR, W., u. C. BO~ESCH: 4iese Z. 180, 4 (1950); vgl. auch MitteilungsblatV
der GDCh-Fachgruppe Lebensmittelchemie 11/12, 104 (1950). - - 2 TILL~A~S, J., H. RI~'FART U. A. K~HN: Z. Unters. Lebensm. 60, 375 (1930). - - 3 LORENZ, ]q. V. : Landwirtschaftl. Vers.StaL 55, 183 (1901); 66, 203 (1907); Chem. Ztg. 82, 707 (1908); vgl. auch diese Z. 48, 330 (1909). - - a WoY, 1~. : Chem. Ztg. 21, 442 u. 469 (1897). - - s LIND~nR, P.: Z. ~ngew. Chem. 35, 346 (1922).
ProL Dr. Dr. W. DIEMAII% Frankfurt a.M., Paul-Ehrlich-StraBe 40.
Aus dem Untersuchungslaboratorium der Badischen Anilin- und Soda-Fabrik, Ludwigshafen a. Rh. (Leiter: Dr. B. WV~ZSeI~ITT).
Die Analyse von Mono~ithylenglykol, Di~ithylenglykol und Diiithylenglykolmonoiithyliither nebeneinander*.
Von BERNHARD WURZSCHMITT.
Mit 2 Textabbildungen.
(Eingegangen am 24. Februar 1951.)
Die yon WAGEXqAAI~ zum l~achweis yon F e t t e n (Glycerin) angegebene Methode 1 be ruh t darauf , dab a l ipha t i sehe Verb indungen , die an zwei oder mehr b e n a c h b a r t e n Koh lens to f f a tomen H y d r o x y l g r n p p e n t r agen , in waBrig-alkal ischer LSsung m i t Kupfe r su l f a t bzw. - h y d r o x y d 15sliehe kupfe rha l t ige K o m p l e x e zu b i lden vermSgen. D a diese Bed ingung yon den 3 in lgrage s t ehenden Verb indungen :
CI-I~--OH HO--CH~ CH2--OH I ] J CH2--OH CH~ CH2
\ o / Mono~thylenglykol (Glykol) Di~thylenglykol
H0--CH 2 CH2--0 • C2H~
l I CH2 CH2
\ o / Diathylenglykolmonoathylather (~thylglykol).
nur vom Mono~Lthylenglykol erf i i l l t wird, mul~te sich dieses neben den be iden anderen Verb indungen e inwandfre i nachweisen bzw. b e s t i m m e n lassen. Ein ige Versuche be s t a t i g t en diese Vermutung . Die Pr t i fung wurde in folgender Weise ausgef i ihr t :
• tIerrn Prof. Dr. W. GEIL3IARrI~ ztlm 60. Geburtsta,g gewidmet.