Embed Size (px)
Citation preview

Z. Anal. Chem. 253, 195--201 (1971) © by Springer-Verlag 1971
195
Bestimmung von Quecksilberspuren in Reinstsilber nach Anreicherung des Hg- DTA-Komplexes an Silberjodid
E. JACKWERTH, E. D6Rr~G und J. LOHM~a~
Institut ffir Spektrochemie und angewandte Spektroskopie, Dortmund
Eingegangen am 25. Juli 1970
Determination o/Traces o/Mercury in High-Purity Silver alter Enrichment o/the Hg-EDTE-Complex on Silver Iodide. Investigations on the sorption of chelate complexes on silver halogenides have shown that in solutions of silver traces of divalent mercury are co-precipitated in presence of EDTE. Based on this principle a method was developed for the enrichment of traces of mercury in high-purity silver. By this method about 70 mg of AgJ are precipitated in the solution of 5--10 g of silver after the addition of 5 tLMol of EDTE. The AgI collector containing the mercury, is dissolved in a solution of sodium thioeyanate, and the mercury enriched to 80--900]0 is determined photometrically using dithizone as a reagent. Samples containing 5 × 10-5°/0 of rig could be analys- ed with a relative standard deviation of 0.04 (40]0). The limit of detection is 6 × 10-e°/o of Hg.
Zusammenfassung. Aus Untersuchungen fiber die Sorption yon Chelatkomplexen an Silberhalogeniden geht hervor, dab Spuren yon zweiwertigem Qnecksflber aus einer silberhaltigcn L6sung schon nach Znsatz geringer Mengen Jodid praktisch vollstiindig mitgerissen werden, wenn die Sflberjodidf~llung in Gegcnwart von ~DTA durchgeffihrt wird. Auf diesem Prinzip wurde ein Verfahren zur Anreieherung yon Quecksflberspuren in Reinst- sflber aufgebaut. Hierbei werden aus der ProbenlSsung mit 5--10 g Ag+ nach Zusatz yon 5 FMol J~DTA etwa 70 mg AgJ ausgef~llt. Der das Quecksflber enthaltende AgJ-Spurenfi~ngcr wird in NatriumthiocyanatlSsung gelSst und das je nach Silbereinwaage zu 80--900/0 angcreicherte Quecksflber mit Dithizon photometrisch be- stimmt. Ffir Analysenmaterial mit 5 • 10-5°/0 Hg betri~gt die relative Standardabweichung 0,04 (4°/0). Die Naehweisgrenze liegt ftir 10 g-Einwaagen bei 6 • 10-e°/o Hg.
I. Sorptionsverhalten von Hg ~+ und Hg(II)- Komplexonkomplexen an Silberhalogenid- NiederscblKgen
Neben einer Reihe anderer Schwermetallionen werden auch Spuren yon zweiwertigem Queeksflber, wie wit in friiheren Untersuchungen gefunden haben, bei der Ausf/~lhing der Matrix Sflber in Form der Sflber- halogenide AgC1, AgBr und AgJ durch Adsorption an der elektrisch geladenen Oberfl/iche des ~qieder- schlags mitgerissen [3]. Abweichend vom Verhalten der meisten dicser adsorbierten Metallionen wird aber beim Hg(II)-Kation eine quantitative Sorption lediglich im Bereich um den J~quivalenzpunkt der F/illungsreaktion beobachtet (Abb.1).
Solange die Silberhalogenid-Suspension noch Ag +- oder schon Halogeuld-Ionen im Ubersehul3 enth/ilt, sind Niederschlag und Spur gleichgerichtet geladen, und die Adsorption des Quecksilbers wird weitgehend verhindert: Bei SilberfibersehuB und positiver Ober- fl/~cheuladung bleibt das Quecksflber als Hg ~+- Kation in L6sung; nach vollstiindiger F/iilung des Silbers und bei ansteigendem Halogenidfiberschul~
13"
bflden sich in zunehmendem Ma~e Halogenomercurat- Anionen, die ebenfalls yon dem nun negativ gelade- nen Niederschlag nieht mitgerissen werden. Im untersuchten Konzentrationsbereich grit dies, wie die Abb.1 zeigt, bevorzugt ffir die Bromide und Jodidc; die Chlorokomplexbfldung des Quccksflbers macht sieh erst bei wesentlich hSherer Chloridkonzentration in diesem Sinne bemerkbar.
In den Diagrammen der Abb.1 sowie in den folgen- den Abb.2--6 wurde als Abszisse die im Verlauf der
1oc
.,.. 8o
4c
~ 20
I /
o ,4gc/ / AgBr /
• AgJ
2 4 6 8 1o mMol/VeX Abb. 1. Sorption vo~ Hg 2+ im Verlaufe der Fgllung yon AgC1, AgBr and AgJ
196 E. Jackwerth, E. DSring und J. Lohmar:
Matrixf~llung (10 mMol Ag+) verwendete F~llungs- mittelmenge gew~hlt, w~hrend auf der Ordinate der im zugeh5rigen Filtrat wiedcrgefundene prozentnale Anteil an Spurenelement (eingesetzt 250~g = 1,25 ~Mol Itg 2+) aufgetragen wurde. Der ~quivalenz- punkt der F~llungsreaktion ist jeweils durch die gestriehelte Linie angedeutet. Die F£11ungsversuche wurden in 0,25M salpetersaurer LSsung dureh- geffihrt. Soweit in dieser Arbeit bei den sparer be- schriebsnen Versuchen zus~tzlich Chelatbildner ver- wendet wurden, haben wir einheitlieh 10 ~Mol der jeweiligen Verbindung eingesetzt. In diesen Fallen waren die LSsungen auf etwa pH 4--5 eingestellt. Jedes Diagramm ist aus einer Folge yon Einzel- versuchen entstanden. N~here Angaben fiber die Versuchsanordnung siehe [4].
Der Abb. 1 ist zu entnehmen, dab Spuren Queok- sflber in Silberpr~paraten angereiehert werden kSnnen, wenn es gelingt, die Matrix Sflber quantita- tiv auszuf~llen und gleichzeitig das Quecksilber mi~ fiberschfissigem F~llungsmittel zu nioht adsorbier- baren Halogenokomplex-Anionen zu maskieren. Kom- plexbfldung und Adsorption des I-Ig2+-Ions an negativ geladenem Sllberhalogenid stehen jedoch so welt miteinander im Gleichgewicht, dab man eine hin- reichende Komplexierung der Queeksflberspuren auch bei Bromid und Jodid erst mit relativ hoher ttalogenidkonzentration erreicht. Um Qnecksflber auf diese Weise dureh vollst~ndiges Ausf~llen der Sflbermatrix anzureiehern, ist ein so grol3er Halo- genidfiberschuB erforderlich, dal~ die nacbfolgende Bestimmung des Quecksilbers im Spurenkonzentrat erfahrungsgem~l~ erhebliche Sehwierigkeiten macht.
Eine weitere MSglichkeit, Queeksflberspuren im Ffltrat einer Silberf~llung anzureichern, wird dadurch erhalten, dal~ man die Pseudohalogenide NaCN oder NaSCN als F~llungsmittel ffir die Matrix verwendet. Daneben wird der ProbenlSsung ein Chelatbildner zugesetzt, der das Spurenelement in ein genfigend stabiles Komplex-Anion fiberffihrt. J~thylendiamin-
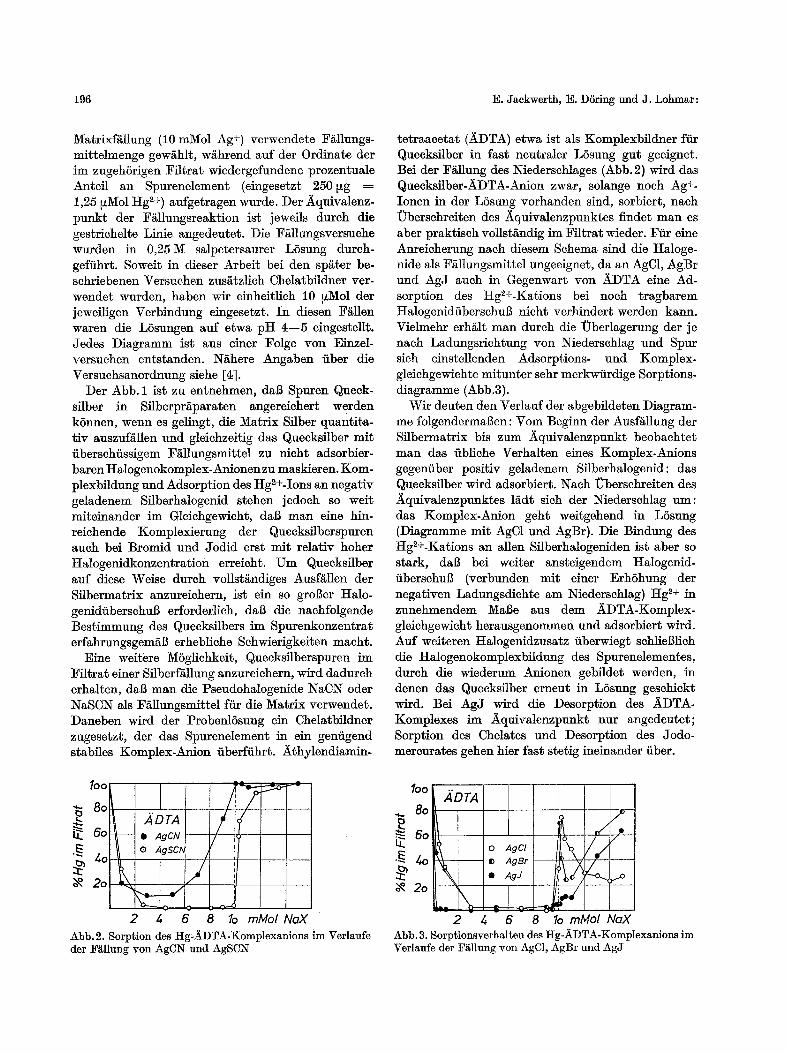
tetraacetat (~DTA) etwa ist als Komplexbildner ffir Queeksflber in fast neutraler LSsung gut geeignet. Bei der F~llung des Niederschlages (Abb. 2) wird das Quecksilber-~DTA-Anion zwar, solange noch Ag+. Ionen in der L5sung vorhanden sind, sorbiert, naeh Ubersehreiten des Aquivalenzpunktes finder man es aber praktisch vollst&ndig im Ffltrat wieder. Ffir eine Anreicherung nach diesem Schema sind die Haloge- nide als Fallungsmittel ungeeignet, da an AgC1, AgBr und AgJ aueh in Gegenwart von ~DTA eine Ad- sorption des Hg2+-Kations bei noch tragbarem HalogenidfibcrschuB nicht verhindert werden kann. Vielmehr erh~tlt man durch die Uberlagerung der ]e nach Ladungsrichtung yon Niederschlag und Spur sich einstellenden Adsorptions- und Komplex- gleichgewichte mitunter sehr merkwfirdige Sorptions- diagramme (Abb.3).
Wit deuten den Verlauf der abgebildeten Diagram- me folgendermaBen: Vom Beginn der Ausf~llung der Silbermatrix bis zum ~quivalenzpunkt beobachtet man das fibliche Verhalten eines Komplex-Anions gegenfiber positiv geladenem Sflberhalogenid: das Quecksflber wird adsorbiert. Nach Uberschreiten des ~quivalenzpunktes ladt sich der Niederschlag um: das Komplex-Anion geht weitgehend in L6sung (Diagramme mit AgC1 und AgBr). Die Bindung des Hg2+-Kations an allen Sflberhalogeniden ist aber so stark, da$ bei welter ansteigendem HMogenid- iiberschuB (verbunden mit einer ErhShung der negativen Ladungsdiehte am Niederschlag) Hg 2+ in zunehmendem Ma6e aus dem ADTA-Komplex- gleiehgewieht herausgenommen und adsorbiert wird. Auf weiteren Halogenidzusatz fiberwiegt schlie$1ich die Halogenokomplexbildung des Spurenelementes, durch die wiederum Anionen gebildet werden, in denen das Quecksilber erneut in LSsung geschickt wird. Bei AgJ wird die Desorption des ~DTA- Komplexes im J~quivalenzpunkt nur angedeutet; Sorption des Chelates und Desorption des Jodo- mereurates gehen hier fast stetig ineinander fiber.
100
ft. 6o ~ • AgcN .~ 0 AgSCN
N 20 -~
2 4 6 8 1o mMolNaX Abb.2. Sorption des Hg-~DTA-Komplexanions im Verlaufe der Y~llung yon AgCN und AgSCN
1oo I
• - 4 0
~ 2 o
A'DTA
2
7
0 AgCI I°kc{ /_~ / AgBr
• AgJ 1o
4 6 8 1o mMol NaX Abb. 3. Sorptionsverhalten des Hg-J~DTA-Komplexanions im Verlaufe der F~llung yon AgC1, AgBr und AgJ
Bestimmung von Quecksilberspuren in Reinstsilber nach Anreichertmg des tIg/~DTA-Komplexes an Silberjodid 197
/ o o i
• ,,.., 8 0
~. 6o ¸
-~ 40
20
AgCI
2 4 6 8 1o mMolNaCI Abb.4. Sorptionsverhalten der Quecksilber(II)-Komplexo- nate bei der Fi~llung yon Silberchlorid
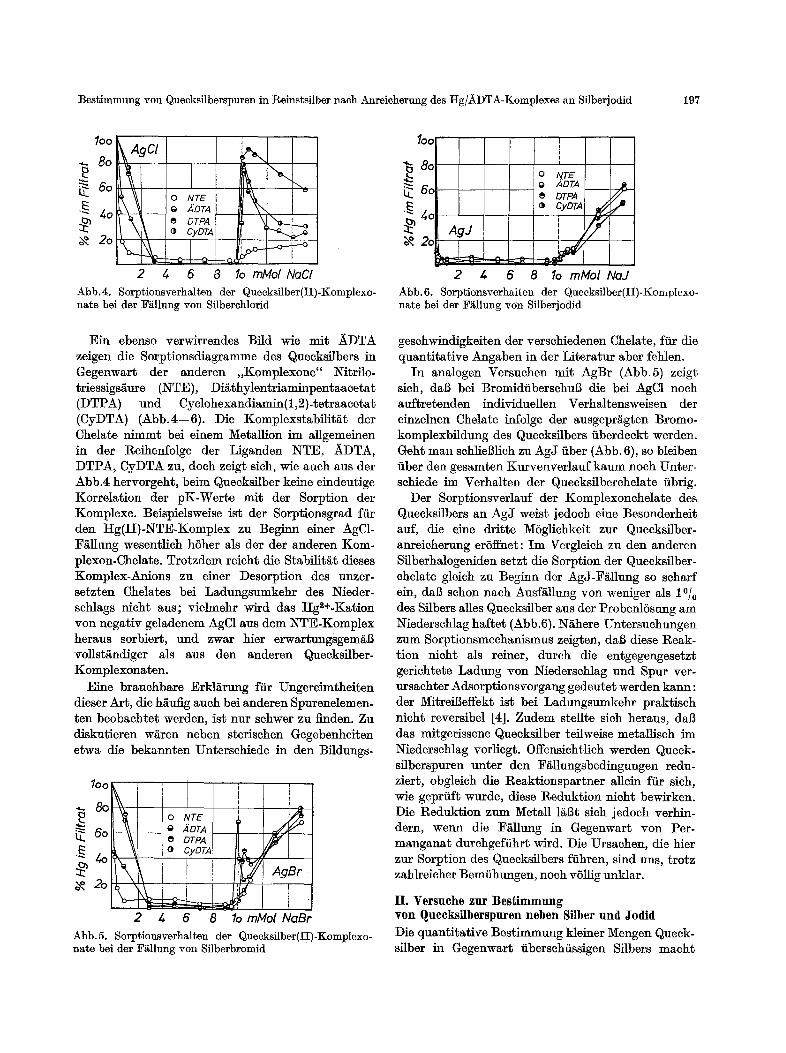
Ein ebenso verwirrendes Bfld wie mit )~DTA zeigen die Sorptionsdiagramme des Queeksflbers in Gegenwart der anderen ,,Komplexone" Nitrflo- triessigs/~ure (NTE), Di~thylentriaminpentaaeetat (DTPA) und Cyelohexandiamin(1,2)4etraacetat (CyDTA) (Abb.4--6). Die Komplexstabilit/it der Chelate nimmt bei einem MetalUon im allgemeinen in der Reihenfolge der Liganden NTE, ~DTA, DTPA, CyDTA zu, doeh zeigt sieh, wie auch aus der Abb.4 hervorgeht, beim Quecksilber keine eindeutige Korrelation der pK-Werte mit der Sorption der Komplexe. Beispielsweise ist der Sorptionsgrad ffir den Hg(II)-~TE-Komplex zu Beginn einer AgC1- F~llung wesentlieh h6her als der der anderen Kom- plexon-Chelate. Trotzdem reieht die Stabilit/~t dieses Komplex-Anions zu einer Desorption des unzer- setzten Chelates bei Ladungsumkehr des Nieder- schlags nieht aus; vielmehr wird das Hg~+-Kation yon negativ geladenem AgC1 aus dem NTE-Komplex heraus sorbiert, und zwar bier erwartungsgem/iB vollst/~ndiger als aus den anderen Quecksflber- Komplexonaten.
Eine brauehbare Erkl~rung ffir Ungereimtheiten dieser Art, die h~uilg aueh bei anderen Spurenelemen. ten beobaehtet werden, ist nur schwer zu finden. Zu diskutieren wi~ren neben sterisehen Gegebenheiten etwa die bekannten Untersehiede in den Bfldungs-
1 O o i I
0 NTE
e DTPA ~Y"
~ 2o
2 4 6 8 10mMolNaBr Abb.5. Sorptionsverhalten der Quecksilber(I!)-Komplexo- nate bei der F~llung yon Silberbromid
/o¢ t I
6C ~ ADTA /.~ e DTPA @ CyDTA/~
AgJ 1
. . , , 4 ~ ___------.~- -....,~
2 4 6 8 1o mMol NaJ Abb.6. Sorptionsverhalten der Quecksilber(II)-Komplexo- nate bei der F~llung yon Silberjodid
geschwindigkeiten der verschiedenen Chelate, ffir die quantitative Angaben in der Literatur aber feh]en.
In analogen Versuchen mit AgBr (Abb.5) zeigt sich, dab bei Bromidfibersehu8 die bei AgC1 noch auftretenden individuellen Verhaltensweisen der einzelnen Chelate infolge der ausgepr~gten Bromo- komplexbfldung des Queeksilbers fiberdeckt werden. Geht man sehlieSlieh zu AgJ fiber (Abb. 6), so bleiben fiber den gesamten Kurvenverlauf kaum noeh Unter- sehiede im Verhalten der Quecksilberehelate fibrig.
Der Sorptionsverlauf der Komplexonchelate des Queeksflbers an AgJ weist jedoeh eine Besonderheit auf, die elne dritte MSgliehkeit zur Quecksilber- anreieherung erSffnet: Im Vergleich zu den anderen Sflberhalogeniden setzt die Sorption der Queeksflber- ehelaf~ gleieh zu Beginn der AgJ-F~llung so seharf ein, dab sehon naeh Ausf£11ung yon weniger als 1°/o des Sflbers alles Queeksilber aus der ProbenlSsung am Niederschlag halter (Abb.6). N~here Untersuchungen zum Sorptionsmechanismus zeigten, dab diese Reak- tion nieht als reiner, dutch die entgegengesetzt gerichtete Ladung yon NiederseMag und Spur ver- ursaehter Adsorptionsvorgang gedeutet werden kann: der MitreiBeffekt ist bei Ladungsumkehr praktisch nicht reversibel [4]. Zudem stellte sieh heraus, dab das mitgerissene Queeksflber teilweise metalliseh im Niederschlag vorliegt. Offensichtlieh werden Queek- silberspuren unter den F/~l]ungsbedingungen redu- ziert, obgleich die Reaktionspartner allein ffir sich, wie geprfift wurde, diese Reduktion nieht bewirken. Die Reduktion zum Metall liiSt sieh jedoch verhin- dern, wenn die Fiillung in Gegenwart yon Per- manganat durehgeffihrt wird. Die Ursaehen, die hier zur Sorption des Quecksilbers ffihren, sind uns, trotz zahlreieher Bemfihungen, noeh vSllig unklar.
II. ¥ersuehe zar Best immung yon Queeksilberspuren neben Silber und Jodid Die quantitative Bestimmung kleiner Mengen Queek- sflber in Gegenwart fiberschfissigen Silbers maeht

198 E. Jackwerth, E. D6ring und d. Lohmar:
mit den verschiedensten Analysenmethoden Sehwie- rigkeiten. Elektrochemische Verfahren scheiden zu- meist wegen der praktisch gleichen Normalpotentiale beider Elemente aus. Allerdings konnten Murray u. McNeely [6] dureh gesehicktes Ausnutzen yon Adsorptionseffekten an der Elektrodenoberfl/~che eine polarographisehe Quecksflberbestimmung bei bis zu 75fachem SilberiiberschuB durchf/ihren. Ebenso werden photometrische Verfahren fiir Queeksflber in der Regel dureh das/~hnliche Reaktionsverhalten des Sflbers gestSr~. Eine der wenigen Ausnahmen ist die Photometrie des Dithizonkoml01exes. Zwar reagieren auch bier je nach Analysenbedingungen beide Ele- mente mit Dithizon; naeh einer Arbeit yon Fischer u. Leopoldi [1] gibt es jedoch MSglichkeiten, wenige N[ikrogramm Quecksilber neben der 500- bis 1000fachen Silbermenge ohne StSrung mit Dithizon zu bestimmen; Angaben von Va§£k u. Sedivee [8] zufolge lassen sich dariiber hinausgehende Silber- mengen mit Thiocyanat maskieren. Trotzdcm ist das NachweisvermSgen aller Bestimmungsverfahren ftir Quecksilber zu gering, um Spuren in Silberpr/~paraten im interessanten Konzentrationsbereich unterhalb 10-4°/0 ohne Voranreicherung zu erfassen. Das gilt auch fiir die verschiedenen atomspektroskopisehen Methoden. _~hnliche Schwierigkeiten ergeben sieh ffir die Trennung der Elemente bei grol3en Konzentra- tionsunterschieden. Zwar gelingt die Abtrennung einiger Milligramm Quecksflber aus Grammeinwaagen Sflber durch Zementation mit Kupfer, wie Maude u. Wilkinson [5] fanden, andere yon den Autoren ffir diesen Zweck eingesetzte Trennverfahren, wie Kat- ionen- und Anionenaustauseh, F/fllungsreaktionen sowie die Dithizonatextraktion, fiihrten jedoeh nicht zum Ziel. Zur Anreicherung yon Quecksilber im ppm- Bereich und darunter sind Verfahren unseres Wissens bisher nicht beschrieben worden.
Unter den ira vorangegangenen Kapitel dieser Arbeit aufgezeigten MSglichkeiten zur Quecksflber- anreicherung ist ftir eine praktische Anwendung be- sonders die Beobachtung yon Interesse, dal~ Hg(II)- Spuren trotz eines grol~en Sflberiiberschusses bereits yon sehr wenig in der ProbenlSsung ausgef/~lltem Silberjodid mitgerissen werden, wenn man die F/~llung in Gegenwart eines geeigneten Chelatbildners dureh- ftihrt. Es bietet sich an, AgJ auf diese Weise als Spurenf/inger zur Anreicherung von Quecksflber in Sflberpr~paraten zu verwenden. Eine besondere Schwierigkeit entsteht aber dadurch, dat~ die Queck- silberspuren an Sflberjodid gebunden anfallen, einer Verbindtmg, die im allgemeinen nur unter Bedingun- gen aufgeschlossen werden kann, unter denen das
enthaltene Quecksflber infolge der F1/ichtigkeit seiner Salze vollst/~ndig verlorengeht. Ein blol~es Erhitzen bzw. Auskochen des Niederschlages mit S/~ure reicht zum AblSsen der angereieherten Queeksilberspuren nicht aus, da offensichtlieh ein merklicher Anteil im Innern der Niederschlagspartikel eingeschlossen bleibt.
Sflberjodid kann aber, wie wir sehon frfiher mit- geteflt haben, zur stSrungsfreien Bestimmung einer ganzen Reihe yon Spurenelementen in konzentrier- tern Thioeyanat gelSs~ werden [2]. Obgleieh das genaue Verhalten yon Quecksilberspuren unter diesen Be- dingungen noch nieht geprfift wurde, liel~en die bereits zitierten Untersuchungen yon Va~£k u. Sedivee [8] erwarten, dab innerhalb bestimmter Konzentrations- bereiche eine photometrischc Quecksilberbestimmung in Silberjodid unter Verwendung yon Dithizon mSglich ist. Vor der Ausarbeitung des Anreiche- rungsverfahrens war es aber notwendig zu wissen, wie grol3 der tats/~cbliche EinfluB der Parameter Ag+ und J - aus dem Spurenf/~nger sowie des zum LSsen verwendeten Thiocyanats auf die Quecksilber- bestimmung ist, und welche Mengen bei der Analyse toleriert werden kSnnen.
a) Einflu[3 wechselnder Mengen Thiocyanat au] die Quecksilberbestimmung in Gegenwart von Ag+ und J - Versuchsanordnung. Zu einer Reihe yon LSsungen mit je 5,0 ~tg Hg 2+ warden wechselnde Zusi~tze an 9 M NaSCN- LSsung, je 2 ml 0,1 l~ ~DTA-LSsung, 10 ml Acetatpuffer und l ml 10°/0ige HydroxylammoniumchloridlSsung ge- geben; dann wurde mit Wasser auf je 50,0 ml erg~nzt. Die LSsungen wurden mit je 10,0 ml Dithizon in Tetrachlor- kohlenstoff I rain geschfittelt. Anschliel3end wurde die der Quecksilberkonzentration im organischen Extrakt pro- portionale Extinktionsabnahme (Dithizonverbrauch) bei 620 nm photometrisch gemessen (Zeiss-Elko II, Filter 1 62, 2 cm-Kiivetten). Als Vergleich diente die unverbrauchte DithizonlSsung. N~here Angaben zu den l~eagentien und MeBbedingungen s. in der folgenden Analysenvorschrift.
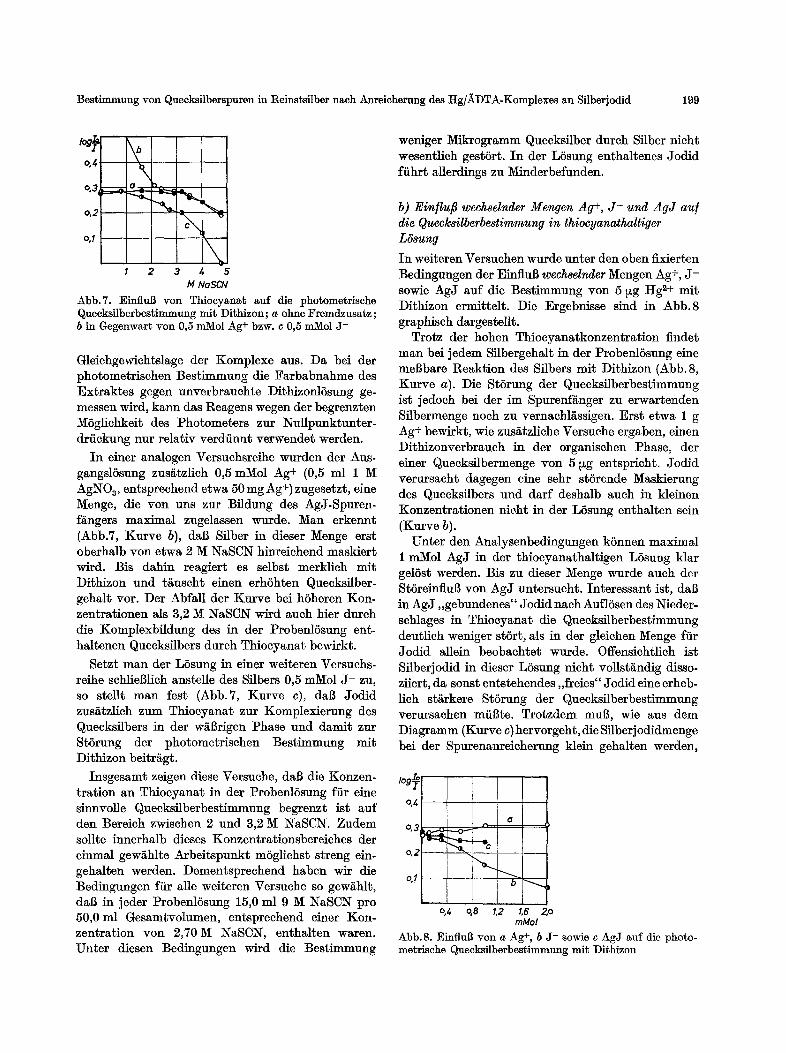
In Abb.7 (Kurve a) ist als Ergebnis dieser Ver- suche die Abh/~ngigkeit der Extinktion des Dithizon- extraktes yon der Thiocyanatkonzentration der ProbenlSsung dargestellt. Die Extinktionswerte wer- den im wesentlichen bestimmt durch die Lage des Gleiehgewichts zwischen Dithizon- und Thiocyanato- komplex des Queeksilbers in organiseher bzw. w/il~- tiger Phase. Innerhalb iiblicher Mel3wertschwankun- gen bleibt die Extinktion nach Aussage der Abbildung bis zu 3,2 M NaSCN konstant, f/illt dann aber, wohl infolge der sich durchsetzenden Thioeyanato-Kom- plexbildung des Quecksilbers in der w~13rigen LSsung, stark ab. Entsprechend wirken sieh auch _;lmderungen in der Dithizonmenge bzw. -konzentration auf die
Bestimmung yon Quecksilberspuren in Reinstsilber nach Anreicherung des Hg/ADTA-Komplexes an Silberjodid 199
0.~
°.3 ..--a ..4. m = ~
0.2 " ~ %'-
o.1
1 2 3 h 5 H NaS~
Abb.7. EinfluB yon Thiocyanat auf die photometrische Quecksilberbestimmung mit Dithizon; a ohne Fremdzusatz; b in Gegenwart yon 0,5 mMol Ag+ bzw. c 0,5 mMol J-
Gleichgewichtslage der Komplexe aus. Da bei der photomegrisehen Bestimmung die Farbabnahme des Extraktes gegen unverbrauehte Dithizonl6sung ge- messen wird, kann das Reagens wegen der begrenzten MSglichkeit des Photometers zur NuUpunktunter- drfiekung nur relativ verdfinnt verwendet werden.
In einer analogen Versuchsreihe wurden der Aus- gangslSsung zus~tzlieh 0,5 mMol Ag+ (0,5 ml 1 M AgNOa, entspreehend etwa 50 mg Ag +) zugesetzt, eine lVIenge, die yon uns zur Bfldung des AgJ-Spuren- f~ngers maximal zugelassen wurde. Man erkennt (Abb.7, Kurve b), dal~ Sflber in dieser Menge erst oberhalb yon etwa 2 M NaSCN hinreiehend masklert wird. Bis dahin reagiert es selbst merklich mit Dithizon und t~uscht einen erhShten Queeksilber- gehalt vor. Der AbfaU der Kurve bei hSheren Kon- zentrationen als 3,2 M NaSCN wird auch bier dureh die Komplexbfldung des in der Probenl5sung ent- haltenen Queeksflbers dutch Thiocyanat bewirkt.
Setzt man der LSsung in einer weiteren Versuchs- reihe schliel31ich anstelle des Sflbers 0,5 mMol J - zu, so stellt man lest (Abb.7, Kurve c), dal~ Jodid zus~tzlich zum Thiocyanat zur Komplexierung des Quecksilbers in der w~rigen Phase und damit zur StSrung der photometrisehen Bestimmung mit Dithizon beitr£gt.
Insgesamt zeigen diese Versuche, da~ die Konzen- tration an Thioeyanat in der ProbenlSsung ffir eine sinnvolle Queeksflberbestlmmung begrenzt ist auf den Bereich zwisehen 2 und 3,2 M I~aSCN. Zudem sollte innerhalb dieses Konzentrationsbereiehes der einmal gew~hlte Arbeitspunkt mSglichst streng ein- gehalten werden. Dementspreehend haben wir die Bedingungen fiir alle weiteren Versuehe so gewi~hlt, da~ in jeder Probenl6sung 15,0 m] 9 IV[ NaSCN pro 50,0 ml Gesamtvolumen, entsprechend einer Kon- zentration yon 2,70 M NaSCN, enthalten waren. Unter diesen Bedingungen wird die Bestimmung
weniger Mikrogramm Queeksflber dureh Silber nicht wesentlieh gestSrt. In der LSsung enthaltenes Jodid ffihrt allerdings zu Minderbefunden.
b) Ein/lufl wechselnder Mengen Ag+, J - und AgJ auf die Queclcsilberbestimmung in thioeyanathaltiger L6sung
In weiteren Versuehen wurde unter den oben fixierten Bedingungen der EinfluB weeh~elnder Mengen Ag+, J - sowie AgJ auf die Bestimmung yon 5 ~g Hg 2+ mit Dithizon ermittelt. Die Ergebnisse sind in Abb.8 graphiseh dargestellt.
Trotz der hohen Thiocyanatkonzentration findet man bei jedem Silbergehalt in der ProbenlSsung eine meBbare Reaktion des Sflbers mit Dithizon (Abb. 8, Kurve a). Die StSrung der Queeksilberbestimmung ist jedoch bei der im Spurenfanger zu erwartenden Silbermenge noeh zu vernaeM~ssigen. Erst etwa 1 g Ag+ bewirkt, wie zus~tzliehe Versuehe ergaben, einen Dithizonverbraueh in der organisehen Phase, der einer Queeksflbermenge yon 5 ~g entsprieht. Jodid verursaeht dagegen eine sehr st6rende Masklerung des Queeksi[bers und darf deshalb aueh in kleinen Konzentrationen nieht in der L6sung enthalten sein (Kurve b).
Unter den Analysenbedingungen kSnnen maximal 1 mMol AgJ in der thioeyanathaltigen LSsung klar gelSst werden. Bis zu dieser Menge wurde such der StSreinflu~ yon AgJ untersueht. Interessant ist, dal3 in AgJ,,gebundenes" Jodid nach AuflSsen des Nieder- sehlages in Thioeyanat die Queeksflberbestimmung deutlich weniger stSrt, als in der gleiehen ~enge ffir Jodid allein beobaehtet wurde. Offensiehtlich ist Silberjodid in dieser LSsung nicht vollst~ndig disso- ziiert, da sonst entstehendes ,,freies" Jodid eine erheb- lich st~rkere StSrung der Quecksilberbestimmung verursaehen inflate. Trotzdem mul~, wie aus dem Diagramm (Kurve c) hervorgeht, die Sflberj odidmenge bei der Spurenanreieherung klein gehalten werden,
0.3
0.2
o,! b
q~ q8 1.2 1,6 2,0 mMol
Abb. 8. EinfluB yon a Ag+, b J- sowie e AgJ auf die photo- metrische Quecksilberbestimmung mit Dithizon

200 E. Jackwerth et al. : Bestimmung yon Quecksilberspuren in Reinstsilber naeh Anreieherung des Hg/~DTA-Komplexes
um einen systematischen Fehler bei der Queeksilber- best immung mSglichst zu verhindern. Die nach der folgenden Analysenvorsehrfft jeweils auszuf/£11ende Menge yon etwa 70 mg AgJ (0,3 mMol) erfiillt diese Bedingung weitgehend.
IH. Versuehe zur Anreieherung yon queeksilberspuren aus Silbermetall
Allgemeine Angaben. Die Untersuehungen wurden an Ein- waagen yon 5 bzw. 10 g Silbergranalien durehgeffihrt, l~ach Zusatz des Spurenelements [Hg(~Oa)e-LSsung mit 1 ~g/ml Hg 2+] wurde das Metall unter leiehtem Erw~rmen in Salpeter- s~Lure gelSst.
Vorversuehe ergaben, dab Hg2+-Spuren zwar beim Eindampfen einer matrix/reien LSsung auf einer etwa 150°C warmen Heizplat te zum groBen Teil verflfich- t igt werden kSnnen; w/~hrend des LSsevorgangs yon Silbermetall und beim Eindampfen der Proben- 15sung treten dagegen keine merkliehen Queeksilber- verluste auf. Diese Gefahr entsteht erst dann, wenn der auskristallisierende AglqO~-Rfickstand 1/~ngere Zeit trocken auf der heiBen Heizplatte stehen bleibt. Dabei h/ingt das AusmaB der Flfiehtigkeit s tark yon der eingesetzten Sflbermenge ab, wobei die Hg 2+- Spuren beim Eindampfen der LSsung erst yon einem grSBeren Salzrfiekstand vollstiindig zurfickgehalten werden. Unter den Bedingungen unserer Arbeits- vorsehrfft ist mi t Queeksilberverlusten bei Einwaagen yon 5 bzw. 10 g Sflber nieht zu reehnen.
Die in unserem Anreieherungsveffahren ausgenutz- te Tatsaehe, dab sich der XDTA-Komplex yon zwei- wertigem Queeksflber neben einem mehr als 10~fachen Ag+-Ubersehug in der ProbenlSsung bfldet, ha t seine Ursaehe in dem erhebliehen Untersehied der Kom- plexbfldungskonstanten beider Chelate (pI~ttg¥~- = 21,8; pKAgy,- ---- 7,2; ¥ 4 - __ Anion der J~thylen- diamintetraessigs/~ure). Die Zahlenwerte der Kon- s tanten sind jedoeh s tark yore pH-Wer t der LSsung abh/~ngig, wobei die pK-Werte der Chelate mit anstei- gender Aeidit/it kleiner werden [7]. Das verlangt, dab die ProbenlSsung naeh dem Aufnehmen des AgNO3- Riiekstandes mit Wasser so welt s/iurefrei ist, dab der Hg-XDTA-Komplex noeh in hinreiehendem MaBe gebfldet und yon AgJ mitgerissen werden kann. Eindampfen zur Troekne allein geniigt nieht, da immer so vie1 Salpetersgure im Kristallri iekstand eingesehlossen bleibt, dab die w/~Brige LSsung des Salzes ftir den sieh ansehlieBenden Anreieherungs- prozeB zu sauer ist. Andererseits da f t der Eindampf- riiekstand wegen der Flfichtigkeit des Queeksflbers nieht zu s tark erhitzt werden. Das dadureh erforder- liehe Neutralisieren der ProbenlSsung gelingt naeh
unserer Erfahrung am besten mit Ammoniak; auf Zusatz auch yon sehr verdiinnter Natronlauge f~llt z.B. an der Eintropfstelle leieht Sflberhydroxid aus, das sieh in dem umgebenden, sehwaeh sauren Medium kaum wieder 15st. Gute Analysenergebnisse werden erhalten, wenn die ProbenlSsung vor der F&llung des Spurenf/~ngers mi t verdiinnter Ammoniak15sung auf etwa p H 3 eingestellt wird. HShere pH-Wer te fiibren, wohl infolge Amminkomplexbildung, zu Minder- befunden.
Bei der Ausf/illung des AgJ-SpurenfKngers ent- s teht eine weitere Schwierigkeit dadurch, dab Silber- jodid, wie aueh andere in Wasser sehwerlSsliehe Silberniedersehl/~ge, yon konzentrierten Sflbernitrat- 15sungen merklieh gelSst werden. U m aus LSsungen untersehiedlieher AgNO3-Konzentration etwa gleieh groBe AgJ-Mengen auszuf/~llen, muB das ~'~llungs- mittel Nag deshalb unter Umst/~nden sehr verschie- den dosiert werden.
Unter Beriieksichtigung der insgesamt fiir die Vor- bereitung der Analysenprobe sowie fiir die Anreiehe- rung und Bestimmung der Queeksflberspuren disku- tierten Sehwierigkeiten ergibt sieh folgende Arbeits- vorsehrfft, die, um reproduzierbare Analysenergeb- nisse zu erhalten, mSgliehst genau befolgt werden soUte:
A rbeitsvorschrift 5 g (bzw. 10 g) Silbermetall werden im 100 ml-Beeherglas zur Verhinderung yon Passivit/itseffekten mit etwa 3 ml Wasser versetzt und nach Znsatz yon 5 (10) ml 65°/0iger Salpeter- s~ure unter schwaehem Erw~rmen gelSst. W/~hrend des LSse- vorgangs wird das Gef/~13 dureh ein Uhrglas bedeekt. Die LSsung wird auf der Heizplatte (150°C) eingedampft, bis der entstehende Kristallbrei gerade beginnt, troeken zu werden. Nach dem Abkfihlen wird der Rfickstand in 20 ml Wasser gelSst. Die LSsung wird mit 2 M AmmoniaklSsung gegen Universalindieatorpapier auf etwa pH 3 eingestellt (Ammoniakverbraueh weniger als 1 ml). l~ach Zusatz yon 2ml 0,1°/0iger KaliumpermanganatlSsung sowie 0,5ml 0,01 M .~DTA-LSsung wird die ProbenlSsung mit insgesamt etwa 35 (20)ml Wasser in einen 100 ml-Schliffkolben ge- spfilt. Zur F~llung des Spurenfgngers werden dann 3 (16) ml 0,1M I~atriumjodidlSsung zugegeben (Endvolumen bei etwa 60 ml). Die Suspension wird 3 rain geschiittelt, dann zentrffugiert. Die klare fiberstehende LSsung wird de- kantiert.
Der zurfickbleibende AgJ-l~iedersehlag wird (ohne ihn auszuwaschen) in 15,0 ml 9 M Natriumthiocyanatl5sung unter Zusatz yon 3,0ml 3a/0iger Hydroxylammonium- chloridl5sung gelSst und mit 20,0 ml Wasser in einen Schfitteltriehter gespiilt. Nach Zusatz yon 2,0ml 0,1 IV[ •DTA-L5sung sowie 10,0 ml Puffer pH 4,6 (a) wird die L5sung mit i0,0 ml Dithizonl5sung (b) i rain geschiittelt. Die Dithizonphase wird dureh wenig Filterflockenmasse filtriert und soforb gegen reine DithizonlSsung photometriert (Zeiss-Elko II, l~ilter 1 62, 2 cm-Kfivetten). Die Dithizon-

Grubi~sch und Schukoff: Invers-voltammetrische Bestimmung yon Nickel(II)-Spuren am h~ngenden Quecksilbertropfen 201
15sung is~ als Vergleich nut kurze Zeit farbs~abil; deshalb sollte bei jeder Analyse gegen eine neu eingefiillte Vergleiehs- 15sung pho$ometriert werden.
Zur Erfassung des Blindwertes haben wir die zum LSsen der Silbereinwaage verwendete S~uremenge unter Zusatz yon 80 mg AgNOa p.a. bis fast zur Troekne eingedampft. Der Riickstand wurde entsprechend der Arbeitsvorschrfft weiterbehandelt.
Besondere Reagentien. a) Puffer pH4,6: LSsung, die je 1 M an Natriumaeetat und Essigsiure ist; b) DithizonlSsung: 100 mg Dithizon werden in 500 ml Tetrachlorkohlenstoff gelSs~. Die LSsung wird unter einer w~Brigen LSsung yon Hydroxylammoninmchlorid und ~DTA aufbewahrt. Zur Analyse wird jeweils ein Teil mi~ Tetrachlorkohlenstoff so welt verdiinn~, dal~, gegen CCla in 2 cm-Kiivetten gemessen, eine Extinktion zwischen 0,98 und 1,05 angezeigt wird. Die Farbstabilitiit dieser LTsung muB vor der Analyse kon- trolliert werden.
Kenndaten des A nalysenver/ahrens U n t e r den Bedingungen der Arbe i t svorschr f f t werden yon den in 5 g-Si lbere inwaagen en tha l t enen Queek- sf lberspuren 906/0, bei 1 0 g - E i n w a a g e n 81°/0 ange-
Tabelle
Silbereinwaage Relative Hg-Gehalt (g) Standardabweichung der untersuchten
Pr~iparate (%)
5 0,033 1. i0 - i 10 0,039 5 • 10 -5
re ieher t . Diese W e r t e fiir die Ausbeu te s ind fiber e inen wei ten Hg-Konzen t r a t i onsbe re i ch k o n s t a n t und kTn- nen als sys t ema t i sche r Feh le r ohne Bee in t r~ch t igung der Genan igke i t des Verfahrens in die E ich funk t ion m i t au fgenommen werden.
Als re la t ive S t a n d a r d a b w e i c h u n g (bereehnet aus je 12 vol ls t~ndigen Analysen) wurden die in der Tabel le zusammenges te l l t en W e r t e e rmi t te l t .
Die aus den s ta t i s t i schen S t reuungen yon 25 Bl ind- wer ten des gesamten Anreicherungs- u n d Best im- mungsver fahrens er rechnete Nachweisgrenze ergab ffir 10 g -E inwaagen einen W e r t yon c ----- 6 • 10-6°/c Hg.
Literatur
1. Fischer, H., Leopoldi, G.: Metal1 Erz 88, 154 (1941). 2. Graffmann, G.: Dissertation, Bochum 1969. 3. Jackwerth, E., Graffmann, G.: diese Z. 24:1, 96 (1968). 4. - - -- diese Z. 251, 81 (1970). 5. Maude, B.M., Wilkinson, K . L . : Rep. U.K. Atomic
Energy Auth. AERE-M 2008, 1968. 6. Murray, R.W. , McNeely, R. C.: Anal. Chem. 89, 1661
(1967); vgl. diese Z. 242, 35 (1968). 7. P~ibil, R.: Komplexone in der chemisehen Analyse.
Berlin: VEB-Verlag der Wissenschaften 1961. 8. Vat,k, V., ~edivee, V.: Chem. Listy 45, 10 (1951); vgl.
diese Z. 187, 223 (1952/53).
Priv.-Doz. Dr. E. Jackwerth Institut ffir Spektrochemie und angewandte Spektroskopie D-4600 Dortmund, Bunsen-Kirchhoff-Stral~e 11
Z. Anal. Chem. 253, 201--205 (1971) O by Springer-Verlag 1971
Invers-voltammetrische Bestimmung yon Niekel(II)-Spuren am h ingenden Queeksilbertropfen*
H. G~UBITSCH und J . SCHV~OFr**
Institut ffir anorganisch-chemische Technologic und analytische Chemic der Technischen Hochschule in Graz, 0sterreich
Eingegangen am 9. Juli 1970
Anodic Stripping Voltammetry of Nickel(II) Traces Using a Hanging Mercury.Drop Electrode. A process was deve loped which made possible the de t e rmina t ion of smal les t quant i t ies of n ickel ( I I ) using the above m e t h o d and an e lec t ro ly te consist ing of 0.1 M K N O 3 and 0.02 NI KSCN. I n the concen t ra t ion range of 5 × 10 -6 to 5 × 10 -s M nickel ( I I ) an excel lent l inear re la t ionship be tween the anodic dissolut ion currents of nickel enr iched on the mercu ry d rop and the n ickel ( I I ) concent ra t ion in the solut ion was es tabl ished, under the condit ion, however, t h a t the nickel concent ra t ion in the mercu ry is no t too high; otherwise the m e t h o d is no longer reproducible .
* Herrn Prof. Dr. A. v. Wacek zum 75. Geburtstag. ** Auszug aus der Dissertation yon J. Schukoff, Technische Hochschule in Graz, 1969.