Embed Size (px)
Citation preview

Z. Anal. Chem. 245, 233--239 (1969) 233
Bestimmung von Spurenverunreinigungen in Gallium und Galliumarsenid durch Neutronenaktivierungsanalyse*
K. H. NE]~B, H. ST6CKERT, R. BRAUN und H. P. BLEICH
Kerntechnische Lahoratorien der Siemens AG, Erlangen
Eingegangen am 30. 0ktober 1968
Determination o] Trace Impurities in Gallium and Gallium-Arsenide by Neutron Activation Analysis. In neutron activation of gallium and gallium arsenide very high specific activities of the radionuelides 72Ga and 78As are formed. Their rather short haft-life values of 14.1 and 26.5 h respectively permit relatively undisturbed deter- ruination of all foreign elements with long-lived radionuclides after a certain degree of decay of the matrix activities. These elements include Ag, Cd, Co, Cr, Fe, Hg, Iqi, Zn and others. ~owever, for the determination of elements with short-lived radionuclides, such as Mn, Cu, and Go, it is necessary to radiochemically process the highly radioactive sample (activity 1 to 10 Ci) behind suitable shielding as soon as possible after irradiation. Chemical separating schemes have been developed for the determination of both long-lived and short.lived radionuclides. These can be varied easily according to the number and nature of the impurity elements to be determined. The sensitivity attainable under optimal irradiation conditions lies between 2 • 10 -a and 2 • 10 -8 ppm. The authors discuss possible disturbance of the analysis due to side reactions and contamination of the liquid gallium by impurities in the packing material during irradiation.
Zusammen]azsung. Bei der NeutronenbestraMung yon Gallium und Galliumarsenid werden recht hohe spezi- fische Aktivit/~ten der Radionuklide 7~Ga und 78As gebildet. Ihre ziemlieh kurzen Halbwertszeiten yon 14,1 bzw. 26,5 h ermSglichen eine relativ ungestSrte Bestimmung aller Fremdelemente mit langlebigen Radionukliden nach weitgehendem Abklingen der Matrixaktivit/~ten; zu diesen Elementen gehSren u. a. Ag, Cd, Co, Cr, Fe, I-Ig, i~i, Zn. Fiir die Bestimmung yon Elementen mit kurzlebigen Radionukliden, wie Mn, Cu, Ge u. a. mug dagegen die hochradioaktive Probe (Aktivit/~t 1--10 Ci) m6glichst frfihzeitig naeh Bestrahlungsende hinter entsprechend starken Abschirmungen aufgearbeitet werden. Sowohl fiir die Bestimmung der langlebigen als auch der kurzlebigen Radionukhde wurden chemische Trennungsg/inge ausgearbeitet, die leicht in bezug auf Zahl und Art der zu bestimmenden Vernnreinigungselemente variiert werden k6nnen. Die bei Einsatz optimaler Bestrahlungsbedingungen erreichbaren unteren Bestimmungsgrenzen liegen zwischen 2.10 -a und 2.10 -8 ppm. M6gliche StSrungen der Bestimmung dutch ~qebeureaktionen sowie dutch Kontamination des w/~hrend der Bestrahlung fliissigen Galliums dureh im Verpackungsmaterial vorhandene Veruureinigungen werden diskutiert.
Gallium und Arson geh6ren zu den Elementen, die aufgrund ihrer relativ groBen Aktivierungsquer- schnitte yon etwa 3 bzw. etwa 5 barn bei der Be- strahlung mit thermischen Neutronen recht hohe spezifische Aktivit/~ten bilden. Das ist yon Vorteil, wenn geringe Gehalte der beiden Elemente in ande- ren Substanzen durch Neutronenaktivierungsanalyse bestimmt werden solien. Fiir die aktivierungsanalyti- sche Bestimmung yon Spuren anderer Elemente in Gallium und Galliumarseuid bedeutet das jedoch, da$ bei der NeutronenbestralMung sehr hohe Matrix. aktivitaten gebfldet werden. Die relativ kurzen Halb- wertszeiten der beiden haupts~ehliehen RadionukSde 7zGa und 7~As yon 14,1 bzw. 26,5 h ermSgliehen es, die langlebige Radioisotope bildenden Elemente nach
* Vortrag bei der Tagung ,,Analytische Probleme der Rein- darstellung yon Halblei~ern", Wiesbaden 24./25.9.1968.
weitgehendem Abklingen der Matrixaktivit/~ten zu bestimmen. Eine derartige Analyse bietet keine wesentlich gr6Bere Schwierigkeit, als eine Akti- vierungsanalyse anderer, weniger gut aktivierbarer Matrices. Allerdings werden dabei die Elemente, die ausschlieBlieh kurzlebige Radionuklide bilden, wie z.B. Kupfer, nieht erfaBt. Zu deren Bestimmung muB vielmehr die bestrahlte Substanz so bald wie m6glich nach Bestrahlungsende aufgearbeitet werden. Bei eincm reaktorfernen Laboratorium wie dem unseren bedeutet das, dab die hoehradioaktiven Proben mit spezifischen Aktivit~ten yon etwa 1 Ci/g in schweren Transportbeh/~ltern vom Kernreaktor zum Laboratorium gebraeht werden miissen. Es be- deutet aber auch vor allem, dab zumindest ein Toil der chemischen ~u an diesen Proben nur hinter Abschirmungen ausgeffihrt werden k6rmen.

234 K.H. hreeb, It. StSckert, 1~. Braun und H. P. Bleich:
Bei den vorliegenden Aktivitgten werden etwa 15 cm starke Bleiwgnde als Strahlungsschutz ben6- tigt (sog. SemiheiBe Zellen), alle Handhabungen in diesem Stadium mfissen mit Manipnlatoren und Fernzangen vorgenommen werden. Das hat zur Folge, dab die anwendbaren Abtrennungsveffahren den speziellen Gegebenheiten des Zellenarbeitens anzupassen sind. AuBerdem mfissen ffir die ersten Arbeitssehritte m6glichst wirksame Trennverfahren eingesetzt werden; es ist yon Vorteil, wenn die abge- trennten Pr/~parate bereits nach einem oder zwei Trennschritten soweit frei yon St6raktivit/it sind, da2 sie ~us der Zelle herausgenommen and mit den bequemeren und zeitsparenderen Techniken des normalen Arbeitens welter gereinigt werden k6nnen. Szab6 u. 1%ausch [3] haben als erste Abtrennungs- schritte extraktive Verfahren vorgeschlagen. Ffir die Auftrennung einer gr6Beren Zahl yon Elementen sind jedoch, wie aueh aus den Darlegungen yon Gebauhr [1] hervorgeht, vielfach F/~llungen yon Vorteil ; beson- ders solche aus sauren LSsungen weisen oft eine fiir die ersten Abtrennungen ausreichende Selektivit/~t auf. Aueh die weiteren Reinigungsschritte mfissen eine sehr gute Trennsehiirfe aufweisen, da bei den hoeh- aktiven Matrices Dekontaminierungsfaktoren von 10s--10 ~~ die Voraussetzung ffir die Erreichung guter Bestimmungsempfindlichkeiten sind. Das grit aueh dann, wenn im 7-Spektrum keine Peakkoinzidenz zwischen Matrixaktivitgt und gesuchter Aktivitgt auftritt; sehon wenn die 7-Energie des gesuchten Radionuklides niedriger liegt als die der Matrixaktivi- tgt, k6nnen durch den Compton-Untergrnnd nnd seine statistischen Schwankungen wesentliche Verschlech- terungen der Bestimmungsempfindlichkeiten hervor- gerufen werden. Die dutch die hohen Matrixaktivitgten bedingten arbeitstechnischen Erschwernisse lassen es als zweck- m~Big erscheinen, den Kreis der unmittelbar nach Bestrahlungsende zu bestimmenden Elemente m6g- lichst klein zu halten und die Elemente mit ls lebigen Radionukliden in einer zweiten Probe nach weitgehendem Abldingen der Matrixaktivit~t zu bestimmen. Die Aufteflung in kurz- und langlebige Radionuklide wird dabei nach rein praktischen Gesichtspunkten vorgenommen; ein bestimmtes Ele- ment wird der Gruppe zugeordnet, in der es unter den gegebenen Voraussetzungen am einfachsten und mit der besten Empfindlichkeit bestimmt werden kann.
Auswahl der zu bestimmenden RadionukUde Bei Aktivierungsanalysen mit angestrebter hoher Nachweisempfindlichkeit muB natfirlich auf mSgliehe
St6rungen durch andere Kernreaktionen Rficksicht genommen werden. In besonderem MaBe gilt das ffir die Elemente, die den Matrixelementen im periodi- schen System benaehbart sind, im vorliegenden Fall ~lso ftir Germanium und Zink. Von den drei dureh (n, ~)-Reaktionen entstehenden, ftir die Aktivierungs- analyse interessanten Radioisotopen des Germaniums k6nnen zwei auch durch StSrreaktionen gebildet werden, und zwar 71Ge fiber die Reaktionskette
69Ga ~ n ' r 70Ga ~ - -~ ~OGe ~ n ' ~ 71Ge ~11~
sowie TSGe dutch (n, p)-Reaktion aus 75As. Trotz vermutlieh sehr geringer Ausbeute dieser Reaktio- nen, fiber die in der Literatur nut rage Angaben vor- liegen, k6nnen StSrungen im unteren ppb-Bereieh nicht zuverl/issig ausgeschlossen werden. Ffir die Bestimmungen wnrde daher das Radionuklid 77Ge herangezogen, das weder aus Gallium noch aus Arsen gebildet werden kann. Xhnlich liegen die Verh/~ltnisse bei Zink. Das kurz- ]ebige Radionuklid ~mZn, das an sich bei geeigneter Arbeitsweise die bessere Bestimmungsempfindlich- keit erm6glicht, wird aus 6~Ga durch (n, p)-Reaktion in betr/ichtlicher Ausbeute gebildet, wodurch Zn- Gehalte im ppm-Bereich vorget/iuseht werden [2]. Die aktivierungsanalytische Bestimmung yon ppb- Gehalten Zn ist daher nur fiber das langlebige ~Zn m6glieh. Bei den anderen Elementen wurde jeweils das emp- findlichste, fiber (n, 7)-Reaktionen gebildete y-strah- lende Radioisotop gemessen. Lediglich bei Nickel wurde wegen der ungfinstigen Eigensehaften der 1%adioisotope 6~Ni bzw. ~sNi das dureh (n, p)-Reak- tion gebildete 5sCo bestimmt. Die y-Peaks der im Co- Endpr/~parat nebeneinander auftretenden Radio- isotope ~sCo und ~~ liegen soweit getrennt (0,8t bzw. 1,17 MeV), dab ledig]ieh bei gr6Berem Uber- schu~ an 6~ eine geringfiigige, jedoch unvermeid- liche Verschlechterung der Nachweisgrenze ffir ~sCo zu beffirchten ist.
Vorbereitung der Bestrahlungsproben
Der grol]e Vorzug der Aktivierungsanalyse hinsicht- lieh Blindwertfreiheit l~Bt sich natfirlich nut auf- rechterhalten, wenn dafiir gesorgt ist, dab die Probcn auch vor oder wghrend der Bestrahlung keine zuss lichen Verunreinigungen aufnehmen. Eine derartige Gefahrenquelle ist die Kontamination der Proben dttrch Verurtreinigungen aus den als Verpaekungs- material verwandten Quarzampullen. Auch bei ober- fl/~ehlieh sehr sorgf/~ltig gereinigten Quarzampullen ist eine Verschmutzung der Proben dadurch mSglich,

Bestimmung yon Spurenverunreinigungen in Gallium und Galliumarsenid 235
daB infolge Kernrfickstoges bei der (n, ~)-Reaktion die Bindungsverh~ltnisse oberfl~chlicher Verunreini- gungen veriindert, bzw. Verunreinigungen aus den oberen Zonen des Quarzglases an die Oberfl~che ~ransportiert werden k6nnen; beide Reaktionen bringen also radioaktive Verunreinigungen in Kon- takt mit dem Probenmaterial. Bei festen Proben wie GaAs ist das nieht allzu stSrend, da man deren Ober- fl~chen naeh der Bestrahlung und vor Beginn der Analyse dureh sorgfi~ltiges Ab~tzen reinigen kann. Gallinm dagegen ist w~thrend der Bestrahlung fliissig und nimmt evil. Vertmreinigungen der Ampnllen- oberfl~ehe in sieh auf, so dab eine nachtr~gliche Oberfls der Probe sinnlos ist. Abhilfe liiBt sieh praktisch nut durch die Verwendung yon hoehreinem Quarzglas (z. B. Suprasil) als Ampullen- material schaffen, das natfirlich vor Einpacken der Proben noeh einer sorgfi~ltigen Oberfl~chenreinigung unterworfen werden mug. Zur Feststellung einer evtl. verbliebenen Restkontamination werden in gleicher Weise vorbehandelte Leerampullen mitbe- strahlt und der gleichen Behandlung hinsichtlich des IterauslSsens des Ga]liums unterzogen wie die mit Proben beschickten Ampullen.
Wegen der Unterteilung in kurz- und langlebige Radionuklide sind ffir eine l~lbersichtsanalyse zwei Proben notwendig. Diejenige zur Bestimmung der Elemente mit langlebigen Radionukliden Mrd 3 bis 4 Tage lang bei einer Neutronenflugdichte yon etwa 6 �9 101an �9 cm -~ �9 sec -1 bestrahlt (Forschungsreaktor Karlsruhe), die Probe ffir die Elemente mit km'z- lebigen Radionuk]iden dagegen 3 - - 4 h bei einer Neutronenflugdichte yon etwa 1 �9 10 lan �9 cm -~ �9 see -1 (Forsehungsreaktor Mfinchen). Zur Eichung der Be- stimmungen werden Standardproben der betreffenden Verunreinigungselemente mitbestrahlt. Bei der rela- t iv grogen Zahl der zu bestimmenden Elemente wi~re es allerdings sehr aufwendig, bei jeder Probenbe- strahlung auch alle Standardsubstanzen mitzube- strahlen und aufzuarbeiten. Daher wnrde diese umfangreiche Eiehung nur bei den ersten Bestrah- lungen vorgenommen; bei den folgenden Bestrah- lungen wurden jeweils vier bis ffinf dieser Standards mitgegeben, davon einer bei jeder Bestrahlung, and zwar Zink bei den Langzeit- and Kupfer bei den Kurzzeitaktivierungen. Der dadnreh gewahrte An- sehlug yon einer Bestrahlung zur n~chsten ist m6g- lich, wenn immer die gleiehe Reaktorposition benutzt werden kann; das Verfahren liefert, wie entsprechende Versuche zeigten, bei den hier angestrebten Genauig- keiten yon etwa 200/6 durchaus zufriedenstellende Ergebnisse.
AuRrennung der kurzlebigen Radionuklide
In dem in der Abb. 1 gezeigten Trennungsgang sind schematisch die wieh~igsten Sehri~te fiir die Auf- trennung der kurzlebigen Radionuklide angegeben. Alle innerhalb der gestriehelten Umrandung angeffihr- ten Arbeiten mfissen dabei hinter Abschirmungen vor- genommen werden. In fiberwiegendem Mage werden Fallungen eingesetzt, und zwar vorzngsweise aus relativ stark sauren L6sungen, dureh die eine sehr selek- give Abtrennung des gewiinschten Elementes yon den Matrixaktivitaten errreicht wird. Daher sind in fast allen Fiillen die betreffenden Elemente bereits nach der ersten F$llungsoperafion soweit frei yon 72Ga bzw. 76As, dab sie aus der Abschirmung herausge- nommen und auBerhalb in gewohnter Weise weiter verarbeitet werden k6nnen. Andernfalls is~ eine Um- fs in Anwesenheit yon Rfickhaltetri~gern zweck- m~Big, die einfaoh and zuverli~ssig zu einer weiteren starken Absenkung der MatrLxaktivit~t ffihr~. Eine gewisse Abweiehung von dieser Arbeitsweise ist beim Mangan notwendig. Bei der Ausf~]lung des MnO~ aus saurer L6sung mit Bromat werden relativ groge 72Ga- and 76As-Aktiviti~ten mitgerissen, so dab abzusehen ist, dab mit einer Umf~llung kein aus- reiehend reines Pri~parat erhalten wird. Andererseits erfordert die relativ kurze Halbwertszeit des ~6Mn yon 2,5 he ine schnelle Isolierung. Als sehr gfinstiges Verfahren hierffir hag sieh der Anionenaustausch erwiesen; aus 8 N salzsaurer L6sung wird Ga quanti- tat iv und As zum grSBten Tell am Austauscherharz adsorbiert, w~hrend Mn prak~iseh quantitativ in der L6sung verbleibt. Zur Zeitersparnis wird der Ionen- austausch nicht als Siiulenoperation ausgeffihrt, sondern dureh einfaehes Rfihren der Probenl6sung und ansehhegendes Filtrieren. Das Filtrat ist prak- tiseh frei yon Fremdaktivits so dab alas aus ihm dureh alkalische Hydroxidfs isolierte Mn-PrKpa- rag direkt zur Aktiviti~tsmessung gegeben werden kann.
Das mit dem Cu-Cd-Niederschlag in re]ativ starkem MaBe mitgef~llte 76As l~$t sich weitestgehend durch Destillation enffernen. Die anschliegende Cu-Cd- Trennung ist praktisch immer notwendig, da die intensivsten y-Linien des 64Cu and des nsCd koinzi- dieren and auch fiber die Ha]bwertszeit eine Unter- seheidung beider Radionuklide nur mit betrs Einbugen an Empfindhchkeit m6glieh ist. Die mit dem angeffihrten Trennungsgang erreichbare Trennsehiirfe ]~gt sieh wohl am besten mit den folgenden Zahlen demonstrieren: Eine 500 mg schwere Ga-Probe bzw. eine 200 mg schwere GaAs-Probe weist nach der Kurzzeitbestrahlung zum Zeitpunkt

236 K. It. Neeb, H. St6ckert, R. Braun und II. P. Bleieh:
I + j~ 10 mg W-, Ge-, Mn-, Cu-, Cd-Tr~ger I in Sall~etersSure 15sen, weitgehedd einengen
Umfiillun~ I ~Fil~rat mi~ Sehwefels~iure weRgehend emengen /
1 Nd ~i
IUm..~ ~ ~ . ' ~ - - q - NaBrOa, erhigzen
~~~ung in8~ I~C1 ]~:;onenaus~ausch Bre~at mi~ ~atI," HaSO , zerst6ren an Dowex-l,mi~ 8 lg HCI waschen, q- Wasser + gesSgK H~S-Wasser Austauscher u mi~ NaaS-Wasser waschen
~ ~ ~ ' ~ ~ Haup~menge ~Ga, ~As verwerfen
+ Ga s+ + As s+ q- Natronlauge + ttaO s
CuS + CdS
mig Salzs~ure + .N~I:[~ �9 HzSO ~ + KBr versetzen, As abdestillieren, R~cks~and + Wasser + I~H~0H + KCN -}- H~S
1 / PxlS
in Salzs~ure 16sen mig Salzs~ure schwach ans~uem -}- NaOH q- As 8+ + H~S |
mit Salzs~. e-Salpeters~ure 16sen + Na0H + As 3+ + H~S
1 Abb. 1. Trennungsgang fiir die kurzlebigen Radionuklide
der Auf t rennungen eine Aktivi~/~ yon 101~ bis 1011 Zerf. /min auf. Alle nach den Abt rennungen erhal~e- nen Endprs sind dagegen prakt isch frei yon 72Ga- und ~6As-Aktivit~t, so dab noch Verunreini- gungsaktivi t / i ten yon unter 100 I m p / m i n y-spektro- metr isch ohne St6rungen erfaBt werden k6nnen.
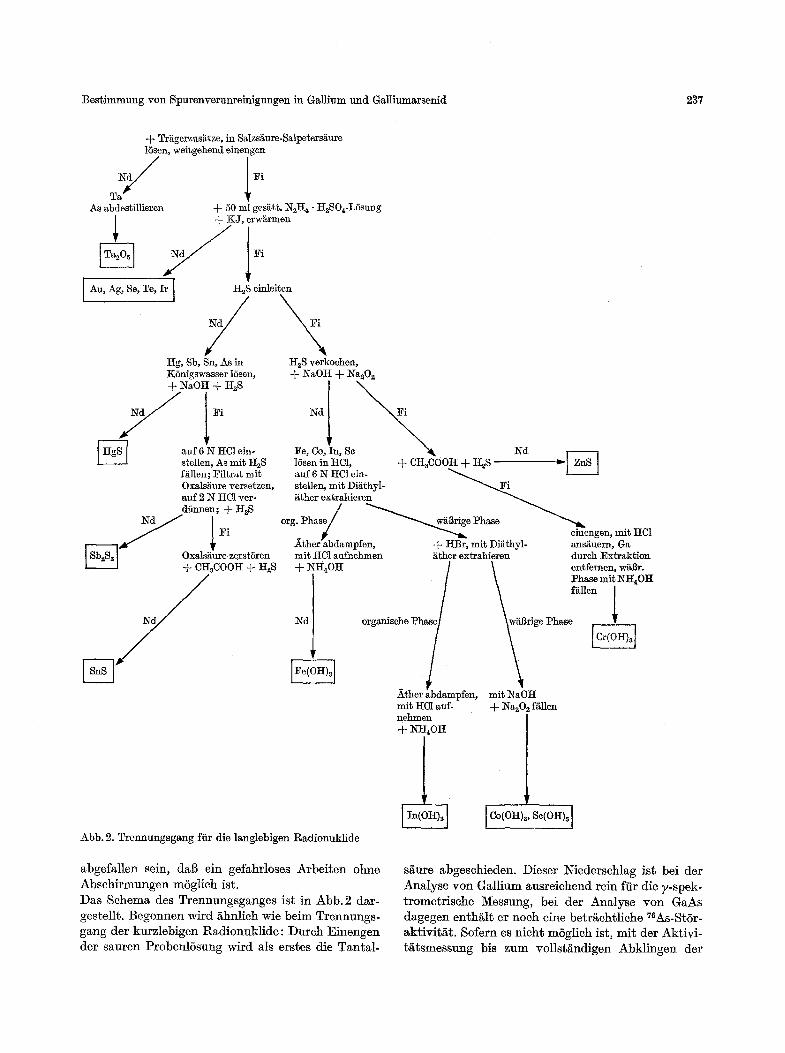
A u f t r e n n u n g der langlebigen Radionuklide ~iit den Arbei ten zur Auf t rennung der langlebigen Radionuklide wird einige Tage naeh Bestrahlungs- ende begonnen. Es ist dazu nichg no twen~g , dag die Matrixaktivit/~ten bei Beginn der Arbei ten voll- st/~ndig abgeklungen sind, sie miissen nu t soweit
Bestimmung yon Spurenverunreinigungen in Gallium und Galliumarsenid 287
4- TrSgerzus~tze, in SalzsSure-Salpet~_xs~ure lSsen, weitgehend einengen
As abdestillieren -t- 50 ml gesgtt. N2H~- H~S0~-L6sung 4- KJ, erwiirmen
Hg, Sb, Sn, As in Kbnigswasser Ibsen, 4- NaOH 4- H~S
/ I auf 6 N HC1 ein- stellen, As mit It,S f/~llen; Filtrat mit Oxalsiiure versetzen, auf2 lq HC1 ver- diinnen; 4- H2S
0xalsgure. zarstSren 4- CH~COOH 4- HaS
HaS verkoehen, 4- ~a0H -t- Na~0a
Fe, Co, In, Se Nd [ ~ Ibsen in HC], 4- CI-IaC00]=[ 4- tt~S ~t Zn~ ] auf 6 N HC1 ein- ~ ! I stellen, rait Dis " - ~ i ~ther extrahieren
~. ~. _ ~ einengen, mit~ tICl Ather abdampfen, 4- ~Br, mit Di~thyl- ans~uern, Ga mit HC1 aufnehmen ~ther extrahieren durch Extrak~ion + l~HaO~[ \ entfernen, wKBr.
\ Phase mit NH~0H \ fallen 1
Nd ~ g r i g e Phase
mit Na0H 4- l~az0 z f~llen
Abb. 2. Trennungsgang ffir die langlebigen Radionuklide
organisehe Ph
~ther abdampfen, mit HC1 auf- nehmen +17H OH
[ Co(OHh, Se(O~h]
abgefallen sein, dab ein gefahrloses Arbeiten ohne Abschirmungen mbglich ist. Das Schema des Trennungsganges ist in Abb. 2 dar- gestellt. Begonnen wird iihnlich wie beim Trennungs- gang der kurzlebigen Radionuklide: Durch Einengen der sauren Probenlbsung wird als erstes die Tantal-
s/iure abgeschieden. Dieser Iqiederschlag ist bei der Analyse yon Gallium ausreichend rein ftir die y-spek- trometrisehe Messung, bei der Analyse yon GaAs dagegen enths er noeh eine betrs 76As-Stbr- aktivit/it. Sofern es nicht mbglich ist, mit der Aktivi- t/itsmessung bis zum vollst/~ndigen Abklingen der

238 Neeb, St6ckert, Braun und Bleich: Bestimmung von Spurenverunreinigungen in Gallium und Galliumarsenid
As-Aktivit/tt zu warren, empfiehlt sich als Reini- gungsschritt ein Abdestillieren des As als AsC13 in Anwesenheit yon As-Tr/tger. Die anschlieBende kombinierte F/tllung mit Hydrazin und K J fiihrt zu einer gemeinsamen Abseheidung der Edelmetalle sowie des Se und Te. Im allgemeinen wird in diesem Niederschlag wegen der sehr guten Aktivierbarkeit des Goldes die l~SAu-Aktivit/tt weir fiberwiegen, auch wenn nur sehr geringe Au-Geha]te vorliegen. Zur Bestimmung der anderen Radionuklide ist daher die Aufnahme eines zweiten ?-Spektrums naeh dem Abklingen des *gSAu (Halbwertszeit 2,7 d) notwendig. In diesem zweiten 7-Spektrum lassen sieh die bei hoehreinen Proben oft in der N/the der Nach- weisgrenzen liegenden Gehalte der anderen vier Ele- mente meist ohne weitere Auftrennung 7-spektro- metrisch bestimmen. Ist jedoch eines der vier Ele- mente in starkem ~bersehuB vorhanden, so kann eine weitergehende Auftrennung notwendig werden. M6g- lichkeiten hierzu sind gegeben durch die F/tllung des Ag als HaIogenid aus leieht oxydierender L6sung, die Abtrennung des Selens dureh oxydative Destillation als Bromid oder die F/tllung des Ir als Cs~[IrC16]. Welehe der angegebenen Reaktionen angewandt wird, bzw. welehe Kombination yon Reaktionen, h/tngt in erster Linie yon der radioehemischen Zusammen- setzung dieses Niederschlages ab. Zu der Auftrennung der It2S-Fallung brauehen keine weiteren Anmerkungen gemach$ werden; die Grund- regel ist auch hier, dab die ehemischen Auftrennungen nur soweit vorgenommen werden, wie es zur Er- reiehung guter Iqachweisgrenzen bei y-spektrometri- scher Auswertung notwendig ist. Aus dem Ffltrat der H~S-F/tllung werden mit Natron- lauge -- Na~02 die schwerlSslichen Hydroxide aus- gefallt; Cr un4 Zn gehen bei dieser Arbeitsweise in das Filtrat und k6nnen dort leieht dureh H2S-F/~llung aus essigsaurer LSsung voneinander getrenn$ werden. Aus der sam'en L6sung des Hydroxidniederschlages werden Fe und In als Chlorid bzw. Bromid mit Ather extrahiert. Die dabei in der w/tBrigen Phase ver- bleibenden Radioisotope der Elemente Co und Se k6nnen bei nieht zu groBem Aktivit/ttsuntersehied y-spektrometrisch nebeneinander bestimmt werden; im anderen Falle ist ihre Auftrennung dureh F/illung des Co mit a-Nitroso-fl-naphthol ohne Sehwierig- keiten m6glich.
A k t i v i t / i t s m e s s u n g e n
Alle Aktivit/ttsmessungen warden mit einem 512 Kanal-?-Spektrometer durchgeffihrt. Als Detektor wurde dabei wegen der niedrigen vorliegenden y-Akti-
vit/tten ein NaJ(T1)-Bohrloehdetektor verwandt. Die wesentlich bessere Energieaufl6sung des Ge(Li)-De- tektors 1/tl]t sich bei der Analyse hoehreiner Proben leider noch nicht ausniitzen, da dessen gegeufiber dem NaJ(TI)-Kristall geringere Z/thtausbeute die Nach- weisgrenzen merklich versehlechtern wfirde. Bei weniger reinen Proben ,nit Verunreinignngsgeha]ten fiber etwa 1 ppm 1/tBt sich der Ge(Li)-Detektor da- gegen in vielen F/tllen mit groBem Vorteil einsetzen. Naeh unseren bei anderen Analysen gemachten Er- fahrungen kann durch Verwendung dieses Detektors ein wesent]icher Teil der chemischen Trennarbeit ein- gespart werden. Ffir die Auswertung der y-Spektren wurden jeweils einige Kan/tle im Photopeak addiert und mit der ent- sprechenden Auswertung der betreffenden Standard- probe verglichen. Bei Vorliegen eines Elementes unter der Nachweisgrenze, kenntlich am Fehlen des be- treffenden Photopeaks im y-Spektrum, wurde die ffinf- fache Standardabweichung des Spektrenuntergrundes an dieser Stelle als Nachweisgrenze eingesetzt.
E m p f i n d l i c h k e i t u n d G e n a u i g k e i t
In der Tabelle sind die unter den angegebenen Be- dingungen erzielbaren unteren Bestimmungsgrenzen angegeben, zusammen mit dem betreffenden Radio- nuklid und dessen zur Auswertung herangezogener
Tabelle. Bestimmungsemp]indlichkeit bei der Aktivierungs- analyse yon Gallium und Galliumarsenid. (Probeneinwaage 500 mg)
Element Bestimmtes Gemessene Untere ]3estim- Radionuklid y - L i n i e mungsgrenze
[MeV] [ppm]
Ag n~ 0,66 Au l~SAu 0,41 Cd n~Cd 0,34 Co s~ 1,17 Cr 5*Cr 0,32 Cu ~4Cu 0,51 Fe 59Fe I,i0 Ge 77Ge 0,22 Hg ~-~ 0,28 In */4mln 0,19 Ir *~21r 0,31 Mn 56Mn 0,84 Ni 5sCo 0,81 Sb '24Sb 0,60 Sc 46Sc 0,89 Se 7~Se 0,27 Sn **3Sn 0,39 Ta *S2Ta 1,2 Te l~5mTe 0,11 W xs:w 0,47 Zn ~sZn 1,11
4 10 -4 2 10 -6 4 10 -4 1 10 -4 4 10 -5 5 10 -5 2 10 -~ 2 10 -3 I 10 -5 4 10 -4
2 10 -5
6 10 -4 2 �9 10 -3 2 10 -4
1 10 -4 1 i0 -a
8 10 -4 4 i0 -4
6 10 -4 1 10 -4
1 10 -4

E. Schuster und K. WohUeben: Bestimmung yon leichten Elementen in Silicium und Selen 239
y-Linie. Diese Grenzen liegen bei den meisten ange- ffihrten Elementen im Bereich 10-a--10 -4 ppm; der ungfinstigste Fall sind Fe, Ge und Ni mit Bestim- mungsgrenzen yon 2" 10 -a ppm, der gfinstigste Fall ist das Gold mit einer solchen yon 2" 10 -6 ppm. Die angeffihrten Werte wurden nicht aus den spezifischen Aktivits der Standardproben berechnet, sondern experimentell ermittelt . Allerdings konnten wegen mangelnder Reinheit der zur Verffigung stehenden Proben die angegebenen Grenzen nicht bei allen Elementen erreicht werden. In derartigen F~llen wurde die untere Bestimmungsgrenze ausgehend yon der Probe mit dem uiedrigsten Gehalt an dem be- treffenden Element berechnet. Die Fehlerbreite der fiber der ~qachweisgrenze liegen- den Gehaltsangaben kann mit etwa 200/0 veranschlagt werden; dieser Wef t wurde in Voruntersuchungen mit radioaktiven Tracern ermittelt. Er ist zum grol~en Tell auf Schwankungen in der chemischen Ausbeute zurfickzuffihren und kann bei der Durchffihrung yon
Ausbeutebestimmungen auf 5--10~ reduziert wer- den. Wegen des relativ grol3en Arbeitsaufwandes wird man bei derartigen ]~bersichtsanalysen jedoch nur dann Ausbeutebestimmnngen vornehmen, wenn die damit verbundene Genauigkeitssteigerung wirklich notwendig ist.
Wir danken Herrn Dr. Sirfl, Forschungslaboratorium Mfin- chen der Siemens AG.. und Herrn Dr. Raab, Forschungs- laboratorium Erlangen der Siemens A.G. ffir die t~berlassung der Analysenproben sowie Herrn Prof. Dr. Gebauhr ffir anregende Diskussionen.
Literatur 1. Gebauhr, W.: diese Z. 245, 209 (1969). 2. Lerch, P., u. L. Kreienbfihh Chimia (Aarau) 15, 519
(1961); vgl. diese Z. 198, 373 (1963). 3. Szab6, E., u. E. Rausch: Acta Chim. Acad. Sci. Hung.
54, 231 (1967).
I)r. K. H. l~eeb Kerntechn. Lab. der Siemens A.G. 8520 Erlangen, Giinther Scharowsky-Strate 2
Z. Anal. Chem. 245, 239--244 (1969)
Bestimmung von leichten Elementen in Silicium und Selen durch Ionenaktivierungsanalyse*
E. SCHUSTER und K. WOHLL~BEN
Kern~echnisehe Labora~orien und Forschungslaboratorium Erlangen der Siemens A.G.
Eingegangen am 30. 0ktober 1968
Determination o] Light Elements in Silicon and Selenium by Ion-Activation Analysis. Elements with low atomic number either cannot be determined at all by activation with thermal neutrons or only with very poor detection limit. In recent years, irradiation with ions (protons, deuterons, tr i t ium ions, 3He- and ~He-ions) for activation analysis of the light elements has therefore gained increasing importance. There are basic differences between ion and neutron activation, which are discussed. Because of their relatively small range in matter , ions are particularly suited for analyzing surfaces as well as thin deposited films and corrosion layers. Since the activation depth can be established in advance by appro- priate selection of the analysis parameters, the distribution of foreign mat te r in the matr ix material can also be analyzed. Proton and deuteron reactions are employed to determine the presence of oxygen, nitrogen and carbon, but other elements such as boron and aluminium can also be detected by ion activation. The detection limit attain- ed in analyzing the elements mentioned is within the range of 1 to 100 ppb with an ion bombarding energy of 3.0 MeV, a flux density of 5 ~A/cm 2 and an irradiation period which corresponds to the half-life of the element to be activated. A report is given on the determination of carbon in silicon and oxygen in thin-deposited selenium films and the results are discussed.
* Vortrag bei der Tagung ,,Analytische Probleme der Reindarstellnng yon Halbleitern", Wiesbaden, 24.--25. 9. 1968.





























