Embed Size (px)
Citation preview

Regulation der plastidären Genexpression
durch kernkodierte Faktoren:
Untersuchungen zur Rolle der plastidären Transkriptionskinase
(PTK) in der chloroplastidären Genexpression in A. thaliana
Dissertation
zur Erlangung des Grades eines Doktors der Naturwissenschaften
der Fakultät für Biologie und Biotechnologie
an der Internationalen Graduiertenschule Biowissenschaften
der Ruhr‐Universität Bochum
angefertigt
in der Arbeitsgruppe
Pflanzliche Zellphysiologie und Molekularbiologie
vorgelegt von
Hacer Türkeri
aus Niğde/Türkei
Bochum
Februar, 2010

Regulation der plastidären Genexpression
durch kernkodierte Faktoren:
Untersuchungen zur Rolle der plastidären Transkriptionskinase
(PTK) in der chloroplastidären Genexpression in A. thaliana
Dissertation
zur Erlangung des Grades eines Doktors der Naturwissenschaften
der Fakultät für Biologie und Biotechnologie
an der Internationalen Graduiertenschule Biowissenschaften
der Ruhr‐Universität Bochum
angefertigt
in der Arbeitsgruppe
Pflanzliche Zellphysiologie und Molekularbiologie
vorgelegt von
Hacer Türkeri
aus Niğde/Türkei
Bochum
Februar, 2010
Erstgutachter: Prof. Dr. Gerhard Link
Zweitgutachter: Prof. Dr. Franz Narberhaus
Tag der mündlichen Prüfung: 13.04.2010

Regulation of Plastid Gene Expression by
Nucleus‐encoded Factors:
Role of the Plastid Transcription Kinase (PTK) in the Control of
Chloroplast Gene Expression in Arabidopsis thaliana
Dissertation
To obtain the degree Doctor Rerum Naturalium (Dr. rer. nat.)
at the Faculty of Biology and Biotechnology, Ruhr‐University Bochum
International Graduate School Biosciences
Ruhr‐University Bochum
Laboratory of
Plant Cell Physiology and Molecular Biology
submitted by
Hacer Türkeri
from Niğde, Türkei
Bochum
Februar, 2010
accepted on the recommendation of
Prof. Dr. Gerhard Link, first supervisor
Prof. Dr. Franz Narberhaus, second supervisor

Canım anneme ve babama..

Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. Gerhard Link für die Bereitstellung des
Arbeitsplatzes, die Überlassung dieses interessanten Forschungsthemas und die exzellente
fachliche Betreuung. Seine hilfreichen Anregungen und motivierenden Worte haben
maßgeblich zum Gelingen dieser Arbeit beigetragen.
Bei Herrn Prof. Dr. Franz Narberhaus bedanke ich mich für die freundliche Übernahme des
Zweitgutachtens.
Allen derzeitigen und ehemaligen Mitgliedern der Arbeitsgruppe „Pflanzliche Zellphysiologie
und Molekularbiologie“, Dr. Jennifer Schweer, Dipl. Biol. Andrea Kolpack, Dipl. Biol. Sylvia
Bock, Thomas Pieta, Brigitte Link und Dr. Heike Loschelder möchte ich ganz herzlich für die
nette Arbeitsatmosphäre und die schöne Zeit im und außerhalb des Labors danken.
Ein besonderer Dank geht an Frau Brigitte Link für ihre exzellente technische Unterstützung,
die zum Gelingen dieser Arbeit von großer Bedeutung war.
Frau Dr. Jennifer Schweer danke ich für ihre stetige Hilfsbereitschaft und die Zusammen‐
arbeit bei den Phosphorylierungsstudien.
Für das freundliche Korrekturlesen bedanke ich mich ganz herzlich bei Frau Dr. Meriyem
Aktas.
Bei Dipl. Biol. Jan Gleichenhagen möchte ich mich für die freundliche Hilfe bei den CD‐
Messungen bedanken.
Bedanken möchte ich mich auch bei all meinen Freunden dafür, dass sie immer für mich da
waren.
Ve son olarak canım aileme, hayatım boyunca sevgilerini ve desteklerini benden
esirgemedikleri için sonsuz teşekkürler ediyorum.

Inhaltsverzeichnis
I
Inhaltsverzeichnis
ABBILDUNGS‐ UND TABELLENVERZEICHNIS V
ABKÜRZUNGSVERZEICHNIS VII
GENBEZEICHNUNGEN XI
1 EINLEITUNG 1
1.1 EVOLUTION, ENTWICKLUNG UND FUNKTION DER CHLOROPLASTEN 1
1.2 ORGANISATION UND EXPRESSION DES PLASTIDENGENOMS 2
1.3 TRANSKRIPTION BEI PROKARYOTEN UND EUKARYOTEN 4
1.4 PLASTIDÄRE TRANSKRIPTION 5
1.4.1 PLASTIDÄRE PROMOTORELEMENTE 5
1.4.2 PLASTIDÄRE RNA‐POLYMERASEN 6
1.4.3 PLASTIDÄRE SIGMAFAKTOREN 10
1.4.4 DIE PLASTIDÄRE TRANSKRIPTIONSKINASE‐PTK 10
1.5 CK2‐PROTEINKINASE IN PFLANZEN 12
2 ZIELSETZUNG 16
3 MATERIAL UND METHODEN 18
3.1 MATERIAL 18
3.1.1 CHEMIKALIEN UND VERBRAUCHSMATERIALIEN 18
3.1.2 ENZYME UND KONJUGATE 21
3.1.3 PUFFER UND LÖSUNGEN 24
3.1.4 PFLANZENMATERIAL 25
3.1.5 BAKTERIENSTÄMME 24
3.1.6 PLASMIDE UND KLONE 25

Inhaltsverzeichnis
II
3.1.7 NÄHRMEDIEN 28
3.1.8 OLIGONUKLEOTIDE 28
3.1.9 INTERNETADRESSEN 29
3.1.10 COMPUTERGESTÜTZTE BILDBEARBEITUNG 29
3.2 METHODEN 30
3.2.1 DNA‐ISOLIERUNG 33
3.2.1.1 ISOLIERUNG GEMOMISCHER DNA 30
3.2.1.2 ISOLIERUNG VON PLASMID‐DNA 30
3.2.2 RNA‐ISOLIERUNG 30
3.2.3 MARKIERUNG VON NUKLEINSÄUREN 30
3.2.3.1 MARKIERUNG DER 5´‐ENDE VON DNA‐FRAGMENTEN 30
3.2.3.2 DIG‐MARKIERUNG VON RNA DURCH IN VITRO‐TRANSKRIPTION 31
3.2.4 KAPILLAR‐TRANSFER VON RNA (NORTHERN‐BLOT) 31
3.2.5 RNA/RNA‐HYBRIDISIERUNG 31
3.2.6 AMPLIFIZIERUNG VON NUKLEINSÄUREN 31
3.2.6.1 POLYMERASE‐KETTENREAKTION (PCR) 31
3.2.6.2 REVERSE‐TRANSKRIPTION‐PCR (RT‐PCR) 32
3.2.7 ELUTION VON DNA‐FRAGMENTEN 32
3.2.8 IN VITRO‐MUTAGENESE 32
3.2.9 DNA‐SEQUENZIERUNG 32
3.2.10 FLORAL DIP 32
3.2.11 ANZUCHT UND SELEKTION VON PFLANZEN 33
3.2.12 EXPRESSION REKOMBINANTER PROTEINE 33
3.2.12.1 BAKTERIELLE ÜBEREXPRESSION MIT HILFE DES PQE‐SYSTEMS 33
3.2.12.2 BAKTERIELLE ÜBEREXPRESSION MIT HILFE DES PMAL‐SYSTEMS 34
3.2.13 REINIGUNG REKOMBINANTER PROTEINE 33
3.2.13.1 ISOLIERUNG UND RENATURIERUNG VON PROTEINEN AUS INCLUSION BODIES 34

Inhaltsverzeichnis
III
3.2.13.2 REINIGUNG NATIVER PROTEINE 34
3.2.14 BIS‐TRIS‐POLYACRYLAMID‐GELELEKTROPHORESE (BIS‐TRIS‐PAGE) 35
3.2.15 NATIVE‐PAGE 35
3.2.16 ELEKTROPHORETISCHER TRANSFER VON PROTEINEN AUF NITROCELLULOSE‐MEMBRANEN
(WESTERNBLOT) UND IMMUNODETEKTION 35
3.2.17 FÄRBUNG VON PROTEINEN 36
3.2.18 ZIRKULAR‐DICHROISMUS‐SPEKTROSKOPIE 36
3.2.19 GELBINDUNGSTEST (EMSA) 36
3.2.20 RADIOAKTIVER KINASETEST 37
4 ERGEBNISSE 38
4.1 KLONIERUNG DER CDNA FÜR DIE CHLOROPLASTIDÄRE CK2 AUS ARABIDOPSIS
THALIANA (ATCK2) 38
4.2 VERGLEICHENDE SEQUENZANALYSE DER CPCK2 AUS VERSCHIEDENEN PFLANZEN 39
4.3 BAKTERIELLE ÜBEREXPRESSION UND AUFREINIGUNG DER CPCK2‐PROTEINE 42
4.4 VERGLEICHENDE FUNKTIONSANALYSE DER CPCK2 AUS SENF UND ARABIDOPSIS 43
4.5 ÜBEREXPRESSION UND AUFREINIGUNG VON SIGMAFAKTOREN ZUR IN VITRO
PHOSPHORYLIERUNG DURCH CPCK2 44
4.6 IN VITRO PHOSPHORYLIERUNG DER REKOMBINANTEN SIGMAFAKTOREN DURCH CPCK2 46
4.7 FUNKTIONELLE KONSEQUENZEN DER SIGMAFAKTOR‐PHOSPHORYLIERUNG 48
4.8 REDOXREGULATION DER AKTIVITÄT VON CPCK2 AUS ARABIDOPSIS IN VITRO 51
4.9 ZIRKULAR‐DICHROISMUS‐SPEKTROSKOPIE 55
4.10 IN VIVO FUNKTIONSANALYSE DER CPCK2 AUS ARABIDOPSIS THALIANA 56
4.10.1 MOLEKULARE CHARAKTERISIERUNG DER T‐DNA‐INSERTIONSMUTANTE VON CPCK2 56
4.10.2 PHÄNOTYPISCHE CHARAKTERISIERUNG DER CPCK2 KONOCKOUT‐MUTANTE 58
4.11 ANALYSE DER TRANSKRIPTAKKUMULATION PLASTIDÄRER GENE IN DER CPCK2‐MUTANTE 61
4.12 IN VIVO ANALYSEN ZUR REDOXREGULATION DER CPCK2‐AKTIVITÄT 62

Inhaltsverzeichnis
IV
5 DISKUSSION 65
5.1 REKOMBINANTE CPCK2 (PTK) AUS ARABIDOPSIS THALIANA 65
5.2 AUTOPHOSPHORYLIERUNG DER ATCK2 67
5.3 EINFLUSS DER PHOSPHORYLIERUNG DURCH CPCK2 AUF DIE SIGMAFAKTOR‐AKTIVITÄT 69
5.4 IN VITRO ANALYSE ZUR REDOXREGULATION DER ATCK2‐AKTIVITÄT 71
5.5 IN VIVO FUNKTIONSANALYSE DER CPCK2 AUS ARABIDOPSIS THALIANA 74
5.6 IN VIVO ANALYSEN ZUR REDOXREGULATION DER CPCK2‐AKTIVITÄT 76
6 ZUSAMMENFASSUNG 78
7 SUMMARY 81
8 LITERATUR 84
WISSENSCHAFTLICHE PRÄSENTATIONEN 104
LEBENSLAUF 107
ERKLÄRUNG 108

Abbildungs‐ und Tabellenverzeichnis
V
Abbildungs‐ und Tabellenverzeichnis
Abb. 1.4.2 Plastidäre RNA‐Polymerasen und Promotorelemente
Abb. 1.5.1 Schematische Darstellung von CK2α
Abb. 1.5.2 Pflanzliche Substrate und Substratklassen der CK2
Abb. 1.5.3 Lokalisation der CK2‐Untereinheiten auf den Chromosomen von Arabidopsis
Abb. 4.1 Nukleotidsequenz (5´ 3´) der cDNA (TAIR‐Datenbank) und abgeleitete Aminosäuresequenz der ATCK2 (At2g23070)
Abb. 4.2 Multipler Vergleich der cpCK2‐Sequenzen aus verschiedenen Pflanzen
Abb. 4.3 Rekombinante cpCK2 (ohne TP) aus Arabidopsis
Abb. 4.4 Nachweis der Kinase‐Aktivität von ATCK2 im Vergleich zu SACK2
Abb. 4.5.1 Verwendete rekombinante Sigmafaktoren
Abb. 4.5.2 Expression und Reinigung rekombinanter Sigmafaktoren
Abb. 4.6 In vitro Phosphorylierung der rekombinanten Sigmafaktoren durch SACK2 und ATCK2
Abb. 4.7 EMSA‐Experimente mit rekombinanten Sigmafaktoren 1 und 6 aus Arabidopsis
Abb. 4.8.1 Schematische Darstellung des rekombinanten cpCK2‐Proteins aus Arabidopsis
Abb. 4.8.2 Aktivitätstest der Cystein‐Mutanten von ATCK2
Abb. 4.8.3 Einfluss von Redox‐Reagenzien auf die Aktivität von ATCK2 und ihrer Cystein‐Mutanten
Abb. 4.8.4 Analyse der Dimerisierung von ATCK2
Abb. 4.9 CD‐Spektren der rekombinanten ATCK2 und ihrer Cystein‐Mutanten
Abb. 4.10.1 Charakterisierung der T‐DNA‐Insertionsmutante für cpck2 aus Arabidopsis
Abb. 4.10.2.1 Phänotypen der Wildtyp‐ und knockout‐Pflanzen

Abbildungs‐ und Tabellenverzeichnis
VI
Abb. 4.10.2.2 Phänotyp der komplementierten Pflanzen
Abb. 4.10.2.3 Phänotypen der Pflanzen unter Langtagbedingungen
Abb. 4.11 Northern‐Blot‐Analysen der cpck2‐knockout‐Linie im Vergleich zu Wildtyp (Col‐0)‐ und komplementierten Pflanzen (P35S:cpCK2)
Abb.4.12.1 Phänotypen der cpCK2 Cystein‐Mutantenlinien
Abb.4.12.2 Northern‐Blot‐Analyse der cpCK2 Cystein‐Mutantenlinien
Abb. 5.1 Aminosäurensequenz‐Vergleich von ATCK2 und SACK2 ohne ihre Transitpeptide
Abb. 5.4 Strukturmodell plastidärer ATCK2
Tabelle 3.1.4 Zusammenfassung der in dieser Arbeit verwendeten/hergestellten Pflanzenlinien
Tabelle 3.1.6 In dieser Arbeit verwendete/erstellte Plasmide und Klone
Tabelle 3.1.8 Liste der verwendeten Oligonukleotide (bezogen von der Firma MWG‐Biotech)

Abkürzungsverzeichnis
VII
Abkürzungsverzeichnis
A Adenin
Abb. Abbildung
amp Ampicillin
AS Aminosäure
A. thaliana Arabidopsis thaliana
ATCK2 chloroplastidäre CK2α aus Arabidopsis
ATP Adenosintriphosphat
ATSIG Sigmafaktor aus Arabidopsis thaliana
bp Basenpaare
bzw. beziehungsweise
C Cytosin
C1 ATCK2 Cystein‐Mutante (1. Cystein gegen Serin ausgetauscht)
C2 ATCK2 Cystein‐Mutante (2. Cystein gegen Serin ausgetauscht)
C3 ATCK2 Cystein‐Mutante (3. Cystein gegen Serin ausgetauscht)
C4 ATCK2 Cystein‐Mutante (4. Cystein gegen Serin ausgetauscht)
C2/3 ATCK2 Cystein‐Mutante (2. und 3. Cystein gleichzeitig gegen Serin
ausgetauscht)
C2/4 ATCK2 Cystein‐Mutante (2. und 4. Cystein gleichzeitig gegen Serin
ausgetauscht)
C3/4 ATCK2 Cystein‐Mutante (3. und 4. Cystein gleichzeitig gegen Serin
ausgetauscht)
cAMP cyklisches Adenosinmonophosphat
CD circular dichroism

Abkürzungsverzeichnis
VIII
cDNA copy DNA
CK2 Casein Kinase 2
CK2α Casein Kinase 2 alpha‐Untereinheit
CK2β Casein Kinase 2 beta‐Untereinheit
cpCK2α chloroplastidäre Casein Kinase 2 alpha
C‐terminal carboxyterminal
Cys Cystein
Da Dalton
d. h. das heißt
∆cpck2 cpck2α knockout‐Mutante
DNA Desoxyribonukleinsäure
DNase Desoxyribonuklease
dNTP Desoxynukleosidtriphosphat (dATP, dCTP, dGTP, dTTP)
E. coli Escherichia coli
EDTA Ethylendiaminotetraessigsäure
G Guanin
GTP Guanosintriphosphat
h Stunde
kan Kanamycin
kb Kilobasenpaare
kDa Kilodalton
M Molar
min Minute

Abkürzungsverzeichnis
IX
mRNA Boten (messenger)‐RNA
NADH NADH‐Dehydrogenase
nt Nukleotide
N‐terminal aminoterminal
NTP Nukleosidtriphosphat (ATP, CTP, GTP, UTP)
p.a. pro analysi
PCR Polymerase‐Kettenreaktion (polymerase chain reaction)
PEP plastidenkodierte RNA‐Polymerase (plastid‐encoded RNA Polymerase)
PTK plastidäre Transkriptionskinase
RNA Ribonukleinsäure
RNase Ribonuklease
rRNA ribosomale RNA
s. siehe
SACK2 chloroplastidäre CK2α aus Sinapis alba (Senf)
SASIG Sigmafaktor aus Sinapis alba
SIG Sigmafaktor
SLF Sigma‐ähnlicher‐Faktor (sigma like factor)
Sulf Sulfadiazin
T Thymin
TAC Membran‐gebundener PEP‐Polymerase Komplex (transcriptionally
active chromosome)
TP plastidäres Transitpeptid
tRNA Transfer‐RNA
WT Wildtyp

Abkürzungsverzeichnis
X
w/v Gewicht/Volumen
z. B. zum Beispiel

Genbezeichnungen
XI
Genbezeichnungen
actin2 Gen für das Protein Aktin2
Atcpck2α Gen für die chloroplasten‐lokalisierte CK2α‐Untereinheit aus Arabidopsis
thaliana
atpB Gen für die β‐Untereinheit der ATP‐Synthase
psaA Gen für die Untereinheit A des Photosystems I
psbA Gen für das D1‐Protein des Photosystems II
rpoA Gen für die α‐ Untereinheit der PEP
rpoB Gen für die β‐Untereinheit der PEP
rpoC1 Gen für die β´‐Untereinheit der PEP
rpoC2 Gen für die β´´‐Untereinheit der PEP
rpoT1 Gen für die mitochondrial‐lokalisierte NEP
rpoT2 Gen für die plastiden‐lokalisierte NEP
rpoT3 Gen für die NEP, die in Plastiden und Mitochondrien lokalisiert ist
trnK Gen für die tRNA für die Aminosäure Lysin

Einleitung
1
1 Einleitung
1.1 Evolution, Entwicklung und Funktion der Plastiden
Plastiden sind die charakteristischen Organellen pflanzlicher Zellen. Sie besitzen eine
doppelte Hüllmembran und ihre eigene DNA mit Proteinbiosyntheseapparat. Die
evolutionäre Herkunft der Plastiden lässt sich mit der Endosymbiontentheorie erklären.
Demnach stammen Plastiden wie auch Mitochondrien aus freilebenden Prokaryoten ab, die
durch Phagozytose in eine Ureukaryote aufgenommen wurden (Schimper, 1883;
Mereschkowsky, 1905; Gray, 1989). Durch die Aufnahme eines photoautotrophen
Cyanobakteriums in eine eukaryotische Urzelle entstand der Vorläufer heutiger
Pflanzenzellen (McFadden, 1999 und 2001). Im Laufe der Evolution ging ein Teil der Gene
des Endosymbionten verloren und ein Teil wurde in den Zellkern transferiert (Martin und
Herrmann, 1998; Martin et al., 2002). Aus diesem Grund sind die Plastiden trotz Besitz des
eigenen Genexpressionssystems genetisch semiautonom. Viele ihrer Proteine sind im
Zellkern kodiert und werden nach der Synthese im Cytoplasma durch eine spezielle
Importmaschinerie in der Chloroplastenhülle in das Organell importiert (Herrmann et al.,
1992; Heins et al., 1998; Soll, 2002; Schünemann, 2004). Die Vorläufer kernkodierter
plastidärer Proteine enthalten spezifische Signal‐Sequenzen in ihrem N‐terminalen Bereich,
die als Transitpeptid bezeichnet werden und über den Import ins Stroma oder noch weiter
ins Innere des Chloroplasten entscheiden (Soll und Tien, 1998).
Es gibt verschiedene Typen der Plastiden und abhängig von ihrem Entwicklungs‐ und
Funktionszustand besitzt eine pflanzliche Zelle jeweils nur einen Plastidentyp. Sie
differenzieren sich aus einem Plastiden‐Vorläufer, den so genannten Proplastiden, zu
gewebespezifischen Plastidenarten und erfüllen wichtige Funktionen. Beispiele hierfür sind
in Speichergeweben Amyloplasten, Proteinoplasten und Elaioplasten (allgemein
Leukoplasten), die zur Speicherung von Stärke, Proteinen und Fetten dienen. Chromoplasten
akkumulieren Carotinoide in Früchten und Blütenblättern und geben diesen ihre spezielle
Farbe. In den Zellen der grünen Gewebe befinden sich die bekanntesten und wichtigsten

Einleitung
2
Vertreter der Plastiden, die Chloroplasten. Sie sind die Orte der Photosynthese, in denen
energiereiche organische Substanzen aus Kohlendioxid und Wasser unter Verwendung von
Lichtenergie synthetisiert werden.
Abhängig von der Lichteinwirkung sowie entwicklungsbedingten Veränderungen können
Plastiden zum Teil ineinander umgewandelt werden. So können sich in Speichergeweben aus
farblosen Leukoplasten durch Lichteinwirkung Chloroplasten entwickeln. Weiterhin
entstehen z. B. während der Seneszenz aus Chloroplasten im Blattgewebe die
Chromoplasten oder Gerontoplasten.
1.2 Organisation und Expression des Plastidengenoms
Das genetische Material der Plastiden, das Plastom, ist je nach Art 120 bis 160 kb groß
(Palmer, 1985 und 1991; Whitfeld und Bottomley, 1993) und kodiert durchschnittlich ca. 130
Gene, davon kodieren etwa 100 für Proteine und der Rest setzt sich aus rRNA‐ und tRNA‐
Genen zusammen. Daneben existieren offene Leserahmen (ORFs: open reading frames), die
möglicherweise für bisher unbekannte Genprodukte kodieren (Sugiura, 1992 und 1995). Die
Kopienzahl der Plastiden‐DNA beträgt – abhängig von Entwicklungsstadium und Pflanzenart–
zwischen 10 und 300 pro Organell (Bendich, 1987; Sugiura, 1992).
Die Zustandsform des Plastoms wurde bisher immer als zirkulär beschrieben (Herrmann et
al., 1975; Bendich, 1987). Die Untersuchungen an Mais (Zea mays) haben jedoch gezeigt,
dass die plastidäre DNA auch in linearer und verzweigter Form vorliegen kann (Bendich,
2004; Oldenburg und Bendich, 2004). Das Plastiden‐Genom wird durch zwei gegenläufige
Sequenzwiederholungen (inverted repeats) in eine kleine (SSC, small single copy) und eine
große (LSC, large single copy) Einzelkopienregion aufgeteilt. Die Anordnung der Gene auf
dem Plastom ist zwar relativ konserviert, jedoch zeigen einige höhere Pflanzen, wie
Leguminosen und Koniferen, Ausnahmen von dieser Anordnung (zur Übersicht siehe: Link,
1996; Stern et al., 1997).
Der Anteil der plastomkodierten Proteine beträgt ca. 5% der Gesamtproteine, die in
Plastiden lokalisiert sind (Martin et al., 2002). Dazu gehören hauptsächlich Bestandteile des

Einleitung
3
Photosyntheseapparates sowie Komponenten der Transkriptions‐ und Translations‐
maschinerie (ribosomale Proteine, Untereinheiten der RNA‐Polymerase) sowie
Komponenten des NADH‐Dehydrogenase‐Komplexes. Wie bereits erwähnt, wird der
überwiegende Anteil der plastidären Proteine (etwa 95 %) vom Zellkern kodiert (Palmer,
1991; Sugiura, 1992; Tzudzuki et al., 1994).
Ein Großteil der plastidären Gene liegt, wie bei Prokaryoten, in polycistronischen
Transkriptionseinheiten vor (Sugita und Sugiura, 1996). Im Gegensatz zu Prokaryoten
besitzen sie jedoch nicht‐kodierende Bereiche, die so genannten Introns (Lambowitz et al.,
1999; Toro, 2003). Diese werden nach der RNA‐Synthese durch Spleißen herausgeschnitten
und die kodierenden Abschnitte, Exons, werden zusammengefügt (Goldschmidt‐Clermont et
al., 1990 und 1991).
Die Expression des Plastidengenoms wird abhängig von Umweltbedingungen und
Entwicklungszustand der Plastiden auf unterschiedlichen Ebenen reguliert; der trans‐
kriptionellen, der posttranskriptionellen und der translationellen (Krupinska, 1992; Krupinska
und Falk, 1994; Mayfield et al., 1995).
Die plastidären Gene besitzen unterschiedliche Promotorelemente, die sich in ihrer Stärke
und in der Art der RNA‐Polymerase, von der sie erkannt werden, unterscheiden (s. 1.4.1).
Diese Besonderheiten stellen wichtige Kontrollpunkte auf der transkriptionellen Ebene dar.
Außerdem können die Kopienzahl der Gene, die Topologie der Plastiden‐DNA und DNA‐
Methylierung die Bindungsaktivität der RNA‐Polymerasen an den Promotor beeinflussen und
somit die Transkription regulieren (Bendich, 1987; Stirdivant et al., 1985; Kobayashi et al.,
1990; Rapp und Mullet 1991). Es stellte sich heraus, dass es offenbar ganz überwiegend
kernkodierte Faktoren sind, die die Genexpression in Plastiden regulieren (Für weitere
Details zur Plastidentranskription, vgl. 1.3 bis 1.5)
Die RNA‐Stabilität (Rott et al., 1998; Schuster und Gruissem, 1991), Prozessierung von
polycistronischen Plastiden‐Transkripten (Westhoff und Herrmann, 1988), mRNA‐Edierung
(Bock et al., 1996; Kudla et al., 1992; Maier et al., 1996) und das Spleißen von Transkripten
(Barkan, 1989) stellen die wichtigsten posttranskriptionellen Regulationsmechanismen auf

Einleitung
4
RNA‐Ebene dar. Die Bildung einer „Stem/Loop“‐Struktur in der untranslatierten Region am
3´‐Ende der mRNA (3'UTR) spielt eine Rolle für die Stabilität der mRNA. Die 3´‐ und 5´‐UTR
Bereiche enthalten Erkennungssequenzen, sowohl für Endonukleasen als auch für die
kernkodierten RNA‐Bindeproteine, die dem Schutz der mRNA vor dem Abbau durch
Endonukleasen dienen (Klaff und Gruissem, 1991; Baginsky et al., 2007).
Auch für die Regulation auf der translationellen Ebene sind kernkodierte Faktoren von
großer Bedeutung (Danon, 1997). Die Initiation der plastidären Translation erfolgt auf zwei
verschiedenen Wegen: Zum einen durch Bindung der Ribosomen an die so genannte Shine
Dalgarno‐Sequenz, die innerhalb der ersten 20 Nukleotide vor dem Translationsstart
vorliegt. Jedoch besitzen nur ca. 40% der Plastidengene dieses für Prokaryoten typische
Sequenzelement (Sugiura et al., 1998). Zum anderen kann die Initiation durch Bindung an
weitere cis‐Elemente, die innerhalb der 5´‐UTR der Transkripte lokalisiert sind, erfolgen
(Danon, 1997; Choquet und Wollman, 2002). Einige kernkodierte RNA‐Bindeproteine binden
spezifisch an 5'‐UTR Sequenzen wie die des psbA‐Genes in Spinat (Alexander et al., 1998;
Baginsky und Link, 2005). Die Bindung zumindest einiger dieser Proteine wird durch ihren
Phosphorylierungsgrad reguliert (Danon und Mayfield, 1994). In Chlamydomonas reinhardtii
wurde ein psbA‐spezifischer Translationsfaktor, RB47, identifiziert, der die Translation des
Gens in Abhängigkeit vom plastidären Redoxzustand sowie von der Lichtqualität und ‐
quantität reguliert (Yohn et al., 1998b; Yohn et al., 1998a).
1.3 Transkription bei Prokaryoten und Eukaryoten
In Prokaryoten wird die Synthese aller RNA‐Spezies (mRNA, tRNA und rRNA) von einer
einzigen DNA‐abhängigen RNA‐Polymerase katalysiert, die aus mehreren Untereinheiten
besteht. Das RNA‐Polymerase Holoenzym aus E. coli ist ein 480 kDa großer Proteinkomplex
und setzt sich aus einem core‐Enzym (α2ββ´) und Sigmafaktor (σ) zusammen. Der Sigma‐
faktor sorgt für die hohe Affinität zu den Promotoren, den DNA‐Sequenzen vor der
Initiationsstelle von Transkriptionseinheiten, und ist entscheidend für die Transkriptions‐
initiation. Nach der Initiation dissoziiert der σ‐Faktor aus dem core‐Enzym und so beginnt die
Elongation der RNA‐Synthese.

Einleitung
5
Bakterielle Promotorregionen enthalten zwei konservierte Sequenzen (consensus sequence),
die aus 6 Nukleotiden bestehen; die – 10‐Region (TATAAT) und die – 35‐Region (TTGACA),
die 10 bzw. 35 bp stromaufwärts des Transkriptionsstartpunktes liegen (Pribnow, 1975a;
1975b; Schaller et al., 1975).
Weitere Arten von DNA‐abhängigen RNA‐Polymerasen kennt man aus Phagen. Dazu gehören
die T3‐, T7‐ und SP6‐Polymerasen. Diese Enzyme sind in den bakteriellen Wirten der Phagen
aktiv und bestehen aus einer einzigen Untereinheit. Sie erkennen spezifische Promotor‐
sequenzen und benötigen keinen Sigmafaktor (Allison et al., 1996; Kapoor et al., 1997).
In Eukaryoten erfolgt die Transkription von Kerngenen durch drei verschiedene RNA‐
Polymerasen. Die RNA‐Polymerase I und III übernehmen die Synthese von rRNA und tRNA.
Für die Transkription von proteinkodierenden Genen, also die Synthese von mRNA‐
Vorläufer, ist die RNA‐Polymerase II zuständig. Dieses Enzym ist ebenfalls für die Expression
kernkodierter Proteine verantwortlich, die in Plastiden bzw. in Mitochondrien lokalisiert
sind.
Die Promotoren der RNA‐Polymerase II weisen ein konserviertes Sequenzelement auf.
Dieses als TATA‐Box bezeichnete Element liegt bei ca. 30 bp stromaufwärts vom
Transkriptionsstart und dient der Promotorerkennung. Neben der TATA‐Box besitzen die
eukaryotischen Promotoren eine große Zahl von cis‐regulatorischen Sequenzelementen, die
entweder positiv (enhancer) oder negativ (silencer) wirken können (zur Übersicht siehe:
Roeder, 1996). Eine spezifische Transkriptionsinitiation und anschließende RNA‐Synthese
können nur durch die Bindung von speziellen Transkriptionsfaktoren an diesen Elementen
und RNA‐Polymerase II erfolgen.
1.4 Plastidäre Transkription
1.4.1 Plastidäre Promotorelemente
Als Konsequenz ihrer evolutionären Herkunft weisen viele plastidäre Promotoren
Ähnlichkeiten zu den prokaryotischen Promotorelementen aus (zur Übersicht siehe: Link,
1994). Diese „typisch“ bakteriellen Sequenzelemente (– 35‐ und – 10‐Region) im Plastom

Einleitung
6
werden als consensus type‐Promotor bezeichnet und von einer bakterien‐ähnlichen RNA‐
Polymerase erkannt (zur Übersicht siehe: Hess und Börner, 1999). Es existieren jedoch
plastidäre Gene, deren Promotorbereiche von diesem Konsensus‐Typ abweichen.
Beispielsweise konnte im 5´‐Bereich des rps16‐Gens aus Senf und des rpl32‐Gens aus Tabak
keine konservierte – 35‐Region identifiziert werden (Neuhaus et al., 1989; Vera et al., 1996).
Dem atpB‐Gen aus Spinat dagegen fehlt eine typische – 10‐Region (Gruissem und Zurawski,
1985). Zusätzlich zu den –10‐ und –35‐Elementen konnten in einigen plastidären Promotoren
eukaryotische cis‐Elemente gefunden werden. Die Promotorbereiche der Gene psbA und
rps16 aus Senf weisen Sequenzregionen auf, die der eukaryotischen TATA‐Box ähnlich sind
(Link und Langridge, 1984; Neuhaus et al., 1989) und zur spezifischen Promotorerkennung
benötigt werden (Eisermann et al., 1990). Neben den Prokaryoten‐ähnlichen consensus type‐
Promotoren existiert eine weitere Gruppe von Promotoren im Plastom, die in drei Klassen,
Typ Ia, Typ Ib und Typ II, unterteilt wird (Kapoor et al., 1997; Kapoor und Suguira, 1999; Liere
et al., 2004). Diese werden bevorzugt von einer Polymerase erkannt, die keine Ähnlichkeit
zum bakteriellen Enzym zeigt (zur Übersicht siehe Hess und Börner, 1999). Es wurden weiter
plastidäre Promotoren identifiziert, die von beiden Polymerasen genutzt werden können, da
sie die Promotormotive beider RNA‐Polymerasen besitzen. Zu diesen Genen zählen bspw.
das rps16‐Gen und das ycf3‐psaAB‐Cluster (Summer et al., 2000).
1.4.2 Plastidäre RNA‐Polymerasen
Wie bereits oben angedeutet, wird die plastidäre Transkription von mindestens zwei
unterschiedlichen RNA‐Polymerasen katalysiert; von einem Plastom‐kodierten Enzym, PEP
(plastid encoded polymerase), und einer kernkodierten Polymerase, NEP (nuclear encoded
polymerase). Strukturell stellt NEP ein T3‐/T7‐Phagen‐ähnliches Enzym mit einer einzigen
katalytischen Untereinheit dar, während PEP ein Enzym aus mehreren Untereinheiten,
ähnlich den bakteriellen Polymerasen ist (Abb. 1.4.2). Die PEP katalysiert die Transkription
der Gene für Komponenten des Photosyntheseapparates. Die „Haushaltsgene“ werden
dagegen von beiden RNA‐Polymerasen transkribiert (zur Übersicht siehe: Maliga, 1998; Hess
und Börner, 1999). Für die Synthese der rpo‐Gene (rpoA, rpoB, rpoC1 und rpoC2), die für
Untereinheiten der PEP kodieren, ist die NEP zuständig. Das bedeutet, PEP entsteht zeitlich

Einleitung
7
nach der NEP und ist später das dominierende Enzym in Chloroplasten (Serino und Maliga,
1998; Liere und Maliga, 1999; Hess und Börner, 1999). Es konnte gezeigt werden, dass in
Proplastiden hauptsächlich die NEP katalytisch aktiv ist, dagegen weist in Plastiden die PEP
eine höhere Aktivität auf (Emanuel et al., 2004 und 2006).
Die Existenz einer NEP wurde erstmals durch die Untersuchungen an einer parasitisch
lebenden Pflanze, Epifagus virginiana, nachgewiesen (dePamphilis und Palmer, 1990;
Morden et al., 1991; Wolfe et al., 1992; Krause et al., 2003). Obwohl dem Plastom dieser
Pflanze das komplette rpoBC‐Operon fehlt, konnten plastidäre Transkripte nachgewiesen
werden. Untersuchungen an Mutanten, bei denen verschiedene rpo‐Gene deletiert waren,
deuteten ebenfalls auf die Existenz einer weiteren RNA‐Polymerase, die möglicherweise vom
Kerngenom kodiert wird (Allison et al., 1996; Hajdukiewicz et al., 1997; Serino und Maliga,
1998; De Santis‐Maclossek et al., 1999; Krause et al., 2000).
Diese kernkodierte Polymerase konnte erstmals aus Spinatchloroplasten isoliert werden
(Lerbs‐Mache, 1993). Es handelte sich dabei um ein 110 kDa großes monomeres Polypeptid,
welches die Eigenschaften einer mitochondrialen RNA‐Polymerase aufwies. Diese wiederum
ist ebenfalls eine kernkodierte, aus einer einzigen Untereinheit bestehende RNA‐
Polymerase, die Homologie zu den RNA‐Polymerasen aus T3‐ und T7‐Bakteriophagen besitzt
(Schinkel und Tabak, 1989). Durch die Isolierung einer entsprechenden cDNA aus Arabidopsis
thaliana wurde die Annahme bestätigt, dass in Plastiden eine kernkodierte phagenähnliche
RNA‐Polymerase existiert. Für das abgeleitete, 113 kDa große Protein (RpoT3) konnte der
Import in Chloroplasten nachgewiesen werden (Hedtke et al., 1999). Eine cDNA für eine
plastiden‐lokaliesierte, T7‐ähnliche RNA‐Polymerase konnte auch aus Mais isoliert werden
(Chang et al., 1999). In A. thaliana wurde noch ein weiteres Gen (rpoT2) für eine
phagenähnliche RNA‐Polymerase gefunden, deren Transitpeptid den Import sowohl in
Chloroplasten als auch in Mitochondrien ermöglicht (Hedtke et al., 2000). Aus Nicotiana
tabacum wurden insgesamt sechs rpoT‐Gene identifiziert, deren Genprodukte in den
Mitochondrien bzw. Plastiden oder in beiden Organellen lokalisiert sind (Hedtke et al.,
2002).

Einleitung
8
NEP‐Promotoren vom Typ Ia sind vollständig definiert durch eine YRTA‐Box (hierbei Y = C
oder T, R = A oder G). Solche vom Typ Ib enthalten als zusätzliches cis‐Element die sog. GAA‐
Box, die weiter stromaufwärts des Transkriptionsstartpunktes liegt. Es gibt auch NEP‐
Promotoren, die keines dieser Elemente aufweisen und als nonconsensus beschrieben
werden (Typ II) (Kapoor et al., 1997; Sriraman et al., 1998) (s. Abb. 1.4.2 B).
Die plastiden‐kodierte RNA‐Polymerase, PEP, wurde viel früher als die NEP beschrieben und
charakterisiert. Aus Plastiden wurden durch unterschiedliche Reinigungsmethoden RNA‐
Polymerase‐Aktivitäten isoliert. Als erstes wurde ein hochmolekularer Protein‐DNA‐
Komplex, welcher die endogene DNA transkribieren kann, identifiziert. Dieser
membrangebundene Komplex wurde als TAC (transcriptionally active chromosome)
bezeichnet (Hallick et al., 1976; Briat et al., 1979; Reiss und Link, 1985; Bülow et al., 1987).
Weiterhin wurde eine „lösliche“ nicht an DNA gebundene RNA‐Polymerase isoliert, die in der
Lage ist, in vitro exogen zugegebene DNA zu transkribieren (zur Übersicht siehe: Igloi und
Kössel, 1992). Aus Senf‐Plastiden konnten sogar zwei verschiedene lösliche RNA‐Polymerase‐
Aktivitäten identifiziert werden (Pfannschmidt und Link, 1994 und 1997). Diese wurden als
Form A (PEP‐A) und Form B (PEP‐B) bezeichnet. Die beiden Formen unterscheiden sich
sowohl strukturell als auch funktionell voneinander. PEP‐A ist ein etwa 750 kDa großer
Proteinkomplex, der sich aus 13‐15 Untereinheiten zusammensetzt. PEP‐B ist dagegen ein
etwa 420 kDa großer Proteinkomplex und besteht aus fünf Untereinheiten. Während die
Aktivität von PEP‐A gegenüber Rifampicin, einem Hemmstoff prokaryotischer RNA‐
Polymerasen, resistent ist, lässt sich die PEP‐B‐Aktivität durch Rifampicin inhibieren. PEP‐B
überwiegt in Etioplasten, PEP‐A hingegen in Chloroplasten. Es konnte in vitro gezeigt
werden, dass die Form A durch Phosphorylierung teilweise in Form B umgewandelt werden
kann (Pfannschmidt et al., 2000). Im Folgenden wird die –mengenmäßig dominierende‐ PEP‐
A Form vereinfachend als PEP bezeichnet.
PEP ist ein multimerer Komplex mit den core‐Untereinheiten α2ββ´β´´. Ähnlich wie die
bakterielle RNA‐Polymerase, benötigt sie für eine spezifische Transkriptionsinitiation
Sigmafaktoren. Darüber hinaus enthält der PEP‐Komplex weitere kernkodierte
Komponenten mit regulatorischer Funktion, u. a. eine Ser‐/Thr‐Kinase, die als PTK (plastid
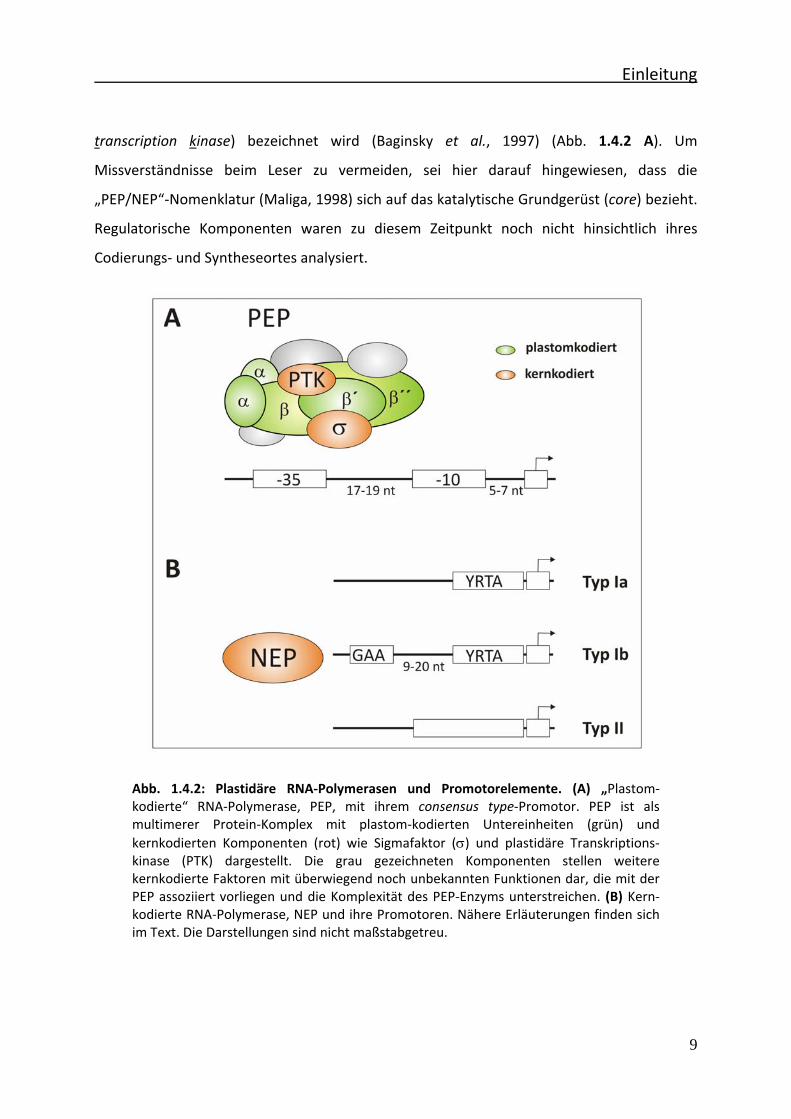
Einleitung
9
transcription kinase) bezeichnet wird (Baginsky et al., 1997) (Abb. 1.4.2 A). Um
Missverständnisse beim Leser zu vermeiden, sei hier darauf hingewiesen, dass die
„PEP/NEP“‐Nomenklatur (Maliga, 1998) sich auf das katalytische Grundgerüst (core) bezieht.
Regulatorische Komponenten waren zu diesem Zeitpunkt noch nicht hinsichtlich ihres
Codierungs‐ und Syntheseortes analysiert.
Abb. 1.4.2: Plastidäre RNA‐Polymerasen und Promotorelemente. (A) „Plastom‐kodierte“ RNA‐Polymerase, PEP, mit ihrem consensus type‐Promotor. PEP ist als multimerer Protein‐Komplex mit plastom‐kodierten Untereinheiten (grün) und kernkodierten Komponenten (rot) wie Sigmafaktor (σ) und plastidäre Transkriptions‐kinase (PTK) dargestellt. Die grau gezeichneten Komponenten stellen weitere kernkodierte Faktoren mit überwiegend noch unbekannten Funktionen dar, die mit der PEP assoziiert vorliegen und die Komplexität des PEP‐Enzyms unterstreichen. (B) Kern‐kodierte RNA‐Polymerase, NEP und ihre Promotoren. Nähere Erläuterungen finden sich im Text. Die Darstellungen sind nicht maßstabgetreu.

Einleitung
10
1.4.3 Plastidäre Sigmafaktoren
Erste Hinweise auf die Existenz „Sigma‐ähnlicher“ Faktor(en) in Plastiden lieferten bereits
Arbeiten von Surzycki und Schellenbarger (1976) an Chlamydomonas. Spätere Arbeiten
ergaben einen biochemisch gereinigten Transkriptionsfaktor (S‐Faktor), der sich jedoch nicht
als Sigma‐Faktor erwies (Jolly und Bogorad, 1980). Erst Untersuchungen an hochgereinigten
plastidären RNA‐Polymerase‐Präparationen mit Antikörpern gegen die Hauptsigmafaktoren
aus E. coli und Anabaena sp. (Lerbs et al., 1988; Troxler et al., 1994) lieferten direkte
Hinweise: In beiden Fällen zeigten die jeweiligen Antikörper Kreuzreaktionen mit
Polypeptiden der RNA‐Polymerasen.
Aus Senfplastiden konnten etwa gleichzeitig drei Proteine biochemisch gereinigt werden, die
in vitro mit dem core‐Enzym der RNA‐Polymerase aus E. coli zusammenwirken und
Promotorbindungs‐ und Initiationsspezifität verleihen (Bülow und Link, 1988; Tiller et al.,
1991). Diese Proteine wurden daher Sigma‐ähnliche Faktoren, kurz SLFs (sigma like factors),
genannt und entsprechend ihrer Molekulargewichte als SLF29, SLF52 und SLF67 bezeichnet.
Sie konnten sowohl aus Etioplasten, als auch aus Chloroplasten isoliert werden (Tiller et al.,
1991; Tiller und Link, 1993a).
Die Gene für die Sigmafaktoren befinden sich im Kerngenom der Pflanze und die
Genprodukte werden posttranslational in die Plastiden importiert (zur Übersicht siehe:
Allison, 2000). In A. thaliana konnten insgesamt sechs Sigmafaktoren identifiziert werden,
die als ATSIG1‐6 bezeichnet werden (Tanaka et al., 1997; Isono et al., 1997; Kanamaru et al.,
1999; Allison, 2000; Fujiwara et al., 2000; Hakimi et al., 2000). Aus Reis (Tozawa et al., 1998),
Weizen (Morikawa et al., 1999) und Mais (Lahiri et al., 1999; Tan und Troxler, 1999) konnten
ebenfalls Gene für die Sigmafaktoren identifiziert werden.
1.4.4 Die plastidäre Transkriptionskinase – PTK
Anhand der Untersuchungen an Senf‐Plastiden konnte gezeigt werden, dass der
Phosphorylierungszustand des plästidären Transkriptionsapparates die RNA‐Polymerase‐
Aktivität in Enzympräparaten aus Etioplasten bzw. Chloroplasten modulieren kann (Tiller und

Einleitung
11
Link, 1993b). Besonders die Sigmafaktoren scheinen dabei ein wichtiges Ziel für die
Modifikation durch Phoshorylierung zu sein. Biochemisch gereinigte PEP‐Präparationen aus
Senf‐Plastiden wiesen eine Kinaseaktivität auf, die in der Lage ist, Sigmafaktoren sowie auch
einige andere Komponenten des PEP‐Komplexes zu phosphorylieren. Diese Kinaseaktivität
wurde als plastidäre Transkriptionskinase, PTK, bezeichnet (Baginsky et al., 1997). Sie kann
entweder als „freier“ Kinase‐Komplex oder mit der PEP assoziiert vorkommen. Die
Phosphorylierung der PEP‐Komponenten, insbesonders der Sigmafaktoren, durch PTK führt
zu Reduktion der plastidären Transkriptionsaktivität (Baginsky et al., 1999). Die Aktivität der
PTK selbst wird über ihren Phosphorylierungs‐ und Redoxzustand reguliert. Reduziertes
Glutathion (GSH) inhibiert die PTK‐Aktivität. Andere Redoxreagenzien wie DTT, β‐
Mercaptoethanol und oxidiertes Glutathion (GSSG) haben dagegen keinen Einfluss auf die
Phosphorylierungsaktivität der PTK. Die Inaktivierung der PTK durch GSH führt dazu, dass
keine Komponenten der PEP phosphoryliert werden können und somit eine in vitro
Transkription möglich wird (Baginsky et al., 1999). Phosphorylierung der PTK durch eine
exogene Kinase wirkt ebenfalls inhibierend auf ihre Kinaseaktivität, was wiederum in vitro zu
einer ungehinderten Transkription durch PEP führt. Außerdem wurde beobachtet, dass GSH
auf die phosphorylierte und somit inaktivierte PTK eine reaktivierende Wirkung ausübt.
Diese Beobachtungen deuten darauf hin, dass die PTK möglicherweise als ein Vermittler bei
der Regulation der plastidären Genexpression durch intrazelluläre Redoxsignale fungiert.
Aufgrund der biochemischen Charakterisierung (Baginsky et al., 1997 und 1999) wurde die
PTK in die Familie der Proteinkinase CK2 (aus der „Fehlbezeichnung“ Casein Kinase 2
abgeleitetes Akronym) eingeordnet (Ogrzewalla et al., 2002). Hierbei handelt es sich um
Serin‐/Threonin‐Kinasen, die zur CMGC‐Familien der eukaryotischen Proteinkinasen
gehören. Die CMGC‐Familie umfaßt neben der CK2 auch die CDKs, MAP‐Kinasen und GSK‐3
(zur Übersicht siehe: Hunter, 1991; Hanks und Quinn, 1991; Stone und Walker, 1995). Alle
Mitglieder dieser Gruppe sind Serin‐/Threonin‐Kinasen, die nicht durch second messenger‐
Moleküle wie Ca2+/Calmodulin oder zyklische Nukleotide reguliert werden und von großer
physiologischer Bedeutung sind.
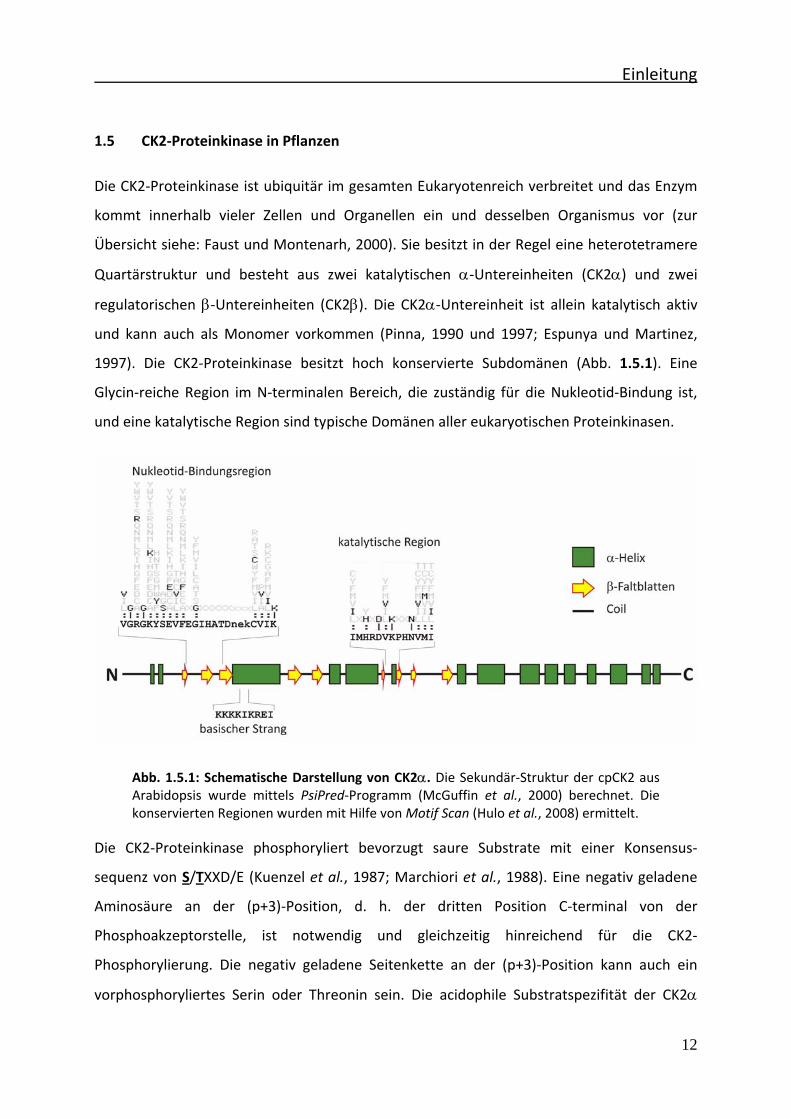
Einleitung
12
1.5 CK2‐Proteinkinase in Pflanzen
Die CK2‐Proteinkinase ist ubiquitär im gesamten Eukaryotenreich verbreitet und das Enzym
kommt innerhalb vieler Zellen und Organellen ein und desselben Organismus vor (zur
Übersicht siehe: Faust und Montenarh, 2000). Sie besitzt in der Regel eine heterotetramere
Quartärstruktur und besteht aus zwei katalytischen α‐Untereinheiten (CK2α) und zwei
regulatorischen β‐Untereinheiten (CK2β). Die CK2α‐Untereinheit ist allein katalytisch aktiv
und kann auch als Monomer vorkommen (Pinna, 1990 und 1997; Espunya und Martinez,
1997). Die CK2‐Proteinkinase besitzt hoch konservierte Subdomänen (Abb. 1.5.1). Eine
Glycin‐reiche Region im N‐terminalen Bereich, die zuständig für die Nukleotid‐Bindung ist,
und eine katalytische Region sind typische Domänen aller eukaryotischen Proteinkinasen.
Abb. 1.5.1: Schematische Darstellung von CK2α. Die Sekundär‐Struktur der cpCK2 aus Arabidopsis wurde mittels PsiPred‐Programm (McGuffin et al., 2000) berechnet. Die konservierten Regionen wurden mit Hilfe von Motif Scan (Hulo et al., 2008) ermittelt.
Die CK2‐Proteinkinase phosphoryliert bevorzugt saure Substrate mit einer Konsensus‐
sequenz von S/TXXD/E (Kuenzel et al., 1987; Marchiori et al., 1988). Eine negativ geladene
Aminosäure an der (p+3)‐Position, d. h. der dritten Position C‐terminal von der
Phosphoakzeptorstelle, ist notwendig und gleichzeitig hinreichend für die CK2‐
Phosphorylierung. Die negativ geladene Seitenkette an der (p+3)‐Position kann auch ein
vorphosphoryliertes Serin oder Threonin sein. Die acidophile Substratspezifität der CK2α

Einleitung
13
wird mit dem sog. basischen Strang in Verbindung gebracht (Meisner et al., 1989), da diese
Region in keiner anderen eukaryotischen Proteinkinase vorhanden ist und nur für CK2α
typisch ist (vgl. Abb. 1.5.1).
Die CK2 ist eine konstitutiv aktive Protein‐Kinase und hat ein sehr breites Substratspektrum.
Bisher wurden mehr als 300 Substrate für CK2 beschrieben, die u. a. Enzyme der DNA‐
Replikation und der DNA‐Transkription, Transkriptionsfaktoren, Signaltransduktionsproteine
und Translationsfaktoren beinhalten (Meggio und Pinna, 2003).
Ein charakteristisches Merkmal der Proteinkinase CK2 ist, dass sie GTP genauso effektiv wie
ATP als Phosphatgruppendonor verwenden kann. Diese Eigenschaft wird als duale
Cosubstratspezifität bezeichnet und diagnostisch zur Identifizierung der CK2‐Aktivität
benutzt. Darüber hinaus inhibieren Polyanionen wie Heparin spezifisch die CK2‐Aktivität, da
sich eine polyanionische Substanz wegen ihres negativen und sauren Charakters kompetitiv
zum Substrat, jedoch nicht zu ATP verhält (Hathaway und Traugh, 1982).
In Pflanzen wurde die Proteinkinase CK2 erst Anfang der 90er Jahre intensiv hauptsächlich in
Mais und Arabidopsis erforscht. In beiden Pflanzen konnten mehrere cDNA‐Sequenzen für α‐
und β‐Untereinheiten isoliert werden (Dobrowolska et al., 1991; Mizoguchi et al., 1993;
Collinge und Walker, 1994; Riera et al., 2001). Funktionelle Untersuchungen der CK2 in
Pflanzen haben gezeigt, dass die CK2 u. a. eine Rolle bei der lichtregulierten Genexpression
spielt (Lee et al., 1999). Bisher wurde eine Reihe von pflanzlichen Substraten beschrieben;
z.B. Kern‐Transkriptionsfaktoren in Arabidopsis wie GBF1 (G‐Box bindender Faktor, Klimczak
et al., 1992 und 1995), HY5 (Hardtke et al., 2000) sowie das LHY (late elongated hypokotyl)‐
und CCA1 (circadian clock‐associated 1)‐Protein (Sugano et al., 1998 und 1999). Abbildung
1.5.2 gibt eine Übersicht über die Substrate und Substratklassen von CK2 in Pflanzen. MFP1
und die β‐Untereiheit der CF0CF1‐ATPase sind in Chloroplasten lokalisiert und werden
möglicherweise von der cpCK2 phosphoryliert.

Einleitung
14
Abb. 1.5.2: Pflanzliche Substrate und Substratklassen der CK2. CF0CF1: Die β‐Untereinheit der chloroplastidären ATP‐Synthase (Kanekatsu et al., 1998; Reiland et al., 2009). MFP1: coiled‐coil Protein in Chloroplasten (Samaniego et al., 2006), Calreticulin: Calcium‐Bindeprotein (Pinna und Meggio, 1997), eIF5: Translationsinitiationsfaktor aus A.thaliana (Dennis et al., 2009). GBF1, HY5, LHY und CCA1 wurden im Text erläutert.
Ein wichtiger Fortschritt in der CK2‐Forschung in Pflanzen war allerdings die Kristallisation
der CK2α‐Untereinheit aus Mais (Niefind et al., 1998; Battistutta et al., 2000). Dies
ermöglichte eine detaillierte Einsicht in die Gesamt‐Struktur des Proteins.
Eine CK2‐Aktivität in Chloroplasten wurde erstmals in Spinat detektiert (Kanekatsu et al.,
1995). Bald darauf wurde aus Senf‐Plastiden die PTK‐Aktivität, die ebenfalls eine CK2‐
typische Aktivität aufweist, isoliert und charakterisiert (Baginsky et al., 1997 und 1999).
Dabei konnten eine „freie“ und eine im PEP‐Transkriptionskomplex „gebundene“ Form der
plastidären Transkriptionskinase detektiert werden. Die Suche nach der cDNA für eine
chloroplasten‐lokalisierte CK2α hat eine vollständige cDNA ergeben, deren Genprodukt ein
putatives Transitpeptid für Chloroplasten‐Import aufweist (Ogrzewalla et al., 2002). Die
Funktionalität dieses Transitpeptides konnte in vitro bestätigt werden und das abgeleitete
Protein wurde als cpCK2α (chloroplastidäre CK2α) bezeichnet. Für funktionelle
Untersuchungen wurde die cpCK2α ohne Transitpeptid bakteriell überexprimiert. Das
rekombinante Protein zeigte Phosphorylierungsaktivität. Ein direkter Vergleich der
Aktivitäten von cpCK2α und PTK wies Ähnlichkeiten der beiden Enzyme zueinander und zu
anderen cytosolischen CK2‐Proteinkinasen auf, wie z.B. die duale Cosubstratspezifität und

Einleitung
15
Inhibierung durch Heparin. Das rekombinante Protein ließ sich wie die PTK auch durch GSH
inhibieren (Ogrzewalla et al., 2002).
Datenbank‐Analysen mit der Aminosäuresequenz von cpCK2α aus Senf haben gezeigt, dass
eine Reihe von Pflanzen ‐darunter Arabidopsis thaliana‐ cpCK2α‐Orthologe besitzen
(Loschelder et al., 2004). Laut Genomanalysen besitzt Arabidopsis vier Gene für CK2α‐ und
vier für die β‐Untereinheit. Abbildung 1.5.3 zeigt die Positionen dieser Untereinheiten auf
den Chromosomen von A. thaliana. Eine extensive in vivo Studie über CK2‐Untereinheiten
und ihre subzelluläre Lokalisierung in A. thaliana hat gezeigt, dass nur eine der vier CK2α‐
Untereinheiten, At2g23070, in Chloroplasten lokalisiert ist (Salinas et al., 2006). Die
restlichen drei α‐Untereinheiten befinden sich im Nukleus, während die β‐Untereinheiten im
Nukleus und/oder Cytosol lokalisiert sind. Es konnte keine CK2β in Arabidopsis‐
Chloroplasten detektiert werden (Salinas et al., 2006). Das Fehlen einer β‐Untereinheit wird
offenbar durch Interaktion des α‐Polypeptids mit anderen Bestandteilen des
Transkriptionskomplexes und/oder durch Vorhandensein anderer regulatorischer
Determinanten im Protein selbst kompensiert. In folgenden Kapiteln dieser Arbeit wird
vereinfachend von cpCK2, d. h. ohne α‐Spezifikation, gesprochen.
Abb. 1.5.3: Lokalisation der CK2‐Untereinheiten auf den Chromosomen von Arabidopsis. Die Abbildung wurde mittels Chromosom Map Tool (TAIR) erstellt (basiert auf Daten von Salinas et al., 2006). Das Gen für die Chloroplasten‐lokalisierte CK2α‐Untereinheit (At2g23070) liegt als Einzel‐Kopie auf Chromosom 2 vor.

Zielsetzung
16
2 Zielsetzung
Die Regulation der Genexpression in den Plastiden ist ein sehr komplexer Prozess, an dem
viele kern‐kodierte Faktoren beteiligt sind. Die Wechselwirkungen dieser Faktoren
miteinander und mit der basalen Transkriptionsmaschinerie stellen wichtige Aspekte der
Regulation der Organellen‐Genexpression dar. In Vorarbeiten wurde eine PEP‐assozierte Ser‐
/Thr‐Proteinkinase aus Senf‐Plastiden als zentraler Spieler der plastidären Genexpression
identifiziert (Baginsky et al., 1997 und 1999). Diese als PTK (plastidäre Transkriptionskinase)
bezeichnete Kinase ist in der Lage, durch Phosphorylierung von Sigmafaktoren und anderen
PEP‐Komponenten die Organellen‐RNA‐Synthese zu regulieren. In vitro Experimente zeigten,
dass PTK einer Redox‐Regulation durch reduziertes Glutathion (GSH) unterliegt und somit
auch in vivo als möglicher Vermittler zwischen zellulären Redoxsignalen und der plastidären
Genexpression in Frage kommen kann (Baginsky et al., 1999; Baena‐González et al., 2001).
Eine cDNA für diese Chloroplasten‐lokalisierte Proteinkinase aus Senf konnte isoliert werden
und das Genprodukt wurde wegen seiner Ähnlichkeit zu der nucleo‐/cytosolischen Casein
Kinase 2 (CK2) als cpCK2 (chloroplastidäre CK2) bezeichnet (Ogrzewalla et al., 2002). Das
rekombinante cpCK2‐Protein zeigte große Ähnlichkeiten zu PTK hinsichtlich seiner
biochemischen Eigenschaften und Empfindlichkeit gegenüber CK2‐spezifischen Inhibitoren
einschließlich der GSH‐Sensitivität (Ogrzewalla et al., 2002). Datenbank‐Analysen mit der
cpCK2 aus Senf zeigten, dass eine Reihe von Pflanzen einschließlich Arabidopsis thaliana ein
einzelnes Gen für die Chloroplasten‐lokalisierte CK2/PTK besitzen (Loschelder et al., 2004).
Die Modellpflanze A. thaliana bietet die Möglichkeit, die Funktion von Genen bzw.
Genprodukten mit Hilfe von Mutanten „klassisch‐“ oder „rückwärts‐“ genetisch zu
untersuchen. Ziel der vorliegenden Arbeit war es diese Strategien und Techniken zur Klärung
der Funktion und Regulation der chloroplastidären CK2 in der Pflanze und in vitro zu
untersuchen.
In einem ersten experimentellen Schritt galt es, die cDNA für cpCK2 aus A. thaliana (ATCK2)
zu clonieren, zu analysieren und für bakterielle Überexpression einzusetzen. Das
rekombinante Protein aus A. thaliana sollte im Vergleich zu dem bereits vorhandenen

Zielsetzung
17
cpCK2‐Protein aus Sinapis alba (Senf) (SACK2) auf seine katalytische Aktivität und
biochemischen Eigenschaften untersucht werden. Dies gilt insbesondere für die „Benutzung“
von Sigmafaktoren als phosphorylierbare Substrate durch ATCK2 und die funktionellen
Konsequenzen für die Sigma‐Aktivität. Zur Klärung dieses Aspekts bieten sich Promotor‐
Bindungsstudien wie z. B. EMSA (Electrophoretic Mobility Shift Assays) an.
Nächstes Ziel der Arbeit war die Untersuchung der Redox‐Regulation der ATCK2‐Aktivität. Im
Vordergrund standen sowohl in vitro als auch in vivo mutationale Analysen mit ATCK2, um
regulatorische Cystein(e) zu identifizieren.
Ein zentraler Aspekt der Funktionsanalyse der chloroplastidären CK2 in planta betrifft die
Regulation der „Zielgene“. Zu diesem Zweck wurden die Expressionsmuster ausgewählter
Chloroplasten‐Gene in einer knockout‐Mutante, die eine T‐DNA‐Insertion im Exon 1 des
Atcpck2‐Gens aufweist, untersucht.

Material und Methoden
18
3 Material und Methoden
3.1 Materialien
3.1.1 Chemikalien und Verbrauchsmaterialien
Acrylamid/Bisacrylamid (29:1) 40% Roth
Acrylamid/Bisacrylamid (37,5:1) 30% Roth
Agarose GERBU Biotechnik
Ampicillin Sigma‐Aldrich
Ammoniumpersulfat (APS) Sigma‐Aldrich
Amylose‐Resin New England Bioloabs
ATP Roche
γ‐32P‐ATP Hartmann Analytics
Bacto‐Agar Difco
Bacto‐Trypton Becton, Dickinson and Co.
BCIP Biomol
2‐β‐Mercaptoethanol Sigma‐Aldrich
Bromphenolblau Serva
α‐Casein Sigma‐Aldrich C8032 (lyophilisiert und
80 % dephosphoryliert)
Desoxynukleosidtriphosphate (dNTPs) Promega
Dialyse‐Membranen Biomol, Typ 8 und 20
Diamid Sigma‐Aldrich
DIG‐UTP Roche

Material und Methoden
19
DNeasy Plant Mini Kit Qiagen
Ficoll 400 Sigma‐Aldrich
γ‐32P‐GTP Hartmann Analytics
Gel Extraction Kit Qiagen
Heparin Sigma‐Aldrich H7005
Hefeextrakt Difco
Imidazol Sigma‐Aldrich
Isopropyl‐β,D‐thiogalactopyranosid (IPTG) GERBU Biotechnik
Kanamycin GERBU Biotechnik
Lambda (λ‐) DNA Invitrogen
Lysozym GERBU Biotechnik
Nitrocellulose‐Membran Porablot NCL, Macherey & Nagel
Ni‐NTA‐Agarose Qiagen
Nitroblue Tetrazolium (NBT) GERBU Biotechnik
N‐Lauroylsarcosin Novagen (Protein Refolding Kit)
PCR Purification Kit Qiagen
Poly d[(I‐C)] Roche
Positive geladene Nylonmembran Roche
ProbeQuant G‐50 Micro Columns GE Healthcare
Protein Refolding Kit Novagen
Protein‐Standards für SDS‐PAGE BioRad, Precission Protein Standards
QuikChange II Site‐Directed Mutagenesis Kit Stratagene
Random Primer Promega

Material und Methoden
20
Reduziertes DTT Sigma‐Aldrich, D9779
Reduziertes Glutathion (GSH) Sigma‐Aldrich, G4251
Rifampicin GERBU Biotechnik
RNA‐Marker Promega
Sulfadiazin GERBU Biotechnik
TBB (4,5,6,7‐Tetrabromobenzotriazol) Sigma‐Aldrich
TEMED Sigma‐Aldrich
Alle sonstigen Chemikalien wurden in p. a. Qualität von handelsüblichen Firmen bezogen.
3.1.2 Enzyme und Konjugate
AMV Reverse Transkriptase Stratagene
Anti‐CK2α (Monoclonal, mouse) Sigma, C5367
Anti‐Digoxigenin‐AP Roche
Anti‐Mouse IgG Alkaline Phosphatase Conjugate Promega
CIAP (calf intestinal alkaline phosphatase) Promega
CDP‐Star Roche
DNaseI Otto Nordwald KG
E. coli RNA core‐Polymerase Epicentre Biotechnologies
GoTaq® Flexi DNA Polymerase Promega
Pfu‐Polymerase Promega
PfuTurbo DNA Polymerase Promega
Proteinase K Merck
Protease Inhibitor Coctail Sigma, P8849
Recombinant RNasin® Ribunuclease Inhibitor Promega

Material und Methoden
21
Restriktionsendonukleasen Promega
SP6‐RNA‐Polymerase Promega
Taq‐DNA‐Polymerase Promega
T3‐RNA‐Polymerase Promega
T4‐DNA‐Ligase Promega
T4‐Polynucleotide Kinase Promega
T7‐RNA‐Polymerase Promega
3.1.3 Puffer und Lösungen
AP‐Puffer 100 mM Tris/HCl pH 8,0, 100 mM NaCl, 5 mM MgCl2
BCIP‐Lösung 5 % (w/v) BCIP in Dimethylformamid
Bis‐Tris‐Gel 8 bzw. 10 % Polyacrylamid (Stocklösung: 40 %, 29:1),
350 mM Bis‐Tris/HCl pH 6.6, 0,035 % APS und 0,12 %
TEMED
Blockierungslösung 2 % (w/v) Magermilchpulver in TBS‐Puffer
CD‐Puffer 25 mM Natriumphosphatpuffer pH 8.0
CK2‐Puffer (10X) 200 mM Tris/HCl pH 7,5, 500 mM KCl, 100 mM MgCl2
Coomassie‐Entfärber 20 % Methanol, 10 % Essigsäure
Coomassie‐Färbelösung 50 % Methanol, 12 % Essigsäure, 0,2 % Coomassie
Brilliant‐Blue G250
Dialyse‐Puffer mit Glycerin 50 mM Tris/HCl pH 8,0, 100 mM NaCl, 0,1 mM EDTA, 5
mM β‐Mercaptoethanol, 50 % Glycerin
DNA‐Probenpuffer (5x) 25 % Ficoll, 0,5 % SDS, 0,1 % Xylencyanol, 0,1 % Brom‐
phenolblau, 10 mM EDTA
Elutionspuffer (pQE‐System) 50 mM Tris/HCl pH 8,0, 300 mM NaCl, 250 mM Imidazol

Material und Methoden
22
Elutionspuffer (pMAL‐System) 10 mM Maltose in Säulenpuffer
EMSA‐Gel (4%) 4 % Acrylamid/Bisacrylamid (37,5:1), 1X TBE‐Puffer, 2 %
APS und 0,2 % TEMED
EMSA‐Laufpuffer 1X TBE‐Puffer
IB‐Waschpuffer 20 mM Tris/HCl pH 7,5, 10 mM EDTA, 1 % Triton‐X‐100
IB‐Solubilisierungspuffer 50 mM CAPS pH 11, 0,3 % N‐Lauroylsarcosin, 1mM DTT
Lysozym‐Lösung 10 mg Lysozym in 1 ml dH2O
NBT‐Lösung 5 % (w/v) NBT in 70 % (v/v) Dimethylformamid
Ponceau S‐Lösung 1 % Ponceau S, 5 % Essigsäure
Probenpuffer (5x, non‐red) 300 mM Tris/HCl, 10 % SDS, 0,2 % Bromphenolblau
Probenpuffer (5X, red) 300 mM Tris/HCl, 10 % SDS, 0,2 % Bromphenolblau,
250mM DTT
Retardationspuffer (1x) 30 mM Tris/HCl pH 7, 0,5 mM β‐Mercaptoethanol,
80mM (NH4)2SO4, 0,5 mM EDTA
RNA‐Gel (denaturierend) 1,2 % Agarose, 1X MOPS‐Puffer, 6,7 % Formaldehyd
(entionisiert)
RNA‐Ladepuffer 50 % Formamid, 6% Formaldehyd (entionisiert), 1X
MOPS‐Puffer, 10“ Glycerin, 0,05 % Bromphenolblau
Säulenpuffer 20 mM Tris/HCl, 200 mM NaCl, 1 mM EDTA, 1mM DTT
SDS‐MOPS‐Laufpuffer (10X) 500 mM MOPS, 500 mM Tris/HCl, 10 mM EDTA, 1% SDS
SSC‐Puffer (20X) 3 M NaCl, 0,3 M Natriumcitrat
TBE‐Puffer (5x) 500 mM Tris, 450 mM Borsäure, 5 mM EDTA, pH 8.3
TBS‐Puffer 50 mM Tris/HCl pH 8,0, 150 mM NaCl
TBS‐TT‐Puffer 50 mM Tris/HCl pH 8,0, 150 mM NaCl, 0,05 % Tween,
0,2 % Triton X‐100
Material und Methoden
23
Transferpuffer 25 mM Tris, 192 mM Glycin, 20 % (v/v) Methanol
Waschpuffer (pQE‐System) 50 mM Tris/HCl pH 8,0, 300 mM NaCl, 20 mM Imidazol
Waschpuffer (pMAL‐System) 0,1‐1 mM Maltose in Säulenpuffer
3.1.4 Pflanzenmaterial
Tabelle 3.1.4: Zusammenfassung der in dieser Arbeit verwendeten/hergestellten Pflanzenlinien
Pflanze Genotyp Referenz
Arabidopsis thaliana
L Heynh. Cv. Columbia Wildtyp
NASC European
Arabidopsis Stock Centre
Arabidopsis thaliana
cpck2‐Knockout‐
Mutante
T‐DNA‐Insertion in Exon 1 des
Atcpck2α (At2g23070) Gens
Linien‐ID: 615F11,
GABI‐Kat, Universität
Bielefeld
Arabidopsis thaliana
komplementierte
cpck2‐Knockout‐
Mutante
T‐DNA‐Insertion in Exon 1 des
Atcpck2α (At2g23070) Gens
AtcpCK2 Wildtyp‐cDNA
im Genom inseriert
Transformation in die
cpck2‐knockout‐Linie,
diese Arbeit
Arabidopsis thaliana
cpck2 C1 Mutante
T‐DNA‐Insertion in Exon 1 des
Atcpck2α (At2g23070) Gens
AtcpCK2 Cys158‐Ser Mutanten‐
konstrukt im Genom inseriert
Transformation in die
cpck2‐knockout‐Linie,
diese Arbeit
Arabidopsis thaliana
cpck2 C2 Mutante
T‐DNA‐Insertion in Exon 1 des
Atcpck2α (At2g23070) Gens
AtcpCK2 Cys182‐Ser Mutanten‐
konstrukt im Genom inseriert
Transformation in die
cpck2‐knockout‐Linie,
diese Arbeit

Material und Methoden
24
Arabidopsis thaliana
cpck2 C3 Mutante
T‐DNA‐Insertion in Exon 1 des
Atcpck2α (At2g23070) Gens
AtcpCK2 Cys240‐Ser Mutanten‐
konstrukt im Genom inseriert
Transformation in die
cpck2‐knockout‐Linie,
diese Arbeit
Arabidopsis thaliana
cpck2 C4 Mutante
T‐DNA‐Insertion in Exon 1 des
Atcpck2α (At2g23070) Gens
AtcpCK2 Cys313‐Ser Mutanten‐
konstrukt im Genom inseriert
Transformation in die
cpck2‐knockout‐Linie,
diese Arbeit
3.1.5 Bakterienstämme
Escherichia coli:
DH5α deoR, endA1, gyrA96, usdR17 (rk – mk+), recA1, supA1, supE44, thi‐1, Δ(lacZYA
argFV169), Φ80lacZ ΔM15, F– (Stratagene, Hanahan 1983)
M15 NalS, StrS, RifS, Lac –, Ara –, Gal –, Mtl –, F –, RecA+, Uvr+, Lon+, pREP5 (Qiagen)
TB1 F –ara Δ(lac‐proAB) [] rpsL(StrR) thi hsdR (New England Biolabs)
XL1Blue recA1, endA1, gyrA96, thi‐1, hsdR17, supE44, relA1, lac [F´ proAB lacIqZ ΔM15
Tn10 (Tet R )] (Stratagene, Bullock et al., 1987)
BL21RIL F– ompT gal dcm lon hsdSB(rB‐ mB‐) λ(DE3 [lacI lacUV5‐T7 gene 1 ind1 sam7
nin5]) ( Stratagene)
Agrobacterium:
Agrobacterium tumefaciens GV3101::pMP90RK , genR, kanR (Koncz & Shell, 1986)
Material und Methoden
25
3.1.6 Plasmide und Klone
Tabelle 3.1.6: In dieser Arbeit verwendete/erstellte Plasmide und Klone
Plasmid/Klon Charakteristika Referenz
pBlueScript® KS(‐) Klonierungsvektor mit Blau/Weiss‐
Selektion, T3/T7‐Promotoren Stratagene
pQE30 Überexpressionsvektor, N‐terminale
Fusion mit sechs Histidinen Qiagen
pMALc2x
Überexpressionsvektor, N‐terminale
Fusion mit Maltose‐Bindeprotein
(MBP)
New England Biolabs
pGEMT‐easy Klonierungsvektor mit Blau/Weiss‐
Selektion, T7/SP6‐Promotoren Promega, Mannheim
pBinAR Pflanzentransformationsvektor, 35S ‐
Promotor, kan R Höfgen und Willmitzer,
1990
pBS/Hinf120 5´‐Ende des Cloroplast‐psbA‐Gens mit
Promotor (120 bp)
A. Homann,
Dissertation 2002
pSA364/B0,5 Intronfragment aus trnK‐Gen aus S.
alba (460 bp)
Neuhaus und Link,
1987
pUCE1.45 5´‐kodierende Region des trnK‐Gens
mit Promotor
Neuhaus, Dissertation
1987
pBlueScript/atpB 5´‐Ende des atpB‐Gens mit Promotor Schweer et al., 2009
pQE/Ck2a1000 cpCK2‐cDNA (ohne TP) aus S. alba in
pQE30
Ogrzewalla,
Dissertation 2001
pQE‐Sig1 Sig1‐cDNA (ohne TP) aus S. alba in
pQE30
Kreutzenbeck,
Diplomarbeit 2000
Material und Methoden
26
pQE‐Sig2 Sig2‐cDNA ( ohne TP) aus S. alba in
pQE30
Berndt, Diplomarbeit
2000
pMALc2x/ATSIG1 Sig1‐cDNA (ohne TP) aus A. thaliana
in pMALc2x
A. Kolpack,
Dissertation 2010
pBlueScript/ATSIG6 Sig6‐cDNA (ohne TP) aus A. thaliana H. Loschelder,
Dissertation, 2007
pMALc2x/ATSIG6 Sig6‐cDNA (ohne TP) aus A. thaliana
in pMALc2x diese Arbeit
pMALc2x/ATCK2 cpCK2‐cDNA (ohne TP) aus A. thaliana
in pMALc2x diese Arbeit
pMALc2x/ATCK2 C1 AtcpCK2‐cDNA (ohne TP) mit einer
Punktmutation an Cys158 diese Arbeit
pMALc2x/ATCK2 C2 AtcpCK2‐cDNA (ohne TP) mit einer
Punktmutation an Cys182 diese Arbeit
pMALc2x/ATCK2 C3 AtcpCK2‐cDNA (‐TP) mit einer Punkt‐
mutation an Cys240 diese Arbeit
pMALc2x/ATCK2 C4 AtcpCK2‐cDNA (ohne TP) mit einer
Punktmutation an Cys313 in diese Arbeit
pMALc2x/ATCK2 C2/3 AtcpCK2‐cDNA (ohne TP) mit einer
Doppelmutation an Cys182 und 240 diese Arbeit
pMALc2x/ATCK2 C2/4 AtcpCK2‐cDNA (ohne TP) mit einer
Doppelmutation an Cys182 und 313 diese Arbeit
pMALc2x/ATCK2 C3/4 AtcpCK2‐cDNA (ohne TP) mit einer
Doppelmutation an Cys240 und 313 diese Arbeit
Material und Methoden
27
pBinAR/ATCK2 + TP AtcpCK2‐cDNA (mit TP) in pBinAR diese Arbeit
pBinAR/ATCK2 + TP C1 AtcpCK2‐cDNA (mit TP) mit einer
Punktmutation an Cys158 in pBinAR diese Arbeit
pBinAR /ATCK2 + TP C2 AtcpCK2‐cDNA (mit TP) mit einer
Punktmutation an Cys182 in pBinAR diese Arbeit
pBinAR ATCK2α +TP C3 AtcpCK2‐cDNA (mit TP) mit einer
Punktmutation an Cys240 in pBinAR diese Arbeit
pBinAR ATCK2 + TP C4 AtcpCK2‐cDNA (mit TP) mit einer
Punktmutation an Cys313 in pBinAR diese Arbeit
pSPT‐452a (psbA) psbA‐Gen‐Sonde für Northern‐Blot‐
Hybridisierung
K. Liere, Dissertation
1995
pGEM‐T Easy/actin2 actin2‐Gen‐Sonde für Northern‐Blot‐
Hybridisierung Schweer et al., 2006
pGEM‐T Easy/atpB atpB‐Gen‐Sonde für Northern‐Blot‐
Hybridisierung Schweer et al., 2006
pGEM‐T Easy/psaA psaA‐Gen‐Sonde für Northern‐Blot‐
Hybridisierung
Loschelder,
Dissertation 2007
3.1.7 Nährmedien
LB‐Medium: 0,5 % NaCl, 0,5 % Hefe‐Extrakt, 1 % Pepton (Bertani et al., 1951)
LBamp‐Medium: LB‐Medium mit 50 µg/ml Ampicillin
LBkan‐Medium: LB‐Medium mit 50 µg/ml Kanamycin
LBamp,kan‐Medium: LB‐Medium mit 100 µg/ml Ampicillin und 50 µg/ml Kanamycin
LBamp,cam‐Medium: LB‐Medium mit 50 µg/ml Ampicillin und 34 µg/ml
Chloramphenicol

Material und Methoden
28
LBcam, kan, rif‐Medium: LB‐Medium, 50 μg/ml Ampicillin, 50 μg/ml Kanamycin, 50
µg/ml Rifampicin
LBpMAL‐Medium: LB‐Medium mit 0,2 % Glukose und 100 µg/ml Ampicillin
MS‐Medium: Murashige und Skoog, 1962
MS‐Medium mit Sulf: MS‐Medium mit 11,25 µg/ml Sulfadiazin
3.1.8 Oligonukleotide
Tabelle 3.1.8: Liste der verwendeten Oligonukleotide (bezogen von der Firma MWG‐Biotech). Schnittstellen für Restriktionsenzyme bzw. die ausgetauschten Sequenzen wurden unterstrichen.
Bezeichnung Sequenz (5´ 3´) Verwendung
ATCK2_VL1
ATCK2_VL2
ATGGCCTTAAGGCCTTGTACTGGA
CAAATGTTTTCGAGATCACTGGCTG
RT‐PCR, zur
Transkript‐
Detektion
ATCK2_VL_BamHI
ATCK2_VL_SalI
GAATTCGGATCCATGGCCTTAAGGCCTTGTACTGGA
CTGCAGGTCGACCAAATGTTTTCGAGATCACTGGCT
RT‐PCR, zur
Volllängen‐
Klonierung
ATCK2_BamHI_for
ATCK2_SalI_rev
GAATTCGGATCCGCTTCTCTTTACCGTCAACATCTCCGG
CTGCAGGTCGACTCACTGGCTGCGCGGCGTACGGCTGC
RT‐PCR, zur
Klonierung
(ohne TP)
AtCYS1_for1
AtCYS1_rev1
AtCYS2_for
AtCYS2_rev
AtCYS3_for
AtCYS3_rev
GCTACTGATAACGAGAAATCCGTTATCAAGATTTTGAAGCC
GGCTTCAAAATCTTGATAACGGATTTCTCGTTATCAGTAGC
GATTAAGATTCTGCAGAACCTCTCCGGTGGGCCG
CGGCCCACCGGAGAGGTTCTGCAGAATCTTAATC
GGCGTTGGATTTCTCCCATTCACGTGGAATCATGC
GCATGATTCCACGTGAATGGGAGAAATCCAACGCC
Mutagenese‐
PCR, zum
Austausch
der Cystein‐
Resten gegen
Serin

Material und Methoden
29
AtCYS4_for
AtCYS4_rev
GGAGTCTTGGGTCTATGTTTGCTGGAATGATATTCCGC
GCGGAATATCATTCCAGCAAACATAGACCCAAGACTCC
CK2HO2
CK2HO9
CK2HO10
CCATTGAACAGCAAGGGACT
TCGCCTTTTTCGTCCATTAG
GTGCGTTGTGCAGGATTAGA
Genomische
PCR, zum
Homozygotie‐
Test
3.1.9 Internetadressen
CPHmodels: http://www.cbs.dtu.dk/services/CPHmodels/
CloroP: http://www.cbs.dtu.dk/services/ChloroP/
ExPASy tools: http://www.expasy.ch/tools/
PYMOL v. 0.99: http://pymol.sourceforge.net/
Predotar: http://urgi.versailles.inra.fr/predotar/predotar.html
ProtParam: http://www.expasy.ch/tools/protparam.html
SWISS‐MODEL: http://www.expasy.ch/swissmod/SWISS‐MODEL.html
Restriction Mapper: http://www.restrictionmapper.org/
TAIR: http://www.arabidopsis.org
CLUSTALW2: http://www.ebi.ac.uk/Tools/clustalw2/index
BLAST: http://blast.ncbi.nlm.nih.gov/Blast.cgi
National Center for Biotechnology Information (NCBI): http://www.ncbi.nlm.nih.gov
3.1.10 Computergestützte Bildbearbeitung
Die Detektion radioaktiver Signale erfolgte mittels Phospho‐Imager. Die Daten wurden mit
FUJI FLA3000 RGB (auf IP stage) eingelesen und mit Hilfe des AIDA (v.4.04)‐Programms
bearbeitet. Eingescannte Röntgenbilder sowie alle Abbildungen in dieser Arbeit wurden mit

Material und Methoden
30
Hilfe der Programme Adobe Photoshop und Corel DrawX4 digital erstellt und inhaltsgetreu
bearbeitet.
3.2 Methoden
Alle nicht gesondert aufgeführten Methoden wurden nach Ausubel et al. (1989‐2001),
Maniatis et al. (1982) und Sambrook et al. (1989) oder nach den Angaben des jeweiligen
Produktherstellers durchgeführt.
3.2.1 DNA‐Isolierung
3.2.1.1 Isolierung genomischer DNA
Gesamt‐DNA‐Isolierung aus Arabidopsiskeimlingen wurde nach der CTAB‐Methode (Rogers
und Bendich, 1985) bzw. mit dem DNeasy Plant Mini Kit (Qiagen, Hilden) durchgeführt.
3.2.1.2 Isolierung von Plasmid‐DNA
Zur Isolierung von Plasmid‐DNA wurde entweder die Methode gemäß Birnboim und Doly
(1979) oder der NucleoSpinR‐Kit (Macherey und Nagel) verwendet.
3.2.2 RNA‐Isolierung
Gesamt‐RNA‐Isolierung aus der Pflanze erfolgte gemäß Chomczynski und Sacchi (1987). Für
die Northern‐Blot‐Untersuchungen wurden von jeder Mutantenlinien drei unabhängige
RNA‐Isolierungen durchgeführt.
3.2.3 Markierung von Nukleinsäuren
3.2.3.1 Markierung der 5´‐Ende von DNA‐Fragmenten
Die radioaktive Markierung der DNA‐Fragmente wurde unter Verwendung von T4‐
Polynukleotid‐Kinase (NEB) und γ‐32P‐ATP (Hartmann Analytics) nach Rao (1994)
durchgeführt. Nicht eingebaute Nukleotide wurden mittels ProbeQuant G‐50 Micro Columns
(GE Healthcare) abgetrennt.

Material und Methoden
31
3.2.3.2 DIG‐Markierung von RNA durch in vitro‐Transkription
Zur Herstellung von Sonden für Northern‐Analysen wurde zuerst das entsprechende Plasmid
durch Restriktion linearisiert. Unter Verwendung von T7‐ oder SP6‐RNA‐Polymerase und
Digoxigenin‐11‐UTP wurde die cRNA den Herstellerangaben entsprechend (DIG User´s
Manual, Roche Diagnostics) in vitro transkribiert. Anschließend wurde die DNA‐Matrize
durch DNaseI‐Behandlung (15 min, 37 °C) entfernt.
3.2.4 Kapillartransfer von RNA (Northern‐Blot)
Für Northern‐Blot‐Analyse wurde 1 µg Gesamt‐RNA pro Spur in denaturierenden
Agarosegelen (1,5%, s. 3.1.3) elektrophoretisch aufgetrennt und anschlißend durch Kapillar‐
Blot mit 10X‐SSC‐Puffer auf eine positiv geladene Nylonmembranen transferiert.
3.2.5 RNA/RNA Hybridisierung
Die Hybridisierung mit DIG‐markierten RNA‐Transkripten (über Nacht bei 68 °C) und
anschließender Chemilumineszenz‐Detektion erfolgte nach Herstellerangaben (DIG User’s
Manual, Roche Diagnostics). Zur Chemilumineszenz‐Detektion wurden Anti‐Digoxigenin‐AP
und CDP‐Star verwendet.
3.2.6 Amplifizierung von Nukleinsäuren
3.2.6.1 Polymerase‐Kettenreaktion (PCR)
PCR‐Amplifikation von DNA‐Fragmenten wurde in 50 μl‐Ansätze mit je 50 – 100 ng DNA
(Matrize), 1 U DNA‐Polymerase (Taq‐, Pfu‐, Pfu Ultra Polymerase), 0,75 mM MgCl2 im Falle
der Taq‐Polymerase, 50 pmol der entsprechenden Oligonukleotide, 50 mM KCl, 10 mM
Tris/HCl pH 9.0, 0,1 % Triton X‐100 und 1 mM dNTPs durchgeführt. Die Reaktionsansätze
wurden 30 sec auf 95 °C erhitzt, anschließend erfolgten 35 Zyklen des folgenden Programms:
30 sec 95 °C, 60 sec 55 – 65 °C, 90 sec 72 °C. Die Synthese im letzten Zyklus wurde bei 72 °C
auf 10 Minuten verlängert.

Material und Methoden
32
3.2.6.2 Reverse Transkription‐PCR (RT‐PCR)
Mit dieser Methode wurde die Gesamt‐RNA aus Arabidopsis thaliana mit dem Enzym AMV
(Avian Myeloblastosis Virus) Reverse Transcriptase (Promega, Mannheim) in cDNA
umgeschrieben. Durch anschließende PCR (3.2.6.1) wurde aus der cDNA doppelsträngige
DNA amplifiziert. Es wurde nach den Angaben des Herstellers verfahren.
3.2.7 Elution von DNA‐Fragmenten
Die Reinigung von Produkten aus Agarose‐Gelen erfolgte mit dem Gel Extraction Kit (Qiagen,
Hilden).
3.2.8 In vitro‐Mutagenese
Die Ortsgerichtete‐Mutagenese von Plasmid DNA mit dem Konstrukt wurde mit Hilfe des
QuickChange II Site‐Directed Mutagenesis Kit (Stratagene, Heidelberg) durchgeführt. Es
wurde bei der Erstellung der Oligonukleotide, welche die entsprechende Mutation
enthielten, bei dem PCR‐Protokoll und anschließender Transformation in E. coli XL1Blue‐
Zellen nach Angaben des Herstellers verfahren.
3.2.9 DNA‐Sequenzierung
Alle Sequenzierungen wurden in Auftragsarbeiten an der Fakultät für Chemie (Ruhr‐
Universität Bochum; Lehrstuhl für molekulare Neurobiochemie) mit dem
Kapillarelektrophoresegerät 3130cl Genetic Analyser (Applied Biosystems, Darmstadt)
durchgeführt. Für die Auswertung wurde das Programm Sequence Scanner 1.0 (Applied
Biosystems, Darmstadt) verwendet.
3.2.10 Floral Dip
Für die Herstellung komplementierter ∆cpck2‐Pflanzen sowie der cpck2‐Cystein‐Mutanten
wurden die entsprechenden Konstrukte hinter dem CaMV 35S Promotor in den pBinAR
Vektor (Höfgen and Willmitzer, 1990) kloniert und in Rhizobium radiobacter (Agrobacterium
tumefaciens GV3101) transformiert. Die T‐DNA Konstrukte wurden über floral dip Methode

Material und Methoden
33
(Clough und Bent, 1998) in das Genom der Arabidopsis ∆cpck2‐Mutante eingebracht. Die
Selektion der transformierten Pflanzen erfolgte mit Kanamycin. Die Insertion wurde mit Hilfe
der PCR‐Analysen nachgewiesen. Von jeder transgenen Pflanze wurden drei unabhängige
Nachkommen‐Linien auf ihren visuellen und molekularen Phänotyp untersucht.
3.2.11 Anzucht und Selektion von Pflanzen
Zur Anzucht der A. thaliana Linien wurden zuerst die Samen für 3 min in 70% (v/v) Ethanol
und für weitere 5 min in 5% (v/v) Na‐Hypochlorit sterilisiert. Nach fünfmal Waschen mit
sterilem Wasser wurde das Saatgut in 0,1% (v/v) sterilem Agar aufgenommen und auf MS‐
Medium in Petri‐Schalen ausplattiert. Es folgte eine Stratifikation für zwei Tage bei 4 °C. Die
Petri‐Schalen mit Samen wurden anschließend im Phytotron unter Kurztag‐ (8h Licht, 100
µmol/m2s, 24°C, 16h Dunkelheit bei 20°C) oder Langtagbedingungen (16h Licht, 60
µmol/m2s, 24°C, 8h Dunkelheit bei 20°C) angezogen. Ab 14 Tage wurden die Pflanzen auf
Erde gesetzt
3.2.12 Expression rekombinanter Proteine
3.2.12.1 Bakterielle Überexpression mit Hilfe des pQE‐Systems
Die Expression von cpCK2 sowie Sigmafaktor 1 und 2 aus Senf (SACK2, SASIG1 und SASIG2)
erfolgte im E. coli‐Stamm M15 durch pQE‐Expressionsvektoren. Diese Vektoren ermöglichen
die Expression eines Fusionsproteins mit sechs aufeinanderfolgenden N‐terminalen
Histidinen. Die Überexpression der rekombinanten Proteine erfolgte in LB‐Medium
(+Antibiotika), das 1 % ig mit einer Übernachtkultur angeimpft und bei 37 °C bis zu einer
OD580 0,5‐0,7 angezogen wurde. Die Expression der Fusionsproteine wurde durch Zugabe
von 1 mM IPTG induziert. Die Expressionskulturen wurden anschließend entweder 3h bei
37°C (zur Isolierung der inclusion bodies) oder 2 h bei 25 °C (für Reinigung nativer Proteine)
inkubiert. Nach der Inkubation wurden die induzierten Bakterienzellen für 20 min bei 4 °C
und 8000 rpm geerntet. Pellets wurden bis zur weiteren Aufarbeitung bei – 80 °C gelagert.

Material und Methoden
34
3.2.12.2 Bakterielle Überexpression mit Hilfe des pMAL‐Systems
Zur Überexpression mittels pMAL‐Systems (New England Biolabs) wurden die cDNAs für das
cpCK2‐Protein (AtCK2) sowie für Sigmafaktor 1 und 6 aus Arabidopsis (AtSIG1 und AtSIG6) in
den pMALc2x‐Vektor kloniert. Dieses System bietet die Möglichkeit, das entsprechende
Protein als MBP‐Fusionsprotein zu synthetisieren. Überexpression wurde in E. coli TB1‐ bzw.
in BL21RIL‐Zellen durchgeführt. Die Überexpression und Aufreinigung der rekombinanten
Proteine erfolgten nach vorgegebenen Herstellerangaben (pMALTM Protein Fusion and
Purification System, NEB).
3.2.13 Reinigung rekombinanter Proteine
3.2.13.1 Isolierung und Renaturierung von Proteinen aus inclusion bodies
Die Reinigung von Proteinen aus inclusion bodies wurde mit Hilfe des Protein Refolding‐Kits
der Firma Novagen durchgeführt. Hierzu wurden die sedimentierten Bakterienzellen (s.
3.2.12.1) zunächst in 20 ml IB‐Waschpuffer resuspendiert. Der Zellaufschluss erfolgte mittels
Lysozym (Endkonzentration 100 µg/ml) und anschließende Ultraschall‐Behandlung (10‐mal
für 15 sec mit Kühlungspausen). Die inclusion bodies wurden über einen
Zentrifugationsschritt sedimentiert (10000 x g, 10 min, 4 °C) und mit 20 ml IB‐Waschpuffer
gewaschen. Nach erneuter Zentrifugation (10000 x g, 10 min, 4 °C) wurde die Konzentration
der inclusion bodies wurde mit IB‐Solubilisierungspuffer auf 10‐20 mg/ml eingestellt. Die
Protein‐Lösung wurde für 2‐mal 3 h gegen 50 Volumina Dialysepuffer mit DTT bei 4 °C
dialysiert. Nach der Dialyse mit Dialysepuffer mit Glycerin wurden die Proteinproben bei ‐
20°C gelagert.
3.2.13.2 Reinigung nativer Proteine
Die Isolierung nativer Proteine erfolgte mittels Qiaexpressionist‐Kit von Qiagen (Handbook
June 2003, Protokoll 9 und 12).

Material und Methoden
35
3.2.14 Bis‐Tris‐Polyacrylamid‐Gelelektrophorese (Bis‐Tris‐PAGE)
Zur Auftrennung der Proteine wurde das kontinuierliche Bis‐Tris‐Gelsystem (6‐10%
Acrylamid/Bisacrylamid in 1x Gel‐Puffer) verwendet (s. 3.1.3). Zur Herstellung der Mini‐Gele
(8 cm x 10 cm x 1 mm) wurde das Mini‐Gel‐System der Firma BioRad verwendet. Als
Laufpuffer wurde 1x SDS‐MOPS‐Laufpuffer verwendet (3.1.3). Die Proben wurden mit
Probenpuffer (mit oder ohne DTT) versetzt und aufgetragen.
3.2.15 Native‐PAGE
Zur Auftrennung der DNA‐Protein‐Komplexe bei der EMSA (3.2.18) wurden 4% ige native
TBE‐Gele mit 0,5X‐TBE‐Laufpuffer verwendet (s. 3.1.3).
3.2.16 Elektrophoretischer Transfer von Proteinen auf Nitrocellulose‐Membranen
(Westernblot) und Immunodetektion
Nach der Bis‐Tris‐PAGE wurden die Proteine mit Hilfe einer Transferapparatur der Firma
BioRad (Mini Trans‐Blot Electrophoretic Transfer Cell) elektrophoretisch unter Verwendung
von Transferpuffer (s. 3.1.3) auf eine Nitrocellulose‐Membran übertragen. Nach dem
Transfer konnten die rekombinanten Proteine mit Antikörper selektiv detektiert werden.
Dazu wurde die Membran zuerst in Blockierungslösung für 1h inkubiert. Anschließend
erfolgte eine Inkubation (1h) mit dem monoklonalen CK2α‐Antikörper (1:1000) in
Blockierungslösung. Durch 3‐maliges Waschen der Membran für je 10 min (2 x mit TBS‐TT‐
Puffer und 1 x mit TBS‐Puffer) wurde überschüssiger und ungebundener Antikörper entfernt.
Anschließend wurde die Membran für 1h in einer 1:5000‐Verdünnung des Sekundär‐
Antikörpers (Anti‐Mouse Alkaline Phosphatase) in Blockierungslösung inkubiert. Es folgten
erneut drei Waschschritte für jeweils 10 min mit 2 x TBS‐TT und 1 x mit TBS. Detektion
erfolgte bei RT im AP‐Puffer durch Zugabe von NBT (0,33 mg/ml) und BCIP (0,165 mg/ml).

Material und Methoden
36
3.2.17 Färbung von Proteinen
Zur Färbung von Proteinen in Bis‐Tris‐Gele wurde Coomassie‐Blau‐Färbung verwendet. Die
auf Nitrocellulose transferierten Proteine wurden durch eine reversible Färbung mit Ponceau
S sichtbar gemacht (Salinovich und Montelaro, 1986).
3.2.18 Zirkular‐Dichroismus‐Spektroskopie
Zur Überprüfung der Sekundärstrukturen der rekombinanten CK2‐Cysteinmutanten im
Vergleich zu Wildtyp‐CK2 wurde die Zirkular‐Dichroismus‐Spektroskopie (circular dichroism,
CD, Greenfield, 2006; Martin und Schilstra, 2007) mit Jasco J‐715 CD‐Spektrometer
durchgeführt. Dazu wurden die Proteinlösungen gegen CD‐Puffer (s. 3.1.3) dialysiert und auf
eine Konzentration von 150 μg/ml eingestellt. Die Messungen wurden mit einer 1 mm‐
Küvette bei 20 °C durchgeführt. Die Scanning‐Geschwindigkeit betrug 100 nm/min. Die
Daten von jeweils 10 Spektren pro Protein von 190 bis 320 nm wurden gemittelt. Dabei
wurde das Spektrum des CD‐Puffers (ebenfalls gemittelte aus 10 Spektren) als Hintergrund
abgezogen. Die Auswertungen erfolgten mittels Jasco Spectra Manager.
3.2.19 Gelbindungstest (EMSA)
Zur Untersuchung der Promotorbindung von RNA‐Polymerase‐Sigmafaktor‐Komplexen
wurden EMSA‐Analysen (electrophoretic mobility shift assay) durchgeführt (Fried und
Crothers, 1981; Garner und Revzin, 1981). Hierzu wurden zuerst die Promotorfragmente
radioaktiv markiert (3.2.3.1). Die Standard‐Reaktionsansätze (50 µl) enthielten 25 ng Sigma‐
Protein, 2,5 ng 5´‐radioaktiv‐markiertes psbA‐ oder atpB‐Promotorfragment, 100 ng E. coli
core‐RNA‐Polymerase und 3 μg Poly d[(I‐C)] als unspezifische Kompetitor‐DNA in 1X‐
Retardationspuffer (3.1.3). Zur Kompetition wurden nicht‐markierte psbA‐, atpB‐ und trnK‐
Promotorfragmente (50 ng) verwendet. Als nicht‐spezifischer Kompetitor wurde ein DNA‐
Fragment ohne Promotor, Bam0.5, (100 ng) eingesetzt. Nach 15‐minütiger Inkubation bei
Raumtemperatur wurden die Proben in 4% igen nativen Polyacrylamid‐Gelen (3.1.3)
getrennt. Die Gele wurden getrocknet und die freie DNA sowie die Protein‐DNA‐Komplexe
wurden mittels Phospho‐Imager sichtbar gemacht.

Material und Methoden
37
3.2.20 Radioaktiver Kinasetest
Zur radioaktiven Detektion der Kinase‐Aktivität wurde 32P‐markierte ATP oder GTP
verwendet. Ein Standard‐Reaktionsansatz (50 µl) enthielt 1 x CK2‐Puffer, 10 µM ATP, ca. 10
µCi/µmol γ‐32P‐ATP/GTP, 1 µg partiell dephosphoryliertes Casein oder 5 µg Sigmafaktoren als
Substrat und 2,5‐5 µg cpCK2 (modifiziert nach Baginsky et al., 1997). Nach einer Inkubation
von 15 min (für Casein‐Phosphorylierung) bzw. 30 min (für Sigmafaktor‐Phosphorylierung)
bei 30 °C wurden die Reaktionen durch Zugabe von SDS‐Probenpuffer gestoppt und die
Reaktionssansätze wurden nach 5 minütiger Inkubation bei 95 °C über Bis‐Tris‐PAGE (großes
Gel) aufgetrennt. Das Gel wurde ca. für 1 h auf einem Gel‐Trockner getrocknet und über
Nacht auf eine Phospho‐Imager‐Platte aufgelegt. Phosphorylierte Proteine wurden mittels
FUJI FLA3000 RGB sichtbar gemacht.

Ergebnisse
38
4 Ergebnisse
4.1 Klonierung der cDNA für die chloroplastidäre CK2 aus Arabidopsis thaliana (ATCK2)
In früheren Arbeiten wurde eine plastidäre CK2 aus der Crucifere Sinapis alba (Senf) kloniert
und als bakteriell exprimiertes, rekombinantes Enzym erfasst (Ogrzewalla et al., 2002). Ziel
der vorliegenden Arbeit war es, die Rolle von cpCK2 in der Modellpflanze A. thaliana durch
kombinierte biochemische und molekular‐genetische Ansätze zu untersuchen. Zu diesem
Zweck sollte zuerst das homologe Gen/Protein isoliert werden. Für die bakterielle
Überexpression (s. 4.3) wurde die cDNA für die chloroplastidäre CK2 aus A. thaliana (Ecotyp
Columbia, Col‐0) ohne Transitpeptidsequenz mit den Primern ATCK2_BamHI_for und
ATCK2_SalI_rev (s. 3.1.8) amplifiziert. Die Amplifikation der Konstrukte für die in vivo
Mutanten‐Komplementation bei Arabidopsis (s. 4.9.2) erfolgte mit Transitpeptidsequenz
mittels Primer‐Paar ATCK2_VL_BamHI und ATCK2_VL_SalI (s. 3.1.8).
Abbildung 4.1 zeigt die cDNA‐Nukleotidsequenz und die abgeleitete Aminosäuresequenz der
ATCK2 (Synonym: cpCK2). Die Volllängen‐cDNA hat eine Länge von 1523 bp und kodiert für
einen offenen Leserahmen mit 432 Aminosäuren. Das Genprodukt besitzt am N‐Terminus
ein 55 Aminosäuren langes Signalpeptid für den Import in den Chloroplasten. Die 5´‐ und 3´‐
nichtkodierenden Regionen sind 27 bp bzw. 197 bp lang. Das errechnete Molekulargewicht
des Proteins ohne Transitpeptid beträgt ca. 44 kDa.
1 ctgaataagagttaaaaaaagacattaatggccttaaggccttgtactggattcacaatc 1 M A L R P C T G F T I 61 tcctctcttcgcaacgcttccgccgccaacaacaatctcttctctctcctctccttctct 12 S S L R N A S A A N N N L F S L L S F S 121 tcgtcgtctccggcgaagaggaatcttcttctctcttcgctccaggacaatctccgtcgt 32 S S S P A K R N L L L S S L Q D N L R R 181 ttcgcatccagcgcttctctttaccgtcaacatctccggaatcaacaacaacaacatcag 52 F A S S A S L Y R Q H L R N Q Q Q Q H Q 241 caacaacagcaatcgcgcgtgaaagaaaaatcagaaacgctggctcagaagatcggtaaa 72 Q Q Q Q S R V K E K S E T L A Q K I G K 301 tctatccggcgtgccggtgcgccgtcgaaagctagggtttacgctgatgtcaacgttgtt 92 S I R R A G A P S K A R V Y A D V N V V 361 agacctaaggactattgggattacgagtcccttgctgttcaatggggtgtacaggatgat 112 R P K D Y W D Y E S L A V Q W G V Q D D 421 tatgaggtggtgaggaaggtaggaaggggaaaatacagtgaggtctttgaaggcattcat 132 Y E V V R K V G R G K Y S E V F E G I H

Ergebnisse
39
481 gctactgataacgagaaatgtgttatcaagattttgaagcctgtgaagaagaagaagatt 152 A T D N E K C V I K I L K P V K K K K I 541 aagagagagattaagattctgcagaacctctgcggtgggcctaatattgtaaaattgctc 172 K R E I K I L Q N L C G G P N I V K L L 601 gacattgttagagatcagcagtctaagacgcccagcttgatatttgaacatgtgaacaat 192 D I V R D Q Q S K T P S L I F E H V N N 661 aaagatttcaaagttctctatccaactctctcagactatgatgttcgatattacattttt 212 K D F K V L Y P T L S D Y D V R Y Y I F 721 gaacttctaaaggcgttggatttctgccattcacgtggaatcatgcacagagatgtgaaa 232 E L L K A L D F C H S R G I M H R D V K 781 ccacataatgtcatgattgatcacgagcagcgaaaacttcgtttaattgactggggtttg 252 P H N V M I D H E Q R K L R L I D W G L 841 gcagaattctatcatcctgggaaagaatacaatgtccgtgttgcttcaaggtactttaag 272 A E F Y H P G K E Y N V R V A S R Y F K 901 ggtccagagctgctggtggatttacaggactatgattattccttagacctatggagtctt 292 G P E L L V D L Q D Y D Y S L D L W S L 961 gggtgtatgtttgctggaatgatattccgcaaggagccgttcttttatggccatgacaac 312 G C M F A G M I F R K E P F F Y G H D N 1021 tatgatcaattggtcaaaattgcgaaggtgcttggcacagatgaactcaacgcctaccta 332 Y D Q L V K I A K V L G T D E L N A Y L 1081 aacaaataccgtatagagttggaccctaatctaacatccctcgtgggcagacatagccgg 352 N K Y R I E L D P N L T S L V G R H S R 1141 aagccatggacaaagttcatcaattctgaaaaccagcacttagcagttcctgaggctgtt 372 K P W T K F I N S E N Q H L A V P E A V 1201 gattttgtggacaagttgctgagatatgatcaccaagaaaggccaacagctaaagaagca 392 D F V D K L L R Y D H Q E R P T A K E A 1261 atggcgcatccatatttctatccgattagaaatgcagagagcagccgtacgccgcgcagc 412 M A H P Y F Y P I R N A E S S R T P R S 1321 cagtgatctcgaaaacatttgcttgttcaatcttctgatttttcatttctttacggattc 432 Q - 1381 aaatttgttataaatcgatctgtcattgtttcattctctcaagtgtgcgtgtgttactgt 1441 tttgctctttctcagtgtttcattcaactcatttgaattttattatataaaaaaaaatat 1501 ccacaagacgggtttcaaaaggt
Abb. 4.1: Nukleotidsequenz (5´ 3´) der cDNA (TAIR‐Datenbank) und abgeleitete Aminosäuresequenz der ATCK2 (At2g23070). Kleine Buchstaben stellen die cDNA‐Sequenz dar und die großen Buchstaben zeigen die Aminosäuresequenz. Die Transitpeptid‐Sequenz wurde unterstrichen. Start‐ und Stop‐Kodons sind fettgedruckt. Die 5´‐ und 3´‐UTRs sind kursiv dargestellt.
4.2 Vergleichende Sequenzanalyse der cpCK2 aus verschiedenen Pflanzen
Datenbank‐Analysen haben bereits gezeigt, dass eine Reihe von Pflanzen ein ATCK2/cpCK2‐
Orthologes Enzym besitzen (Loschelder et al., 2004). Inzwischen sind noch mehr Sequenzen
anderer Pflanzenspezies in der Datenbank für eine putative cpCK2 zu finden. Abbildung 4.2
zeigt ein multiples Sequenz‐Alignment von cpCK2‐Proteinen aus ausgewählten höheren
Pflanzen. Die Sequenzen wurden entsprechend ihrer Ähnlichkeit zu Arabidopsis cpCK2

Ergebnisse
40
geordnet. Die Längen der Transitpeptide wurden mit Hilfe des ChloroP‐Programms ermittelt
und in der Abbildung grün dargestellt. Die Sequenz‐Homologie ist im N‐terminalen Bereich,
d.h. im Bereich der Transitpeptide am geringsten.
Sequenz- identität(%) Ath 100 MALR-------PCTGFTISSLRNASAANNNLFSLLSFSSSSPAKRN----LLLSSLQD-- 47 Sal 88 MAFR-------PIG-YTISSLRKASAANDLRFSFLSIYSSFPAKKN----LLLS------ 42 Bna 87 -------------------------------------SSPSPAKKK----SLLS------ 13 Vvi 77 MAVR---------------PLRFIISHRRLLAFSLRLPSSSSSHLSQSPPSLLV------ 39 Ptr 76 MAIRPFLLQQKLTTILNRQRQKKKNNINKTLFSSLLFIRNNNNNNNNNNPFLFSSPQSSS 60 Mac 74 ------------MAFGPLGSLSLLSRHLQSLAPLLRLPSPPSP----------------- 31 Lpe 74 -------------------------------------MSAAPP----------------- 6 Sbi 74 -------------------------------------MIGAPPP---------------- 7 Hvu 73 -------------------------------------MSDAPP----------------- 6 Zma 73 -------------------------------------MIGASP----------------- 6 Osa 73 -------------------------------------MTDAPPP---------------- 7 Psi 71 -------------------------MAGRPLASHFHILQGATPSFYTSLGQYPLPPQLSP 35 Ath 100 ---NLRRFASSASLYRQHLRNQQQQHQQQQQSRVKE-------KSETLAQKIGKSIR--- 94 Sal 88 ---FLRGFSS---LHRRQQ----------QQSRVN--------KSETLAQKIGKSIR--- 75 Bna 87 ---FLRGFAS---LYR-------------QQPRVN--------KSETLAQKIGKSIR--- 43 Vvi 77 ----FHRLASSPAPLSHPQKSPPPPPPPPSHHRFALS-----SLPETLAQRIGKSVR--- 87 Ptr 76 ISTLLRQFASSTTAFSHQK-----QGQQEIQHRLPHRFRSLVPPPDTLAQKIGKSIR--- 112 Mac 74 -----HHLATYCLAGLHWISGAAGQGSSPSRLRPSAVA----AVAGALAQKIGKAVR--- 79 Lpe 74 ----SKRPP---AS--FVFSTAAAVLIAALASSLIAL------SPRQAPPAAALRP---- 47 Sbi 74 ----WNRLP---TGI-AKPAAAAAVVVAALASSFLALP----RPRAAAPPPAGSGP---- 51 Hvu 73 ----RKRPP---ASS-VARASAAAVVVAALASSFIAL------SPRCTPAAAGSGP---- 48 Zma 73 ----WNRLP---SGI-AKPAAAAAVVVAALASSFLALP----RPR-AAPPPAGSGP---- 49 Osa 73 ----RSRPHPPSSSV-AVPAAAAAVIAAALASSFLALL----QPPRRAPVAAGSRV---- 54 Psi 71 LTGLWSRLRICSASCGSKPKVSRTATVPARVSMVTAVE----ASPASKSAVAAAAAKLAQ 91 . . -----N-terminales Segment------------ Ath 100 -------RAGAPSKARVYADVNVVRPKDYWDYESLAVQWGVQDDYEVVRKVGRGKYSEVF 147 Sal 88 -------RAGAPSKARVYADVNVIKPKDYWDYESLAVQWGAQDDYEVVRKVGRGKYSEVF 128 Bna 87 -------RAGAPSKARVYADVNVIKPKDYWDYESLAVQWGSQDDYEVVRKVGRGKYSEVF 96 Vvi 77 -------RPGAPSKARVYADINVIRPKEYWDYESLTVQWGEQDDYEVVRKVGRGKYSEVF 140 Ptr 76 -------RPGAPSKARVYADINVIRPKDYWDYESLTVQWGEQDDYEVVKKVGRGKYSEVF 165 Mac 74 -------RPGAPSRARVYADINVHRPKDYWDYESLTVQWGEQDDYEVVRKVGRGKYSEVF 132 Lpe 74 ----------IMSKARVYTDVNVVRPKEYWDYEALTVQWGEQDDYEVVRKVGRGKYSEVF 97 Sbi 74 ----------LMSKARVYTDVNVLRPKEYWDYEALTVQWGEQDDYEVVRKVGRGKYSEVF 101 Hvu 73 ----------SMSKARVYTDVNVVRPKEYWDYEALTVQWGEQDDYEVVRKVGRGKYSEVF 98 Zma 73 ----------LMSKAKVYTDVNVLRPKEYWDYEALTVQWGEQDDYEVVRKVGRGKYSEVF 99 Osa 73 ----------GMSKARVYADVNVLRPKEYWDYEALTVQWGEQDDYEVVRKVGRGKYSEVF 104 Psi 71 KIGKSIRRPGVTSKARVYADVNVVRPKEYWDYEALTVQWGDQEDYEVVRKVGRGKYSEVF 151 *:*:**:*:** :**:*****:*:**** *:*****:*********** Ath 100 EGIHATDNEKCVIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQQSKTPSLIFE 207 Sal 88 EGIHATDNEKCVIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQQSKTPSLIFE 188 Bna 87 EGIHATDNEKCVIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQQSKTPSLIFE 156 Vvi 77 EGVHCTDNEKCIIKILKPVKKKKIK----ILQNLCGGPNIVKLLDIVRDQQSKTPSLIFE 196 Ptr 76 EGVHCTDNEKCIIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQQSKTPSLIFE 225 Mac 74 EGVHATNNEKCIIKILKPVKKKKIKREIKILQNLCGGPNIIKLLDIVRDQQSKTPSLIFE 192 Lpe 74 EGINVTNSEKCVIKILKPVKKKKIKREIKILQNICGGPNIIKLLDIVRDQHSKTPSLIFE 157 Sbi 74 EGINVNNNEKCIIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQHSKTPSLIFE 161 Hvu 73 EGFSVNNSEKCVIKILKPVKKKKIKREIKILQNLCGGPNIIKLLDIVRDQHSKTPSLIFE 158 Zma 73 EGINVNNYEKCIIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQHSKTPSLIFE 159 Osa 73 EGINVNNNEKCIIKILKPVKKKKIKREIKILQNLCGGPNIVKLLDIVRDQHSKTPSLIFE 164 Psi 71 EGVNSINNERCIIKILKPVKKKKIKREIKILQNLSEGPNIVKLLDIVRDQQSKTPSLIFE 211 **. : *:*:************* ****:. ****:*********:*********

Ergebnisse
41
Ath 100 HVNNKDFKVLYPTLSDYDVRYYIFELLKALDFCHSRGIMHRDVKPHNVMIDHEQRKLRLI 267 Sal 88 HVNNKDFKVLYPTLSDYDVRYYIYELLKALDFCHSRGIMHRDVKPHNVMIDHEQRKLRLI 248 Bna 87 HVNNKDFKVLYPTLSDYDVRYYIYELLKALDFCHSRGIMHRDVKPHNVMIDHEQRKLRLI 216 Vvi 77 YVNNTDFKVLYPTLSDYDIRYYIYELLKALDYCHSQGIMHRDVKPHNVMIDHEQRKLRLI 256 Ptr 76 YVNNTDFKVLYPTLSDFDIRYYIYELLKALDYCHSQGIMHRDVKPHNVMIDHEQRKLRLI 285 Mac 74 YVNNTDF--------------------KALDYCHSQGIMHRDVKPHNVMIDHQHRKLRLI 232 Lpe 74 YVNNTDFKVLYPTLTDYDIRYYLYELLKALDYCHSQGIMHRDVKPHNVMIDHELRKLRLI 217 Sbi 74 FVNNTDFKVLYPTLTDYDIRYYIYELLKALDYCHSQGIMHRDVKPHNVMIDHELRKLRLI 221 Hvu 73 YVNNTDFKVLYPTLTDYDIRYYLYELLKALDHCHSQGIMHRDVKPHNVMIDHDLRKLRLI 218 Zma 73 FVNNTDFKVLYPTLTDYDIRYYIYELLKALDYCHSQGIMHRDVKPHNVMIDHELRKLRLI 219 Osa 73 YVNNTDFKVLYPTLTDYDIRYYIYELLKALDYCHSQGIMHRDVKPHNVMIDHELRKLRLI 224 Psi 71 YVNNTDFKVLYPTLSDFDIRYYIYELLKALDYCHSQGIMHRDVKPHNVMIDHEHRKLRLI 271 .***.** ****.***:****************: ****** Ath 100 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDLWSLGCMFAGMIFRKEPFFY 327 Sal 88 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLMDYDYSLDLWSLGCMFAGMIFRKEPFFY 308 Bna 87 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLMDYDYSLDLWSLGCMFAGMIFRRNPSFM 276 Vvi 77 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDLWSLGCMFAGMIFRKEPFFY 316 Ptr 76 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDLWSLGCMFAGMIFRKEPFFY 345 Mac 74 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 292 Lpe 74 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 277 Sbi 74 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 281 Hvu 73 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 278 Zma 73 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 279 Osa 73 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 284 Psi 71 DWGLAEFYHPGKEYNVRVASRYFKGPELLVDLQDYDYSLDMWSLGCMFAGMIFRKEPFFY 331 ******************************** *******:*************::* * Ath 100 GHDNYDQLVKIAKVLGTDELNAYLNKYRIELDPNLTSLVGRHSRKPWTKFINSENQHLAV 387 Sal 88 GHDNYDQLVKIAKVLGTDELNTYLNRYRIELDPNLASLVGRHSRKPWSKFINSENQHLAV 368 Bna 87 ------------------DMTTMIN----------------------------------- 283 Vvi 77 GHDNYDQLVKIAKVLGTDELNAYLNKYRLELDPHLAALVGRHSRKPWTKFVNADNQHLAV 376 Ptr 76 GHDNYDQLVKIAKVLGTDELNAYLNRYRIELDLHLAALVGRHSRKPWSKFINVDNQHLAV 405 Mac 74 GHDNYDQLVKIAKVLGTDGLNTYLNKYRLELDPQLEALVGRHSRKPWTRFINGDNQHLAV 352 Lpe 74 GHDNHDQLVKIAKVLGTDGLNAYLKKYHIELDPHLEHLVGRHSRKPWSKFINADNQHLVS 337 Sbi 74 GHDNHDQLVKIAKVLGTDGLNAYLNKYHIELDPQLEALVGRHSRKPWAKFINADNQHLVS 341 Hvu 73 GHDNHDQLVKIAKVLGTDSLNAYLKKYHLELDPQLEHLVGRHSRKPWSKFINADNQHLVS 338 Zma 73 GHDNHDQLVKIAKVLGTDGLNAYLNKYHIELDPQLEALVGRHSRKPWSKFMNADNQHLVS 339 Osa 73 GHDNHDQLVKIAKVLGTEALNAYLNKYHIELDPQLEALVGRHSRKPWSKFINADNQHLVS 344 Psi 71 GHDNYDQLVKIAKVLGTDELNAYLNKYRIELDPHLEALVGRHSRKPWSKFINADNQHLVV 391 :.: :: Ath 100 PE-AVDFVDKLLRYDHQERPTAKEAMAHPYFYPIRNAESSR-TPRSQ 432 Sal 88 PE-AVDFVDKLLKYDHQERPTAKEAMAHPYFNPIRNAESSRSTPRAQ 414 Bna 87 ----------------------------------------------- Vvi 77 PE-SIDFLDKLLRYDHQERPTAKEAMAHPYFYPIRNAESSRSRAS-- 420 Ptr 76 PE-AVDFLDKLLRYDHQERPTAKEAMAHPYFYPIRNAESSRTRT--- 448 Mac 74 PEVAVDFVDKLLQYDHQDRPTAKEAMAHAYFIPVRNAESSGRART-- 397 Lpe 74 PE-AIDFLDKLLRYDHQDRLTAREAMAHPYFLQVKAAENSRSRPQ-- 381 Sbi 74 PE-AIDFLDKLLRYDHQDRLTAREAMAHPYFLQVRAVENSRSRPQ-- 385 Hvu 73 PE-AIDFLDKLLRYDHQDRLTAREAMAHPYFLQVRAAENSRARPQ-- 382 Zma 73 PE-AIDFLDKLLRYDHQDRLTAREAMAHPYFLQVRAVENSRTRPQ-- 383 Osa 73 PE-AVDFLDKLLRYDHQDRLTAREAMAHPYFLQVRAAENSRARPQ-- 388 Psi 71 PE-AVDFLDKLLRYDHQERPTAKEAMAHPYFYHVRSAEASRTRAQ-- 435
Abb. 4.2: Multipler Vergleich der cpCK2‐Sequenzen aus verschiedenen Pflanzen. Ath: Arabidopsis thaliana (Accession NP_179889), Sal: Sinapis alba (Accession CAD12663), Bna: Brassica napus (Accession EL590122), Vvi: Vitis vinifera (Accession XP_002284333), Ptr: Populus trichocarpa (Accession XP_002303393), Mac: Musa acuminata (Accession ABF70009), Lpe: Lolium perenne (Accession BAD98469), Sbi: Sorghum bicolor (Accession XP_002463842), Hvu: Hordeum vulgare (Accession BAF03601), Zma: Zea mays (Accession ACG36153), Osa: Oryza sativa (Accession EEC76225), Psi: Picea sitchensis

Ergebnisse
42
(Accession ABK24612). Das Alignment wurde mit ClustalW2 durchgeführt. Putative Transitpeptide sind grün dargestellt. Konservierte CK2α‐Subdomänen wie Nukleotid‐Binderegion (blau) und katalytische Region (rot) wurden ebenfalls hervorgehoben.
Die nukleo‐/cytosolischen CK2α‐Sequenzen besitzen keine Verlängerung am N‐Terminus
und beginnen mit dem CK2‐typischen SKARVY‐Motif (Pinna, 1997) im N‐terminalen Segment
(s. Abb. 4.2).
4.3 Bakterielle Überexpression und Aufreinigung der cpCK2‐Proteine
Um eine vergleichende biochemische und funktionelle Charakterisierung der
chloroplastidären CK2 aus Senf und Arabidosis durchführen zu können, mussten die Proteine
zuerst in ausreichender Menge aufgereinigt werden. Der Überexpressionsklon von cpCK2
aus Senf (SACK2) lag bereits aus Vorarbeiten vor. Die Expression und Aufreinigung des
rekombinanten SACK2‐Proteins erfolgte mittels PQE‐System wie in Ogrzewalla et al. (2002)
beschrieben. Zur bakteriellen Überexpression des Arabidopsis cpCK2‐Proteins (ATCK2)
wurde das pMAL‐Expressionssystem gewählt, da sich die ATCK2‐Expression im Gegensatz zu
SACK2 nicht im pQE‐System induzieren ließ (s. Abb. 4.3 A). Die cDNA von ATCK2 wurde ohne
Transitpeptid in den pMALc2x‐Vektor kloniert. Dieses System ermöglicht die Überexpression
rekombinanter Proteine mit einer N‐terminalen E. coli eigenen MBP (maltose binding
protein)‐Fusion. Die Überexpression in E. coli TB1‐Zellen wurde mit 0,3 mM IPTG induziert
und nach einer Inkubation für 2 h bei Raumtemperatur wurden die Zellen geerntet.
Abbildung 4.3 A zeigt den Induktionstest mit IPTG in pQE30 und pMALc2x. Die Induktion
erfolgte nur im pMAL‐System und die induzierte Probe zeigte bei ca. 85 kDa eine
Überexpressionsbande, welche der Größe des MBP:ATCK2‐Fusionsproteins entspricht. Das
Protein war löslich und konnte über Amylose‐Resin unter nativen Bedingungen in großer
Menge aufgereinigt werden (Abb. 4.3 B). Die Abspaltung des MBP‐Tags durch Faktor‐Xa‐
Protease wurde erfolgreich durchgeführt (Abb. 4.3 C). Dazu wurde das Fusions‐Protein ohne
und mit Faktor‐Xa (1 µg Faktor‐Xa pro 50 µg Protein) für 2 h bei Raumtemperatur inkubiert.
Nach der SDS‐PAGE wurden die Proteine auf Nitrocellulose‐Membran transferiert und mit
einem monoklonalen CK2α‐Antikörper detektiert. Die Probe mit Faktor‐Xa zeigt zusätzlich zu
der 85 kDa‐Bande eine Bande bei ca. 44 kDa, welche dem cpCK2‐Protein ohne MBP‐Tag
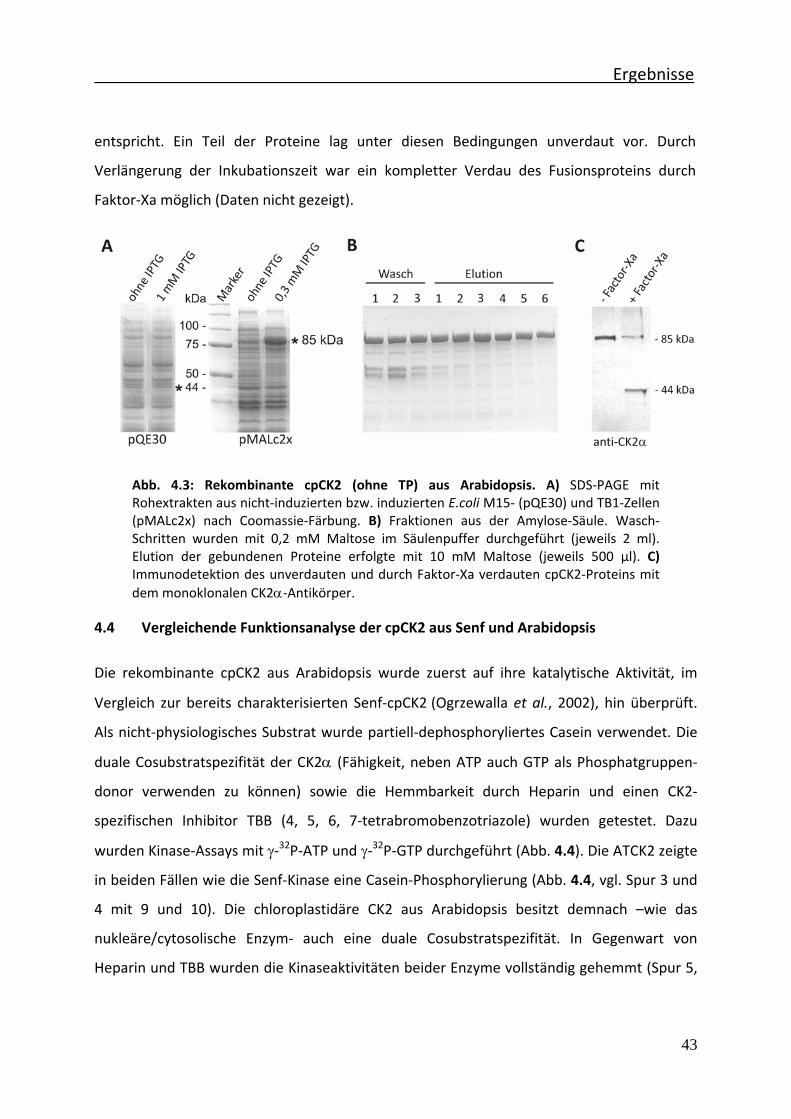
Ergebnisse
43
entspricht. Ein Teil der Proteine lag unter diesen Bedingungen unverdaut vor. Durch
Verlängerung der Inkubationszeit war ein kompletter Verdau des Fusionsproteins durch
Faktor‐Xa möglich (Daten nicht gezeigt).
Abb. 4.3: Rekombinante cpCK2 (ohne TP) aus Arabidopsis. A) SDS‐PAGE mit Rohextrakten aus nicht‐induzierten bzw. induzierten E.coli M15‐ (pQE30) und TB1‐Zellen (pMALc2x) nach Coomassie‐Färbung. B) Fraktionen aus der Amylose‐Säule. Wasch‐Schritten wurden mit 0,2 mM Maltose im Säulenpuffer durchgeführt (jeweils 2 ml). Elution der gebundenen Proteine erfolgte mit 10 mM Maltose (jeweils 500 µl). C) Immunodetektion des unverdauten und durch Faktor‐Xa verdauten cpCK2‐Proteins mit dem monoklonalen CK2α‐Antikörper.
4.4 Vergleichende Funktionsanalyse der cpCK2 aus Senf und Arabidopsis
Die rekombinante cpCK2 aus Arabidopsis wurde zuerst auf ihre katalytische Aktivität, im
Vergleich zur bereits charakterisierten Senf‐cpCK2 (Ogrzewalla et al., 2002), hin überprüft.
Als nicht‐physiologisches Substrat wurde partiell‐dephosphoryliertes Casein verwendet. Die
duale Cosubstratspezifität der CK2α (Fähigkeit, neben ATP auch GTP als Phosphatgruppen‐
donor verwenden zu können) sowie die Hemmbarkeit durch Heparin und einen CK2‐
spezifischen Inhibitor TBB (4, 5, 6, 7‐tetrabromobenzotriazole) wurden getestet. Dazu
wurden Kinase‐Assays mit γ‐32P‐ATP und γ‐32P‐GTP durchgeführt (Abb. 4.4). Die ATCK2 zeigte
in beiden Fällen wie die Senf‐Kinase eine Casein‐Phosphorylierung (Abb. 4.4, vgl. Spur 3 und
4 mit 9 und 10). Die chloroplastidäre CK2 aus Arabidopsis besitzt demnach –wie das
nukleäre/cytosolische Enzym‐ auch eine duale Cosubstratspezifität. In Gegenwart von
Heparin und TBB wurden die Kinaseaktivitäten beider Enzyme vollständig gehemmt (Spur 5,

Ergebnisse
44
6, 11 und 12). Kontroll‐Ansätze mit beiden Kinasen allein (Spur 1 und 8) und mit Casein ohne
Kinase (Spur 2) zeigten keine Phosphorylierungssignale.
Abb. 4.4: Nachweis der Kinase‐Aktivität von ATCK2 im Vergleich zu SACK2. Dargestellt ist ein Autoradiogramm mit radioaktiv phosphoryliertem Casein. Jeweils 2,5 µg cpCK2 pro 50 µl Reaktionsansatz wurde für die Phosphorylierung von 1 µg Casein eingesetzt. Rekombinante Arabidopsis cpCK2 (rechts) kann wie Senf‐cpCK2 (links) Casein sowohl mit γ‐32P‐ATP (Spur 9) als auch mit γ‐32P‐GTP (Spur 10) phosphorylieren. Beide Kinasen lassen sich durch Heparin und TBB in den angegebenen Konzentrationen inhibieren.
Da das TBB‐Reagenz in DMSO gelöst war, wurde die Aktivität auch in Gegenwart von DMSO
allein getestet. Diese Kontrolle zeigte, dass das Lösungsmittel offenbar keinen Einfluss auf
die Kinase‐Aktivität hat (Spur 7). TBB ist wie Heparin ein effektiver CK2α‐Inhibitor. Die
Aktivität von ATCK2 wurde auch nach dem Abspalten des MBP‐Tags getestet (Spur 13).
Gemeinsam mit der Negativkontrolle (MBP allein, nicht gezeigt) zeigt dies, dass die
Kinaseaktivität des Fusionsproteins ausschließlich vom cpCK2‐Anteil stammt.
4.5 Überexpression und Aufreinigung von Sigmafaktoren zur in vitro Phosphorylierung
durch cpCK2
Nach der biochemischen Charakterisierung der ATCK2 waren bereits wichtige methodische
Voraussetzungen gegeben um zu klären, ob ATCK2 in der Lage ist, pflanzliche Sigmafaktoren
als Substrat zu erkennen und sie zu phosphorylieren. Zu diesem Zweck wurden homologe
Sigmafaktoren nach Umklonierung der cDNAs in geeignete bakterielle Expressionsvektoren

Ergebnisse
45
ebenfalls rekombinant hergestellt und aufgereinigt. Überexpressionsplasmide für SIG1 und
SIG2 aus Senf (SASIG1 und SASIG2) sowie SIG1 aus Arabidopsis (ATSIG1) standen bereits aus
anderen Arbeiten in unserer Arbeitsgruppe zur Verfügung (Kreutzenbeck, Diplomarbeit
2000; Berndt, Diplomarbeit 2000; Kolpack, Dissertation 2010). Die Überexpression der
SASIG1 und 2 erfolgte mittels pQE‐Expressionssystem in E. coli M15‐Zellen. Dieses System
ermöglicht eine N‐terminale His‐Tag‐Fusion an die entsprechenden Proteine. Die ATSIG1‐
Expression wurde in pMALc2x‐Vektor in E. coli BL21RIL‐Zellen durchgeführt. Im Rahmen der
vorliegenden Arbeit wurde der Sigmafaktor 6 aus Arabidopsis (ATSIG6) ohne Transitpeptid in
den pMAL2cx‐Vektor kloniert und zur Überexpression eingestzt. Dieses System erlaubt eine
N‐terminale MBP‐Fusion an die entsprechenden Proteine. In Abbildung 4.5.1 sind sämtliche
verwendete Rekombinant‐Faktoren schematisch dargestellt.
Abb. 4.5.1: Verwendete rekombinante Sigmafaktoren. SASIG1‐ und SASIG2‐Protein besitzen einen N‐terminalen 6x‐His‐Tag. ATSIG1 und ATSIG6 wurden dagegen als MBP‐Fusionsproteine exprimiert. Die Abspaltungsstelle für die Protease Faktor‐Xa ist abgebildet. Die Molekulargewichte der Proteine sind in Klammern angegeben. Das MBP‐Protein hat ein Molekulargewicht von 42 kDa.
Die Expression von ATSIG6 ließ sich in E. coli TB1‐Zellen nicht induzieren, jedoch war die
Induktion in BL21RIL‐Zellen möglich (Abb. 4.5.2 A). Die Aufreinigung des MBP‐
Fusionsproteins erfolgte mittels Amylose‐Resin (Abb. 4.5.2 B). Der rekombinante, gut
lösliche Faktor konnte für die weiteren Analysen in großer Menge isoliert werden.
Die aufgereinigten SASIG1‐, SASIG2‐ und ATSIG1‐Proteine wurden mittels SDS‐PAGE
überprüft (s. Abb. 4.5.2 C).

Ergebnisse
46
Abb. 4.5.2: Expression und Reinigung rekombinanter Sigmafaktoren. A) Induktionstest von ATSIG6 in E. coli TB1‐ und BL21RIL‐Zellen. Rohextrakte aus nicht‐induzierten (‐IPTG) und mit 0,3 mM IPTG induzierten Zellen (+IPTG) wurden im SDS‐Gel aufgetrennt und die Proteine wurden mit Coomassie‐Färbung visualisiert. B) SDS‐PAGE der Reinigung von ATSIG6 mittels Amylose‐Resin. Aufgetragen sind die Wasch‐ bzw. Elutionsfraktionen. C) Aufgereinigte SIG1 und SIG2 aus Senf (His:SASIG1 bzw. His:SASIG2) und SIG1 aus Arabidopsis (MBP:ATSIG1).
4.6 In vitro Phosphorylierung der rekombinanten Sigmafaktoren durch cpCK2
Aus Vorarbeiten war bereits bekannt, dass die Phosphorylierung der Sigmafaktoren durch
PTK eine regulatorische Wirkung auf die plastidäre Transkription ausübt (Baginsky et al.,
1997). Die in vitro Analysen zeigten, dass das rekombinante „Gegenstück“ der PTK aus Senf
(cpCK2) die clonierten Senf Sigmafaktoren 1‐3 phosphorylieren kann (Ogrzewalla et al.,
2002; Ogrzewalla, Dissertation 2001). Um zu überprüfen, ob die rekombinante cpCK2 aus
Arabidopsis ebenfalls in der Lage ist, die Sigmafaktoren zu phosphorylieren, wurden in vitro
Kinase‐Assays mit γ‐32P‐ATP durchgeführt. Die rekombinanten Faktoren, SASIG1, SASIG2,
ATSIG1 und ATSIG6, wurden als Substrat in einem Phosphorylierungsansatz mit cpCK2 aus
Senf bzw. aus Arabidopsis eingesetzt (Abb. 4.6). Als Kontrollen wurden SASIG1 (Spur 1) und
ATSIG6 (Spur 9) ohne cpCK2 unter Phosphorylierungsbedingungen inkubiert. Bei beiden
Ansätzen wurde kein Phosphorylierungssignal detektiert. Während der Reaktionsansatz mit
SACK2 allein ohne Substrat (Spur 2) kein Signal zeigte, war beim gleichen Ansatz mit ATCK2
eine Phosphorylierungsbande auf der Höhe der rekombinanten Kinase, bei ca. 87 kDa,
erkennbar (Spur 10). Diese Bande, die am ehesten als Autophosphorylierungssignal
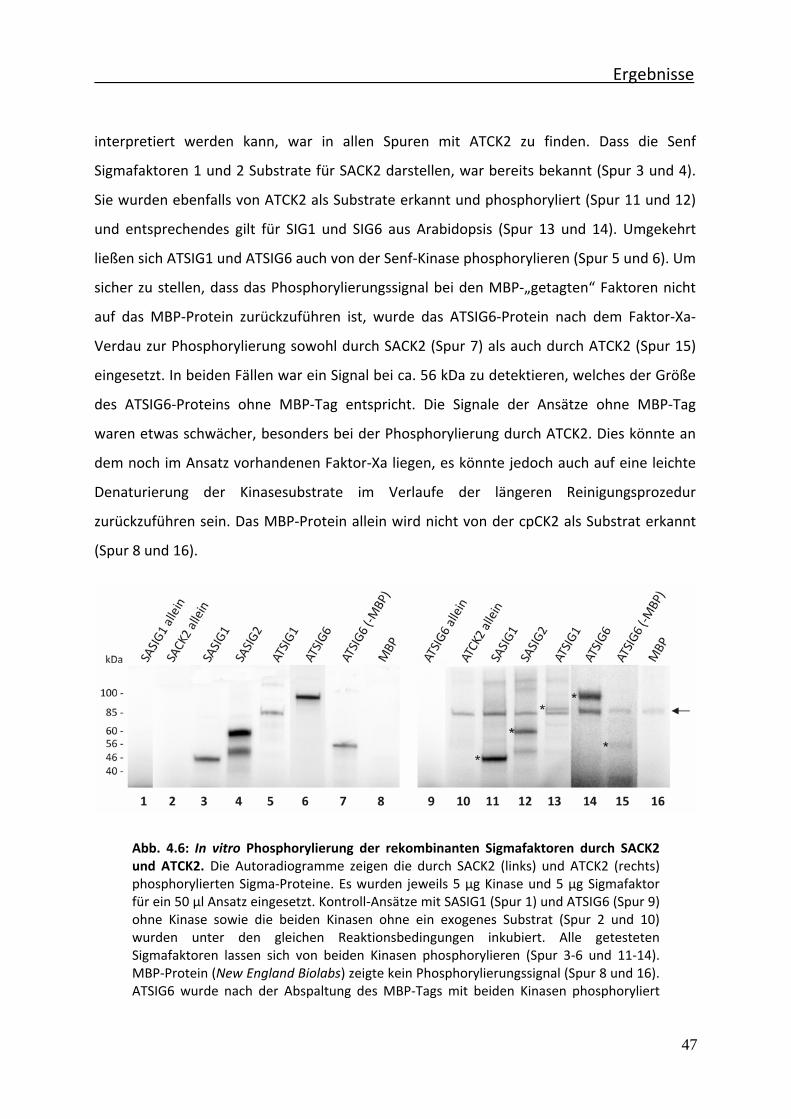
Ergebnisse
47
interpretiert werden kann, war in allen Spuren mit ATCK2 zu finden. Dass die Senf
Sigmafaktoren 1 und 2 Substrate für SACK2 darstellen, war bereits bekannt (Spur 3 und 4).
Sie wurden ebenfalls von ATCK2 als Substrate erkannt und phosphoryliert (Spur 11 und 12)
und entsprechendes gilt für SIG1 und SIG6 aus Arabidopsis (Spur 13 und 14). Umgekehrt
ließen sich ATSIG1 und ATSIG6 auch von der Senf‐Kinase phosphorylieren (Spur 5 und 6). Um
sicher zu stellen, dass das Phosphorylierungssignal bei den MBP‐„getagten“ Faktoren nicht
auf das MBP‐Protein zurückzuführen ist, wurde das ATSIG6‐Protein nach dem Faktor‐Xa‐
Verdau zur Phosphorylierung sowohl durch SACK2 (Spur 7) als auch durch ATCK2 (Spur 15)
eingesetzt. In beiden Fällen war ein Signal bei ca. 56 kDa zu detektieren, welches der Größe
des ATSIG6‐Proteins ohne MBP‐Tag entspricht. Die Signale der Ansätze ohne MBP‐Tag
waren etwas schwächer, besonders bei der Phosphorylierung durch ATCK2. Dies könnte an
dem noch im Ansatz vorhandenen Faktor‐Xa liegen, es könnte jedoch auch auf eine leichte
Denaturierung der Kinasesubstrate im Verlaufe der längeren Reinigungsprozedur
zurückzuführen sein. Das MBP‐Protein allein wird nicht von der cpCK2 als Substrat erkannt
(Spur 8 und 16).
Abb. 4.6: In vitro Phosphorylierung der rekombinanten Sigmafaktoren durch SACK2 und ATCK2. Die Autoradiogramme zeigen die durch SACK2 (links) und ATCK2 (rechts) phosphorylierten Sigma‐Proteine. Es wurden jeweils 5 µg Kinase und 5 µg Sigmafaktor für ein 50 µl Ansatz eingesetzt. Kontroll‐Ansätze mit SASIG1 (Spur 1) und ATSIG6 (Spur 9) ohne Kinase sowie die beiden Kinasen ohne ein exogenes Substrat (Spur 2 und 10) wurden unter den gleichen Reaktionsbedingungen inkubiert. Alle getesteten Sigmafaktoren lassen sich von beiden Kinasen phosphorylieren (Spur 3‐6 und 11‐14). MBP‐Protein (New England Biolabs) zeigte kein Phosphorylierungssignal (Spur 8 und 16). ATSIG6 wurde nach der Abspaltung des MBP‐Tags mit beiden Kinasen phosphoryliert

Ergebnisse
48
(Spur 7 und 15). Sterne zeigen die Phosphorylierungsbanden der Sigmafaktoren. Pfeil deutet auf die Autophosphorylierungs‐bande der ATCK2 an.
Diese Ergebnisse zeigen, dass nicht nur die Senf Sigmafaktoren sondern auch die Sigma‐
Proteine aus Arabidopsis als Substrate für cpCK2 fungieren, unabhängig davon, aus welcher
der beiden Pflanzen die Rekombinant‐Kinase stammt.
4.7 Funktionelle Konsequenzen der Sigmafaktor‐Phosphorylierung
Nachdem gezeigt war, dass mehrere Sigmafaktoren in vitro Substrate der cpCK2 darstellen,
wurde als nächstes getestet, ob diese Phosphorylierung eine funktionelle Bedeutung für die
Aktivität der betreffenden Faktoren hat. Zu diesem Zweck wurden EMSA‐Experimente mit 32P‐markierten Promotorfragmenten und core‐Polymerase aus E. coli durchgeführt (Abb.
4.7). Zur Bildung einer bindungsfähigen RNA‐Polymerase (Holoenzym) wurden die
rekombinanten Sigmafaktoren 1 und 6 aus Arabidopsis eingesetzt. Die Faktoren wurden vor
der Inkubation mit dem core‐Enzym und den radioaktiv‐markierten Promotorfragmenten
durch ATCK2 in Gegenwart von ATP phosphoryliert (ATSIG1‐P/ATSIG6‐P). Die nicht‐
phosphorylierten Faktoren wurden ebenfalls mit ATCK2, jedoch ohne ATP, inkubiert
(ATSIG1/ATSIG6). Als Promotoren wurden psbA‐ (Homann und Link, 2003) und atpB‐
Fragmente (Schweer et al., 2010), die die 5´‐Region des jeweiligen Gens mit den Promotoren
enthalten, verwendet. Die nicht‐markierten psbA‐, atpB‐ sowie trnK‐Promotorfragmente
(Neuhaus und Link, 1987) wurden als Kompetitoren in 20‐fachem molaren Überschuss
eingesetzt. Ein 500 bp DNA‐Fragment aus dem Intronbereich des trnK‐Gens aus Senf
(Bam0.5; Neuhaus und Link, 1987) diente als nicht‐spezifischer Kompetitor. Um eine
unspezifische Bindung der RNA‐Polymerase an die DNA zu verhindern, wurde Poly d[(I‐C)]
(60 ng/µl) dem Ansatz zugegeben. Beide Sigmafaktoren zeigten für sich allein keine DNA‐
Bindungsaktivität (Spur A3, A14, B2 und B15), auch nicht im phosphorylierten Zustand (B9
und B16). Die core‐Polymerase allein war auch nicht in der Lage, spezifisch an die DNA zu
binden (Spur A2 und B3).

Ergebnisse
49
Abb. 4.7: EMSA‐Experimente mit rekombinanten Sigmafaktoren 1 und 6 aus Arabidopsis. Bindungsstudien mit radioaktiv‐markiertem psbA‐ (A) bzw. atpB‐Promotor‐fragmenten (B), E. coli core‐RNA‐Polymerase und nicht‐phosphorylierten (ATSIG1/ATSIG6) bzw. durch ATCK2 vorphosphorylierten (ATSIG1‐P/ATSIG6‐P) Sigmafaktoren. Dargestellt sind die Autoradiogramme der nativen Polyacrylamidgele. Die Kontrollen beinhalten die radioaktiv‐markierten Promotorfragmente allein (Spur A1 und B1), core‐Enzym ohne Sigmafaktor (Spur A2 und B3) sowie die Faktoren allein ohne core‐Polymerase (Spur A3, A14, B2, B9, B15 und B16). Nicht‐markierte psbA‐, atpB‐ und trnK‐Promotorfragmente wurden als Kompetitoren im vollständigen System (Promotor + core‐Polymerase + Sigmafaktor) eingesetzt (Spuren A5‐A7, A16‐A18, A21‐A23, B5‐B7, B11‐B13 und B19‐B21). Ein DNA‐Fragment ohne Promotor, Bam0.5, wurde als nicht‐spezifischer Kompetitor (SpurA8, A13, A19, A24, B8, B14 und B22) verwendet.
Die Rekonstitution des Holoenzyms sowohl mit nicht‐phosphoryliertem (Spur A4) als auch
mit phosphoryliertem ATSIG1‐Protein (Spur A9) führte zu einer Bindung des psbA‐
Promotorfragments. Die Phosphorylierung hatte keinen Einfluss auf die Bindungsaktivität
des ATSIG1‐Proteins an den psbA‐Promotor (vgl. Spur A4 mit A9). Die Spezifität der Bindung
wurde durch Kompetition mit nicht‐markiertem psbA‐Promotor und Bam0.5‐Fragment
analysiert (A5, A8, A10 und A13). Es zeigte sich im Falle des psbA‐Fragments eine deutliche
Abschwächung der Bindungssignale (A5 und A10), während das Bam0.5‐Fragment keine

Ergebnisse
50
kompetitive Wirkung aufwies (A8 und A13). Um die Präferenzen von ATSIG1 für andere
plastidäre Promotoren zu untersuchen, wurden Kompetition‐Experimente mit atpB‐ und
trnK‐Promotorfragmente durchgeführt (Spur A6, A7, A11 und A12). Im Falle von ATSIG1
wurden die Bindungssignale durch nicht‐markierte atpB‐ und trnK‐Promotorfragmente
deutlich abgeschwächt (Spur A6 und A7). Dies bedeutet, dass die core‐Polymerase mit
ATSIG1 an diese beiden Promotoren ebenfalls binden kann. Die Phosphorylierung des
ATSIG1‐Proteins ändert anscheinend seine Präferenz für den atpB‐Promotor, denn die
Bindung ließ sich hier nicht so effektiv mit nicht‐markiertem atpB‐Promotor kompetitieren
(Spur A11). Die Bindung von ATSIG1 an den trnK‐Promotor war durch Phosphorylierung
unbeeinflusst (vgl. Spur A7 mit A12).
Der Sigmafaktor 6 aus Arabidopsis war ebenfalls in der Lage, sowohl im nicht‐
phosphorylierten (ATSIG6) als auch im phosphorylierten Zustand (ATSIG6‐P) an den psbA‐
Promotor zu binden (vgl. Spur A15 mit A20). Während sich das Bindungssignal bei ATSIG6
nicht durch atpB‐ und trnK‐Promotoren schwächen ließ, war die Intensität im Falle von
ATSIG6‐P durch beide Promotorfragmente ersichtlich beeinträchtigt (vgl. Spur A17 und A18
mit A22 und A23). Diese Ergebnisse zeigen deutlich, dass das ATSIG6‐Protein nur im
phosphorylierten Zustand die atpB‐ und trnK‐Promotoren effizient binden kann.
Die oben beschriebenen Promotor‐Bindestudien wurden auch mit radioaktiv‐markiertem
atpB‐Promotorfragment durchgeführt (Abb. 4.7 B). Im Vergleich zu den Experimenten mit
dem psbA‐Promotor waren hier die Bindungssignale bei ATSIG1 und ATSIG1‐P ziemlich
schwach. Phosphoryliertes ATSIG6‐Protein wies eine stärkere Bindung an den atpB‐
Promotor auf als das nicht‐phosphorylierte Protein (vgl. Spur B17 und B18). Das
Bindungssignal ließ sich mit nicht‐markiertem atpB‐Promotor (Spur B20), ebenso wie mit
psbA‐ und trnK‐Fragmente (Spur B19 und B21), jedoch nicht mit Bam0.5, kompetitieren
(Spur B22).
Diese Experimente zeigen deutlich, dass die Phosphorylierung durch rekombinante cpCK2
aus Arabidopsis die Bindungsaktivität der Sigmafaktoren für unterschiedliche Promotoren
regulieren kann.

Ergebnisse
51
4.8 Redoxregulation der Aktivität von cpCK2 aus Arabidopsis in vitro
Frühere Arbeiten gaben Hinweise darauf, dass die Aktivität der plastidären
Transkriptionskinase und ihres rekombinanten Pendants, cpCK2, einer Redoxregulation
unterliegen (Baginsky et al., 1999, Baena‐González et al., 2001, Ogrzewalla et al., 2002). Für
diese Art von Regulation sind die SH‐Gruppen und somit die Cysteine in Proteinen von
großer Bedeutung.
Um die strukturellen und mechanistischen Determinanten, die in die Redoxregulation des
Enzyms involviert sind, zu untersuchen, wurden alle vier Cysteine von ATCK2 einzeln und
doppelt gegen Serin ausgetauscht. Dazu wurde die cDNA der ATCK2 ohne Transitpeptid‐
sequenz im pBSKS‐Vektor mit den entsprechenden Primern (s. 3.8.1) mutiert. Die Konstrukte
wurden anschließend zur bakteriellen Überexpression in den pMALc2x‐Vektor kloniert. Die
Abbildung 4.8.1 zeigt die Positionen der Cystein‐Reste im rekombinanten ATCK2‐Protein. Die
hochkonservierten funktionellen Subdomänen der CK2α‐Untereinheiten, wie
Nukleotidbindungs‐ (NB) und die katalytische Region (K), sind ebenfalls abgebildet. Die
Cystein‐Mutanten wurden je nach der Position des mutierten Cysteinrests als C1, C2, C3, C4,
C2/3, C3/4 und C2/4 bezeichnet.
Abb. 4.8.1: Schematische Darstellung des rekombinanten cpCK2‐Proteins aus Arabidopsis. Die Abbildung gibt eine Übersicht über die Positionen der mutierten Cystein‐Reste sowie die Subdomänen von ATCK2. Bei der Nummerierung der Aminosäuren wurde das 55 Aminosäuren lange Transitpeptid berücksichtigt. C: Cystein, K: katalytische Region, MBP: Maltose‐Bindeprotein, NB: Nukleotidbindungsregion.
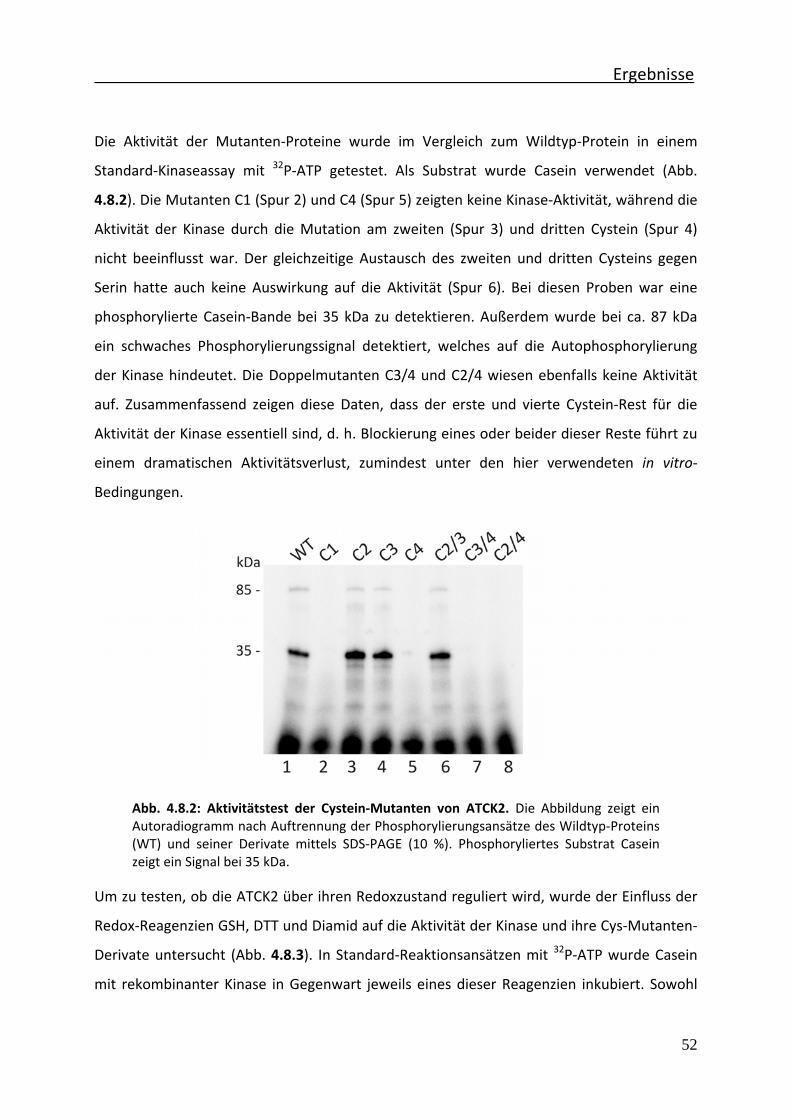
Ergebnisse
52
Die Aktivität der Mutanten‐Proteine wurde im Vergleich zum Wildtyp‐Protein in einem
Standard‐Kinaseassay mit 32P‐ATP getestet. Als Substrat wurde Casein verwendet (Abb.
4.8.2). Die Mutanten C1 (Spur 2) und C4 (Spur 5) zeigten keine Kinase‐Aktivität, während die
Aktivität der Kinase durch die Mutation am zweiten (Spur 3) und dritten Cystein (Spur 4)
nicht beeinflusst war. Der gleichzeitige Austausch des zweiten und dritten Cysteins gegen
Serin hatte auch keine Auswirkung auf die Aktivität (Spur 6). Bei diesen Proben war eine
phosphorylierte Casein‐Bande bei 35 kDa zu detektieren. Außerdem wurde bei ca. 87 kDa
ein schwaches Phosphorylierungssignal detektiert, welches auf die Autophosphorylierung
der Kinase hindeutet. Die Doppelmutanten C3/4 und C2/4 wiesen ebenfalls keine Aktivität
auf. Zusammenfassend zeigen diese Daten, dass der erste und vierte Cystein‐Rest für die
Aktivität der Kinase essentiell sind, d. h. Blockierung eines oder beider dieser Reste führt zu
einem dramatischen Aktivitätsverlust, zumindest unter den hier verwendeten in vitro‐
Bedingungen.
Abb. 4.8.2: Aktivitätstest der Cystein‐Mutanten von ATCK2. Die Abbildung zeigt ein Autoradiogramm nach Auftrennung der Phosphorylierungsansätze des Wildtyp‐Proteins (WT) und seiner Derivate mittels SDS‐PAGE (10 %). Phosphoryliertes Substrat Casein zeigt ein Signal bei 35 kDa.
Um zu testen, ob die ATCK2 über ihren Redoxzustand reguliert wird, wurde der Einfluss der
Redox‐Reagenzien GSH, DTT und Diamid auf die Aktivität der Kinase und ihre Cys‐Mutanten‐
Derivate untersucht (Abb. 4.8.3). In Standard‐Reaktionsansätzen mit 32P‐ATP wurde Casein
mit rekombinanter Kinase in Gegenwart jeweils eines dieser Reagenzien inkubiert. Sowohl

Ergebnisse
53
Wildtyp‐ATCK2 (WT) als auch aktive Cystein‐Mutanten (C2, C3 und C2/3) ließen sich durch
GSH komplett inhibieren (Spur A2‐A5 in Abb. 4.8.3). Ein Kontrollansatz mit WT‐Kinase zeigt
die Aktivität ohne Redox‐Reagenzien (Spur A1). Obwohl DTT ebenso wie GSH ein Reduktant
ist, zeigt es keinen Einfluss auf die Kinase‐Aktivität (Spur A6‐A7). Diamid ist ein
Oxidationsmittel, welches die exponierten Cysteine in Proteinen oxidiert (Kosower und
Kosower, 1995). WT‐ und Mutanten‐Proteine reagieren unterschiedlich auf die Diamid‐
Behandlung (Abb. 4.8.3 B). Während das WT‐Protein schon bei 2 mM Diamid‐Konzentration
komplett inhibiert wurde (Spur B3), waren die Mutanten C2 und C2/3 noch aktiv bei dieser
Konzentration (Spur B5 und B8). Noch höhere Konzentrationen von Diamid hatten ebenfalls
keine Auswirkung auf die Aktivität bei diesen Mutanten (Spur B6, B7, B9 und B10). Im
Unterschied dazu war die C3‐Mutante wie WT sensitiv gegenüber Diamid (Spur B11‐B13).
Abb. 4.8.3: Einfluss von Redox‐Reagenzien auf die Aktivität von ATCK2 und ihrer Cystein‐Mutanten. A) Kinasetests mit rekombinanten Proteinen in Gegenwart von 10 mM GSH (links) und 40 mM DTT (rechts). Ein Ansatz mit WT‐ATCK2 ohne Redox‐Reagenzien dient als Kontrolle (Spur A1). B) Effekt von Diamid auf die Aktivität. Phosphorylierungsexperimente wurden in Gegenwart der angegebenen Diamid‐Konzentrationen durchgeführt.
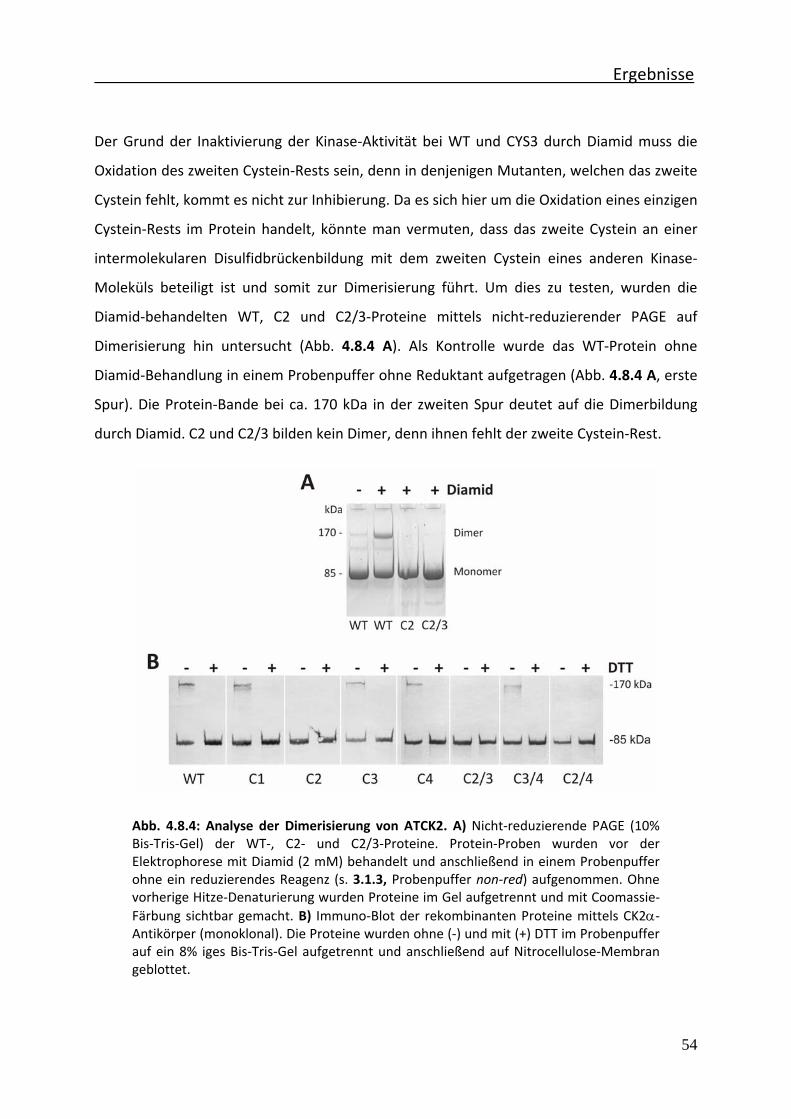
Ergebnisse
54
Der Grund der Inaktivierung der Kinase‐Aktivität bei WT und CYS3 durch Diamid muss die
Oxidation des zweiten Cystein‐Rests sein, denn in denjenigen Mutanten, welchen das zweite
Cystein fehlt, kommt es nicht zur Inhibierung. Da es sich hier um die Oxidation eines einzigen
Cystein‐Rests im Protein handelt, könnte man vermuten, dass das zweite Cystein an einer
intermolekularen Disulfidbrückenbildung mit dem zweiten Cystein eines anderen Kinase‐
Moleküls beteiligt ist und somit zur Dimerisierung führt. Um dies zu testen, wurden die
Diamid‐behandelten WT, C2 und C2/3‐Proteine mittels nicht‐reduzierender PAGE auf
Dimerisierung hin untersucht (Abb. 4.8.4 A). Als Kontrolle wurde das WT‐Protein ohne
Diamid‐Behandlung in einem Probenpuffer ohne Reduktant aufgetragen (Abb. 4.8.4 A, erste
Spur). Die Protein‐Bande bei ca. 170 kDa in der zweiten Spur deutet auf die Dimerbildung
durch Diamid. C2 und C2/3 bilden kein Dimer, denn ihnen fehlt der zweite Cystein‐Rest.
Abb. 4.8.4: Analyse der Dimerisierung von ATCK2. A) Nicht‐reduzierende PAGE (10% Bis‐Tris‐Gel) der WT‐, C2‐ und C2/3‐Proteine. Protein‐Proben wurden vor der Elektrophorese mit Diamid (2 mM) behandelt und anschließend in einem Probenpuffer ohne ein reduzierendes Reagenz (s. 3.1.3, Probenpuffer non‐red) aufgenommen. Ohne vorherige Hitze‐Denaturierung wurden Proteine im Gel aufgetrennt und mit Coomassie‐Färbung sichtbar gemacht. B) Immuno‐Blot der rekombinanten Proteine mittels CK2α‐Antikörper (monoklonal). Die Proteine wurden ohne (‐) und mit (+) DTT im Probenpuffer auf ein 8% iges Bis‐Tris‐Gel aufgetrennt und anschließend auf Nitrocellulose‐Membran geblottet.

Ergebnisse
55
Western‐Blot‐Analysen verstärkten die Annahme, dass das zweite Cystein an einer
intermolekularen Disulfidbrückenbildung beteiligt sein könnte (Abb. 4.8.4 B). In diesem Fall
wurden WT und alle Cystein‐Mutanten nicht mit Diamid behandelt, sondern sie wurden zur
Reduktion möglicher intermolekularen Disulfidbrücken in Probenpuffer ohne und mit DTT
(50 mM) aufgenommen und anschließend im Bis‐Tris‐Gel (8 %) aufgetrennt. Nach dem
Blotten der Proteine auf eine Nitrocellulose‐Membran erfolgte die Immunodetektion mit
monoklonalem CK2α‐Antikörper. Auf dem Blot ist deutlich zu erkennen, dass DTT im
Probenpuffer zu Reduzierung der Dimer‐Bande (170 kDa) bei WT, C1, C3, C4 und C3/4 führt.
Bei den Mutanten C2, C2/3 und C2/4 kommt es nicht zu Dimerbildung, weil der zweite
Cystein‐Rest nicht mehr vorhanden ist.
Zusammenfassend zeigen diese Ergebnisse, dass Cysteine der ATCK2 regulatorische
Funktionen haben können. Cys1 und Cys4 sind wichtig für die Aktivität (vgl. Abb 4.8.2) und
Cys2 ist an der Dimerisierung (intermolekularen Wechselwirkung) der Kinase beteiligt. Die
Dimerisierung führt in dem hier verwendeten in vitro System offenbar zur Inaktivierung der
Kinase (vgl. Abb. 4.8.3 B). Es kann davon ausgegangen werden, dass im authentischen
PEP/cpCK2‐Transkriptionskomplex in vivo andere Interaktionspartner für intermolekulare
(reversible) Disulfidbrückenbildung und Regulation zur Verfügung stehen.
4.9 Zirkular‐Dichroismus Spektroskopie der Cystein‐Mutanten von ATCK2
Mittels Zirkular‐Dichroismus (CD) Spektroskopie lässt sich überprüfen, ob die Cystein‐
Punkmutanten von ATCK2 in einer annäherend gleichen Faltung wie Wildtyp ATCK2
vorliegen. Mit Hilfe dieser Methode können Änderungen in der Sekundär‐ und
Tertiärstruktur eines Mutanten‐Proteins durch den Vergleich mit CD‐Spektren von Wildtyp‐
Protein verfolgt werden (zur Übersicht s. Kelly et al., 2005). Abb. 4.9 zeigt die CD‐Spektren
von rekombinanten C1‐, C2‐, C3‐, C4‐, C2/3‐ und C2/4‐Proteine im Vergleich zu Wildtyp‐
ATCK2 (WT). Die C1‐, C4‐ und C2/4‐Proteine zeigen Unterschiede in ihrer Spektren im
Vergleich zu WT‐ATCK2 und den restlichen Mutanten. Somit scheint die Mutation an Cys1
und Cys4 die Faltung dieser Proteine zu beeinflussen.
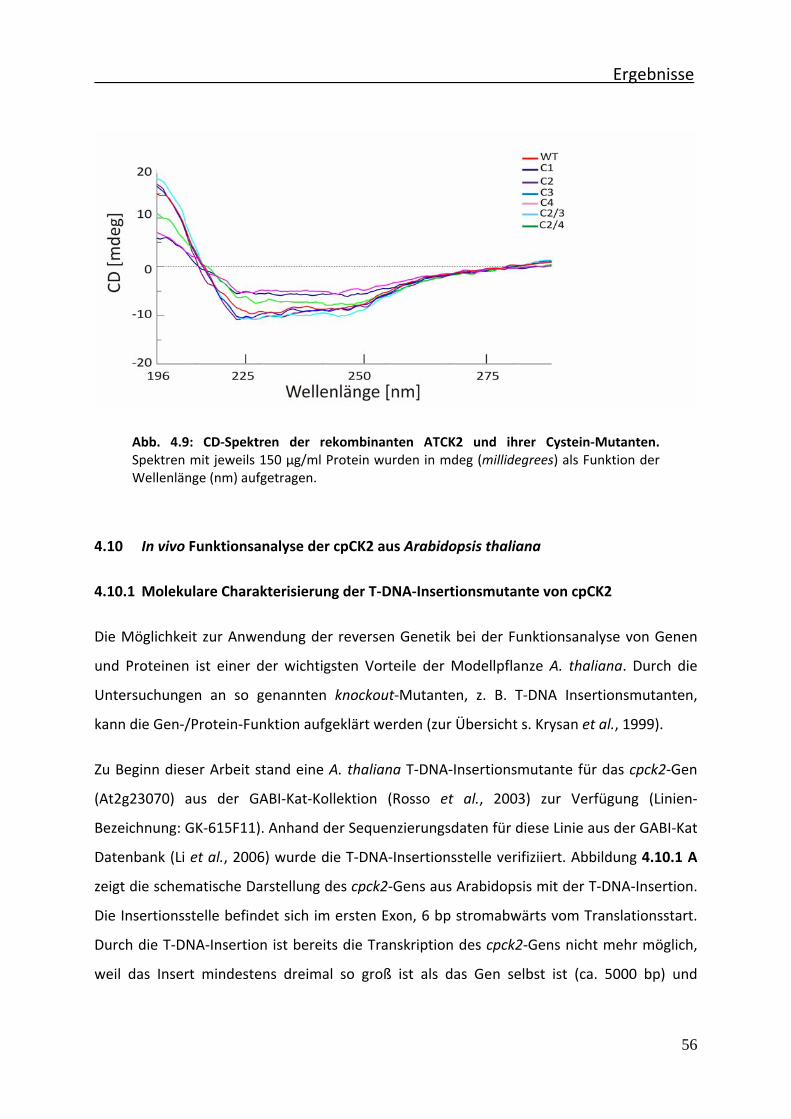
Ergebnisse
56
Abb. 4.9: CD‐Spektren der rekombinanten ATCK2 und ihrer Cystein‐Mutanten. Spektren mit jeweils 150 µg/ml Protein wurden in mdeg (millidegrees) als Funktion der Wellenlänge (nm) aufgetragen.
4.10 In vivo Funktionsanalyse der cpCK2 aus Arabidopsis thaliana
4.10.1 Molekulare Charakterisierung der T‐DNA‐Insertionsmutante von cpCK2
Die Möglichkeit zur Anwendung der reversen Genetik bei der Funktionsanalyse von Genen
und Proteinen ist einer der wichtigsten Vorteile der Modellpflanze A. thaliana. Durch die
Untersuchungen an so genannten knockout‐Mutanten, z. B. T‐DNA Insertionsmutanten,
kann die Gen‐/Protein‐Funktion aufgeklärt werden (zur Übersicht s. Krysan et al., 1999).
Zu Beginn dieser Arbeit stand eine A. thaliana T‐DNA‐Insertionsmutante für das cpck2‐Gen
(At2g23070) aus der GABI‐Kat‐Kollektion (Rosso et al., 2003) zur Verfügung (Linien‐
Bezeichnung: GK‐615F11). Anhand der Sequenzierungsdaten für diese Linie aus der GABI‐Kat
Datenbank (Li et al., 2006) wurde die T‐DNA‐Insertionsstelle verifiziiert. Abbildung 4.10.1 A
zeigt die schematische Darstellung des cpck2‐Gens aus Arabidopsis mit der T‐DNA‐Insertion.
Die Insertionsstelle befindet sich im ersten Exon, 6 bp stromabwärts vom Translationsstart.
Durch die T‐DNA‐Insertion ist bereits die Transkription des cpck2‐Gens nicht mehr möglich,
weil das Insert mindestens dreimal so groß ist als das Gen selbst ist (ca. 5000 bp) und
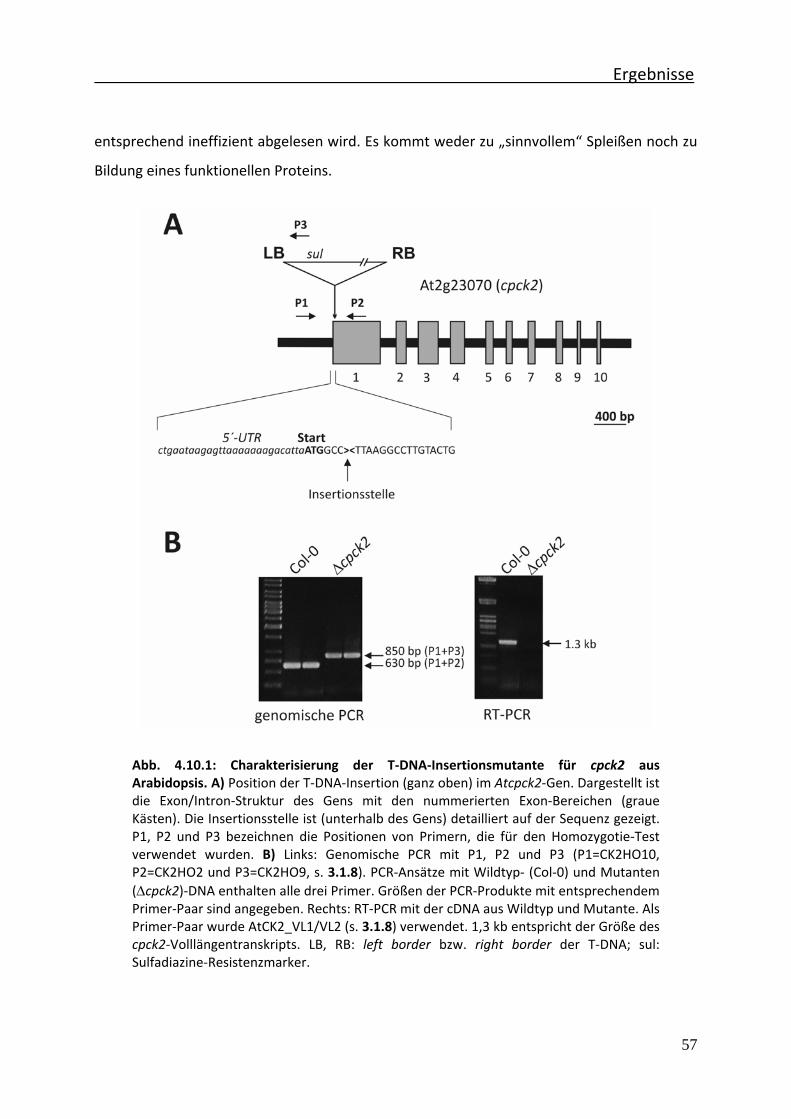
Ergebnisse
57
entsprechend ineffizient abgelesen wird. Es kommt weder zu „sinnvollem“ Spleißen noch zu
Bildung eines funktionellen Proteins.
Abb. 4.10.1: Charakterisierung der T‐DNA‐Insertionsmutante für cpck2 aus Arabidopsis. A) Position der T‐DNA‐Insertion (ganz oben) im Atcpck2‐Gen. Dargestellt ist die Exon/Intron‐Struktur des Gens mit den nummerierten Exon‐Bereichen (graue Kästen). Die Insertionsstelle ist (unterhalb des Gens) detailliert auf der Sequenz gezeigt. P1, P2 und P3 bezeichnen die Positionen von Primern, die für den Homozygotie‐Test verwendet wurden. B) Links: Genomische PCR mit P1, P2 und P3 (P1=CK2HO10, P2=CK2HO2 und P3=CK2HO9, s. 3.1.8). PCR‐Ansätze mit Wildtyp‐ (Col‐0) und Mutanten (∆cpck2)‐DNA enthalten alle drei Primer. Größen der PCR‐Produkte mit entsprechendem Primer‐Paar sind angegeben. Rechts: RT‐PCR mit der cDNA aus Wildtyp und Mutante. Als Primer‐Paar wurde AtCK2_VL1/VL2 (s. 3.1.8) verwendet. 1,3 kb entspricht der Größe des cpck2‐Volllängentranskripts. LB, RB: left border bzw. right border der T‐DNA; sul: Sulfadiazine‐Resistenzmarker.

Ergebnisse
58
Die Samen der 615F11‐Linie lagen als segregierende T1‐Population vor und daher mussten
einzelne Pflanzen aus dieser Population hinsichtlich des Segregationszustandes der T‐DNA
Insertion analysiert werden. Dies erfolgte mittels PCR unter Verwendung von Gen‐ und T‐
DNA‐spezifischer Primer in zwei parallelen PCR‐Ansätzen mit jeweils Wildtyp‐ (Col‐0) bzw.
Mutanten (∆cpck2)‐DNA (Abb. 4.10.1 B). In der PCR‐Reaktion mit der Wildtyp‐DNA entstand
eine 630 bp‐Bande, die der erwarteten Größe des Genom‐spezifischen Primer‐Paars P1/P2
entspricht. Der Ansatz mit der Mutanten‐DNA wies nur ein 850 bp großes P1/P3‐Produkt
und kein P1/P2‐Produkt auf. Das Fehlen des P1/P2‐Produktes im Mutanten‐Ansatz deutet
darauf hin, dass die Mutanten‐Linie homozygot für das mutierte cpck2‐Gen ist, d. h. das Gen
ist auf beiden Chromosomen ausgeschaltet. RT‐PCR‐Analysen mit der cDNA aus Wildtyp und
Mutante zeigten, dass in der Mutante kein cpck2‐Transkript gebildet wird und stellen somit
eine weitere Bestätigung der Homozygotie der Mutanten‐Linie dar.
4.10.2 Phänotypische Charakterisierung der cpck2 knockout‐Mutante
Die knockout‐Mutantenlinie wurde zunächst hinsichtlich morphologischer Auffälligkeiten
unter verschiedenen Bedingungen untersucht. Als erstes wurde getestet, ob die
Mutantenkeimlinge unter photoautotrophen Bedingungen (ohne Saccharose im MS‐
Medium) wachstumsfähig sind. Dazu wurden die Col‐0‐ und Mutanten‐Pflanzen auf MS‐
Medium mit und ohne Saccharose unter Kurztagbedingungen (8h Licht (100 µmol/m2s) / 16h
Dunkel) analysiert (Abb. 4.10.2.1).
Die Mutante zeigte ein verzögertes Wachstum unter mixotrophen Bedingungen (mit
Saccharose im Medium) und hatte wie der Wildtyp grüne Keim‐ und Folgeblätter (Abb.
4.10.2.1, das obere Bild). Photoautotrophe Wachstumsbedingungen waren dagegen letal für
die Mutanten‐Keimlinge (das untere Bild). Die Samen konnten zwar keimen, aber die
Keimlinge waren nicht in der Lage, ohne externe Kohlenstoffquelle weiter zu wachsen.
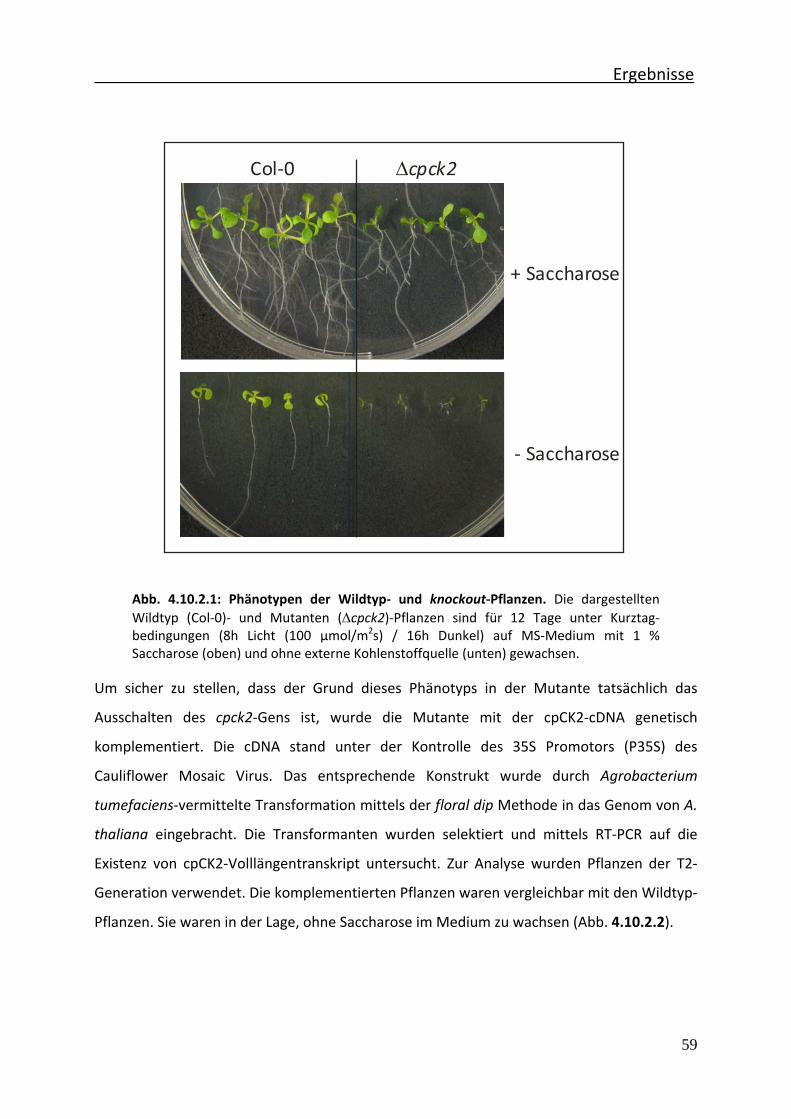
Ergebnisse
59
∆cpck2Col‐0
+ Saccharose
‐ Saccharose
Abb. 4.10.2.1: Phänotypen der Wildtyp‐ und knockout‐Pflanzen. Die dargestellten Wildtyp (Col‐0)‐ und Mutanten (∆cpck2)‐Pflanzen sind für 12 Tage unter Kurztag‐bedingungen (8h Licht (100 μmol/m2s) / 16h Dunkel) auf MS‐Medium mit 1 % Saccharose (oben) und ohne externe Kohlenstoffquelle (unten) gewachsen.
Um sicher zu stellen, dass der Grund dieses Phänotyps in der Mutante tatsächlich das
Ausschalten des cpck2‐Gens ist, wurde die Mutante mit der cpCK2‐cDNA genetisch
komplementiert. Die cDNA stand unter der Kontrolle des 35S Promotors (P35S) des
Cauliflower Mosaic Virus. Das entsprechende Konstrukt wurde durch Agrobacterium
tumefaciens‐vermittelte Transformation mittels der floral dip Methode in das Genom von A.
thaliana eingebracht. Die Transformanten wurden selektiert und mittels RT‐PCR auf die
Existenz von cpCK2‐Volllängentranskript untersucht. Zur Analyse wurden Pflanzen der T2‐
Generation verwendet. Die komplementierten Pflanzen waren vergleichbar mit den Wildtyp‐
Pflanzen. Sie waren in der Lage, ohne Saccharose im Medium zu wachsen (Abb. 4.10.2.2).

Ergebnisse
60
P35S:cpCK2
+ Saccharose ‐ Saccharose
Abb. 4.10.2.2: Phänotyp der komplementierten Pflanzen. Mit P35S:cpCK2‐Konstrukt komplementierte knockout‐Pflanzen sind für 12 Tage unter Kurztagbedingungen auf MS‐Medium mit 1 % Saccharose (links) und ohne Saccharose (rechts) angezogen.
Die knockout‐Mutante wies gelbliche Keim‐ und Folgeblätter unter Langtagbedingungen
(16h Licht (60 μmol/m2s) / 8h Dunkel) auf (Abb. 4.10.2.3). Dieser Phänotyp war bei 12 Tage
alten Pflanzen stärker ausgeprägt als bei 6 Tage. Die komplementierte knockout‐Pflanze
(P35S:cpCK2) zeigte wie Wildtyp grüne Kotyledonen und Folgeblätter unter gleichen
Bedingungen.
6d
12d
∆cpck2Col‐0 P35S:cpCK2
Abb. 4.10.2.3: Phänotypen der Pflanzen unter Langtagbedingungen. Dargestellt sind die 6 (6d) bzw. 12 Tage (12d) alten Wildtyp (Col‐0)‐, knockout (∆cpck2)‐ und komplementierte (P35S:cpCK2) Pflanzen, die unter Langtagbedingungen auf MS‐Medium mit 1 % Saccharose angezogen wurden.
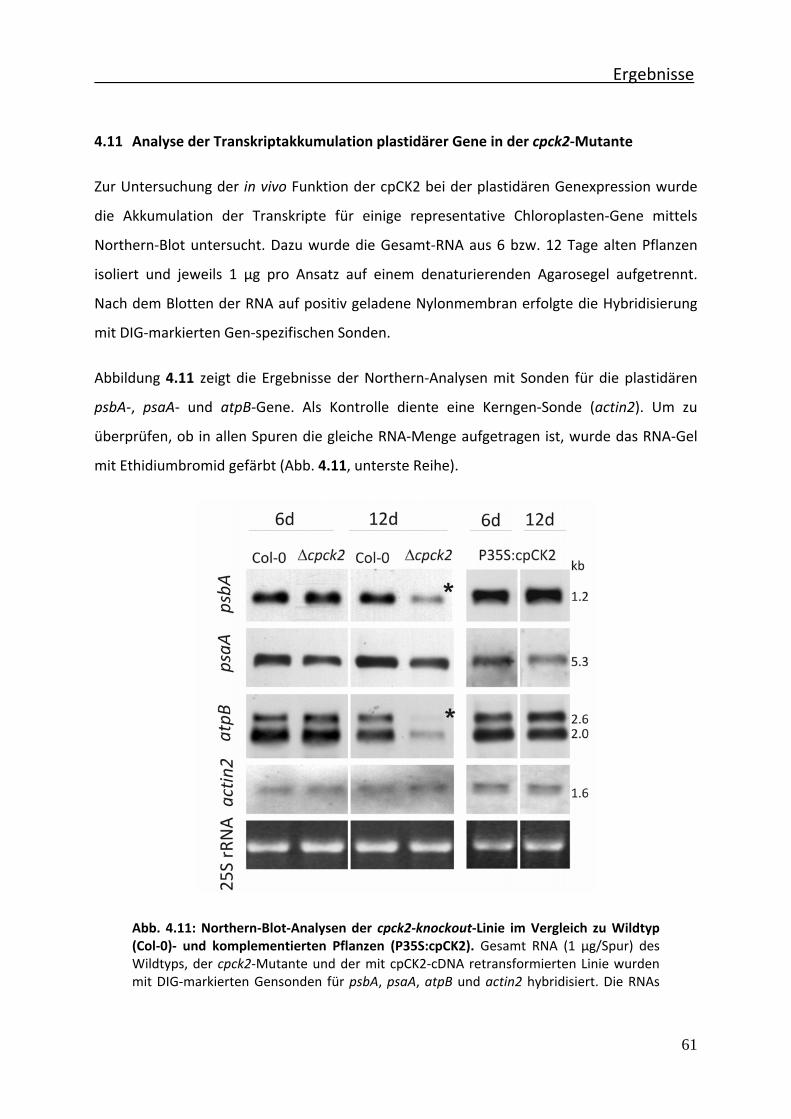
Ergebnisse
61
4.11 Analyse der Transkriptakkumulation plastidärer Gene in der cpck2‐Mutante
Zur Untersuchung der in vivo Funktion der cpCK2 bei der plastidären Genexpression wurde
die Akkumulation der Transkripte für einige representative Chloroplasten‐Gene mittels
Northern‐Blot untersucht. Dazu wurde die Gesamt‐RNA aus 6 bzw. 12 Tage alten Pflanzen
isoliert und jeweils 1 µg pro Ansatz auf einem denaturierenden Agarosegel aufgetrennt.
Nach dem Blotten der RNA auf positiv geladene Nylonmembran erfolgte die Hybridisierung
mit DIG‐markierten Gen‐spezifischen Sonden.
Abbildung 4.11 zeigt die Ergebnisse der Northern‐Analysen mit Sonden für die plastidären
psbA‐, psaA‐ und atpB‐Gene. Als Kontrolle diente eine Kerngen‐Sonde (actin2). Um zu
überprüfen, ob in allen Spuren die gleiche RNA‐Menge aufgetragen ist, wurde das RNA‐Gel
mit Ethidiumbromid gefärbt (Abb. 4.11, unterste Reihe).
Abb. 4.11: Northern‐Blot‐Analysen der cpck2‐knockout‐Linie im Vergleich zu Wildtyp (Col‐0)‐ und komplementierten Pflanzen (P35S:cpCK2). Gesamt RNA (1 µg/Spur) des Wildtyps, der cpck2‐Mutante und der mit cpCK2‐cDNA retransformierten Linie wurden mit DIG‐markierten Gensonden für psbA, psaA, atpB und actin2 hybridisiert. Die RNAs

Ergebnisse
62
wurden aus 6 bzw. 12 Tage (6d bzw. 12d) alten Langtag‐Pflanzen isoliert. Signifikante Änderungen in der Intensität von Hybridisierungssignalen sind durch (*) markiert (s. Text). Transkriptgrößen der Gene sind links in kb angegeben. Als Beladungskontrolle diente ein Ethidiumbromid‐gefärbtes Gel, welches in allen Spuren die 25S rRNA in gleicher Intensität zeigt (ganz unten).
Die Mutante (∆cpck2) wies eine deutlich reduzierte psbA‐ und atpB‐Transkriptmenge nach
12 Tagen (Sterne) im Vergleich zum Wildtyp auf. Dieser Verlust ließ sich wieder
komplementieren (Abb. 4.11, s. P35S:cpCK2). Das Signal für das psaA‐Transkript war
hingegen in der komplementierten Pflanze (nicht aber in der ∆cpck2‐Mutante) schwächer als
im Wildtyp, sowohl bei 6 als auch bei 12 Tage alten Keimlingen. Umgekehrt scheint das
psbA‐Transkript in der komplementierten Linie sogar stärker als im Wildtyp vorzuliegen. Die
Transkriptmenge für actin2 war in allen Pflanzen unter diesen Bedingungen vergleichbar.
Dies zeigte, dass die T‐DNA‐Insertion und die Retransformation mit dem P35S:cpCK2‐
Konstrukt keinen Einfluss auf die Kern‐Transkription und/oder RNA‐Stabilität hatten.
4.12 In vivo Analysen zur Redoxregulation der cpCK2‐Aktivität
Die in vitro Experimente mit den Cystein‐Mutanten (s. 4.8) zeigten, dass die Cysteine von
cpCK2 regulatorische Funktionen besitzen. Um zu testen, wie sich die Mutationen an den
Cystein‐Resten auf die in vivo Funktion der Kinase auswirken, wurden cpCK2‐Konstrukte, die
jeweils an einem Cystein‐Rest mutagenisiert waren, mit Transitpeptid‐Sequenz in die cpck2‐
knockout‐Pflanzenlinie eingebracht. Die Nachkommen‐Population wurde mittels RT‐PCR auf
die cpCK2‐Volllängen‐cDNA untersucht. Um sicherzustellen, dass die Kandidaten‐Linien
tatsächlich die mutierten Konstrukte enthalten, wurden PCR‐Produkte sequenziert. Im
Ergebnis wurden homozygote Linien erhalten, die jeweils entweder an C1, C2, C3 oder C4
mutagenisierte cpCK2‐cDNA tragen. Abbildung 4.12.1 zeigt die Phänotypen der 6 bzw. 12
Tage alten Cystein‐Mutantenlinien unter Langtagbedingungen. Diejenigen, die C1‐ bzw. C4‐
Mutanten‐Konstrukte enthalten, hatten gelbe Keim‐ und Folgeblätter wie die parentale
knockout‐ Pflanzenlinie (vgl. Abb. 4.10.2.3).

Ergebnisse
63
P35S:cpCK2 C1
6d
12d
P35S:cpCK2 C2 P35S:cpCK2 C3 P35S:cpCK2 C4
Abb.4.12.1: Phänotypen der cpCK2 Cystein‐Mutantenlinien. Dargestellt sind die mit jeweils p35S:cpCK2 C1‐, C2‐, C3‐ oder C4‐Konstrukten retransformierten Pflanzen, die 6 bzw. 12 Tage unter Langtagbedingungen angezogen sind. Kontrolle mit dem Wildtyp‐Konstrukt (P35S:cpCK2) ist in der Abb. 4.10.2.3 zu sehen.
Die Akkumulation der plastidären psbA‐, psaA‐, atpB‐Transkripte in den Cystein‐
Mutantenlinien wurde mittels Northern‐Blot untersucht. Als Kontrolle diente wiederum das
Kerntranskript actin2. Abbildung 4.12.2 zeigt die Expressionsmuster der untersuchten Gene
bei 6 bzw. 12 Tage alten retransformierten Mutanten‐Linien unter Langtagbedingungen.
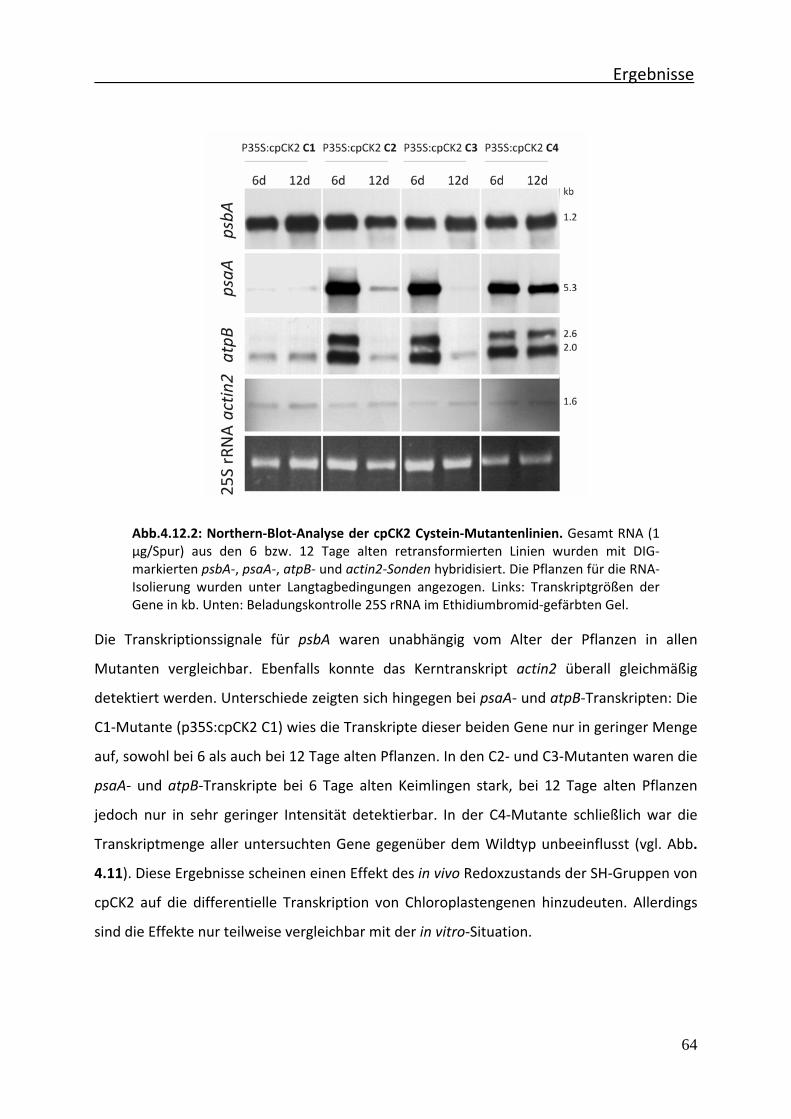
Ergebnisse
64
Abb.4.12.2: Northern‐Blot‐Analyse der cpCK2 Cystein‐Mutantenlinien. Gesamt RNA (1 µg/Spur) aus den 6 bzw. 12 Tage alten retransformierten Linien wurden mit DIG‐markierten psbA‐, psaA‐, atpB‐ und actin2‐Sonden hybridisiert. Die Pflanzen für die RNA‐Isolierung wurden unter Langtagbedingungen angezogen. Links: Transkriptgrößen der Gene in kb. Unten: Beladungskontrolle 25S rRNA im Ethidiumbromid‐gefärbten Gel.
Die Transkriptionssignale für psbA waren unabhängig vom Alter der Pflanzen in allen
Mutanten vergleichbar. Ebenfalls konnte das Kerntranskript actin2 überall gleichmäßig
detektiert werden. Unterschiede zeigten sich hingegen bei psaA‐ und atpB‐Transkripten: Die
C1‐Mutante (p35S:cpCK2 C1) wies die Transkripte dieser beiden Gene nur in geringer Menge
auf, sowohl bei 6 als auch bei 12 Tage alten Pflanzen. In den C2‐ und C3‐Mutanten waren die
psaA‐ und atpB‐Transkripte bei 6 Tage alten Keimlingen stark, bei 12 Tage alten Pflanzen
jedoch nur in sehr geringer Intensität detektierbar. In der C4‐Mutante schließlich war die
Transkriptmenge aller untersuchten Gene gegenüber dem Wildtyp unbeeinflusst (vgl. Abb.
4.11). Diese Ergebnisse scheinen einen Effekt des in vivo Redoxzustands der SH‐Gruppen von
cpCK2 auf die differentielle Transkription von Chloroplastengenen hinzudeuten. Allerdings
sind die Effekte nur teilweise vergleichbar mit der in vitro‐Situation.

Diskussion
65
5 Diskussion
5.1 Rekombinante cpCK2 (PTK) aus Arabidopsis thaliana
Das erste Ziel der vorliegenden Arbeit bestand in der rekombinanten Herstellung und
Charakterisierung der chloroplastidären CK2 aus Arabidopsis thaliana (ATCK2). Dazu wurde
die cDNA für ATCK2 ohne Transitpeptid (TP) zunächst in den pQE30‐Vektor kloniert, da die
Überexpression der cpCK2 aus Senf (SACK2) in Vorarbeiten erfolgreich in diesem System
durchgeführt werden konnte. Induktionstests in E. coli M15‐Zellen zeigten jedoch, dass eine
Überexpression der ATCK2 in diesem Expressionssystem nicht möglich ist (s. Abb. 4.3.1). Das
„reife“ ATCK2‐Protein (ohne TP) weist eine 95% ige Ähnlichkeit zu SACK2 auf. Diese Proteine
unterscheiden sich in der vorhergesagten Länge ihrer Transitpeptide. Laut dem Vorhersage‐
Programm ChloroP (Emanuelsson et al., 1999) besitzt ATCK2 ein 55 Aminosäure langes
Transitpeptid, während für SACK2 66 Aminosäuren vorhergesagt werden (s. Abb. 4.2). Die
rekombinante ATCK2 enthält eine 30 Aminosäure lange Verlängerung am N‐terminalen
Bereich (Abb. 5.1), die zum Teil Homologien zur Transitpeptidsequenz der SACK2 aufweist (s.
Abb. 5.1, grün markierte Sequenz). Auffällig ist, dass diese Extra‐Sequenz reich an Glutamin
(Q)‐Resten ist. Sie könnte somit die lokale Faltung am N‐Terminus beeinflussen, ohne dass
dies das CD‐Spektrum oder die biochemischen Parameter signifikant beeinflusst. Diese Q‐
reiche Extra‐Sequenz könnte der Grund sein, warum sich ATCK2 in Bezug auf ihre
Induzierbarkeit im pQE30‐System anders als SACK2 verhält.
Hingegen konnte die ATCK2 erfolgreich in dem pMALc2x‐Vektor in E. coli TB1‐Zellen
überexprimiert und in ausreichender Menge als MBP‐Fusion isoliert werden. Das pMAL‐
System ist dafür bekannt, dass es sich oft günstig auf die Löslichkeit und/oder Faltung sonst
"schwieriger" Fusionsproteine auswirkt (zur Übersicht s. Waugh, 2005).

Diskussion
66
ATCK2(ohne TP) ASLYRQHLRNQQQQHQQQQQSRVKEKSETLAQKIGKSIRRAGAPSKARVYADVNVVRPKD 60
SACK2(ohne TP) --LHRR----------QQQQSRVN-KSETLAQKIGKSIRRAGAPSKARVYADVNVIKPKD 47 *:*: *******: ******************************::*** Nukleotid-Bindung ATCK2(ohne TP) YWDYESLAVQWGVQDDYEVVRKVGRGKYSEVFEGIHATDNEKCVIKILKPVKKKKIKREI 120 SACK2(ohne TP) YWDYESLAVQWGAQDDYEVVRKVGRGKYSEVFEGIHATDNEKCVIKILKPVKKKKIKREI 107 ************.*********************************************** ATCK2(ohne TP) KILQNLCGGPNIVKLLDIVRDQQSKTPSLIFEHVNNKDFKVLYPTLSDYDVRYYIFELLK 180 SACK2(ohne TP) KILQNLCGGPNIVKLLDIVRDQQSKTPSLIFEHVNNKDFKVLYPTLSDYDVRYYIYELLK 167 *******************************************************:**** katalytische Region ATCK2(ohne TP) ALDFCHSRGIMHRDVKPHNVMIDHEQRKLRLIDWGLAEFYHPGKEYNVRVASRYFKGPEL 240 SACK2(ohne TP) ALDFCHSRGIMHRDVKPHNVMIDHEQRKLRLIDWGLAEFYHPGKEYNVRVASRYFKGPEL 227 ************************************************************ ATCK2(ohne TP) LVDLQDYDYSLDLWSLGCMFAGMIFRKEPFFYGHDNYDQLVKIAKVLGTDELNAYLNKYR 300 SACK2(ohne TP) LVDLMDYDYSLDLWSLGCMFAGMIFRKEPFFYGHDNYDQLVKIAKVLGTDELNTYLNRYR 287 **** ************************************************:***:** ATCK2(ohne TP) IELDPNLTSLVGRHSRKPWTKFINSENQHLAVPEAVDFVDKLLRYDHQERPTAKEAMAHP 360 SACK2(ohne TP) IELDPNLASLVGRHSRKPWSKFINSENQHLAVPEAVDFVDKLLKYDHQERPTAKEAMAHP 347 *******:***********:***********************:**************** ATCK2(ohne TP) YFYPIRNAESSR-TPRSQ 377 SACK2(ohne TP) YFNPIRNAESSRSTPRAQ 365 ** ********* ***:*
Abb. 5.1: Aminosäurensequenz‐Vergleich von ATCK2 und SACK2 ohne ihre Transitpeptide. Das Alignment wurde mit Hilfe von ClustalW (Larkin et al., 2007) durchgeführt. Identische Aminosäuren (*) sind grau markiert. Transitpeptid‐Anteil der SACK2 ist mit grünen Buchstaben dargestellt. Funktionelle Domänen sind fett gedruckt. TP: Transitpeptid, (:): konservierte und (.): schwach konservierte Aminosäureaustausche.
Die mittels unterschiedlicher Expressionssysteme hergestellte ATCK2 und SACK2 wurden auf
ihre katalytische Aktivität hin untersucht. Die rekombinante SACK2 wurde bereits funktionell
charakterisiert und weist die Eigenschaften der katalytischen α‐Untereinheit der CK2‐
Proteinkinase auf (Ogrzewalla et al., 2002). Sie wurde als das rekombinante „Gegenstück“
zur authentischen PTK (Baginsky et al., 1997 und 1999) verifiziert (Ogrzewalla et al., 2002).
Eine vergleichende Funktionsanalyse von ATCK2 mit SACK2 (s. Abb. 4.4) zeigte, dass ATCK2
genauso wie SACK2 katalytisch aktiv ist. Dass es sich bei dieser Aktivität um eine CK2‐
typische Proteinkinase‐Aktivität handelt, konnte durch Experimente mit ATP bzw. GTP als
Cosubstrat und CK2‐spezifischen Inhibitoren wie Heparin und TBB nachgewiesen werden.
Diese Eigenschaften sind charakteristisch für CK2 und werden zur Identifizierung der CK2‐

Diskussion
67
Aktivität benutzt (Niefind et al., 1999; Hathaway und Traugh, 1982; Sarno et al., 2001). Trotz
der N‐terminalen Verlängerung wies das rekombinante ATCK2‐Protein hinsichtlich seiner
biochemischen und funktionellen Eigenschaften keinen Unterschied zu SACK2 auf.
Ein weiterer Vergleich zwischen rekombinanter ATCK2 und SACK2 wurde bezüglich ihrer
Substraterkennung durchgeführt. Aus Vorarbeiten ist bekannt, dass die SACK2 die
rekombinanten Sigmafaktoren 1‐3 aus Senf phosphorylieren kann (Ogrzewalla et al., 2002;
Ogrzewalla, Dissertation 2001). Um die Frage zu klären, ob die Sigmafaktoren ebenfalls
Substrate für ATCK2 darstellen, wurde die Phosphorylierung der rekombinanten Senf‐
Sigmafaktoren 1 und 2 (SASIG1 und SASIG2) und Arabidopsis‐Sigmafaktoren 1 und 6 (ATSIG1
und ATSIG6) untersucht (s. Abb. 4.6). Dabei zeigte sich, dass alle Sigmafaktoren sich durch
beide Kinasen phosphorylieren lassen. Die CK2‐Phosphorylierungsstellen innerhalb von
ATSIG6 wurden sowohl in vivo als auch in vitro mittels Mutagenesestudien analysiert
(Schweer et al., 2010). Es stellte sich dabei heraus, dass in ATSIG6 mehrere
Posphoakzeptorstellen Ziele für cpCK2‐Phosphorylierung sind.
5.2 Autophosphorylierung der ATCK2
In vitro Phosphorylierungsexperimente mit Sigmafaktoren als Substrate zeigten, dass die
ATCK2 –im Gegensatz zu SACK2– eine Autophosphorylierung aufweist (vgl. Abb. 4.6). Die
Autophosphorylierungsbande der ATCK2 bei ca. 87 kDa war ebenfalls in den Experimenten
mit Casein als Substrat zu detektieren, jedoch unter anderen Expositionseinstellungen (vgl.
Abb. 4.4 und 4.8.2). Da in der Abb. 4.4 Casein stöchiometrisch ein viel stärkeres
Phosphorylierungssignal als die ATCK2 aufweist, ist die ATCK2‐Autophosphorylierungsbande
unter diesen Einstellungen nicht sichtbar.
Wo findet diese Autophosphorylierung in ATCK2 statt? Warum zeigt SACK2 keine
autokatalytische Phosphorylierung? Wie bereits in der Abb. 5.1.1 dargestellt wurde, ist die
Aminosäurezusammensetzung der beiden rekombinanten Kinasen bis auf die N‐terminale
Verlängerung der ATCK2 nahezu identisch. Die wenigen Aminosäureaustausche ergeben
auch keine zusätzlichen CK2‐Phosphorylierungsstellen in der ATCK2‐Sequenz. Daher ist es
denkbar, dass die Autophosphorylierungsstelle(n) entweder in der N‐terminalen

Diskussion
68
Verlängerung oder im MBP‐Tag lokalisiert sein könnte(n). Die Kontrollen unter Verwendung
des MBP‐Proteins (New England Biolabs) zeigten jedoch keine MBP‐Phosphorylierung (vgl.
Abb. 4.6). Phosphorylierung des mit Faktor‐Xa geschnittenen ATSIG6‐Proteins deutet
ebenfalls darauf hin, dass MBP nicht phosphoryliert wird. Obwohl einige Vorhersage‐
Programme mehrere potentielle CK2‐Phosphorylierungsstellen innerhalb des MBP‐Proteins
erkennen, werden diese anscheinend in vitro nicht benutzt. Die 30 Aminosäuren lange N‐
terminale Extra‐Sequenz der ATCK2 im Vergleich zu SACK2 wurde auf potentielle CK2‐
Phosphorylierungsstellen mittels der Vorhersage‐Programme NetPhosK (Blom et al., 2004)
und GPS 2.1 (Xue et al., 2008) untersucht. Aufgrund der Ergebnisse beider Programme
wurde Serin an Position 27 als potentielle Phosphoakzeptorstelle für CK2 erkannt (Daten
nicht gezeigt). Es befindet sich zwar an der (n+3)‐Stelle hinter diesem Serin keine saure
Aminosäure (D/E) wie bei einer "klassischen" CK2 Phosphorylierungsstelle; es ist jedoch
bekannt, dass auch die (n+1)‐Position mitentscheidend für die Substraterkennung durch
CK2‐Proteinkinasen sein kann (Meggio und Pinna, 2003).
Aus Vorarbeiten war bekannt, dass die in vitro Phosphorylierung der authentischen PTK aus
Senf‐Chloroplasten durch eine exogene Kinase (katalytische Untereinheit der Proteinkinase
A, PKA) zur Inaktivierung des Enzyms führt (Baginsky et al., 1999). Dies konnte mit der
rekombinanten SACK2 nicht gezeigt werden, da sich diese nicht durch PKA phosphorylieren
ließ (Ogrzewalla, nicht‐veröffentlichte Daten). Eine Autophosphorylierung der SACK2 war in
diesen früheren Arbeiten nicht detektierbar.
Für Präparate nukleärer/cytosolischer CK2 (Holoenzym, α2β2) wird von einer Auto‐
phosphorylierung an der CK2β‐Untereinheit berichtet (Dahmus und Natzle, 1977; Pinna,
1990). Eine regulatorische Funktion dieser Autophosphorylierung für CK2 konnte jedoch
nicht nachgewiesen werden –ebenso wenig wie eine Fremdphosphorylierung durch eine
andere Kinase– (Meggio et al., 1993). Innerhalb der CK2α‐Familie wurde bisher nur von einer
autokatalytischen Tyrosin‐Phosphorylierung berichtet, dessen funktionelle Bedeutung
ebenfalls ungewiss ist (Donella‐Deana et al., 2001).

Diskussion
69
Im Falle der ATCK2 zeigt die Autophosphorylierung auch keinen Einfluss auf ihre katalytische
Aktivität. Sie scheint keine physiologische Bedeutung für die Regulation der cpCK2 zu
besitzen. Aus diesem Grund wurde die Autophosphorylierung der rekombinanten ATCK2
nicht weiter untersucht.
5.3 Einfluss der Phosphorylierung durch cpCK2 auf die Sigmafaktor‐Aktivität
Phosphorylierung ist vielleicht die bestuntersuchte posttranslationale Modifikation, die
Protein‐Aktivitäten direkt regulieren kann. In vielen Signaltransduktionswegen spielt sie eine
wichtige Rolle bei der Regulation der eukaryotischen Genexpression und Zellfunktion (zur
Übersicht s. Brivanlou und Darnell, 2002). Dabei sind Transkriptionsfaktoren wichtige Ziele
für solche Modifikationen. Vielleicht nicht unerwartet, kommt dieser universelle
Regulationsmechanismus nicht zuletzt auch in Plastiden vor: So konnte gezeigt werden, dass
der Phosphorylierungszustand der Sigma‐Transkriptionsfaktoren in Senf‐Chloroplasten und
Etioplasten regulatorisch auf die plastidäre Transkription wirkt (Tiller und Link, 1993b;
Baginsky et al., 1997). Dabei spielt PTK/cpCK2 als terminale (endständige) Transkriptions‐
kinase, die ihrerseits einer Phosphorylierungskontrolle durch vorgeschaltete "Aufwärts"‐
Kinase(n) zu unterliegen scheint, eine wichtige Rolle (Baginsky et al., 1997; Ogrzewalla et al.,
2002).
Nachdem im ersten Schritt der vorliegenden Arbeit gezeigt wurde, dass die Sigmafaktoren
SASIG1, SASIG2, ATSIG1 und ATSIG6 durch cpCK2 aus Senf und Arabidopsis phosphoryliert
werden (vgl. Abb. 4.5.2), ging es im nächsten Schritt darum, den Einfluss dieser
Phosphorylierung auf die Promotorbindungsaktivität der rekombinanten Faktoren zu
studieren. Dies wurde am Beispiel von ATSIG1 und ATSIG6 untersucht. Beide Proteine
wurden durch ATCK2 phosphoryliert und anschließend in DNA‐Bindungsstudien eingesetzt.
Es konnte gezeigt werden, dass ATSIG1 und ATSIG6 durch die Phosphorylierung ihre
Präferenzen für die getesteten Promotoren ändern (vgl. Abb. 4.7). Im Falle von ATSIG1 führt
die Phosphorylierung zu Reduktion der Bindung des Faktors an den atpB‐Promotor. Die
ATSIG1‐Bindung an psbA‐ und trnK‐Promotoren ist jedoch nahezu unbeeinflusst durch
Phoshorylierung. Für Sigmafaktor 1 konnte bisher keine Promotor‐/Gen‐spezifische Funktion

Diskussion
70
zugeordnet werden. Verschiedene Daten deuten darauf hin, dass dieser Faktor eine
generelle Funktion in der plastidären Genexpression ausübt (Tanaka et al., 1997;
Kestermann et al., 1998; Tozawa et al., 1998; Kanamaru et al., 1999; Morikawa et al., 1999).
Die Aktivität dieses Proteins wird möglicherweise über ein Interaktionspartner, das sog.
sigma binding protein 1 (SIB1), reguliert (Morikawa et al., 2002).
Im Gegensatz zu ATSIG1 bindet der phosphorylierte ATSIG6 viel stärker als der nicht‐
phosphorylierte Faktor an den atpB‐Promotor (vgl. Abb. 4.7). Kompetitionsexperimente mit
dem trnK‐Promotor deuten darauf hin, dass die ATSIG6‐Bindung an den trnK‐Promotor
ebenfalls durch Phosphorylierung des Faktors begünstigt wird. Für die Bindung an den psbA‐
Promotor scheint der Phosphorylierungszustand hingegen nicht entscheidend zu sein. In vivo
Mutations‐Analysen mit ATSIG6 zeigen ebenfalls, dass die Phosphorylierung von ATSIG6 für
seine atpB‐spezifische Bindungsaktivität wichtig ist (Schweer et al., 2010).

Diskussion
71
5.4 In vitro Analyse zur Redoxregulation der ATCK2‐Aktivität
Sowohl in vitro als auch in vivo existieren Hinweise darauf, dass die Aktivität der PTK/cpCK2
über ihren Redoxzustand reguliert wird. Es wurde gezeigt, dass reduziertes Glutathion (GSH)
die cpCK2‐Aktivität inhibiert (Baginsky et al., 1999; Baena‐Gonzáles et al., 2001, Ogrzewalla
et al., 2002). Ziel der vorliegenden Arbeit war, den genauen Mechanismus dieser Regulation
zu analysieren. Es ist bekannt, dass bei der Redoxregulation von Proteinen die Cystein‐Reste
eine wichtige Rolle spielen. Deshalb wurden die Cysteine innerhalb der rekombinanten
ATCK2 einzeln bzw. paarweise mutiert und die Auswirkung dieser Mutation(en) auf die
Aktivität der entsprechenden Proteine untersucht (vgl. Abb. 4.8.1). Es zeigte sich, dass Cys1
und Cys4 für die Aktivität der rekombinanten Kinase essentiell sind. Dies könnte drauf
hinweisen, dass Cys1 und Cys4 möglicherweise an einer intermolekularen
Disulfidbrückenbildung beteiligt sind. CD‐spektroskopische Analysen mit den Cystein‐
Mutanten zeigen, dass Cys1‐, Cys4‐ und Cys2/4‐Mutanten eine im Vergeich zum Wildtyp‐
Protein veränderte Faltung aufweisen. Möglicherweise ist diese Änderung auf die Zerstörung
der intermolekularen Disulfidbrücke zurückzuführen. Der Einzel‐Austausch der Cys2‐ oder
Cys3‐Reste bzw. Austausch beider Cys Reste gleichzeitig, hatte hingegen keinen Einfluss auf
die Phosphorylierungsaktivität der Kinase. Somit scheinen diese Cysteinreste keine wichtige
Rolle für die Aktivität zu spielen.
Um weitere Einblicke in die Redoxregulation der ATCK2 zu bekommen, wurde die
Empfindlichkeit der Kinase und ihrer aktiven Cystein‐Mutanten gegenüber Redoxreagenzien
wie GSH, DTT und Diamid getestet (vgl. Abb. 4.8.3). GSH wirkte inhibierend auf Wildtyp‐ und
Mutanten‐Proteine, während DTT keinen Effekt zeigte. Der Effekt von GSH und DTT wurde in
Vorarbeiten ebenfalls mit PTK und rekombinanter cpCK2 aus Senf beobachtet (Baginsky et
al., 1999; Ogrzewalla et al., 2002). Beide Reagenzien sind Reduktanten, d.h. sie üben eine
reduzierende Wirkung auf die Proteine aus. GSH ist, anders als DTT, ein Monothiol‐
Reduktant. Es kann gemischte Disulfide mit den Cystein‐Resten der Zielproteine bilden und
ihre Aktivitäten modulieren (zur Übersicht s. Shelton et al., 2005). Diese Art von
posttranslationaler Modifikation wird als Glutathionylierung bezeichnet.

Diskussion
72
Es stellte sich die Frage, über welchen Mechanismus GSH die Kinase inaktiviert; durch
Reduktion der Disulfide innerhalb ATCK2 oder durch Glutathionylierung?
Die in vitro Analyse mit den Cys‐Mutanten lieferten Hinweise darauf, dass die Mutationen an
Cys1 und Cys4 möglicherweise aufgrund der Zerstörung einer intramolekularen
Disulfidbrücke zu einem Aktivitätsverlust führt. Sollte dies der Fall sein, müsste die Wildtyp‐
Kinase sowohl durch GSH als auch durch Dithiol‐Reduktant DTT (zerstört die Disulfidbrücken)
inaktiviert werden. Dies ist jedoch für DTT nicht der Fall. Eine mögliche Erklärung dafür wäre,
dass vielleicht die Disulfidbrücke innerhalb der ATCK2 zwar zugänglich für GSH, jedoch nicht
für DTT, ist. Monothiol‐ und Dithiol‐Reduktanten können sich in ihrer Wirkung
unterschiedlich verhalten (Trebitsh et al., 2000). Weiterhin ist möglich, dass der
Aktivitätsverlust der C1‐ und C4‐Mutanten nicht allein auf die Zerstörung der Disulfidbrücke
zurückzuführen ist, sondern gleichzeitig auf das Fehlen dieser hochkonservierten
Aminosäure‐Reste. Die Inaktivierung der Wildtyp‐Kinase durch GSH könnte man in diesem
Fall durch Bildung gemischter Disulfide zwischen GSH und ATCK2 erklären. Da die C2‐, C3‐
und C2/3‐Mutanten ebenfalls empfindlich gegenüber GSH reagieren, ist es denkbar, dass
GSH mit Cys1 und/oder Cys4 gemischte Disulfide bilden kann. Um diese These zu kräftigen,
sind jedoch weitere Untersuchungen nötig.
Ein unerwartetes, interessantes Ergebnis lieferten die Experimente mit dem Oxidationsmittel
Diamid. Es wirkte unterschiedlich auf die Aktivität der rekombinanten Proteine. Wildtyp und
C3‐Mutante ließen sich durch Diamid schon bei niedrigen Konzentrationen vollständig
inhibieren, während auch höhere Diamid‐Konzentrationen auf die Aktivität der C2‐ und
C2/3‐Mutanten keinen Einfluss hatten. Die Frage nach dem Grund der Diamid‐„Resistenz“
ließ sich durch einfache gelelektrophoretische und immunologische Analysen beantworten.
Es stellte sich dabei heraus, dass das Cys2 an einer intramolekularen Disulfidbrückenbildung
beteiligt ist (vgl. Abb. 4.8.4 A). Proteine, die eine Mutation am zweiten Cystein trugen,
bildeten keine Dimere. Aus nukleären/cytosolischen CK2‐Proteinkinasen bei Menschen ist
eine Dimerbildung nur für die CK2β‐Untereinheiten bekannt (Gietz et al., 1995; Kusk et al.,
1995; Boldyreff et al., 1996). Es wurden keine Dimere bei CK2α‐Untereinheiten aus Mensch
und Drosophila (Birnbaum et al., 1992) beobachtet. Dass die CK2α‐Untereinheiten Dimere

Diskussion
73
bilden können, wurde bisher ausschließlich für pflanzliche CK2αs beschrieben (Yan und Tao,
1982; Erdmann et al., 1982).
Anders als tierische CK2αs verfügen pflanzliche CK2α‐Untereinheiten, einschließlich ATCK2,
über vier Cysteine (vgl. Abb. 4.8.1). Die C‐terminal gelegenen Cysteine, Cys3 und Cys4, sind
innerhalb aller CK2α‐Untereinheiten aus tierischen und pflanzlichen Organismen hoch
konserviert, während die Cys1 und Cys2 lediglich in pflanzlichen CK2αs vorkommen. Dies
steht im Einklang mit der Dimerbildung der CK2α‐Untereinheiten aus pflanzlichen
Organismen, jedoch nicht aus tierischen.
Betrachtet man alle vier Cysteine der ATCK2 in der 3D‐Struktur, so sieht man, dass sich nur
das zweite Cystein an der Oberfläche des Proteins befindet (s. Abb. 5.4). Dies liefert ein
weiteres Argument dafür, dass am Cys2‐Rest eine Dimerbildung strukturell auch möglich ist.
Dimerisierung als ein Regulationsmechanismus konnte bereits durch zahlreiche Studien
gezeigt werden (Klemm et al., 1998; Williams et al., 2005; Yamaguchi et al., 2006;
Rajakulendran et al., 2009). Man könnte somit postulieren, dass die Aktivität der cpCK2 in
Plastiden durch Bildung von Homodimeren und/oder auch Heterodimeren mit anderen
regulatorischen Interaktionspartnern moduliert werden könnte. Angesichts der großen
Anzahl proteomisch identifizierter Komponenten des plastidären Transkriptionsapparats
(Ogrzewalla et al., 2002, Loschelder et al., 2004) ist diese Erklärung plausibel, wenn auch
nicht experimentell leicht nachprüfbar.

Diskussion
74
Abb. 5.4: Strukturmodell plastidärer ATCK2. Die Abbildung zeigt die Lage der Cysteine innerhalb ATCK2. Die Oberflächen‐Darstellung (rechts) präsentiert den exponiert vorliegenden Cys2‐Rest. Die konservierten Subdomänen sind ebenfalls im gleichen Farbcode wie links dargestellt. Die Struktur wurde unter Verwendung von CPHmodels (Lund et al., 2002) berechnet. Die graphische Darstellung erfolgte mit Hilfe des PyMOL‐Programms (DeLano, 1998). Als Grundlage für die Berechnungen diente die nucleo‐/cytosolische CK2α‐Untereinheit aus Mais (Niefind et al., 1998).
5.5 In vivo Funktionsanalyse der cpCK2 aus Arabidopsis thaliana
Zum Verständnis der Funktion der cpCK2 in der plastidären Genexpresion waren die in
planta Untersuchungen von großem Interesse. Diese Möglichkeit bot die cpck2‐T‐DNA‐
Insertionsmutante aus Arabidopsis thaliana (GABI‐Kat‐Linie). Durch PCR‐Analysen wurde die
Homozygotie der Mutanten‐Linie sowie die Abwesenheit der cpck2‐Transkript in der
Homozygoten‐Pflanze nachgewiesen. Der Effekt dieser Mutation auf den visuellen und
molekularen Phänotyp wurde untersucht. Die Analysen hinsichtlich des visuellen Phänotyps
der cpck2‐Mutante zeigten, dass die Mutantenlinien unter photoautotrophen Bedingungen
keimlingsletal sind (vgl. Abb. 4.10.2.1). Sie waren nicht in der Lage, Photosynthese zu
treiben, daher waren sie auf eine externe Kohlenstoff‐Quelle angewiesen. Dieser Phänotyp
wird durch Retransformation der knockout‐Linie mit dem P35S:cpCK2‐Konstrukt, Wildtyp‐
cDNA hinter dem 35S‐Promotor, wieder kuriert (vgl. Abb. 4.10.2.2), was darauf hindeutet,
dass der letale Phänotyp auf das Fehlen der cpCK2 zurückzuführen ist. Es ist denkbar, dass

Diskussion
75
die cpCK2 eine Funktion in der Entwicklung der Chloroplasten besitzt. Da die Keimlinge kurz
nach der Keimung abstarben, könnte man denken, dass bei ihnen ein Wechsel vom
heterotrophen zum photoautotrophen Wachstum nicht mehr möglich war.
Unter mixotrophen Bedingungen zeigten die Mutanten‐Keimlinge ein leicht verzögertes
Wachstum bei einem Alter von 6 Tagen (vgl. Abb. 4.10.2.3). Nach 12 Tagen waren sie in ihrer
Größe vergleichbar mit dem Wildtyp, hatten jedoch gelbe Keim‐ und Folgeblätter unter
Langtagbedingungen. Dies könnte auf eine Störung in der Synthese oder Anreicherung von
Chlorophyll in den Mutanten hindeuten.
Der Effekt der cpck2‐Mutation auf die plastidären RNA‐Muster für die psbA‐ (codiert für D1‐
Protein von Photosystems II (PSII)), psaA‐ (codiert für A‐Untereinheit von PSI) und atpB‐Gene
(codiert für die β‐Untereinheit der ATP‐Synthase) wurde mittels Northern‐Blot unter
Verwendung von DIG‐markierten Gensonden untersucht. Dabei stellte sich heraus, dass die
knockout‐Mutante eine starke Reduktion in der psbA‐ und atpB‐Transkriptmenge bei 12
Tage alten Pflanzen aufweist. Unter gleichen Bedingungen weist die komplementierte Linie
eine Erhöhung in der psbA‐Transkriptmenge auf, während das psaA‐Transkript durch die
Komplementation reduziert wird. Ein solcher Effekt, d.h. entgegengesetzte Veränderungen
in den Transkriptmengen der plastidären psbA‐ und psaA‐Genen wird oft mit den
Änderungen in der Photosystem‐Stöchiometrie korreliert (zur Übersicht s. Pfannschmidt und
Liere, 2005). Dies erfolgt als eine Langzeit‐Antwort auf die wechselnden Lichtqualitäten an
beiden Photosystemen. D. h. wird PSI bevorzugt belichtet, ändert sich die PS‐Stöchiometrie
zugunsten von PSII und die Expression der PSII‐Gene wird hochreguliert. Unter PSII‐
Belichtung passiert das Umgekehrte. Diese Fein‐Regulation wird über den Redoxstatus des
Plastoquinon‐Pools kontrolliert. Im Falle der komplementierten ∆cpck2‐Linie könnte man
diesen Effekt auf die transiente Expression der cpCK2‐Volllängen‐cDNA unter der Kontrolle
des P35S‐Promotors zurückführen. Möglicherweise ist die starke cpCK2‐Expression in der
komplementierten Linie unter diesen Bedingungen durch einen PSI/PSII‐Shift zu
kompensieren.

Diskussion
76
5.6 In vivo Analysen zur Redoxregulation der cpCK2‐Aktivität
Die Redoxregulation der plastidären Genexpression erfolgt durch redoxaktive Komponenten,
die als Sensor für zelluläre Redoxsignale fungieren, welche z.B. während der Photosynthese
generiert werden. Die Frage, wie diese Signale auf der Ebene der Genexpression übermittelt
werden, bleibt noch ungeklärt. Es ist sehr wahrscheinlich, dass sie in ein anderes Signal
umgewandelt werden, wie z.B. in ein Phosphorylierungssignal durch eine redoxaktive Kinase
(Baginsky et al., 1999, Baena‐Gonzáles et al., 2001; Puthiyaveetil et al., 2008).
Um die Frage näher zu untersuchen, ob die Cysteine der cpCK2 auch in vivo regulatorische
Funktionen ausüben, wurde die ∆cpck2‐Mutantenlinie mit jeweils an einem Cystein
mutierten cpCK2‐Konstrukt (P35S:cpCK2 C1 bis C4) transformiert. Mittels RT‐PCR und
anschließende Sequenzierung der PCR‐Produkte wurde nachgewiesen, dass die
retransformierten Linien nur das jeweilige mutierte cpCK2‐Konstrukt enthalten. Die Analyse
bezüglich des visuellen Phänotyps zeigten, dass C1‐ und C4‐Mutantenlinien – wie die
parentale knockout‐Linie – eine Chlorophylldefizienz aufweisen. Die in vitro Funktions‐
analyse mit den rekombinanten C1‐ und C4‐Mutante hatte bereits gezeigt, dass das Fehlen
eines der beiden Cystein‐Reste zur Inaktivierung der Kinase führt (vgl. Abb 5.4). Der somit
„erwartete“ Befund, dass die mit C1‐ bzw. C4‐Konstrukten retransformierten Linien immer
noch den (defekten) Phänotyp der parentalen ∆cpck2‐Linie zeigen, belegt, dass offenbar in
vivo die gleichen Regulationsmechanismen wie in vitro wirken. Diese Schlussfolgerung gilt
umso mehr, als die C2‐ und C3‐Mutantenlinien übereinstimmend mit den in vitro Analysen
wie Wildtyp grüne Keim‐ und Folgeblätter aufwiesen.
Untersuchungen zur Transkriptakkumulation der plastidären psbA‐, psaA‐ und atpB‐Gene
zeigten, dass die Cys‐Mutantenlinien sogar einen stärkeren molekularen Phänotyp im
Vergleich zu Wildtyp‐, ∆cpck2‐ und komplementierter Linie aufwiesen. Die C1‐Mutantenlinie
zeigte eine starke Reduktion der psaA‐ und atpB‐Transkripte sowohl bei 6 als auch 12 Tage
alten Pflanzen. Die psbA‐Transkriptmenge war dagegen erhöht im Vergleich zum Wildtyp.
Das Cys1 scheint auch in vivo wichtig für die Aktivität/Regulation der cpCK2 zu sein. Dieses
Cystein ist ein hochkonservierter Rest und spezifisch für die pflanzlichen CK2αs. Vielleicht

Diskussion
77
kommt dem Cys1 eine Funktion in der pflanzenspezifischen Regulation des Enzyms zu. C2‐
und C3‐Linien zeigen einen ähnlichen molekularen Phänotyp wie die C1‐Mutantenlinie,
jedoch nur bei 12 Tage alten Pflanzen. Dass der molekulare Phänotyp in den „Fehl“‐
komplementierten Linien stärker ausfällt als in der ∆cpck2‐Linie, könnte man dadurch
erklären, dass die –durch den vorgeschalteten 35S‐Promotor stark exprimierte– nicht‐
funktionelle cpCK2 auf andere Regulationsmechanismen in den Chloroplasten blockierend
wirken könnte.
Im Falle des atpB‐Experiments war nicht nur das 2.6 kb PEP‐Transkript sondern auch das 2.0
kb NEP Transkript betroffen. Dieser z. B. bei einer Sigmafaktor 6‐Mutante nicht beobachtete
gekoppelte Effekt (Schweer et al., 2006) lässt sich am einfachsten dadurch erklären, dass
cpCK2 primär auf das PEP‐System zu wirken scheint (Baginsky und Link, 2005). Das Fehlen
dieses Master‐Regulators in der Mutante könnte jedoch auch als Signal an NEP
weitergeleitet werden und diese zum „Verzicht“ auf die sonst durchgeführte SOS‐
Transkription (Schweer et al., 2006) veranlassen.
Analysen des Chloroplasten‐Phosphoproteoms zeigten, dass die (cp)CK2 eine dominierende
Kinase in den Organellen ist (Rheiland et al., 2009). Als potentielle plastidäre Substrate
wurden TAC (transcriptionally active chromosom)‐Untereinheiten und RNA‐Bindungs‐
proteine beschrieben. Außerdem wurde gezeigt, dass die Substrate der (cp)CK2 nicht auf
eine Rolle in der Genexpression beschränkt sind, sondern dass auch metabolische Enzyme
einschließlich der photosynthetischen ATP‐Synthase ebenfalls Substrate für (cp)CK2
darstellen. Für weitere Untersuchungen wäre es interessant, das Phosphoproteom der
Chloroplasten in der ∆cpck2‐Mutante zu untersuchen.

Zusammenfassung
78
6 Zusammenfassung
Die plastidäre Transkription wird durch mindestens zwei RNA‐Polymerasen katalysiert; die
sog. NEP (nuclear encoded polymerase) und PEP (plastid encoded polymerase). NEP ist
homolog zu den T3‐/T7‐Phagen‐Polymerasen und kommt als funktionelles Monomer vor.
PEP dagegen ähnelt den bakteriellen RNA‐Polymerasen und besteht aus plastidenkodierten
core‐Untereinheiten (α2ββ´β´´) sowie weiteren akzessorischen Proteinen, die vom Kern
kodiert werden und posttranslational in den Plastiden importiert werden. Zu Letzteren
gehören u. a. Sigmafaktoren, die der PEP die Fähigkeit der spezifischen Promotorbindung
und Transkription verleihen. Es gibt Hinweise darauf, dass die Aktivität dieser Faktoren über
ihren Phosphorylierungsgrad reguliert wird. In Vorarbeiten wurde eine Ser‐/Thr‐Kinase, die
ebenfalls als eine Komponente der PEP vorliegen kann, als die vorgeschaltete Proteinkinase
aus Senf‐Plastiden identifiziert und plastidäre Transkriptionskinase (PTK) genannt. Eine
biochemische und molekulare Charakterisierung der PTK zeigte, dass dieses Enzym
äquivalent zur katalytischen α‐Untereinheit der nucleo‐/cytosolischen CK2‐Proteinkinase ist.
Allerdings unterliegt die PTK (cpCK2) im Unterschied zu CK2 einer Redoxregulation durch
reduziertes Glutathion (GSH) und wurde somit als ein potentieller Vermittler zwischen
zellulären Redoxsignalen und plastidärer Genexpression angesehen. In weiteren Vorarbeiten
gelang es, die cDNA für die PTK/cpCK2 aus Senf zu klonieren und bakteriell zu exprimieren.
Anhand von Datenbank‐Analysen zeichnete sich darüber hinaus ab, dass auch andere
Pflanzen, einschließlich Arabidopsis thaliana, ein Gen für die Chloroplasten‐lokalisierte
Transkriptionskinase (cpck2) besitzen.
Um die Hinweise aus Vor‐Untersuchungen experimentell zu erhärten, konzentrierte sich die
vorliegende Arbeit auf die cpCK2 aus der Modellpflanze Arabidopsis (ATCK2). Im
experimentellen Fokus stand dabei die Frage einer "universellen" Rolle dieses
Chloroplastenproteins. Sollte dies zutreffen, wäre das Protein aus Arabidopsis für weitere
Funktionsstudien besonders geeignet. Als Strategie zur Klärung dieser Frage wurde das
betreffende Gen als cDNA kloniert und bakteriell überexprimiert. Ein Vergleich der

Zusammenfassung
79
rekombinanten cpCK2‐Proteine aus Senf und A. thaliana zeigte, dass beide Präparate
tatsächlich in vitro sehr ähnliche, CK2‐typische, Eigenschaften aufweisen.
Eine zentrale Rolle spielten im nächsten experimentellen Schritt die pflanzlichen
Sigmafaktoren als physiologische Substrate der Transkriptionskinase. Es konnte gezeigt
werden, dass die ATCK2 die rekombinanten Sigmafaktoren SASIG1, SASIG2, ATSIG1 und
ATSIG6 in vitro phosphorylieren kann. Durch DNA‐Bindungsstudien konnten die
funktionellen Konsequenzen dieser Phosphorylierung auf die Sigma‐Aktivität am Beispiel von
ATSIG1 und ATSIG6 exemplarisch geklärt werden. Besonders auffällig war dabei die
phosphorylierungsabhängige Bindung von ATSIG6 an den atpB‐Promotor. Dieser Faktor wies
im phosphorylierten Zustand eine stärkere Bindungsaktivität auf als im nicht‐
phosphorylierten Zustand. Somit liefert dieses System ein klares Beispiel für die Bedeutung
der Sigmafaktor‐Phosphorylierung für die Aktivität dieser Enzyme.
Ein weiterer Punkt war die Untersuchung der ATCK2 hinsichtlich ihrer Redoxregulation.
Dabei zeigte sich, dass die Aktivität der ATCK2 –ebenso wie das authentische bzw. klonierte
Enzym aus Senf– einer Redoxregulation durch reduziertes Glutathion unterliegt. Durch
Mutagenesestudien bezüglich der Cystein‐Reste konnte weiterhin erstmals ein neuer
Regulationsmechanismus für cpCK2 durch Disulfidbrückenbildung demonstriert werden. Der
zweite Cystein‐Rest war in vitro für eine Dimerisierung, und damit verbundene Inaktivierung,
von ATCK2 verantwortlich. Eine solche Disulfidbrückenbildung kann in vivo womöglich mit
anderen Komponenten des plastidären Transkriptionsapparats erfolgen.
Um Rückschlüsse auf die in vivo Funktion der cpCK2 zu bekommen, wurde eine T‐DNA‐
Insertionsmutante der ATCK2 (∆cpck2) charakterisiert. Phänotypische Analysen mit der
∆cpck2‐Mutante zeigten, dass die Mutanten‐Nachkommenlinien unter photoauthotrophen
Bedingungen keimlingsletal sind. Transkripte ausgewählter Gene (psbA, psaA und atpB)
zeigten im Vergleich zu Wildtyp signifikante Änderungen in der ∆cpck2‐Mutante, aber auch
in der komplementierten Linie. Diese Ergebnisse unterstreichen die grundsätzliche
Bedeutung der cpCK2 und deuten u. U. auch auf noch weiter vorgelagerte Regulatoren der
Redox‐ und Phosphorylierungsnetzwerke im Chloroplasten.

Zusammenfassung
80
Für die Untersuchungen zur in vivo Redoxregulation der cpCK2 wurden die ∆cpck2‐Linien mit
cpCK2‐Konstrukten, die jeweils an einem der vier Cysteine mutiert wurden, retransformiert.
Dabei zeigte sich, dass die Cysteine auch in vivo eine regulatorische Rolle spielen. Diese
gezielte "Fehl"‐Komplementation führte bei den mit C1‐, C2‐ und C3‐Konstrukten
komplementierten Linien zu dramatischen Veränderungen im plastidären RNA‐Muster.

Summary
81
7 Summary
Transcription of the plastid genes is driven by at least two RNA polymerases, the so‐called
NEP (nuclear encoded polymerase) and PEP (plastid encoded polymerase). NEP is a single
subunit enzyme homologous to the T3/T7 phage polymerases. In contrast, PEP is a bacterial‐
type RNA polymerase composed of multiple plastid encoded core subunits (α, β, β´, β´´). In
addition, it contains a number of accessory proteins that are encoded by the nucleus and
imported posttranslationally into the plastids. The latter include sigma factors which confer
on PEP the ability of specific promoter binding and transcription initiation. Evidence suggests
that the activity of these factors is regulated by their phosphorylation state. In previous
work, a PEP associated Ser‐/Thr‐Kinase was identified as the cognate protein kinase from
mustard plastids and called the plastid transcription kinase (PTK). A biochemical and
molecular characterization of PTK showed that this enzyme is equivalent to the catalytic α‐
subunit of nucleo/cytosolic CK2. However, in contrast to the nucleo/cytosolic enzyme, PTK
(cpCK2) is subject to redox regulation by reduced glutathione (GSH). Therefore, this kinase
can be considered as a potential mediator between cellular redox signals and plastid gene
expression. In other previous work, the cDNA for PTK/cpCK2 from mustard was cloned and
bacterially overexpressed. Database analysis revealed the existence of several orthologs for
mustard chloroplast cpCK2 in other plants including the model plant Arabidopsis thaliana.
The present work has concentrated on the physical and functional characterisation of cpCK2
from Arabidopsis (ATCK2). Initial experimental efforts focused on the question of a
"universal" role of this chloroplast enzym. If this were the case, then the cpCK2 from
Arabidopsis would be particularly suitable for the further functional studies. As a strategy to
clarify this question, the corresponding cDNA was cloned and bacterially overexpressed. A
functional comparison of the recombinant cpCK2 proteins from mustard and A. thaliana
showed that both enzym have in vitro substantially the same CK2‐typical characteristics.
In the next experimental step, the role of plant sigma factors as physiological substrates for
the transcription kinase was adressed. It was shown that indeed ATCK2 can in vitro

Summary
82
phosphorylate the recombinant sigma factors SASIG1, SASIG2, ATSIG1 and ATSIG6. Using
DNA binding studies, the functional consequences of this phosphorylation on the sigma
activity were tested with ATSIG1 and ATSIG6. A particularly notable effect was the
phosphorylation‐dependent binding of ATSIG6 to the atpB promoter. This factor revealed
strong binding activity in its phosphorylated form but not so in its unphosphorylated form.
Thus, this system provides clear evidence for the importance of sigma factor
phosphorylation as a determinant of enzymatic activity of these enzymes.
Another aim of this thesis work was the examination of the redox regulation of ATCK2. It was
shown that the activity of ATCK2 responds to reduced glutathione in much the same way as
does the authentic and recombinant enzyme from mustard. A mutational analysis of the
cysteine residues revealed a –previously not recognized– regulatory mechanism for cpCK2
(ATCK2) via disulfide bond formation. The second cysteine residue was found responsible for
dimerization, and in this way inactivation, of ATCK2 in vitro. It seems conceivable, that
hetero dimerization could occur with other components of the plastid transcription
apparatus in vivo.
To investigate the in vivo function of cpCK2, a T‐DNA insertion mutant of ATCK2 (∆cpck2)
was characterized. The phenotypic analysis of the ∆cpck2 mutant showed that the progeny
of the mutant line are seedling lethal under photoauthotrophic conditions. Compared to
wild type, transcripts of the plastid genes psaA and atpB showed significant changes in the
∆cpck2 mutant, and, furthermore also in a re‐transformed complemented line. Together
these results point to the fundamental importance of cpCK2 and may also help define
further upstream regulators of redox and phosphorylation networks in chloroplasts.
To further investigate the in vivo redox regulation of cpCK2, the ∆cpck2 line was also re‐
transformed with cpCK2 constructs that were each mutated in one of the four cysteines. It
was found that the cysteines indeed play a regulatory role in vivo. These experiments
involving directed mis‐complementation resulted in dramatic changes of the plastid RNA
patterns in the retransformed C1, C2 and C3 lines.

Literatur
83
8 Literatur
Alexander C, Faber N und Klaff P (1998). Characterisation of protein‐binding to the spinat
chloroplast psbA mRNA 5´untranslated region. Nucleic Acids Res 26: 2265‐2272
Allison LA, Simon LD und Maliga P (1996). Deletion of rpoB reveals a second distinct
transcription system in plastids of higher plants. EMBO J 15(11): 2802‐2809
Allison LA (2000). The role of sigma factors in plastid transcription. Biochemie 82:537‐548
Ausubel FM, Brent R, Kingston RG, Moore DD, Seidman JG, Smith JA und Struhl K (1989‐
2001). Current protocols in molecular biology. Green Publishing Associates and Wiley
Interscience, New York.
Baena‐González E, Baginsky S, Mulo P, Summer H, Aro EM und Link G (2001). Chloroplast
transcription at different light intensities. Glutathione‐mediated phosphorylation of the
major RNA polymerase involved in redox‐regulated organellar gene expression. Plant Physiol
127(3): 1044‐52
Baginsky S, Tiller K und Link G (1997). Transcription factor phosphorylation by a protein
kinase associated with chloroplast RNA polymerase from mustard (Sinapis alba). Plant Mol
Biol 34: 181‐189
Baginsky S, Tiller K, Pfannschmidt T und Link G (1999). PTK, the chloroplast RNA
polymersae‐associated protein kinase from mustard (Sinapis alba), mediates redox control
of plastid in vitro transcription. Plant Mol Biol 39: 1013‐1023
Baginsky S und Link G (2005). Redox regulation of chloroplast gene expression. In B
Demming‐Adams, WI Adams, AK Matto, eds, Photoprotection, Photoinhibition, Gene
Regulation and Environment. Springer, Dordrecht, The Netherlands, pp 269‐287
Baginsky S, Grossmann J und Gruissem W (2007). Proteome analysis of chloroplast mRNA
processing and degradation. J Proteom Res 2: 809‐820

Literatur
84
Barkan A (1989). Tissue‐dependent plastid RNA splicing in maize: Transcripts from four
plastid genes are predominantly unspliced in leaf meristems and roots. Plant Cell 1: 437‐445
Battistutta R, Sarno S, De Moliner E, Marin O, Issinger OG, Zanotti G und Pinna L (2000).
The crystal structure of the complex of Zea mays " α subunit with a fragment of human β
subunit provides the clue to the architecture of protein kinase CK2 holoenzyme. Eur J
Biochem 267: 5184‐5190
Bendich AJ (1987). Why do chloroplasts and mitochondria contain so many copies of their
genome? Bioessays 6: 279‐82
Bendich AJ (2004). Circular chloroplast chromosomes: the grand illusion. Plant Cell 16: 1661‐
1666
Bertani G (1951). Studies on lysogenesis. I. The mode of phage liberation by lysogenic
Escherichia coli. J Bacteriol 62: 293‐300
Birnbaum MJ, Wu J, O’Reilly DR, Rivera‐Marrero CA, Hanna DE, Miller LK und Glover CV
(1992). Expression and purification of the alpha and beta subunits of Drosophila casein
kinase II using a baculovirus vector. Protein Expr Purif 3: 142‐150
Birnboim HC und Doly J (1979). A rapid alkaline extraction procedure for screening
recombinant plasmid DNA. Nucleic Acids Research 7: 1513‐1524
Blom N, Sicheritz‐Ponten T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post‐
translational glycosylation and phosphorylation of proteins from the amino acid sequence.
Proteomics 4(6): 1633‐49
Bock R, Hermann M und Kössel H (1996). In vivo dissection of cis‐acting determinants for
plastid RNA editing. EMBO J, 15: 5052‐5059
Boldyreff B, Mietens U und Issinger OG (1996). Structure of protein kinase CK2:
dimerization of the human β‐subunit. FEBS Lett 379: 153‐156

Literatur
85
Briat JF, Laulhere JP und Mache R (1979). Transcription activity of a DNA‐protein complex
isolated from spinach plastids. Eur J Biochem 98: 285‐292
Brivanlou AH und Darnell JE Jr. (2002). Transcription ‐ Signal transduction und the control of
gene expression. Science 295: 813‐818
Bullock WO, Fernandez JM und Short JM (1987). XL 1–Blue: A high efficiency plasmid
transforming recA Escherichia coli strain with beta–galactosidase selection. BioTechniques 5:
376–378
Bülow S, Reiss T und Link G (1987). DNA‐binding proteins of the transcriptionally active
chromosome from mustard (Sinapis alba, L.) chloroplasts. Curr Genet 12: 157‐159
Bülow S und Link G (1988). Sigma‐like activity from mustard (Sinapis alba L) chloroplasts
conferring DNA‐binding and transcription specificity to E coli core RNA polymerase. Plant
Mol Biol 10: 349‐357
Chang CC, Sheen J, Bligny M, Niwa Y, Lerbs‐Mache S und Stern DB (1999). Functional
analysis of two maize cDNAs encoding T7‐like RNA polymerases. Plant Cell 11: 911‐926
Chomczynski P und Sacchi N (1987). Single‐step method of RNA isolation by acid
guanidinium thiocyanate‐phenol‐chloroform extraction. Anal Biochem 162: 156‐159
Choquet Y und Wollman FA (2002). Translational regulations as specific traits of chloroplast
gene expression. FEBS Lett 529: 39.42
Clough SJ und Bent AF (1998). Floral dip: a simplified method for Agrobacterium‐mediated
transformation of Arabidopsis thaliana. Plant J 16: 735‐744
Collinge MA und Walker JC (1994). Isolation of an Arabidopsis thaliana casein kinase II beta
subunit by complementation in Saccharomyces cerevisiae. Plant Mol Biol 25: 649‐658
Dahmus ME und Natzle J (1977). Purification and characterization of Novikoff ascites tumor
protein kinase. Biochemistry 16, 1901‐1908

Literatur
86
Danon A und Mayfield S (1994). Light‐regulated translation of chloroplast mRNAs through
redox potential. Science, 266: 1717‐1719
Danon A (1997). Translational regulation in the chloroplast. Plant Physiol 115: 1293‐1298
Dennis MD, Person MD, Browning KS (2009). Phosphorylation of plant translation initiation
factors by CK2 enhances the in vitro interaction of multifactor complex components. J Biol
Chem 284(31): 20615‐20628
DePamphilis CW und Palmer JD (1990). Loss of photosynthetic and chloro‐respiratory genes
from the plastid genome of a parasitic flowering plant. Nature 348: 337‐339
De Santis‐Maciossek G, Kofer W, Bock A, Schoch S, Maier R,Wanner G, Rüdiger W, Koop
HU und Herrmann R (1999). Targeted disruption of the plastid RNA polymerase genes rpoA,
B and C1: molecular biology, biochemistry and ultrastructure. Plant J 18: 477‐489
Dobrowolska G, Boldyreff B und Issinger, OG (1991). Cloning and sequencing of the casein
kinase 2 α subunit from Zea Mays. Biochim Biophys Acta 1129: 139‐140
Donella‐Deana A, Cesaro L, Sarno S, Brunati AM, Ruzzene M, Pinna LA (2001). Autocatalytic
tyrosine‐phosphorylation of protein kinase CK2 alpha and alpha' subunits: implication of
Tyr182. Biochem J 357(Pt 2): 563‐7
Eisermann A, Tiller K und Link G (1990). In vitro transcription and DNA binding
characteristics of chloroplast und etioplast extracts from mustard (Sinapis alba) indicate
differential usage of the psbA promoter. EMBO J 9: 3981‐3987
Emanuel C, Weihe A, Graner A, Hess WR und Börner T (2004). Chloroplast development
affects expression of phage‐type RNA polymerases in barley leaves. Plant J 38: 460‐472
Emanuel C, von Groll U, Muller M, Börner T und Weihe A (2006). Development‐and tissue‐
specific expression of the RpoT gene family of Arabidopsis encoding mitochondrial and
plastid RNA polymerases. Planta 223: 998‐1009

Literatur
87
Erdmann H, Böcher M und Wagner KG (1982). Two protein kinases from nuclei of cultured
tobacco cells with properties similar to the cyclic nucleotide‐independent enzymes (NI and
NII) from animal tissue. FEBS Lett 137: 245‐248
Espunya MC und Martínez MC (1997). Identification of two different molecular forms of
Arabidopsis thaliana casein kinase II. Plant Science 124: 131‐142
Faust M und Montenarh M (2000). Subcellular localization of protein kinase CK2. A key to ist
function? Cell Tissue Res 301: 329‐40
Fried M und Crothers DM (1981). Equilibria and kinetics of lac repressor‐operator
interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 9(23): 6505‐6525
Fujiwara M, Nagashima A, Kanamaru K, Tanaka K und Takahashi H (2000). Three new
nuclear genes, sigD, sigE und sigF, encoding putative plastid RNA polymerase σ factors in
Arabidopsis thaliana. FEBS Lett 481: 47‐52
Garner MM und Revzin A (1981). A gel electrophoresis method for quantifying the binding
of proteins to specific DNA regions: application to components of the Escherichia coli lactose
operon regulatory system. Nucleic Acids Res. 9(13): 3047‐60
Gietz RD, Graham KC und Litchfield DW (1995). Interactions between the subunits of casein
kinase II. J Biol Chem 270: 13017‐13021
Goldschmidt‐Clermont M, Girard‐Bascou J, Choquet Y, Rochaix JD (1990). Trans‐splicing
mutants of Chlamydomonas reinhardtii. Mol Gen Genet 223: 417‐425
Goldschmidt‐Clermont M, Choquet Y, Girard‐Bascou J, Michel F, Schirmer‐Rahire M,
Rochaix JD (1991). A small chloroplast RNA may be required for transsplicing in
Chlamydomonas reinhardtii. Cell 65: 135‐143
Goldschmidt‐Clermont M (1998) Coordination of nuclear and chloroplast gene expression in
plant cells. Int Rev Cytol 177: 115‐180

Literatur
88
Gray M (1989). The evolutionary origins of organelles. Trends Genet, 5: 294.299
Greenfield NJ (2006). Using circular dichroism spectra to estimate protein secondary
structure. Nat Protoc. 1(6): 2876‐2890
Gruissem W, Zurawski G (1985). Analysis of promoter regions for the spinach chloroplast
rbcL, atpB and psbA genes. EMBO J 4(13A): 3375‐3383
Gruissem W (1989). Chloroplast gene expression: how plants turn their plastids on. Cell 56:
161‐170
Hakimi MA, Privat I, Valay JG und Lerbs‐Mache S (2000). Evolutionary conservation of C‐
terminal domains of primary sigma70 –type transcription factors between plants and
bacteria. J Biol Chem 275: 9215‐9221
Hallick R, Lipper C, Richards O und Rutter W (1976). Isolation of a transcriptionally active
chromosome from chloroplasts of Euglena gracilis. Biochemistry 15: 3039‐3045
Hajdukiewicz PTJ, Allison LA und Maliga P (1997). The two RNA polymerases encoded by
the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco
plastids. EMBO J 16: 4041‐4048
Hanahan D (1983). Studies on transformation of Escherichia coli with plasmids. J Mol Biol
166: 557‐580
Hanks SK und Quinn AM (1991). Protein kinase catalytic domain sequence database:
identification of conserved features of primary structure and classification of family
members. Methods Enzymol 200: 38‐62
Hardtke CS, Gohda K, Osterlund MT, Oyama T, Okada K, Deng XW (2000). HY5 stability and
activity in arabidopsis is regulated by phosphorylation in its COP1 binding domain. EMBO J
19(18): 4997‐5006

Literatur
89
Hathaway GM und Traugh JA (1982). Casein kinases ‐ multipotential protein kinases. Curr
Topics Cell Reg. 21: 101‐127
Hedtke B, Wagner I, Börner T und Hess WR (1999). Inter‐organellar crosstalk in higher
plants: impaired chloroplast development affects mitochondrial gene and transcript levels.
Plant J 19: 635‐644
Hedtke B, Börner T und Weihe A (2000). One RNA polymerase serving two genomes. EMBO
Reports 1: 435‐440
Hedtke B, Legen J, Weihe A, Herrmann RG und Börner T (2002). Six active phage‐type RNA
polymerase genes in Nicotiana tabacum. Plant J 30: 625‐637
Heins L und Soll J (1998). Chloroplast biogenesis: Mixing the prokaryotic and the eukaryotic?
Curr Biol 8: 215‐217
Herrmann RG, Bohnert HJ, Kowallik KV und Schmitt JM (1975). Size, conformation and
purity of chloroplast DNA of some higher plants. Biochim Biophys Acta 378: 305–317
Herrmann RG, Westhof P, Link G (1992). Biogenesis of plastids in higher plant. In: Cell
organelles, (Herrmann R, G, Ed.), Springer‐Verlag, Wien 275‐349
Hess WR und Börner T (1999). Organellar RNA polymerases of higher plants. Int RevCytol
190:1‐59
Höfgen R und Willmitzer L (1990). Biochemical und genetic analysis of different patatin
isoforms expresses in various organs of potato (Solanum tuberosum). Plant Sci 66: 221‐230
Homann A und Link G (2003). DNA‐binding and transcription characteristics of three cloned
sigma factors from mustard (Sinapis alba L.) suggest overlapping and distinct roles in plastid
gene expression. Euro J Biochem 270: 1288‐1300

Literatur
90
Hulo N, Bairoch A, Bulliard V, Cerutti L, Cuche BA, de Castro E, Lachaize C, Langendijk‐
Genevaux PS, Sigrist CJ (2008). The 20 years of PROSITE. Nucleic Acids Res. 36 (Database
issue): D245‐9
Hunter T (1991). Protein phosphorylation. Part A. Protein kinases: assays, purification,
antibodies, functional analysis, cloning, and expression. Protein kinase classification.
Methods Enzymol 200, 3‐37
Igloi GL und Kössel H (1992). The transcriptional apparatus of chloroplasts. Crit Rev Plant Sci
10: 525‐558
Isono K, Niwa Y, Satoh K und Kobayashi H (1997). Evidence for transcriptional regulation of
plastid photosynthesis genes in Arabidopsis thaliana roots. Plant Physiol 114: 623‐630
Jolly SO, Bogorad L (1980). Preferential transcription of cloned maize chloroplast DNA
sequences by maize chloroplast RNA polymerase. Proc Natl Acad Sci U S A77(2): 822‐826
Kanamaru K, Fujiwara M, Seki M, Katagiri T, Nakamura M, Mochizuki N, Nagatani A,
Shinozaki K, Tanaka K und Takahashi H (1999). Plastidic RNA polymerase σ factors in
Arabidopsis thaliana. Plant Cell Physiol 40: 832‐842
Kanekatsu M, Ezumi A, Nakamura T und Ohtsuki K (1995). Chloroplast ribonucleoproteins
(RNPs) as phosphate acceptors for casein kinase II: purification by ssDNA‐cellulose column
chromatography. Plant Cell Physiol 36: 1649‐1656
Kanekatsu M, Saito H, Motohashi K und Hisabori T (1998). The beta subunit of chloroplast
ATP synthase (CF0CF1‐ATPase) is phosphorylated by casein kinase II. Biochem Mol Biol Int
46: 99‐105
Kapoor S, Suzuki JY und Sugiura M (1997). Identification und functional significance of a
new class of non‐consensus‐type plastid promoters. Plant J 11: 327‐338

Literatur
91
Kapoor S und Sugiura M (1999). Identification of two essential sequence elements in the
nonconsensus type II PatpB‐290 plastid promoter by using plastid transcription extracts from
cultured tobacco BY‐2 cells. Plant Cell 11: 1799‐1810
Kestermann M, Neukirchen S, Kloppstech K und Link G (1998). Sequence and expression
characteristics of a nuclear‐encoded chloroplast sigma factor from mustard (Sinapis alba).
Nucleic Acids Res 26: 2747‐2753
Klaff P und Gruissem W (1991). Changes in chloroplast mRNA stability during leaf
development. Plant Cell 3: 517‐529
Klemm JD, Schreiber SL, Crabtree GR (1998). Dimerization as a regulatory mechanism in
signal transduction. Annu Rev Immunol 16: 569‐592
Klimczak LJ, Schindler U und Cashmore AR (1992). DNA binding activity of the Arabidopsis
G‐Box binding factor GBF1 is stimulated by phosphorylation by casein kinase II from Broccoli.
Plant Cell 4: 87‐98
Klimczak LJ, Collinge MA, Farini D, Giuliano G, Walker JC und Cashmore AR (1995).
Reconstitution of Arabidopsis casein kinase II from recombinant subunits and
phosphorylation of transcription factor GBF1. Plant Cell 7: 105‐115
Kobayashi H, Ngernprasirtsiri J und Akazawa T (1990). Transcription regulation and DNA
methylation in plastids during transitional conversion of chloroplasts to chromoplasts. EMBO
J 9: 307‐313
Koncz C, Schell J (1986). The promoter of TL‐DNA gene 5 controls the tissue‐specific
expression of chimaeric genes carried by a novel type of Agrobacterium binary vector.
Molecular and General Genetics 204: 383‐396
Krause K, Maier RM, Kofer W, Krupinska K und Herrmann RG (2000). Disruption of plastid‐
encoded RNA polymerase genes in tobacco: expression of only a distinct set of genes is not
based on selective transcription of the plastid chromosome. Mol Gen Genet 263: 1022‐1030

Literatur
92
Krause K, Berg S und Krupinska K (2003). Plastid transcription in the holoparasitic plant
genus Cuscuta: parallel loss of the rrn16 PEP‐promoter and of the rpoA and rpoB genes
coding for the plastid‐encoded RNA polymerase. Planta 216(5): 815‐23
Krupinska K (1992). Transcriptional control of plastid gene expression during development
of primary foliage leaves of barley grown under a daily light‐dark‐regime. Planta 186: 294‐
303
Krupinska K und Falk J (1994). Changes in RNA polymerase activity during biogenesis,
maturation and scenescence of barley chloroplasts. Comparative analysis of transcripts
synthesized either in run‐on assays or by transcriptionally active chromosomes. J Plant
Physiol 143: 298‐305
Krysan PJ, Young JC, Sussman MR (1999). T‐DNA as an insertional mutagen in Arabidopsis.
Plant Cell 12: 2283‐2290
Kudla J, Igloi G, Metzlaff M, Hagemann R und Kössel H (1992). RNA editing in tobacco
chloroplasts leads to the formation of a translatable psbL mRNA by a C to U substitution
within the initiation codon. EMBO J, 11: 1099‐1103
Kuenzel EA, Mulligan JA, Sommercorn J und Krebs EG (1987). Substrate specificity
determinants for casein kinase II as deduced from studies with synthetic peptides. J Biol
Chem 262: 9136‐9140
Kusk M, Bendixen C, Duno M, Westergaard O und Thomsen B (1995). Genetic dissection of
intersubunit contacts within human protein kinase CK2. J Mo. Biol 253: 703‐711
Lahiri SD, Yao J, McCumbers C und Allison LA (1999). Tissue‐specific and lightdependent
expression within a family of nuclear‐encoded sigma‐like factors from Zea mays. Mol Cell
Biol Res Commun 1: 14‐20
Lambowitz AM, Caprara MG, Zimmerly S und Perlman PS (1999). Group I and group II
ribozymes as RNPs: clues to the past and guides to the future In Gesteland,RF, Cech,TR and

Literatur
93
Atkins,JF (eds), The RNA World Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY,
pp 451‐485
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F,
Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ und Higgins DG (2007). ClustalW and
ClustalX version 2. Bioinformatics 23(21): 2947‐2948
Lee Y, Lloyd AM und Roux SJ (1999). Antisense expression of the CK2 alphasubunit gene in
Arabidopsis. Effects on light‐regulated gene expression and plant growth. Plant Physiol 119:
989‐1000
Lerbs S, Bräutigam E und Mache R (1988). DNA‐dependent RNA polymerase of spinach
chloroplasts: Characterization of sigma‐like and alpha‐like polypeptides. Mol Gen Genet 211:
459‐464
Lerbs‐Mache S (1993). The 110‐kDa polypeptide of spinach plastid DNA‐dependent RNA
polymerase: Single‐subunit enzyme or catalytic core of multimeric enzyme complexes? Proc
Natl Acad Sci USA 90: 5509‐5513
Li Y, Rosso MG, Viehoever P, Weisshaar B (2007). GABI‐Kat SimpleSearch: an Arabidopsis
thaliana T‐DNA mutant database with detailed information for confirmed insertions. Nucleic
Acids Res 35(Database issue): D874‐878
Liere K und Maliga P (1999). In vitro characterization of the tobacco rpoB promoter reveals a
core sequence motif conserved between phage‐type plastid and plant mitochondrial
promoters. EMBO J 18: 249‐257
Liere K, Kaden D, Maliga P und Börner T (2004) Overexpression of phage –type RNA
polymerase PpoTp in tobacco demonstrates its role in chloroplast transcription by
recognizing a distinct promoter type. Ncleic Acids Res 32: 1159‐1165

Literatur
94
Link G und Langridge U (1984). Structure of the chloroplast gene for the precursor of the
Mr32,000 photosystem II protein from mustard (Sinapis alba L.). Nucleic Acids Res 12: 945‐
958
Link G (1994). Plastid differentiation: organelle promoters and transcription factors. In Plant
promoters and transcription factors ‐‐ Results and problems in cell differentiation, Vol. 20. L.
Nover, ed. (Berlin Heidelberg: Springer‐Verlag), pp. 65‐85
Link G (1996) Green life: control of chloroplast gene transcription. BioEssays 18: 465‐472
Loschelder H, Homann A, Ogrzewalla K und Link G (2004) Proteomics‐based sequence
analysis of plant gene expression ‐ the chloroplast transcription apparatus. Phytochemistry
65: 1785‐1793
Lund O, Nielsen M, Lundegaard C, Worning P (2002). CPHmodels 2.0: X3M a Computer
Program to Extract 3D Models. Abstract at the CASP5 conferenceA102
Maier R, Zeltz P, Kössel H, Bonnard G, Gualberto J und Grienenberger J (1996). RNA editing
in plant mitochondria and chloroplasts. Plant Mol Biol, 32: 343.365
Maliga P (1998). Two plastid RNA polymerases of higher plants: an evolving story. Trends
Plant Sci 3: 4‐6
Maniatis T, Fritsch EF und Sambrook J (1982). Molecular Cloning, A Laboratory Manual. Cold
Spring Harbor Laboratory, Cold Spring Harbor, New York
Marchiori F, Meggio F, Marin O, Borin G, Calderan A, Ruzza P und Pinna LA (1988).
Synthetic peptide substrates for casein kinase 2. Assessment of minimum structural
requirements for phosphorylation. Biochim Biophys Acta 971: 332‐338
Martin SR und Schilstra MJ (2007). Circular Dichroism and Ist Application to the Study of
Biomolecules. Methods in Cell Biology, 84: 263‐293

Literatur
95
Martin W und Herrmann RG (1998). Gene transfer from organelles to the nucleus: How
much, what happens, und why. Plant Physiol 118: 9‐17
Martin W, Rujan T, Richly E, Hansen A, Cornelsen S, Lins T, Leister D, Stoebe B, Hasegawa
M und Penny D (2002). Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast
genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus.
Proc Natl Acad Sci USA 19: 12246–12251
Mayfield SP, Yohn CB, Cohen A und Danon A (1995). Regulation of chloroplast gene
expression. Annu Rev Plant Physiol 46: 147‐166
McFadden G (1999). Endosymbiosis and evolution of the plant cell. Curr Opin Plant Biol 2:
513‐519McFadden GI (2001) Chloroplast origin and integration Plant Physiol 125: 50‐53
Meggio F, Boldyreff B, Issinger OG und Pinna LA (1993). The autophosphorylation and
p34cdc2 phosphorylation sites of casein kinase‐2 β‐subunit are not essential for reconstituting
the fully‐active heterotetrameric holoenzyme. Biochem Biophys Acta 1164: 223‐225
Meggio F und Pinna LA (2003). One‐thousand‐and‐one substrates of protein kinase CK2?
FASEB J 17(3): 346‐368
Meisner H, Heller‐Harrison R, Buxton J und Czech MP (1989). Molecular cloning of the
human casein kinase II α subunit. Biochemistry 28: 4072‐4076
Mereschkowsky C (1905). Über die Natur und den Ursprung der Chromatophoren im
Pflanzenreiche. Biologisches Zentralblatt 25: 593‐604, 989‐691
Mizoguchi T, Yamaguchi‐Shinozaki K, Hayashida N, Kamada H und Shinozaki K (1993).
Cloning and characterization of two cDNAs encoding casein kinase II catalytic subunits in
Arabidopsis thaliana. Plant Mol Biol 21: 279‐289
Morden CW, Wolfe KH, DePamphilis CW und Palmer JD (1991). Plastid translation and
transcription genes in a non‐photosynthetic plant: Intact, missing und pseudo genes. EMBO J
10: 3281‐3288

Literatur
96
Morikawa K, Ito S, Tsunoyama Y, Nakahira Y, Shiina T, Toyoshima Y (1999). Circadian‐
regulated expression of a nuclear encoded plastid sigma factor gene (sigA) in wheat
seedlings. FEBS Lett 451: 275‐278
Morikawa K, Shiina T, Murakami S und Toyoshima Y (2002). Novel nuclearencoded proteins
interacting with a plastid sigma factor, Sig1, in Arabidopsis thaliana. FEBS Lett 514: 300‐304
Murashige T und Skoog F (1962). A revised medium for rapid growth and bioassays with
tobacco tissue cultures. Physiol Plant 15(3): 473‐497
Neuhaus H und Link G (1987). Nucleotide sequence and transcriptional organization of the
chloroplast tRNALys(UUU) gene from mustard (Sinapis alba). Curr Genet 11: 251‐257
Neuhaus H, Scholz A und Link G (1989). Structure and expression of a split chloroplast gene
from mustard (Sinapis alba): Ribosomal protein gene rps16 reveals unusual transcriptional
features und complex RNA maturation. Curr Genet 15: 63‐70.
Niefind K, Guerra B, Pinna LA, Issinger OG und Schomburg D (1998). Crystal structure of the
catalytic subunit of protein kinase CK2 from Zea mays at 2.1 Å resolution. EMBO J 17: 2451‐
2462
Niefind K, Pütter M, Guerra B, Issinger OG und Schomburg D (1999). GTP plus water mimics
ATP in the active site of protein kinase CK2. Nat Struct Biol 6: 1100‐1103
Oldenburg DJ und Bendich AJ (2004). Most chloroplast DNA of maize seedlings in linear
molecules with defined ends and branched forms. J Mol Biol 335: 953‐970
Ogrzewalla K, Piotrowski M, Reinbothe S und Link G (2002).The plastid transcription kinase
from mustard (Sinapis alba L.) ‐ a nuclear‐encoded CK2‐type chloroplast enzyme with redox‐
sensitive function. Eur J Biochem 269: 3329‐3337
Palmer JD (1985). Comparative organization of chloroplast genomes. Annu Rev Genet 19:
325–354

Literatur
97
Palmer JD (1991). Plastid chromosomes: Structure and evolution In The Molecular Biology of
Plastids, IK Vasil, ed (San Diego, CA: Academic Press)
Pfannschmidt T und Link G (1994). Separation of two classes of plastid DNA‐dependent RNA
polymerases that are differentially expressed in mustard (Sinapis alba L) seedlings. Plant Mol
Biol 25: 69‐81
Pfannschmidt T und Link G (1997). The A und B forms of plastid DNA‐dependent RNA
polymerase from mustard (Sinapis alba L) transcribe the same genes in a different
developmental context. Mol Gen Genet 257: 35‐44
Pfannschmidt T, Ogrzewalla K, Baginsky S, Sickmann A, Meyer HE und Link G (2000). The
multisubunit chloroplast RNA polymerase A from mustard (Sinapis alba L): Integration of a
prokaryotic core into a larger complex with organelle‐specific functions. Eur J Biochem 267:
253‐261
Pfannschmidt T, Liere K (2005). Redox regulation and modification of proteins controlling
chloroplast gene expression. Antioxid Redox Signal (5‐6): 607‐618.
Pinna LA (1990). Casein kinase 2: an 'eminence grise' in cellular regulation? Biochim Biophys
Acta 1054(3): 267‐84
Pinna LA (1997). Protein kinase CK2. Int J Biochem Cell Biol 4: 551‐554
Pinna LA und Meggio F (1997). Protein kinase CK2 (“casein kinase‐2”) and its implication in
cell division and proliferation. Progr Cell Cycle Res 3: 77‐97
Pribnow D (1975a). Nucleotide sequence of an RNA polymerase binding site at an early T7
promoter. Proc Natl Acad Sci USA 72(3): 784‐788
Pribnow D (1975b). Bacteriophage T7 early promoters: nucleotide sequences of two RNA
polymerase binding sites. J Mol Biol 99(3): 419‐443

Literatur
98
Rajakulendran T, Sahmi M, Lefrançois M, Sicheri F und Therrien M (2009). A dimerization‐
dependent mechanism drives RAF catalytic activation. Nature 461(7263): 542‐545
Rapp JC, Mullet JE (1991). Chloroplast transcription is required to express the nuclear genes
rbcS and cab. Plastid DNA copy number is regulated independently. Plant Mol Biol 4: 813‐23
Rao VB (1994). Direct sequencing of polymerase chain reaction‐amplified DNA. Anal
Biochem 216: 1‐14
Reiland S, Messerli G, Baerenfaller K, Gerrits B, Endler A, Grossmann J, Gruissem W und
Baginsky S (2009). Large‐scale Arabidopsis phosphoproteome profiling reveals novel
chloroplast kinase substrates and phosphorylation networks. Plant Physiol 150(2): 889‐903
Reiss T und Link G (1985). Characterization of transcriptionally active DNA‐protein
complexes from chloroplasts and etioplasts of mustard (Sinapis alba L.). Eur J Biochem 148:
207‐212
Riera M, Peracchia G, de Nadal E, Arino J und Pages M (2001). Maize protein kinase CK2:
regulation and functionality of three beta regulatory subunits. Plant J. 25: 365‐374
Roeder RG (1996). The role of general initiation factors in transcription by RNA polymerase
II. Trends Biochem Sci 21: 327‐335
Rogers SO und Bendich AJ (1985). Extraction of DNA from milligram amounts of fresh,
herbarium and mummified plant tissue. Plant Mol Biol 5: 69‐76
Rosso MG, Li Y, Strizhov N, Reiss B, Dekker K und Weisshaar B (2003). An Arabidopsis
thaliana T‐DNA mutagenized population (GABI‐Kat) for flanking sequence tag‐based reverse
genetics. Plant Mol Biol 53: 247‐259
Rott R, Levy H, Drager RG, Stern DB und Schuster G (1998). 3´‐processed mRNA is
preferentially translated in Chlamydomonas reinhardtii chloroplasts. Mol. Cell. Biol. 18:
4605‐4611.

Literatur
99
Salinas P, Fuentes D, Vidal E, Jordana X, Echeverria M und Holuigue L (2006). An extensive
survey of CK2 alpha and beta subunits in Arabidopsis: multiple isoforms exhibit differential
subcellular localization. Plant Cell Physiol 47(9): 1295‐308
Samaniego R, Jeong SY, de la Torre C, Meier I und Moreno Díaz de la Espina S (2006). CK2
phosphorylation weakens 90 kDa MFP1 association to the nuclear matrix in Allium cepa. J
Exp Bot 57(1): 113‐24
Sambrook J, Fritsch EF und Maniatis T (1989). Molecular cloning. A Laboratory Manual,
Second Edition. Cold Spring Harbor Laboratory, New York
Sarno S, Ghisellini P, Cesaro L, Battistutta R und Pinna LA (2001). Generation of mutants of
CK2α which are dependent on the β−subunit for catalytic activity. Mol Cell Biochem 227: 13‐
19
Schaller H, Gray C und Herrmann K (1975). Nucleotide sequence of an RNA polymerase
binding site from the DNA of bacteriophage fd. Proc Natl Acad Sci USA 72(2): 737‐41
Shelton MD, Chock PB, Mieyal JJ (2005). Glutaredoxin: role in reversible protein S
glutathionylation and regulation of redox signal transduction and protein translocation.7(3‐
4): 348‐66
Schimper A (1883). Über die Entwicklung der Chlorophyllkörner und Farbkörper. Botanische
Zeitung 41: 105‐112, 121‐131, 137‐146, 153‐162
Schinkel AH und Tabak HF (1989). Mitochondrial RNA polymerase: dual role in transcription
and replication. Trends Genet. 51: 149‐154
Schuster G und Gruissem W (1991). Chloroplast mRNA 3' end processing requires a nuclear
encoded RNA‐binding protein. EMBO J 10: 1493‐1502.
Schünemann D (2004). Structure and function of the chloroplast signal recognition particle.
Curr Genet 44: 295‐304

Literatur
100
Schweer J, Loschelder H und Link G (2006) A promoter switch that can rescue a plant sigma
factor mutant. FEBS Lett 580: 6617‐6622
Schweer J, Geimer S, Meurer J und Link G (2009). Arabidopsis mutants carrying chimeric
sigma factor genes reveal regulatory determinants for plastid gene expression. Plant Cell
Physiol 50(7): 1382‐1386
Schweer J, Türkeri H, Link B und Link G (2010). AtSIG6, a plastid sigma factor from
Arabidopsis, reveals functional impact of cpCK2 phosphorylation. Plant J [Epub ahead of
print]
Serino G und Maliga P (1998) RNA polymerase subunits encoded by the plastid rpo genes
are not shared with the nucleus‐encoded plastid enzyme Plant Physiol 117: 1165‐1170
Soll J und Tien R (1998) Protein translocation into and across the chloroplastic envelope
membranes. Plant Mol Biol 38(1‐2): 191‐207
Soll J (2002). Protein import into chloroplasts. Curr Opin Plant Biol (5)6: 529‐535
Sriraman P, Silhavy D und Maliga P (1998) Transcription from heterologous rRNA operon
promoters in chloroplasts reveals requirement for specific activating factors. Plant Physiol
117: 1495‐1499
Stern DB, Higgs DC und Yang J (1997). Transcription and translation in chloroplasts. Trends
Plant Sci. 2: 308‐315
Stirdivant S, Crossland L und Bogorad L (1985). DNA supercoiling affects transcription of two
maize chloroplast genes differently. Proc Natl Acad Sci USA, 82: 4886‐4890
Stone JM und Walker JC (1995). Plant protein kinase families and signal transduction. Plant
Physiol. 108: 451‐457

Literatur
101
Sugano S and Ronis C, Green, RM, Wang ZY und Tobin EM (1998). Protein kinase CK2
interacts with and phosphorylates the Arabidopsis circadian clock‐associated 1 protein. Proc
Natl Acad Sci USA 95: 11020‐11025
Sugano S and Ronis C, Ong, MS, Green RM und Tobin EM (1999). The protein kinase CK2 is
involved in regulation of circadian rhythms in Arabidopsis. Proc Natl Acad Sci USA 96: 12362‐
12366
Sugita M und Sugiura M (1996). Regulation of gene expression in chloroplasts of higher
plants. Plant Mol Biol 32: 315‐326
Sugiura M (1992). The chloroplast genome. Plant Mol Biol 19: 149‐168
Sugiura M (1995). The chloroplast genome. Essays Biochem 30: 49‐57
Sugiura M, Hirose T, Sugita M. (1998). Evolution and mechanism of translation in
chloroplasts. Annu Rev Genet 32: 437‐459
Summer H, Pfannschmidt T und Link G (2000). Transcripts and sequence elements suggest
differential promoter usage within the ycf3‐psaAB gene cluster on mustard (Sinapis alba L.)
chloroplast DNA. Curr Genet 37: 45‐52
Surzycki SJ und Shellenbarger DL (1976). Purification and characterization of a putative
sigma factor from Chalamydomonas reinhardi. Proc Natl Acad Sci U S A 73(11): 3961‐3965
Tan S und Troxler RF (1999) Characterization of two chloroplast RNA polymerase sigma
factors from Zea mays: Photoregulation and differential expression. Proc Natl Acad Sci USA
96: 5316‐5321
Tanaka K, Tozawa Y, Mochizuki N, Shinozaki K, Nagatani A, Wakasa K and Takahashi H
(1997). Characterization of three cDNA species encoding plastid RNA polymerase sigma
factors in Arabidopsis thaliana: Evidence for the sigma factor heterogeneity in higher plant
plastids. FEBS Lett 413: 309‐313

Literatur
102
Tiller K, Eisermann A und Link G (1991). The chloroplast transcription apparatus from
mustard (Sinapis alba L)‐‐Evidence for three different transcription factors which resemble
bacterial σ factors Eur J Biochem 198: 93‐99
Tiller K und Link G (1993a). Sigma‐like transcription factors from mustard (Sinapis alba L)
etioplast are similar in size to, but functionally distinct from, their chloroplast counterparts.
Plant Mol Biol 21: 503‐513
Tiller K und Link G (1993b). Phosphorylation and dephosphorylation affect functional
characteristics of chloroplast and etioplast transcription systems from mustard (Sinapis alba
L). EMBO J 12: 1745‐1753
Toro N (2003). Bacteria and Archaea group II introns: additional mobile genetic elements in
the environment. Environ Microbiol 5: 143‐151
Tozawa Y, Tanaka K, Takahashi H und Wakasa K (1998). Nuclear encoding of a plastid σ
factor in rice and its tissue‐and light‐dependent expression. Nucleic Acids Res 26: 415‐419
Troxler RF, Zhang F, Hu J und Bogorad L (1994). Evidence that σ factors are components of
chloroplast RNA polymerase. Plant Physiol 104: 753‐759
Tsudzuki J, Ito S, Tsudzuki T, Wakasugi T und Sugiura M (1994). A new gene encoding
tRNA(Pro) (GGG) is present in the chloroplast genome of black pine: a compilation of 32
tRNA genes from black pine chloroplasts. Curr Genet 26: 153‐158
Vera A, Hirose T und Sugiura M (1996). A ribosomal protein gene (rpl32) from tobacco
chloroplast DNA is transcribed from alternative promoters: Similarities in promoter region
organization in plastid housekeeping genes. Mol Gen Genet 251: 518‐525
Waugh DS (2005). Making the most of affinity tags. Trends Biotechnol 23(6): 316‐320
Westhoff P und Herrmann R (1988). Complex RNA maturation in chloroplasts: The psbB
operon from spinach. Eur J Biochem, 171: 551.564

Literatur
103
Whitfeld P und Bottomley W (1993). Organisation and structure of chloroplast genes. Annu
Rev Plant Physiol 34: 279‐310
Williams MA, Cave CM, Quaid G, Robinson C, Daly TJ, Witt D, Lentsch AB, Solomkin JS
(2005). Interleukin 8 Dimerization As A Mechanism for Regulation of Neutrophil Adherence‐
Dependent Oxidant Production. Shock, 23 (4): 371–376
Wolfe KH, Morden CW und Palmer JD (1992). Function and evolution of a minimal plastid
genome from a nonphotosynthetic parasitic plant. Proc Natl Acad Sci USA 89: 10648‐10652
Xue Y, Ren J, Gao X, Jin C, Wen L und Yao X (2008). GPS 2.0, a Tool to Predict Kinase‐specific
Phosphorylation Sites in Hierarchy. Mol Cell Proteomics 7: 1598‐1608
Yamaguchi H, Kasa M, Amano M, Kaibuchi K und Hakoshima T (2006). Molecular
Mechanism for the Regulation of Rho‐Kinase by Dimerization and Its Inhibition by Fasudil.
Structure 14: 589‐600
Yan TFJ und Tao M (1982). Purification and characterization of a wheat germ protein kinase.
J Biol Chem 257: 7037‐7043
Yohn C, Cohen A, Danon A und Mayfield S (1998a). A poly(a) binding protein functions in
the chloroplast as a message‐specific translation factor. Proc Natl Acad Sci USA 95: 2238‐
2243
Yohn C, Cohen A, Rosch C, Kuchka M und Mayfield S (1998b). Translation of the chloroplast
psbA mRNA requires the nuclear‐encoded poly (A)‐binding protein RB47. J Cell Biol 142: 435‐
442

Wissenschaftliche Präsentationen
104
Wissenschaftliche Präsentationen
Publikationen
Schweer J, Türkeri H, Link B, Link G (2010). AtSIG6, a plastid sigma factor from Arabidopsis,
reveals functional impact of cpCK2 phosphorylation. Plant J [Epub ahead of print]
Vorträge
H Türkeri, H Loschelder, J Schweer, A Kolpack, B Link and G Link (2006). “Redoxregulation
der Chloroplastentranskription: Funktionsanalyse der rekombinanten chloroplastidären
Transkriptionskinase cpCK2α“. Forschergruppentreffen (FOR 387), Bielefeld.
H Türkeri, H Loschelder, J Schweer, A Kolpack, B Link and G Link (2007). “Redox Regulation of
Chloroplast Transcription: The Plastid Transcription Kinase”. German‐Japanese Workshop at
the University of Konstanz. Evolutionary and Functional Aspects of Redox Regulation in
Photosynthetic Organisms.
H Türkeri, H Loschelder, J Schweer, A Kolpack, B Link and G Link (2007). “Redox Regulation of
Chloroplast Transcription: The Plastid Transcription Kinase (PTK)”. Redox Signal Integration:
From Stimulus to Networks and Genes, Bielefeld.
H Türkeri, J Schweer, A Kolpack, H Loschelder, B Link and G Link (2008). „Regulation of
Chloroplast Transcription: Functional Characterization of the Plastid Transcription Kinase
(PTK) in Arabidopsis thaliana”. 1 international PhD Student Symposium of the SFB 480 Ruhr‐
University, Bochum.
Poster‐Präsentationen
H Loschelder, J Schweer, H Türkeri, B Link, S Rattay, A Kolpack und G Link (2006). Functional
Role of Plastid Sigma Factors: A Sigma 6 Knock‐out from Arabidopsis. Interne Tagung SFB
480, Duisburg.

Wissenschaftliche Präsentationen
105
H Loschelder, H Türkeri, J Schweer, B Link, S Rattay, A Kolpack und G Link (2006).
Funktionsanalyse der Transkription in den Plastiden: Licht‐, Redox‐ und Entwicklungs‐
Regulation. 19. Tagung „Molekularbiologie der Pflanzen“, Dabringhausen.
H Loschelder, J Schweer, H Türkeri, B Link, S Rattay und G Link (2006). Dual temporal Role of
Plastid Sigma Factor 6 in Arabidopsis Development. 3rd International PhD Student
Symposium “Horizons in Molecular Biology”, Göttingen.
J Schweer, H Loschelder, H Türkeri, B Link und G Link (2006). A plant sigma factor, ATSIG6,
mutant may be rescued by a promoter switch during development. 3rd International PhD
Student Symposium “Horizons in Molecular Biology”, Göttingen.
J Schweer, H Loschelder, H Türkeri, B Link, S Rattay, A Kolpack und G Link (2006). Rolle
plastidärer Transkriptionsfaktoren: Ergebnisse und Strategien mit Arabidopsis
Sigmamutanten. Interne Tagung SFB 480, Bochum.
J Schweer, H Loschelder, H Türkeri, B Link, A Kolpack und G Link (2007). A Promotor Switch
that can rescue a Plant Sigma Factor Mutant. FEBS Advanced Lecture Course „Origin and
Evolution of Mitochondria and Chloroplasts”, Acquafredda di Maratea, Italien.
H Türkeri, H Loschelder, A Kolpack, J Schweer, B Link und G Link (2007). Redox Regulation of
Chloroplast Transcription: The Plastid Transcription Kinase“. German‐Japanese Workshop
“Evolutionary and Functional Aspects of Redox Regulation in Photosynthetic Organisms”, in
Konstanz.
H Loschelder, J Schweer, H Türkeri, A Kolpack, B Link und G Link (2007). Dual temporal Role
of Plastid Sigma Factor 6 in Arabidopsis Development. 17. Tagung der Gesellschaft für
Entwicklungsbiologie, Marburg.
A Kolpack, H Türkeri, H Loschelder, J Schweer und G Link (2007). Developmentally regulated
plastid transcription in plants: Interaction partners and signalling mechanisms. 17. Tagung
der Gesellschaft für Entwicklungsbiologie, Marburg.

Wissenschaftliche Präsentationen
106
A Kolpack, H Türkeri, J Schweer, H Loschelder und G Link (2007). Regulation of Chloroplast
Transcription. Interne Tagung SFB 480, Velen.
H Türkeri, J Schweer, A Kolpack, H Loschelder, B Link und G Link (2008). Analyse zur Funktion
der plastidären Transkriptionskinase (PTK) bei der Regulation der Genexpression in
Arabidopsis thaliana. 21. Tagung „Molekularbiologie der Pflanzen“, Dabringhausen.
H Türkeri, J Schweer, A Kolpack, H Loschelder und G Link (2008). Redox Regulation of
Chloroplast Transcription: The Plastid Transcription Kinase. Gordon Research Conference
“Thiol‐based Redox Regulation & Signaling”, 25‐30.05.2008, Italien.
J Schweer, H Türkeri, A Kolpack, H Loschelder und G Link (2008). Multi‐facetted controls in
chloroplast gene regulation: impact of multiple sigma factors and their master regulator.
FESPB 2008 Congress, XVI Congress of the Federation of the European Societies of the Plant
Biology, Tampere, Finnland.
A Kolpack, H Türkeri, J Schweer und G Link (2009). Protein‐Protein Interaktionen in der
plastidären Genexpression: Sigma Faktoren und ihre Interaktoren. 22. Tagung
„Molekularbiologie der Pflanzen“, Dabringhausen.
Workshop‐Teilnahme
Einführung in die Proteinkristallisation (SFB Workshop, 2005).
Massenspektrometrie in der Proteinanalytik (Update) (SFB Workshop, 2005).
GatewayTM‐Technologie: Potenzial und Anwendungen in der molekularen Pflanzenforschung
(SFB Workshop, 2008)

Lebenslauf
107
Lebenslauf
Persönliche Daten
Name Hacer Türkeri
Geburtsdatum 5. Oktober 1976
Geburtsort Niğde/Türkei
Familienstand ledig
Schulausbildung
09/1982–06/1987 Grundschule „Gazi İlkögretim Okulu“, Niğde/Türkei
09/1987–06/1990 Mittelschule „Bahceli Ortaokulu“, Niğde/Türkei
09/1990–06/1993 Lyzeum „Niğde Lisesi“, Niğde/Türkei
Studium
09/1994–07/1998 Biologie‐Studium an der Erciyes Universität in Kayseri/Türkei
Abschluss: Bachelor of Science (mit zusätlicher pädagogischen
Fortbildung)
07/1998–09/2000 Deutschkurs an der Universität Essen (Abschluss: DSH‐Prüfung)
10/1998 Einstufung in das 5. Semester in der Fakultät für Biologie an der Ruhr‐
Universität Bochum
09/2000–05/2006 Biologie‐Studium an der Ruhr‐Universität Bochum
Abschluss mit der Diplomarbeit „Redoxregulation der Chloroplasten‐
Transkription: Funktionsuntersuchungen der clonierten
chloroplastidären Transkriptionskinase (PTK) / cpCK2α"
seit Juni 2006 Doktorandin und Wissenschaftliche Mitarbeiterin in der Arbeitsgruppe
„Pflanzliche Zellphysiologie und Molekularbiologie“
Anfertigung der Dissertation: „Regulation der plastidären
Genexpression durch kernkodierte Faktoren: Untersuchungen zur
Rolle der Plastiden‐Transkriptionskinase (PTK) in der chloroplastidären
Genexpression in A. thaliana“ unter der Betreuung von Prof. Dr. G.
Link
Bochum, den 25.01.2010

108
Erklärung
Hiermit erkläre ich, dass ich die Arbeit selbstständig verfasst und bei keiner anderen Fakultät
eingereicht und dass ich keine anderen als die angegebenen Hilfsmittel verwendet habe. Es
handelt sich bei der heute von mir eingereichten Dissertation um fünf in Wort und Bild völlig
übereinstimmende Exemplare. Weiterhin erkläre ich, dass digitale Abbildungen nur die
originalen Daten enthalten und in keinem Fall inhaltsverändernde Bildbearbeitung
vorgenommen wurde.
Bochum, den
(Hacer Türkeri)







![Biosystemtechnik Einfuehrung 2012 1 [Schreibgeschützt] · Hintergrund - Biosystemtechnik? Immunologie Mikroarrays zur Erforschung der Genexpression Higher expressed in RNA A Similar](https://img.pdfslide.org/doc/110x75/5d56d70488c9934d328ba55d/biosystemtechnik-einfuehrung-2012-1-schreibgeschuetzt-hintergrund-biosystemtechnik.jpg)





















