Embed Size (px)
Citation preview

Evaluation eines Radioimmunoassays zur Bestimmung der freien Metanephrine im Plasma im
Vergleich zu bisher angewandten biochemischen Verfahren in der Diagnostik des
Phäochromozytoms
Dissertation
zur Erlangung des akademischen Grades
Dr. med.
an der medizinischen Fakultät
der Universität Leipzig
eingereicht von:
Ulrich Miehe
06.06.1981, Magdeburg
angefertigt in:
Universität Leipzig, Klinik für Endokrinologie und Nephrologie
Betreuer: Prof. Dr. Christian Koch
Prof. Dr. Jürgen Kratzsch
Beschluss über die Verleihung des Doktorgrades vom: 13.12.2010

2
Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. Christian Koch sowie Herrn Prof. Dr. Jürgen
Kratzsch, die mich bei der Konzeption und Durchführung der Arbeit sowie der Erstellung der
Dissertationsschrift betreut und mir in fachlichen Fragen stets zur Seite gestanden haben.
Bedanken möchte ich mich darüber hinaus bei meinen Eltern sowie bei Dir, Anett. Ohne Eure
ausdauernde und vielseitige Unterstützung wäre die Umsetzung meiner Promotion kaum
möglich gewesen.

3
Bibliographische Beschreibung
Miehe, Ulrich
Evaluation eines Radioimmunoassays zur Bestimmung der freien Metanephrine im Plasma im
Vergleich zu bisher angewandten biochemischen Verfahren in der Diagnostik des
Phäochromozytoms
Universität Leipzig, Dissertation
55 S., 178 Lit., 23 Abb.
Referat:
In den vergangen Jahren ist durch eine verbesserte Bildgebung die Anzahl der zufällig
entdeckten adrenalen Raumforderungen beständig angewachsen. Eine wichtige
Differenzialdiagnose sowohl bei adrenalen Inzidentalomen als auch bei der refraktären
Hypertonie stellt das Phäochromozytom dar. Aus diesem Hintergrund heraus leitet sich der
Wunsch nach Methoden zur sensitiven und spezifischen Bestimmung biochemischer
Parameter in der Diagnostik des Phäochromozytoms ab. Derzeit werden hierzu die freien
Plasmametanephrine durch High Pressure Liquid Chromatography (HPLC) als Mittel der
Wahl angesehen. In der vorliegenden Arbeit wurden bei 113 Patienten mit klinischem
Verdacht auf bzw. zum Ausschluss eines Phäochromozytoms die freien Plasmametanephrine
mit Hilfe eines Radioimmunoassays (RIA) untersucht. Ziel war zum einen die Evaluierung
der freien Metanephrine im Plasma mittels RIA als auch der Vergleich dieser mit anderen
biochemischen Parametern und diagnostischen Verfahren hinsichtlich Sensitivität und
Spezifität.
Der hier verwendete RIA ist relativ zeitnah durchzuführen, kostengünstig und dadurch breit
verfügbar. Er zeigt eine hohe diagnostische Sensitivität und Spezifität bei der Bestimmung der
freien Metanephrine im Plasma und bietet sich somit für den Einsatz in der Routinediagnostik
des Phäochromozytoms an, wodurch verbesserte Aussagen zum Ausschluss bzw. zur
Bestätigung der Anwesenheit des Tumors möglich werden.

4
Inhaltsverzeichnis
1 Einleitung 6
1.1 EPIDEMIOLOGIE 6 1.2 GENETISCHER HINTERGRUND 6 1.3 MALIGNITÄT DES PHÄOCHROMOZYTOMS 11 1.4 GESCHICHTLICHER ABRISS 11 1.5 PHYSIOLOGIE DES KATECHOLAMINMETABOLISMUS 12 1.6 KATECHOLAMINMETABOLISMUS BEIM PHÄOCHROMOZYTOM 14 1.7 LABORDIAGNOSTIK DES PHÄOCHROMOZYTOMS 15
2 Zielstellung 19
3 Patientencharakteristika und Methoden 20
3.1 PATIENTENCHARAKTERISTIKA 20 3.2 METHODEN 22 3.2.1 RADIOIMMUNOASSAY FÜR PLASMAMETANEPHRINE 22 3.2.2 ERMITTLUNG EINES GRENZWERTES FÜR PLASMAMETANEPHRINE MITTELS
RADIOIMMUNOASSAY (RIA) DER FIRMA LDN GMBH & CO. KG 24 3.2.3 RADIOIMMUNOASSAY FÜR SERUM-CHROMOGRANIN A 26 3.2.4 HPLC FÜR PLASMA-, URINMETANEPHRINE UND URINKATECHOLAMINE 27 3.3 DIAGNOSTISCHER ABLAUF 27 3.4 STATISTISCHE VERFAHREN 28
4 Ergebnisse 29
4.1 BESTIMMUNG DER PLASMAMETANEPHRINE MIT RADIOIMMUNOASSAY (RIA) 29 4.2 VERGLEICH DER PLASMAMETANEPHRINE BEI PATIENTEN MIT PHÄOCHROMOZYTOM PRÄ-
UND POSTOPERATIV MIT RIA 31 4.3 VERGLEICH DER RIA-PLASMAMETANEPHRINBESTIMMUNG MIT HPLC 33 4.4 BESTIMMUNG DER URINKATECHOLAMINE MIT HPLC 36 4.5 BESTIMMUNG DER URINMETANEPHRINE MIT HPLC 37 4.6 BESTIMMUNG DES SERUM-CHROMOGRANIN A MITTELS RIA 39 4.7 MEHRFACHBESTIMMUNG DER METANEPHRINE IM PLASMA MIT RIA 40 4.8 PLASMAMETANEPHRINE IN ABHÄNGIGKEIT VOM ALTER 41 4.9 VERGLEICH DER ANALYSIERTEN PARAMETER 42
5 Diskussion 44
5.1 ÜBERBLICK 44 5.2 PLASMAMETANEPHRINE 46 5.2.1 KATECHOLAMINE UND METANEPHRINE IM 24-H-SAMMELURIN 50 5.2.2 CHROMOGRANIN A IM SERUM 51 5.2.3 PRÄKLINISCHE EINFLUSSFAKTOREN 52 5.3 EINSCHRÄNKUNGEN DIESER STUDIE 53 5.4 ÖKONOMISCHE GESICHTSPUNKTE 53
6 Zusammenfassung 56

5
Abkürzungsverzeichnis 58
Abbildungsverzeichnis 59
Literaturverzeichnis 61

6
1 Einleitung
1.1 Epidemiologie
Das Phäochromozytom ist mit einer jährlichen Inzidenz von 2 bis 8 Fällen pro 1 Million
Einwohner ein extrem seltener Tumor (Beard et al. 1983, Hartley et al 1985, Stentstrom et al.
1986, Andersen et al. 1988, Fernandez et al.1994). Es handelt sich hierbei entsprechend der
WHO-Klassifikation endokriner Tumoren aus dem Jahr 2004 um einen Tumor, der von den
Katecholamin produzierenden chromaffinen Zellen des Nebennierenmarks ausgeht. Nach de
Lellis (2004) wird das Phäochromozytom auch als intraadrenales Paragangliom bezeichnet. In
der Fetalzeit sind die chromaffinen Zellen in vielen Körperregionen vorhanden. Sie
degenerieren aber im Verlauf der Entwicklung, so dass lediglich die sympathischen und
parasympathischen Ganglien sowie das Nebennierenmark als Sitz der chromaffinen Zellen
erhalten bleiben. Aus der Embryologie erklärt sich die variable Lokalisation des Tumors. Er
entspringt in circa 85% der Fälle aus den chromaffinen Zellen des adrenalen Gewebes (Reisch
et al. 2006). Das Phäochromozytom tritt jedoch auch in bis zu 15% der Fälle als so genanntes
extraadrenales Paragangliom auf, wenn es seinen Ursprung im extraadrenalen Gewebe hat
(Hernandez et al. 2000, Baguet et al.2004). Insgesamt entstehen 95% aller
Phäochromozytome im Abdomen. Die häufigsten extraadrenalen Lokalisationen sind
paraaortal, in der Gallenblase sowie im Kopf- und Halsbereich (Bravo et al. 2003, Elsayes et
al. 2005).
1.2 Genetischer Hintergrund
Entgegen der früheren Annahme, dass ca. 10% der Phäochromozytome genetischen
Ursprungs sind, wird heutzutage durch verschiedene in den letzten Jahren veröffentlichte
Studien von etwa 20% bis 30% genetisch bedingten Phäochromozytomen ausgegangen
(Neumann HP et al. 2002, Amar et al. 2005). Wie Abbildung 1 zeigt sind verschiedene
hereditäre Erkrankungen bekannt, die mit der Entstehung von Phäochromozytomen assoziiert
sind. Die von-Hippel-Lindau Erkrankung (VHL) ist eine autosomal-dominant vererbte
Krankheit, die durch einen Gendefekt auf dem Locus p25-26 des Chromosoms 3 verursacht
wird (Latif et al. 1993, Neumann et al. 2002). Sie tritt mit einer Häufigkeit von 1 zu 30.000
bis 1 zu 40.000 auf (Maher et al. 1991, Neumann et al. 1991). Das VHL-Gen ist ein typisches
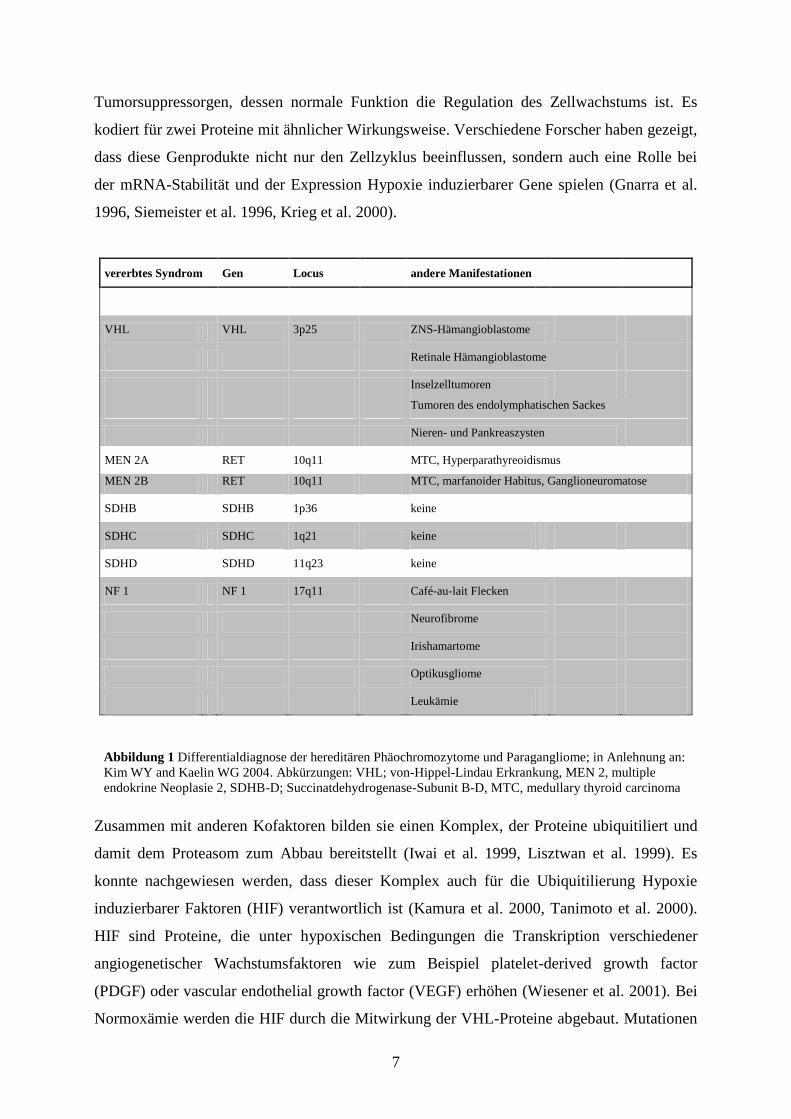
7
Abbildung 1 Differentialdiagnose der hereditären Phäochromozytome und Paragangliome; in Anlehnung an:
Kim WY and Kaelin WG 2004. Abkürzungen: VHL; von-Hippel-Lindau Erkrankung, MEN 2, multiple
endokrine Neoplasie 2, SDHB-D; Succinatdehydrogenase-Subunit B-D, MTC, medullary thyroid carcinoma
Tumorsuppressorgen, dessen normale Funktion die Regulation des Zellwachstums ist. Es
kodiert für zwei Proteine mit ähnlicher Wirkungsweise. Verschiedene Forscher haben gezeigt,
dass diese Genprodukte nicht nur den Zellzyklus beeinflussen, sondern auch eine Rolle bei
der mRNA-Stabilität und der Expression Hypoxie induzierbarer Gene spielen (Gnarra et al.
1996, Siemeister et al. 1996, Krieg et al. 2000).
vererbtes Syndrom Gen Locus andere Manifestationen
VHL VHL 3p25 ZNS-Hämangioblastome
Retinale Hämangioblastome
Inselzelltumoren
Tumoren des endolymphatischen Sackes
Nieren- und Pankreaszysten
MEN 2A RET 10q11 MTC, Hyperparathyreoidismus
MEN 2B RET 10q11 MTC, marfanoider Habitus, Ganglioneuromatose
SDHB SDHB 1p36 keine
SDHC SDHC 1q21 keine
SDHD SDHD 11q23 keine
NF 1 NF 1 17q11 Café-au-lait Flecken
Neurofibrome
Irishamartome
Optikusgliome
Leukämie
Zusammen mit anderen Kofaktoren bilden sie einen Komplex, der Proteine ubiquitiliert und
damit dem Proteasom zum Abbau bereitstellt (Iwai et al. 1999, Lisztwan et al. 1999). Es
konnte nachgewiesen werden, dass dieser Komplex auch für die Ubiquitilierung Hypoxie
induzierbarer Faktoren (HIF) verantwortlich ist (Kamura et al. 2000, Tanimoto et al. 2000).
HIF sind Proteine, die unter hypoxischen Bedingungen die Transkription verschiedener
angiogenetischer Wachstumsfaktoren wie zum Beispiel platelet-derived growth factor
(PDGF) oder vascular endothelial growth factor (VEGF) erhöhen (Wiesener et al. 2001). Bei
Normoxämie werden die HIF durch die Mitwirkung der VHL-Proteine abgebaut. Mutationen

8
im VHL-Gen resultieren jedoch in einem Verlust der Fähigkeit, Proteine zu ubiquitilieren und
damit in einer Hochregulation der HIF (Flamme et al. 1998). Hierdurch entstehen vermehrt
angiogenetisch wirksame Wachstumsfaktoren. Diese Erkenntnisse liefern eine Erklärung für
die Assoziation der VHL mit hochvaskularisierten Tumoren.
Trotz des autosomal-dominanten Erbgangs kommt es erst dann zu unkontrolliertem
Zellwachstum und Tumorentstehung, wenn beide Genkopien des VHL-Gens durch Mutation
oder Verlust von genetischer Information inaktiviert sind. Jedoch verfügen an VHL erkrankte
Patienten zunächst über ein gesundes, vom nicht erkrankten Elternteil geerbtes Allel und ein
mutiertes, vom betroffenen Elternteil geerbtes Allel. Nach der „two-hit“-Hypothese von
Knudson und Strong existiert die Mutation zwar in allen Körperzellen, es entarten jedoch nur
diejenigen Zellen, bei denen das gesunde Allel einer zweiten somatischen Mutation unterliegt
(Knudson et al. 1972, Knudson et al. 1986).
Die VHL kann durch die Abwesenheit (Typ 1) oder Anwesenheit (Typ 2) von
Phäochromozytomen weiter unterteilt werden (Lonser et al. 2003). Ferner wird bei der VHL
Typ 2 an Hand des jeweiligen Risikos für die Entwicklung eines Nierenzellkarzinoms eine
zusätzliche Unterscheidung in Typ 2 A (niedriges Risiko) und Typ 2 B (hohes Risiko)
vorgenommen. Darüber hinaus ist die VHL durch das Auftreten von Hämangioblastomen des
zentralen Nervensystems, Tumoren des endolymphatischen Sackes und der Inselzellen sowie
Pankreaszysten charakterisiert (Eras et al. 2004, Gläsker et al. 2005). Werden in einer an VHL
erkrankten Familie nur Phäochromozytome beobachtet, so wird dies als VHL Typ 2 C
klassifiziert. Insgesamt betrachtet treten Phäochromozytome bei 19 bis 39% der Patienten auf,
dann allerdings oft als Erstmanifestation dieser Krankheit (Neumann et al. 1993, Walther et
al. 1999).
Darüber hinaus stellt die Gruppe der multiplen endokrinen Neoplasien (MEN) eine weitere
hereditäre Erkrankungsform dar, die mit der Entstehung von Phäochromozytomen assoziiert
ist. Sie werden in MEN 1 und MEN 2 unterteilt.
Die MEN 1 ist eine autosomal-dominante Erkrankung, deren Prävalenz auf 1 zu 30.000 bis 1
zu 60.000 geschätzt wird (Katai et al. 1997, Balogh et al. 2007). Chandrasekhareppa et al.
(1997) entdeckten, dass dieses familiäre Tumorsyndrom durch einen Defekt des
Tumorsuppressorgens Menin auf Chromosom 11q13 verursacht wird. Ähnlich dem VHL-Gen
führt erst eine zweite somatische Mutation zum Verlust der Funktion des normalen
Meninallels und somit zum Tumorwachstum. Endokrine Organe, die typischerweise
Neoplasien aufweisen sind Nebenschilddrüsen, Pankreas, Duodenum und Hypophyse
(Schussheim et al. 2001, Tonelli et al. 2005, Lakhani et al. 2007). Der Einfluss des Meningens

9
auf die Entstehung von Nebennierentumoren, Karzinoide und Lipome scheint dagegen eher
gering zu sein, so dass sie bei der MEN 1 eher selten auftreten (Gortz et al. 1999, Heppner et
al. 1999).
Die ebenfalls autosomal-dominant vererbte MEN 2 tritt mit einer Häufigkeit von 1 zu 30.000
auf und wird in die MEN 2 A und MEN 2 B unterteilt (Marini et al. 2006). Das Auftreten von
medullären Schilddrüsenkarzinomen ist ein obligater Bestandteil der MEN 2 A. Ein weiteres
Charakteristikum der Erkrankung ist die Entstehung eines primären Hyperparathyreoidismus
in 15% bis 30% der Fälle (Nguyen et al. 2001, Jimenez et al. 2004, Gujral et al. 2006).
Darüber hinaus werden bei 50% bis 60% der Patienten uni- oder bilaterale
Phäochromozytome diagnostiziert (Gimm et al. 2001, Koch et al. 2001, Peczkowska et al.
2005). Insgesamt stellt die MEN 2 A mit einem Anteil von 70% bis 80% aller MEN die am
häufigsten vorkommende Entität dar (Eng et al. 1996).
Die MEN 2 B ist ebenfalls durch das Auftreten von Phäochromozytomen (50% bis 60% der
Fälle) charakterisiert. Jedoch ist bei den betroffenen Patienten die Kombination einer
Ganglioneuromatose mit medullärem Schilddrüsenkarzinom und marfanoidem Habitus
weitaus häufiger (Lee et al. 2000).
Eine weitere Erscheinungsform der MEN ist das familiäre medulläre Schilddrüsenkarzinom
(FMTC). Bei dieser seltenen Erkrankung sind viele Mitglieder innerhalb einer Familie von
einem medullären Schilddrüsenkarzinom betroffen (Machens et al. 2001). Im Gegensatz zur
MEN 1 und MEN 2 beobachtet man bei jenen Patienten jedoch keine weiteren krankhaften
Vorgänge.
Verschiedene Forschergruppen konnten nachweisen, dass die Ursache der MEN 2 in einer
Mutation des auf Chromosom 10q11.2 liegenden REarranged during Transfection (RET-)
Protooncogens liegt (Mulligan et al. 1993, Huang et al. 2000, Koch et al. 2002, Zhou et al.
2009). Es kodiert für einen membrangebundenen Rezeptor mit Tyrosinkinaseaktivität, der von
den Zellen der ehemaligen Neuralleiste (C-Zellen der Schilddrüse, Nebennierenzellen und
andere) exprimiert wird (Arighi et al. 2005). Verschiedene Liganden können an den Rezeptor
binden und eine komplexe Signalkaskade auslösen, die Einfluss auf die Proliferation,
Migration, Differenzierung und das Überleben der Zellen hat (Koch et al. 2005). Mutationen
auf dem oben genannten Chromosomenabschnitt führen zu einer Aktivitätszunahme des
Rezeptors und damit zur Tumorentstehung (Marx et al. 2005).
Eine weitere hereditäre Erkrankung, die mit der Entstehung von Phäochromozytomen
einhergeht, ist die autosomal-dominant vererbte Neurofibromatose 1 (NF 1). Die Prävalenz
dieser Krankheit liegt bei 1 zu 3.000 bis 1 zu 5.000 (Lammert et al. 2005, Poyhonen et al.

10
2005). Im Jahr 1990 gelang es erstmals, das NF1-Gen auf Chromosom 17q11.2 zu isolieren
(Cawthon et al. 1990, Viskochil et al. 1990, Wallace et al. 1990). Mutationen in diesem
Chromosomenbereich verursachen den Funktionsverlust von Neurofibromin, dem Produkt des
NF1-Gens (John et al. 2000, Serra et al. 2001). Neurofibromin ist ein GTPase aktivierendes
Protein, welches die Zellproliferation durch Beschleunigung der Inaktivierung von
Protooncogenen (z.B. p21ras) hemmt (Buchberg et al. 1990, Xu et al. 1990). Eine durch
Mutationen hervorgerufene Funktionsbeeinträchtigung von Neurofibromin führt zu
beschleunigtem Zellwachstum und Tumorentstehung (Basu et al. 1992, Bollag et al. 1996).
Die extrem vielgestaltige Krankheit ist durch das Auftreten von café-au-lait Flecken,
Neurofibromen, axillären und inguinalen Pigmentveränderungen sowie Lischknötchen
charakterisiert (Yohay 2006, Ferner et al. 2007). Darüber hinaus werden in 1-5% der Fälle
Phäochromozytome beobachtet (Bausch et al. 2006, Erem et al. 2007, Erlic et al. 2009).
In den letzten Jahren ist mit dem so genannten Paragangliomsyndrom (PGL) eine weitere
genetische Ursache für das Auftreten von Phäochromozytomen und familiären
Paragangliomen (extraadrenale Phäochromozytome) entdeckt worden. Durch Genanalysen
identifizierten Forscher 4 verschiedene Loci, die mit der Entstehung dieser Tumoren
assoziiert sind (Astuti et al. 2001, Heutink et al. 1992, Mariman et al. 1995, Niemann et al.
1999). Dementsprechend werden die PGL heute in PGL 1 bis PGL 4 unterteilt.
Im Jahr 2000 wiesen Baysal et al. erstmals nach, dass PGL 1 mit dem Gen der
Succinatdehydrogenase-Subunit D (SDHD) korrespondiert. Weitere Studien folgten, welche
die Übereinstimmung von PGL 3 mit dem SDHC-Gen und PGL 4 mit dem SDHB-Gen
zeigten (Astuti et al. 2001, Baysal et al. 2004). Das verursachende Gen von PGL 2 wurde
bisher noch nicht identifiziert. Insgesamt wurde jedoch nachgewiesen, dass dem gehäuften
Auftreten der genannten Tumoren bei einigen Familien Mutationen des Gens der
Succinatdehydrogenase (SDH) zu Grunde liegen. Die aus den Untereinheiten A bis D
bestehende SDH gehört zum mitochondrialen Komplex 2 der Atmungskette, auch bekannt als
Succinat-Ubichinon-Oxidoreduktase. Als ein wichtiger Bestandteil der Atmungskette stellt sie
die Verbindung zum Zitronensäurezyklus her. In bisher durchgeführten Studien zeigte sich,
dass Mutationen von SDHB, SDHC und SDHD zu einer Beeinträchtigung der Aktivität der
SDH führen (Gimenez-Roqueplo et al. 2001, Gimenez-Roqueplo et al. 2002). Ähnlich wie bei
der VHL-Erkrankung werden hierdurch hypoxiesensible Proteine aktiviert (u. a. HIF, VEGF),
die die Entstehung von stark vaskularisierten Paragangliomen und Phäochromozytomen
fördern (Pollard et al. 2005, Selak et al. 2005). Somit stellen die SDHB, SDHC und SDHD
typische Tumorsuppressorgene dar. Im Gegensatz dazu sind Mutationen der SDHA nicht mit

11
Paragangliomen und Phäochromozytomen assoziiert. Vielmehr werden Veränderungen dieses
Gens beim Leigh-Syndrom - einem kindlichen Enzephalomyopathiesyndrom - beobachtet
(Bourgeron et al. 1995, Birch-Machin et al. 2000).
1.3 Malignität des Phäochromozytoms
Die Prävalenz von malignen Phäochromozytomen wird im Allgemeinen mit circa 10%
angegeben, jedoch ist sie stark davon abhängig, ob eine genetische Grunderkrankung vorliegt
(Mannelli et al. 1999). In mehreren Studien wurde ein signifikant erhöhtes Risiko für das
Auftreten von malignen Phäochromozytomen bei SDHB-Mutationen nachgewiesen (Benn et
al. 2006, Timmers et al. 2007, Solis et al. 2009). Auf der anderen Seite sind die im Rahmen
von MEN 2, VHL, NF 1 und SDHD-Mutationen auftretenden Phäochromozytome zu über
90% benigne (Opocher et al. 2005, Van Houtum et al. 2005, Scholz et al. 2007).
Verschiedene biochemische, morphologische und molekulare Marker wurden untersucht, um
eine Differenzierung zwischen benignem und malignem Verhalten des Tumors vornehmen zu
können (Boltze et al. 2003, Guillemot et al. 2006, Willenberg et al. 2006). Bisher konnte
jedoch durch keinen dieser Parameter ein sicherer Ausschluss von Malignität erfolgen. Die
Bestimmung der Dignität des Phäochromozytoms ist damit weiterhin nur durch den Nachweis
lokaler Invasion oder entfernter Metastasen möglich, die sich bevorzugt in Lymphknoten,
Knochen, Leber und Lunge befinden (Huang et al. 2008). Zurzeit besteht keine Möglichkeit,
ein malignes Phäochromozytom effektiv zu heilen.
1.4 Geschichtlicher Abriss
Im Jahr 1886 beschrieb der deutsche Arzt Felix Fränkel erstmals einen bilateral auftretenden
Nebennierentumor bei einer mit 18 Jahren verstorbenen Frau, die über Palpitationen,
Übelkeit, Kopfschmerzen, Erbrechen und Verwirrtheit geklagt hatte (Fränkel 1886). Erst
kürzlich wurde von Neumann et al. (2007) nachgewiesen, dass diese Frau an einer MEN II
erkrankt war und somit Fränkel der Erstbeschreiber des Phäochromozytoms ist. Bereits 1908
wurde der Begriff Paragangliom als Ausdruck eines extraadrenal in den Ganglien
auftretenden Tumors geprägt (Alezais und Peyron 1908). Der Terminus Phäochromozytom
wurde 1912 von Ludwig Pick eingeführt, nachdem er das charakteristische Färbeverhalten
chromaffiner Zellen entdeckt hatte (Pick 1912). Im Jahr 1926 führten Cesar Roux in der
Schweiz und Charles H. Mayo in den USA die ersten erfolgreichen Operationen an

12
Phäochromozytompatienten durch (Saegesser 1984, Mayo 1924). Sie versuchten den Tumor
durch spezifische klinische Zeichen und explorative Laparotomien zu entdecken. In den
1940er Jahren und Anfang der 1950er Jahre wandten Wissenschaftler erstmals diagnostische
Tests an, die auf Histaminstimulation oder Phentolaminsuppression beruhten (Emlet et al.
1951, Roth und Kvale 1945). Diese Tests zeigten sich dahingehend als nachteilig, als dass sie
zum einen für den Patienten gefährlich waren und zum anderen sowohl falsch-positive als
auch falsch-negative Resultate ergaben. Eine wesentliche Verbesserung in der Diagnostik des
Phäochromozytoms kam im Laufe der 1950er und 1960er Jahre mit der Entwicklung neuer
biochemischer Tests auf. Pisano et al. (1960, 1962) gelang es, Vanillinmandelsäure (VMS)
und Metanephrine im Urin mit Hilfe spektrophotometrischer Messungen nachzuweisen.
Zusätzlich konnten von Euler et al. (1961) erstmals Urinkatecholamine fluorimetrisch
bestimmen. Durch verfeinerte Methoden der Katecholaminmessung und die
computergestützte Bildgebung, die in den 1970er und 1980er Jahren aufkam, gelang es, die
Diagnostik des Phäochromozytoms weiter zu verbessern. Heutzutage werden beim Verdacht
auf das Vorliegen eines Phäochromozytoms die Katecholamine und ihre Metabolite aus dem
Blut oder Urin bestimmt. Dazu wird meistens die HPLC, ein gaschromatographisches
Verfahren oder die Massenspektrometrie verwendet.
1.5 Physiologie des Katecholaminmetabolismus
In den Zellen des Nebennierenmarkes und in adrenergen, postganglionären
Nervenendigungen werden die Katecholamine synthetisiert. Aus Tyrosin entsteht durch
Hydroxylierung und Decarboxylierung Dopamin, welches dann aktiv in die chromaffinen
Granula des Nebennierenmarkes bzw. der postganglionären Neurone aufgenommen und
weiter zu Noradrenalin hydroxyliert wird. Durch Methylierung des Noradrenalins entsteht
Adrenalin. Die Katecholaminsekretion erfolgt exozytotisch, indem die Sekretgranula mit der
Zellmembran verschmelzen und ihren Inhalt nach außen abgeben. Adrenalin wird
hauptsächlich von den Zellen des Nebennierenmarkes in den Blutkreislauf abgegeben und
fungiert als Hormon in den Zielzellen. Dahingegen nimmt Noradrenalin vorwiegend die
Funktion eines Neurotransmitters ein, der von den Axonenden sympathischer
postganglionärer Neurone freigesetzt wird und direkt an den innervierten Zellen wirkt. Jedoch
kann es auch aus dem Nebennierenmark freigesetzt werden und dann hormonelle Funktionen
ausüben.

13
Wie in Abbildung 2 dargestellt, ist der Abbau der Katecholamine sehr komplex und findet in
verschiedenen Geweben des Körpers statt. Ein Weg des Katabolismus, den vor allem
Noradrenalin und in geringem Maß auch Adrenalin durchläuft, wird durch das Enzym
Monoaminooxidase (MAO) und eine Reduktase eingeleitet. Als erstes Zwischenprodukt
entsteht 3,4-Dihydroxyphenylglycol (DHPG). Dieser Abbauschritt findet häufig noch
innerhalb der Zelle statt, in dem die Katecholamine produziert wurden: zum Beispiel in den
sympathischen Nervenendigungen aber auch in den chromaffinen Zellen des
Nebennierenmarks. DHPG wird durch das Enzym Catechol-O-Methyltransferase (COMT)
weiter zu 3-Methoxy-4-Hydroxyphenylglycol (MHPG) verarbeitet, das seinerseits zu VMS
abgebaut wird. Verschiedene Studien haben gezeigt, dass VMS vornehmlich in der Leber
produziert wird (Eisenhofer et al. 1996). Jedoch liegt der Anteil der direkt aus Noradrenalin
und Adrenalin gebildeten VMS nur bei 11%. Dagegen entstehen 87% der aus der Leber
stammenden VMS durch die hepatische Aufnahme und Umwandlung von MHPG und DHPG.
Damit ist nachgewiesen, dass der Katecholaminabbau nicht an einem Ort, sondern in
verschiedenen Organen des Körpers erfolgt.
Eine weitere Möglichkeit des Katecholaminabbaus besteht in der O-Methylierung durch
COMT. Vor allem im Blut zirkulierendes Adrenalin wird über diesen Weg abgebaut, da
Noradrenalin hauptsächlich bereits intraneuronal verstoffwechselt wird. Durch die O-
Abbildung 2 Abbauwege der Katecholamine. In Anlehnung an: Eisenhofer et al. 2003, Abkürzungen: COMT,
Catechol-O-Methyltransferase; MAO, Monoaminooxidase; AR, Aldose Reduktase; ADH,
Alkoholdehydrogenase; DHPGA, 3,4-Dihydroxyphenylglycolaldehyd; MN, Metanephrin; NMN,
Normetanephrin; DHPG, 3,4-Dihydroxyphenylgylcol; MHPG, 3-methoxy-4-hydroxyphenylglycol; VMS,
Vanillinmandelsäure
Adrenalin
MHPG
MN
MHPG
DHPG
DHPGA
VMS
VMS
NMN
VMS
MHPG
Noradrenalin
COMT COMT
COMT
MAO
MAO
ADH
AR
ADH
ADH
MAO

14
Methylierung entsteht aus Adrenalin Metanephrin (MN) und aus Noradrenalin
Normetanephrin (NMN). Das Enzym COMT ist in hohem Maß in der Leber und der Niere
exprimiert, jedoch wird nur ein geringer Teil der zirkulierenden Metanephrine tatsächlich von
ihnen produziert. Über 80% des im Blut vorhandenen NMN und über 90% des MN sind
bereits in den Herkunftsorganen o-methyliert worden. So kommen 91% des zirkulierenden
Plasma-MN und 23% des Plasma-NMN aus der Nebenniere selbst, hingegen nur 6% des
zirkulierenden Noradrenalins beziehungsweise 19% des Adrenalins (Eisenhofer et al. 1995).
Dadurch stellt die Nebenniere die größte natürliche Quelle der Metanephrine im menschlichen
Körper dar. Dies erklärt auch die immense Bedeutung der Metanephrine in der Diagnostik des
Phäochromozytoms. Die entstandenen Zwischenprodukte MN und NMN können entweder
direkt über die Niere ausgeschieden werden oder weiter zu VMS prozessiert werden. VMS ist
somit das Endprodukt beider Katecholaminabbauwege.
Zusätzlich besteht für den Körper mit Hilfe des vor allem im Dünndarm lokalisierten Enzyms
Sulfotransferase 1A3 die Möglichkeit, MHPG, MN, NMN und VMS zu sulfatieren und dann
über die Niere auszuscheiden (Rubin et al. 1996). Dieser Schritt reduziert jedoch die
Geschwindigkeit der Elimination und verlängert die Plasmahalbwertzeiten der einzelnen
Substanzen, so dass ihre Plasmakonzentrationen erheblich ansteigen (Goldstein et al. 2003).
Folglich liegt das Niveau der sulfatierten Metabolite 20fach bis 40fach über dem der freien
Metabolite.
1.6 Katecholaminmetabolismus beim Phäochromozytom
Das Phäochromozytom ist in der Lage, die in gesteigerter Menge produzierten Katecholamine
Noradrenalin und Adrenalin durch die Expression von COMT abzubauen und in MN bzw.
NMN umzuwandeln. Dadurch steigt zum einen die Konzentration von freien Metanephrinen
im Tumor selbst auf das 13.000fache bis 23.000fache des Plasmawertes an (Eisenhofer et al.
1998). Zum anderen wurde auch nachgewiesen, dass bei Phäochromozytompatienten über
94% der erhöhten Plasmametanephrine aus dem Phäochromozytom selbst stammen. Ursache
hierfür ist ein passiver Ausstrom von Katecholaminen aus intrazellulären Speichervesikeln
des Zytoplasmas mit folgendem aktivem Rücktransport oder Abbau zu freien Metanephrinen
und konsekutiver Abgabe in den Blutkreislauf. Dieser Prozess läuft kontinuierlich und
unabhängig von der Katecholaminsekretion des Tumors statt. In der Folge sind Erhöhungen
der freien Metanephrine im Plasma beständiger als die der Katecholamine bei Patienten mit
Phäochromozytom (Eisenhofer et al. 2000). Dadurch ist es möglich, ein Phäochromozytom

15
bei Patienten zu diagnostizieren, die nur einen geringen oder gar keinen Anstieg der Urin-
oder Plasmakatecholaminwerte haben.
Das Phäochromozytom kann neben der COMT auch MAO besitzen und folglich DHPG oder
MHPG sezernieren. Da diese beiden Metabolite jedoch schon beim physiologischen Abbau
der Katecholamine in hohen Mengen anfallen, stellen sie keine sensitiven Marker dar und
sind zur Diagnostik des Phäochromozytoms nicht geeignet. Darüber hinaus erklärt dies auch
den diagnostisch sehr eingeschränkten Nutzen von VMS, da dieses hautsächlich aus
zirkulierendem DHPG und MHPG entsteht.
1.7 Labordiagnostik des Phäochromozytoms
Durch das genaue Wissen sowohl um den physiologischen Katecholaminmetabolismus als
auch den Katecholaminabbau beim Phäochromozytom in Kombination mit verfeinerten
Nachweisverfahren, steht heutzutage ein breites Spektrum an biochemischer Diagnostik zur
Verfügung:
a) Katecholamine im 24-h-Urin:
Die Bestimmung der Urinkatecholamine kann bei der Differenzierung zwischen einem
Tumor oder einer angestiegenen Katecholaminfreisetzung bedingt durch nervale
Prozesse (z. B. Sport, Stress) hilfreich sein. Allerdings ist es durch eine Vielzahl an
physiologischen, pharmakologischen und pathologischen (z.B. Herzinsuffizienz,
Schock, Sepsis) Prozessen, die den Katecholaminspiegel beeinflussen, oftmals
schwierig, den definitiven Ursprung eines Katecholaminanstieges zu ermitteln (Bravo
2004). Darüber hinaus sezernieren Phäochromozytome Katecholamine periodisch, so
dass ein Bestimmen der Werte in einer stummen Phase erfolgen kann und ein falsch-
negatives Ergebnis nicht auszuschießen ist.
b) Bestimmung der totalen Urinmetanephrine im 24-h-Sammelurin:
Bei den totalen Urinmetanephrinen handelt es sich um die Summe aus freien und
Sulfat konjugierten Metanephrinen. Sie sind verhältnismäßig einfach zu bestimmen,
da ihre Konzentration bedingt durch eine langsame Elimination circa 20fach bis
30fach höher ist als die der freien Metanephrine. Der guten Spezifität der
Urinmetanephrine steht eine relativ geringe Sensitivität gegenüber. Ursache hierfür ist

16
die Tatsache, dass die Urinmetanephrine vor ihrer Bestimmung einen
Dekonjugationsschritt durchlaufen, bei dem aus Sulfat gebundenen Metanephrinen
freie Metanephrine gewonnen werden. Die Sulfat gebundenen Metanephrine werden
jedoch in vielen anderen Geweben, vor allem im Gastrointestinaltrakt gebildet
(Eisenhofer 2003c). Dadurch stellen die totalen Urinmetanephrine ein Gemisch von
Katecholaminmetaboliten dar, die aus unterschiedlichen Regionen des Körpers
stammen. Ein geringer Anstieg der durch ein Phäochromozytom gebildeten
Metanephrine ist hierdurch schwerer zu entdecken.
c) Einsatz von VMS im 24-h-Sammelurin:
Die Urin-VMS wird hauptsächlich aus DHPG und MHPG in der Leber gebildet. Diese
Metabolite entstammen vor allem dem Katecholaminabbau aus Nervenendigungen
sympathischer Neurone. Durch die physiologisch hohe Produktion von DHPG und
MHPG erklärt sich die relativ geringe Sensitivität der Urin-VMS (Widimsky 2006).
Ein Vorteil der Untersuchung mit VMS liegt jedoch in der hohen Spezifität des
Markers (Lenders et al. 2002a).
d) Serum-Chromogranin A:
Chromogranin A ist ein säurelösliches Protein, das ebenso wie andere Chromogranine
eine wichtige Funktion bei der Formation sekretorischer Granula übernimmt
(Taupenot et al. 2003). Es scheint vor allem in neuroendokrinen Zellen, unter anderem
auch in denen des Nebennierenmarks und sympathischer Nervenendigungen, bei der
Freisetzung von Hormonen und Katecholaminen von Bedeutung zu sein (Gorr et al.
1989, Bargsten et al. 1992, Watkinson et al. 1992). Durch die vielseitigen Funktionen
und das häufige Vorkommen des Chromogranin A in verschiedenen Geweben, ist es
ein relativ unspezifischer Marker für viele neuroendokrine Tumore (Nobels et al.
1997). Die Spezifität wird bei der Diagnostik des Phäochromozytoms zwischen 74%
und 99% angegeben. Ebenso liegt die Sensitivität des Chromogranin A bei 76% bis
95% (Canale et al. 1994, d´Herbomez et al. 2001, Cotesta et al. 2005, Giovanella et al.
2006). Ein Vorteil der Bestimmung des Chromogranin A liegt in der Tatsache, dass
der Plasmawert mit der Tumormasse des Phäochromozytoms korreliert und somit als
Verlaufsparameter genutzt werden kann (Grossrubatscher et al. 2006, Bilek et al.

17
2008). Ebenso beeinflussen zur Therapie des Phäochromozytoms genutzte
Medikamente wie Clonidin, Phentolamin oder Metoprolol den Wert des
Chromogranin A kaum. Deswegen wird von einigen Autoren empfohlen,
Chromogranin A als additiven Test vor allem bei der endokrinologischen
Differenzierung adrenaler Inzidentalome zu verwenden (Miehle et al. 2005).
Andererseits steigen die Plasmawerte des Chromogranin A bei Patienten mit
Niereninsuffizienz an, da es hauptsächlich renal eliminiert wird (Bernini et al. 2004).
e) Plasmakatecholamine:
Die Bestimmung der Plasmakatecholamine wird heutzutage nur noch selten
durchgeführt, da die Plasmakonzentration der Katecholamine vielen externen
Einflüssen (z.B. Position des Patienten bei der Blutentnahme, Stress) unterliegt.
Darüber hinaus wird die Bestimmung durch eine geringe Plasmahalbwertszeit
erschwert. Des Weiteren sezernieren manche Phäochromozytome nur geringe
Katecholaminmengen oder geben diese nur periodisch ab, so dass sie biochemisch
unauffällig bleiben. Dadurch wird die Sensitivität zwischen 70% und 84% angegeben.
Die Spezifität liegt bei 69% bis 81% (Lenders et al. 2002, Unger et al. 2006).
f) Freie Plasmametanephrine:
Obwohl die Metanephrine schon seit vielen Jahren als Abbauprodukte im
Katecholaminstoffwechsel bekannt sind, ist es erst Mitte der 1990er Jahre gelungen, einen
verlässlichen Test zu entwickeln, der die freien Plasmametanephrine (das ist das
unkonjugierte Nor-, Metanephrin) nachweist. Seitdem wurde in verschiedenen Studien die
hohe Sensitivität und Spezifität der Plasmametanephrine mit Werten zwischen 96% und 100%
bzw. 79% und 89% belegt (Lenders et al. 2002, Hickman et al. 2009). Ein Grund hiefür liegt
in dem speziellen Katecholaminmetabolismus des Phäochromozytoms (siehe oben). Wie
Abbildung 3 illustriert, existiert die Möglichkeit, die Metanephrine als totale Metanephrine
(Summe von NMN und MN) zu messen. Darüber hinaus werden auch die fraktionierten
Metanephrine (getrennte Bestimmung von MN und NMN) analysiert. Des Weiteren können
Plasmametanephrine in der freien oder in der sulfatierten Form gemessen werden. Die
sulfatkonjugierten Metanephrine liegen im Plasma in 20-30fach höherer Konzentration vor,

18
weil sie langsam und ausschließlich renal eliminiert werden. Dadurch steigt ihre
Konzentration im Plasma bei renaler Funktionsbeeinträchtigung sehr schnell an.
Die gebräuchlichste Methode zur Diagnose eines Phäochromozytoms ist derzeit die
Bestimmung der Katecholamine und ihrer Abbauprodukte- den Metanephrinen und VMS- im
24-Stunden-Urin. In den meisten Fällen ist mit Hilfe dieser Methoden eine korrekte Diagnose
möglich. So liegt die Sensitivität für Urinkatecholamine zwischen 74% und 90%, für totale
Urinmetanephrine zwischen 65% und 77%, für VMS im Urin zwischen 47% und 64%. Die
Spezifität wird für Urinkatecholamine zwischen 88% und 99%, für totale Urinmetanephrine
zwischen 93% und 99% und für Vanillinmandelsäure im Urin zwischen 95% und 100%
angegeben (Eisenhofer et al. 1999, Raber et al. 2000, Lenders et al. 2002a, Kudva et al. 2003).
Abbildung 3 Möglichkeiten der Katecholamin- und Metanephrinbestimmung. In Anlehnung an
Eisenhofer et al. 2004. Abkürzungen: COMT, Catechol-O-Methyltransferase; SULT1A3,
Sulfotransferase Isoenzym Typ 1A3
COMT
Adrenalin
sulfatkonju-
gierte Meta-
nephrine
totale Meta-
nephrine
freie Meta-
nephrine
freie Katechol-
amine
SULT1A3
Normetanephrin
Metanephrin
Metanephrin-
sulfat
Normetanephrin-
sulfat
Noradrenalin

19
2 Zielstellung
Die biochemische Diagnostik des Phäochromozytoms stützte sich bis Anfang der 1980er
Jahre auf die Messung verschiedener Parameter im menschlichen Urin. Es wurden vor allem
fluorimetrische, colorimetrische oder gaschromatographische Methoden verwandt. Mit der
Einführung der HPLC wurde es möglich, diese Metabolite auch im Blut zu bestimmen. Das
Ziel dieser Studie war es, die sensitive und zugleich spezifische Messung von freien
Metanephrinen im Blut mit einem RIA (Firma Labor Diagnostika Nord GmbH & Co. KG,
Nordhorn, Deutschland) nachzuweisen und dieses Verfahren gleichzeitig mit bisherigen
Methoden zu vergleichen.
Die Fragestellungen an diese Studie lassen sich stichpunktartig wie folgt zusammenfassen:
1. Ist die Anwendung des RIA zur Bestimmung der freien Metanephrine im Plasma
sensitiv und spezifisch, um in der Routinediagnostik des Phäochromozytoms
eingesetzt werden zu können?
2. Welche Vor- und Nachteile hat der RIA im Vergleich zu bisher angewandten
Methoden?
3. Ist eine Beeinflussung der Plasmakonzentration der freien Metanephrine mittels RIA
durch Grunderkrankungen der Patienten wie arterielle Hypertonie, Niereninsuffizienz
oder Diabetes mellitus möglich?
4. Eignet sich der RIA zum postoperativen Follow-up bei Patienten mit
Phäochromozytom?
5. Gibt es eine Korrelation der Plasmametanephrinwerte mit dem Lebensalter bei
Patienten mit klinisch nicht nachgewiesenem Phäochromozytom?
20
3 Patientencharakteristika und Methoden
3.1 Patientencharakteristika
Zwischen den Jahren 2000 bis 2006 untersuchten wir 163 Patienten, die sich in ambulanter
oder stationärer Behandlung befanden, mit Hilfe eines Radioimmunoassays auf das
Vorhandensein eines Phäochromozytoms. Das mediane Alter lag bei 60 Jahren (Bereich 16-
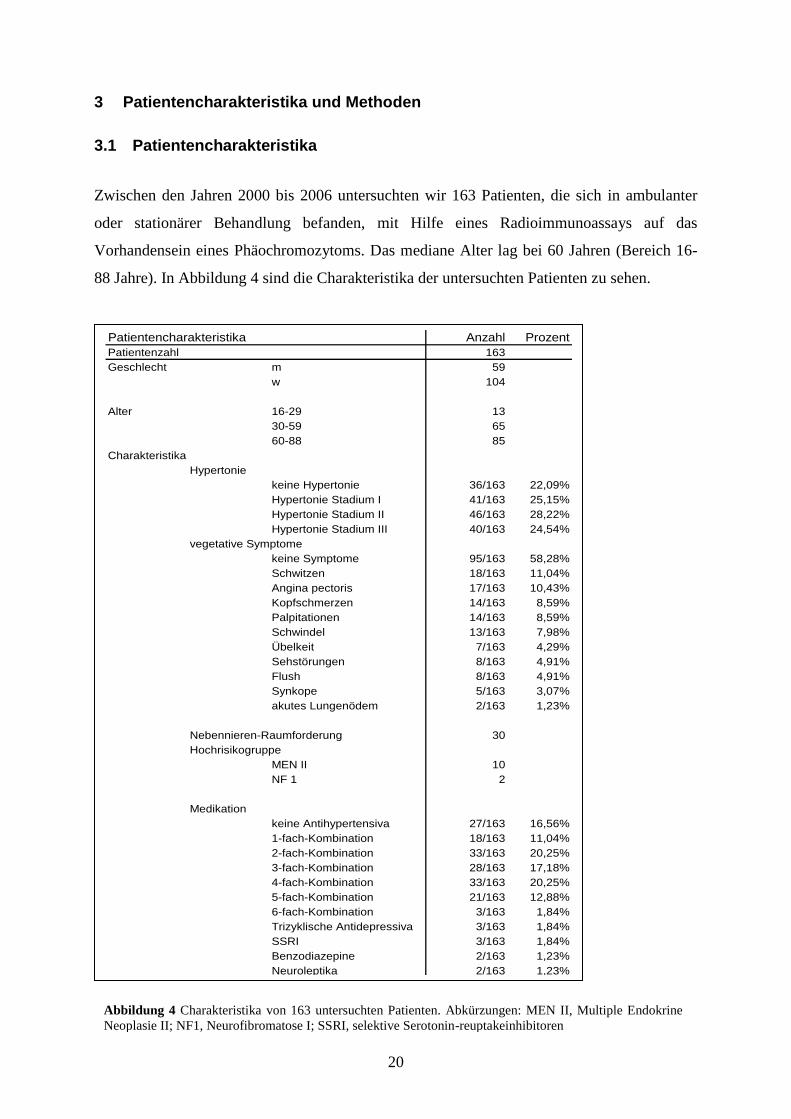
88 Jahre). In Abbildung 4 sind die Charakteristika der untersuchten Patienten zu sehen.
Abbildung 4 Charakteristika von 163 untersuchten Patienten. Abkürzungen: MEN II, Multiple Endokrine
Neoplasie II; NF1, Neurofibromatose I; SSRI, selektive Serotonin-reuptakeinhibitoren
Patientencharakteristika Anzahl Prozent
Patientenzahl 163
Geschlecht m 59
w 104
Alter 16-29 13
30-59 65
60-88 85
Charakteristika
Hypertonie
keine Hypertonie 36/163 22,09%
Hypertonie Stadium I 41/163 25,15%
Hypertonie Stadium II 46/163 28,22%
Hypertonie Stadium III 40/163 24,54%
vegetative Symptome
keine Symptome 95/163 58,28%
Schwitzen 18/163 11,04%
Angina pectoris 17/163 10,43%
Kopfschmerzen 14/163 8,59%
Palpitationen 14/163 8,59%
Schwindel 13/163 7,98%
Übelkeit 7/163 4,29%
Sehstörungen 8/163 4,91%
Flush 8/163 4,91%
Synkope 5/163 3,07%
akutes Lungenödem 2/163 1,23%
Nebennieren-Raumforderung 30
Hochrisikogruppe
MEN II 10
NF 1 2
Medikation
keine Antihypertensiva 27/163 16,56%
1-fach-Kombination 18/163 11,04%
2-fach-Kombination 33/163 20,25%
3-fach-Kombination 28/163 17,18%
4-fach-Kombination 33/163 20,25%
5-fach-Kombination 21/163 12,88%
6-fach-Kombination 3/163 1,84%
Trizyklische Antidepressiva 3/163 1,84%
SSRI 3/163 1,84%
Benzodiazepine 2/163 1,23%
Neuroleptika 2/163 1,23%

21
Die initialen Plasma- und Urinmessungen wurden bei 12 Patienten im Rahmen eines
Screenings auf ein Phäochromozytom durchgeführt, da bei ihnen eine familiäre Anamnese
von MEN II beziehungsweise NF 1 bekannt war oder sie bereits an einem Phäochromozytom
oder Paragangliom operiert worden waren. Darüber hinaus erfolgte die biochemische Analyse
auf Grund eines Hypertonus (refraktär, paroxysmal, neu aufgetreten, Blutdruckspitzen) oder
vegetativen Symptomen. Am häufigsten traten hierbei Schweißausbrüche, Angina pectoris,
Kopfschmerzen und Palpitationen auf. Zusätzlich waren Schwindel, Synkopen, Übelkeit,
Sehstörungen, Flush und ein akutes Lungenödem zu beobachten. Die klassische Trias
bestehend aus Palpitationen, Kopfschmerz und Schwitzen gaben lediglich 4 Patienten an. Die
meisten der Patienten mit Bluthochdruck in unserer Untersuchung erhielten eine
antihypertensive Therapie mit 1 bis 3 Medikamenten (48,47%). Allerdings war bei 34,97%
der Patienten eine 4- bis 6-fach-Kombination nötig, um eine optimale Einstellung des
Hypertonus zu erzielen.
a) Patienten mit Phäochromozytom
Insgesamt wurde in diesen Jahren bei 8 Patienten ein Phäochromozytom diagnostiziert. Die
initiale laborchemische und bildgebende Diagnostik erfolgte bei 7 der 8 Patienten im
Universitätsklinikum Leipzig. Auf Grund der niedrigen Fallzahl wurde eine
Phäochromozytompatientin aus dem Spital Bern Tiefenau/Schweiz mit in die Analyse
eingeschlossen. Mit Ausnahme der Auswertung der Plasmametanephrine mit Hilfe des RIA
wurde die gesamte Diagnostik und Therapie dieser Patientin in der Schweiz durchgeführt.
Von den 8 diagnostizierten Phäochromozytomen waren 7 benigne und 1 maligne. Bei 2
Patienten traten die Tumoren im Rahmen eines familiären Syndroms auf (MEN 2 A,
Neurofibromatose Typ I). Die Patientin mit bekannter Neurofibromatose Typ I und Zustand
nach Adrenalektomie bei Phäochromozytom erlitt ein Rezidiv des Tumors mit Metastasen im
Skelettsystem und in der Lunge im Sinne eines malignen Phäochromozytoms (Koch et al.
2006). Bei der Patientin mit MEN 2 A wurde ebenfalls ein Rezidiv nach vorheriger
beidseitiger Adrenalektomie vor 10 Jahren auf Grund eines Phäochromozytoms
diagnostiziert. Die Primärtumoren waren bei allen 8 Patienten adrenal lokalisiert. Alle
Katecholamin produzierenden Tumoren wurden histologisch untersucht und bestätigt.

22
3.2 Methoden
3.2.1 Radioimmunoassay für Plasmametanephrine
Die RIA KITS wurden von Labor Diagnostika Nord GmbH & Co. KG (Nordhorn,
Deutschland) hergestellt.
Blutproben der Patienten
Es wurden Blutproben von den Patienten in Lithium-Heparin-Röhrchen entnommen. Die
Blutentnahme erfolgte ohne die Verwendung einer Venenverweilkanüle. Auf das Einhalten
einer nächtlichen Diät oder das Absetzen von Medikamenten wurde verzichtet. Das Plasma
konnte durch Zentrifugation (3,000 x g) separiert und bei -20°C bis zur Verwertung
aufbewahrt werden.
Radioimmunoassay für freie Metanephrine und Normetanephrine
Die Proteinmatrix der Plasmaproben wurde durch spezielle Präzipitationsreagenzien entfernt
und die Metanephrine aus dem Überstand entnommen. Da diese keine suffizienten
immunogenen Strukturen aufweisen, die es erlauben Antikörper dagegen herzustellen, wurden
sie zu ihren N-acetyl-derivaten acetyliert.
Der Assay folgt den grundlegenden Prinzipien eines Radioimmunoassays, basierend auf der
Konkurrenz von radioaktiven und nichtradioaktiven Antigenen um eine fixe Anzahl von
Antikörperbindungsstellen. Die Menge an 125-I-markiertem Antigen, gebunden an die
Antikörper, ist umgekehrt proportional zur Analysekonzentration der Plasmaprobe. Wenn das
System im Äquilibrium ist, wird die antigengebundene Radioaktivität separiert, indem man
einen zweiten Antikörper und Polyethylenglykol (PEG) benutzt. Die Messung des nach der
Zentrifugation gewonnenen Präzipitates erfolgt im Gammazähler. Eine Quantifikation
unbekannter Proben wird erreicht, indem man deren Aktivität mit einer Referenzkurve
vergleicht, die durch Messen von Standards mit bekannten Konzentrationen erhalten wurde.
Die in diesem Test verwendeten Antiseren erkennen nur die biologisch relevanten L-Formen
der Metanephrine.

23
Testablauf
a) Vorbereitung der Reagenzien
Die Standards und Kontrollen wurden mit jeweils 1,25 ml destilliertem Wasser verdünnt. 80
µl konzentrierter Acetylierungsreagenz wurden mit 3 ml destilliertem Wasser verdünnt.
b) Präzipitation von Standards, Plasmaproben und Kontrollen
500 µl von Standards, Plasmaproben und Kontrollen wurden in die Präzipitationsröhrchen
gefüllt. Hinzugefügt wurden 50 µl Präzipitator 1 und 50 µl Präzipitator 2. Nach vorsichtigem
Mixen (Vortex) erfolgten eine 60-minütige Inkubation und anschließend die Zentrifugation
bei 3000 x g. Vom Überstand wurde 100 µl für die Bestimmung der MN und 25 µl für die
Bestimmung der NMN entnommen.
c) Radioimmunoassay
100 µl destilliertes Wasser wurden zur Bestimmung der nichtspezifischen Bindungen (NSB)
in jedes Röhrchen pipettiert. Danach wurden 100 µl der Überstände von Standards,
Plasmaproben und Kontrollen in die entsprechenden Röhrchen gefügt. Anschließend erfolgte
die Zugabe von 25 µl Acetylierungspuffer in alle Röhrchen. Danach wurden 25 µl verdünnte
Acetylierungsreagenz in jedes Röhrchen pipettiert. Hierauf wurde sorgfältig gemixt und für
15 Minuten bei Zimmertemperatur inkubiert. Nun wurden 50 µl des 125-I-MN- bzw. 125-I-
NMN-Tracers in alle Röhrchen pipettiert. Hieran schloss sich die Zugabe von 50 µl des MN-
bzw. NMN-Antiserums, das Mixen und einminütige Zentrifugieren bei 500 x g. Dies diente
dem Entfernen aller sich an der Innenseite der Röhrchen befindlichen Tropfen. Nach einer
nächtlichen Inkubationszeit von 15 bis 20 Stunden bei 2 bis 8°C folgten das vorsichtige
Mixen des Präzipitationsreagenz und das Pipettieren von jeweils 1ml in alle präzipierten
Reagenzien. Daraufhin wurde erneut mit dem Schüttler gemixt und 15 Minuten bei 2 bis 8°C
inkubiert. Anschließend erfolgte die Zentrifugation mit 3000 x g über 15 Minuten. Der
Überstand wurde vorsichtig abgegossen oder aspiriert. Die Flüssigkeitsreste wurden
ausgeschlagen, wozu man die Röhrchen 2 Minuten auf dem Kopf stehen ließ. Den Abschluss
des RIA bildete das Zählen der Röhrchen mit dem Gammazähler.

24
Kalkulation der Resultate
Die Kalkulation der Resultate erfolgte mit dem Computerprogramm LBIS 501 von Berthold.
Der Counts-per-minute (CPM)-Mittelwert der nichtspezifischen Bindung wurde vom CPM-
Mittelwert der Nullreferenz (= Standard A), Standard B bis F, Kontrolle 1 und 2 sowie den
Patientenproben subtrahiert. Anschließend wurde eine Standardkurve konstruiert, indem der
prozentuale CPM-Wert jeder Standardprobe im Verhältnis zum CPM-Wert der Nullreferenz
(y-Achse, linear) gegen die entsprechende Konzentration (x-Achse, logarithmisch)
aufgetragen wurde [(B-NSB)/(B0-NSB) in %]. Aus diesen Standardkurven konnte daraufhin
die Metanephrin- und Normetanephrinkonzentration der Kontrollen und Patientenproben
abgelesen werden. Für die Messung wurde der Gamma-Counter LB 2111 der Firma Berthold
verwendet.
Reproduzierbarkeit
Die Reproduzierbarkeit des RIA wurde durch Bestimmung des Inter- und Intraassay-
Variationskoeffizienten ermittelt. Durch wiederholte Messungen zweier Proben mit
unterschiedlichen Konzentrationen von Metanephrin und Normetanephrin konnten
Variationskoeffizienten von 15% bis 4,2% für Metanephrin bzw. 16,9% bis 3,6% für
Normetanephrin ermittelt werden.
Qualitätskontrolle
In jedem Assay inbegriffen waren 2 Kontrollproben, die von LDN GmbH & Co. KG
(Nordhorn, Deutschland) gestellt wurden.
3.2.2 Ermittlung eines Grenzwertes für Plasmametanephrine mittels Radioimmunoassay (RIA) der Firma LDN GmbH & Co. KG
Auf Grund dessen, dass uns in der Anfangsphase unserer Arbeit keine validierten
Referenzwerte für den RIA zur Verfügung standen, orientierten wir uns an den von Unger et
al. (2004) in einer klinischen Studie ermittelten Grenzwerten für Normetanephrin und
Metanephrin. In dieser Studie wurden 119 Patienten untersucht, von denen 17 ein histologisch
nachgewiesenes Phäochromozytom aufwiesen. Es wurden Grenzwerte von 123 pg/ml für
NMN und 96 pg/ml für MN verwendet. Die Sensitivität und Spezifität gaben Unger et al. mit

25
58,8% und 97,1% für Plasma-MN bzw. 94,1% und 97,1% für Plasma-NMN an. Für die
Validierung eigener Grenzwerte, untersuchten wir von 163 Patienten die ersten 50, bei denen
ein Phäochromozytom durch bildgebende Verfahren nahezu ausgeschlossen wurde (wenn
man von solchen Patienten absieht, die Paragangliome/extraadrenale Phäochromozytome
haben, welche (noch) keine erhöhte Katecholaminsekretion zeigen). Zur bildgebenden
Diagnostik wurden CT, MRT, Sonographie und Metajodobenzylguanidin-Szintigraphie
(MIBG) -Szintigraphie verwandt. Die klinischen Charakteristika der Patienten sind in
Abbildung 5 illustriert.
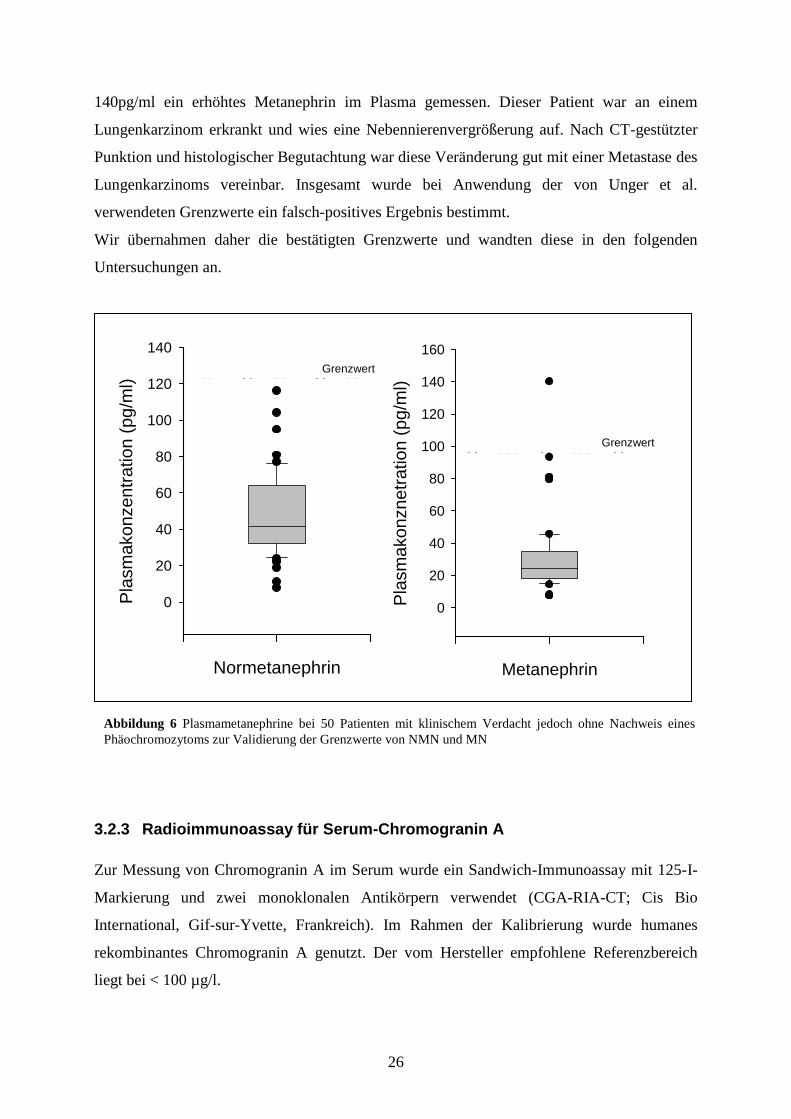
Die NMN-Werte lagen bei allen 50 Patienten unter dem von Unger et al. verwendeten
Grenzwert (s. Abb. 6). Der Median der Plasma-NMN-Werte wurde mit 40,70 pg/ml
gemessen, der Wert des 3. Quartils ergab 64,10 pg/ml. Die MN-Werte waren bei 49 der 50
Patienten unterhalb des Grenzwertes von Unger et al. Der Median der MN wurde mit 23,9
pg/ml gemessen, der Wert des 3. Quartils ergab 34,60 pg/ml. Bei einem Patienten wurde mit
Patientencharakteristika
Anzahl
Geschlecht m 19
w 31
Alter 16-29 7
30-59 21
60-99 22
Blutdruck Normotonie 6
Hypertonie Stadium I 13
Hypertonie Stadium II 18
Hypertonie Stadium III 13
Medikation keine Antihypertensiva 9
1-fach-Kombination 5
2-fach-Kombination 6
3-fach-Kombination 10
4-fach-Kombination 11
5-fach-Kombination 8
6-fach-Kombination 1
Nierenfunktion unauffällig 44
Niereninsuffizienz St. I 3
Niereninsuffizienz St. II 1
Niereninsuffizienz St. III 2
Niereninsuffizienz St. IV 0
Abbildung 5 Charakteristika der Patienten zur Validierung der Grenzwerte
26
140pg/ml ein erhöhtes Metanephrin im Plasma gemessen. Dieser Patient war an einem
Lungenkarzinom erkrankt und wies eine Nebennierenvergrößerung auf. Nach CT-gestützter
Punktion und histologischer Begutachtung war diese Veränderung gut mit einer Metastase des
Lungenkarzinoms vereinbar. Insgesamt wurde bei Anwendung der von Unger et al.
verwendeten Grenzwerte ein falsch-positives Ergebnis bestimmt.
Wir übernahmen daher die bestätigten Grenzwerte und wandten diese in den folgenden
Untersuchungen an.
3.2.3 Radioimmunoassay für Serum-Chromogranin A
Zur Messung von Chromogranin A im Serum wurde ein Sandwich-Immunoassay mit 125-I-
Markierung und zwei monoklonalen Antikörpern verwendet (CGA-RIA-CT; Cis Bio
International, Gif-sur-Yvette, Frankreich). Im Rahmen der Kalibrierung wurde humanes
rekombinantes Chromogranin A genutzt. Der vom Hersteller empfohlene Referenzbereich
liegt bei < 100 µg/l.
Abbildung 6 Plasmametanephrine bei 50 Patienten mit klinischem Verdacht jedoch ohne Nachweis eines
Phäochromozytoms zur Validierung der Grenzwerte von NMN und MN
Normetanephrin
Pla
sm
ako
nze
ntr
atio
n (
pg
/ml)
0
20
40
60
80
100
120
140
Metanephrin
Pla
sm
ako
nzn
etr
atio
n (
pg
/ml)
0
20
40
60
80
100
120
140
160
Grenzwert
Grenzwert

27
3.2.4 HPLC für Plasma-, Urinmetanephrine und Urinkatecholamine
Wir ließen zusätzlich zu dem RIA bei einigen Patienten eine Vergleichsuntersuchung der
Plasmametanephrine mittels HPLC als Goldstandard der Plasmametanephrinmessung zur
weiteren Validierung unserer Ergebnisse durchführen. Die HPLC stellt ein Trennverfahren
dar, bei dem Probengemische untersucht werden, indem man sie in ihre einzelnen
Bestandteile auftrennt. Basis der Auftrennung der Proben ist die unterschiedliche Verteilung
der Probenbestandteile in zwei Phasen (flüssige und stationäre Phase). Das zu analysierende
Gemisch wird mittels der flüssigen Phase unter hohem Druck über die stationäre Phase
transportiert, wobei die Auftrennung durch unterschiedlich starke Wechselwirkungen mit der
stationären Phase erreicht wird. Die Analyse der einzelnen Bestandteile erfolgt nach dem
Austritt aus der stationären Phase mittels eines geeigneten Detektors. Die Bestimmung der
Plasmametanephrine wurde freundlicherweise von Prof. Lenders/Nijmwegen durchgeführt
und erfolgte nach dem bereits von ihm beschriebenen Verfahren (Lenders et al. 1993). Die
Grenzwerte betrugen 600 pmol/l für NMN und 300 pmol/l für MN.
Die Bestimmung der Urinmetanephrine und –metanephrine erfolgte ebenfalls mittels HPLC
der Firma Biorad. Die Testkits wurden von Chromsystems Instruments & Chemicals GmbH
(München) hergestellt.
3.3 Diagnostischer Ablauf
Nach der Validierung eines eigenen Grenzwertes für Plasmanormetanephrin und –
metanephrin mittels RIA bei 50 konsekutiven Patienten mit durch bildgebende Verfahren
nahezu ausgeschlossenem Phäochromozytom, bestimmten wir bei 113 weiteren Patienten die
Metanephrine im Plasma. Ein falsch-positives Ergebnis eines Plasmametanephrintests von
Patienten ohne Phäochromozytom wurde definiert durch Werte von Metanephrin oder
Normetanephrin, die dem Grenzwert entsprachen oder über ihm lagen. Ein falsch-negatives
Testergebnis eines Plasmametanephrintests von Patienten mit Phäochromozytom wurde
definiert durch Werte von Metanephrin und Normetanephrin, die unter dem entsprechenden
Grenzwert lagen.
Wir führten eine weiterführende bildgebende Diagnostik bei denjenigen Patienten durch, bei
denen erhöhte Plasmametanephrinwerte festgestellt wurden (s. Abb. 7). Bei
altersentsprechenden Metanephrinen im Plasma wurden keine zusätzlichen Untersuchungen
eingeleitet.

28
Zur Validierung und Sicherung der Ergebnisse, die durch die Bestimmung der Metanephrine
im Plasma erzielt wurden, untersuchten wir die Patienten zusätzlich auf erhöhte Werte von
Chromogranin A im Serum bzw. von Katecholaminen und Metanephrinen im 24-h-
Sammelurin. Des Weiteren untersuchten wir die Stabilität der Plasmametanephrine, indem
wir zum einen Mehrfachbestimmungen dieser Parameter durchführten und zum anderen der
Frage nachgingen, inwieweit eine Altersabhängigkeit der Werte vorliegt
Abbildung 7 Diagnostisches Prozedere bei 163 Patienten mit Verdacht auf bzw. zum Ausschluss eines
Phäochromozytoms
3.4 Statistische Verfahren
Bei der Auswertung der Daten wurde eine deskriptive Statistik angewandt. Als Kenngrößen
dienten Lagemaße (Median, Quantile) und Streuungsmaße (Variationsbreite/Spannweite).
Eine Ausnahme ist die Untersuchung der Metanephrine in Abhängigkeit vom Lebensalter der
untersuchten Personen, da hier eine Regressionsanalyse nach Spearman durchgeführt wurde.
163 Patienten
50 Patienten zur Validierung eines Referenzwertes
113 Patienten zum Ausschluss eines Phäochromozytoms
erhöhtes MN / NMN unauffälliges MN / NMN
CT / MRT
negativ
positiv
Szintigraphie keine weitere Diagnostik
positiv negativ
OP / Histologie
29
4 Ergebnisse
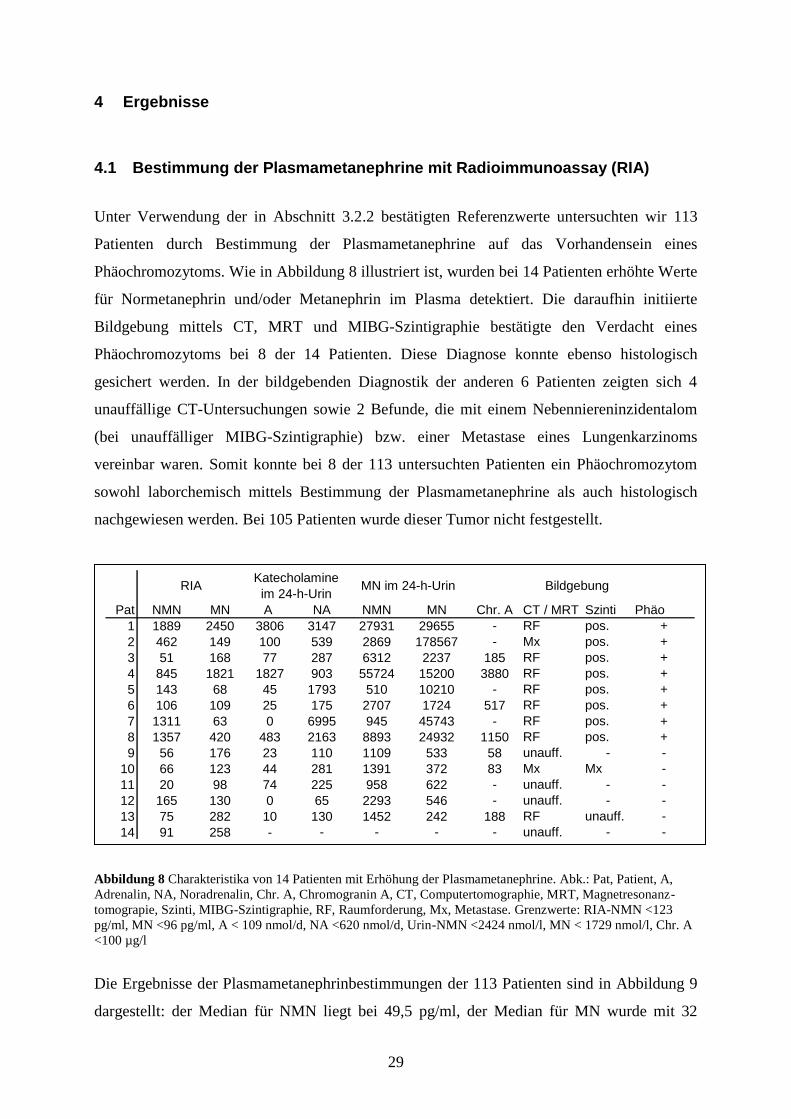
4.1 Bestimmung der Plasmametanephrine mit Radioimmunoassay (RIA)
Unter Verwendung der in Abschnitt 3.2.2 bestätigten Referenzwerte untersuchten wir 113
Patienten durch Bestimmung der Plasmametanephrine auf das Vorhandensein eines
Phäochromozytoms. Wie in Abbildung 8 illustriert ist, wurden bei 14 Patienten erhöhte Werte
für Normetanephrin und/oder Metanephrin im Plasma detektiert. Die daraufhin initiierte
Bildgebung mittels CT, MRT und MIBG-Szintigraphie bestätigte den Verdacht eines
Phäochromozytoms bei 8 der 14 Patienten. Diese Diagnose konnte ebenso histologisch
gesichert werden. In der bildgebenden Diagnostik der anderen 6 Patienten zeigten sich 4
unauffällige CT-Untersuchungen sowie 2 Befunde, die mit einem Nebenniereninzidentalom
(bei unauffälliger MIBG-Szintigraphie) bzw. einer Metastase eines Lungenkarzinoms
vereinbar waren. Somit konnte bei 8 der 113 untersuchten Patienten ein Phäochromozytom
sowohl laborchemisch mittels Bestimmung der Plasmametanephrine als auch histologisch
nachgewiesen werden. Bei 105 Patienten wurde dieser Tumor nicht festgestellt.
Abbildung 8 Charakteristika von 14 Patienten mit Erhöhung der Plasmametanephrine. Abk.: Pat, Patient, A,
Adrenalin, NA, Noradrenalin, Chr. A, Chromogranin A, CT, Computertomographie, MRT, Magnetresonanz-
tomograpie, Szinti, MIBG-Szintigraphie, RF, Raumforderung, Mx, Metastase. Grenzwerte: RIA-NMN <123
pg/ml, MN <96 pg/ml, A < 109 nmol/d, NA <620 nmol/d, Urin-NMN <2424 nmol/l, MN < 1729 nmol/l, Chr. A
<100 µg/l
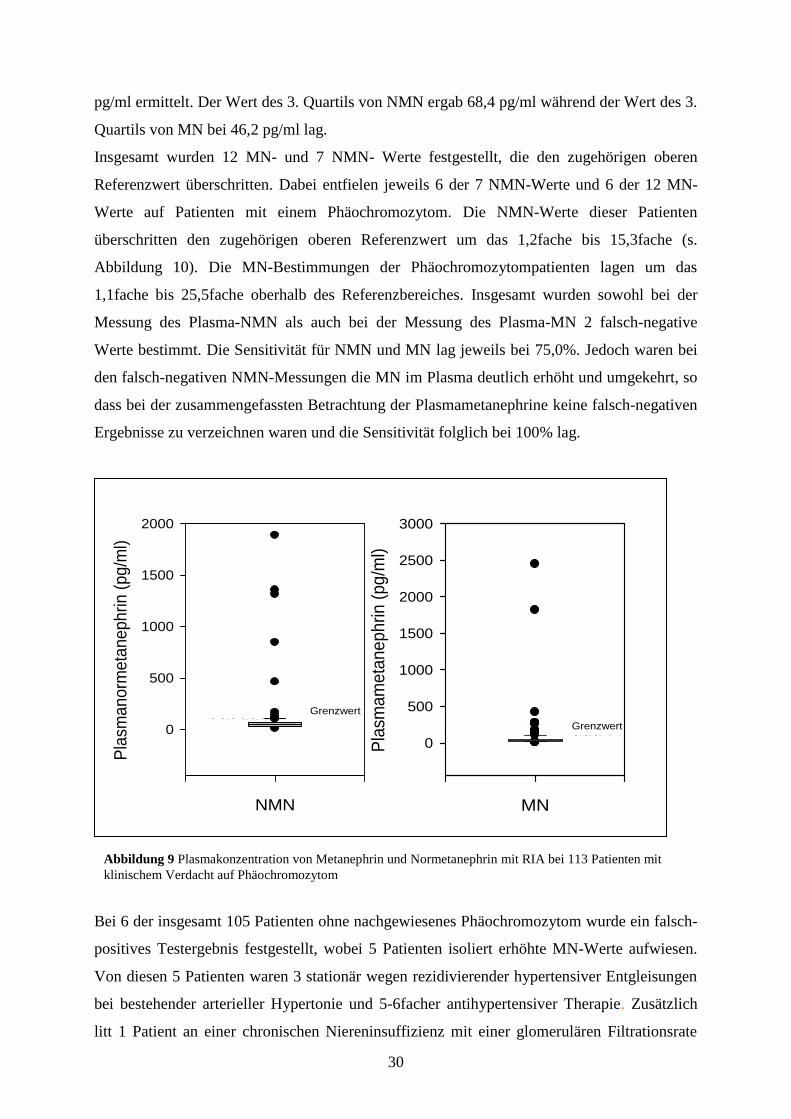
Die Ergebnisse der Plasmametanephrinbestimmungen der 113 Patienten sind in Abbildung 9
dargestellt: der Median für NMN liegt bei 49,5 pg/ml, der Median für MN wurde mit 32
Pat NMN MN A NA NMN MN Chr. A CT / MRT Szinti Phäo
1 1889 2450 3806 3147 27931 29655 - RF pos. +
2 462 149 100 539 2869 178567 - Mx pos. +
3 51 168 77 287 6312 2237 185 RF pos. +
4 845 1821 1827 903 55724 15200 3880 RF pos. +
5 143 68 45 1793 510 10210 - RF pos. +
6 106 109 25 175 2707 1724 517 RF pos. +
7 1311 63 0 6995 945 45743 - RF pos. +
8 1357 420 483 2163 8893 24932 1150 RF pos. +
9 56 176 23 110 1109 533 58 unauff. - -
10 66 123 44 281 1391 372 83 Mx Mx -
11 20 98 74 225 958 622 - unauff. - -
12 165 130 0 65 2293 546 - unauff. - -
13 75 282 10 130 1452 242 188 RF unauff. -
14 91 258 - - - - - unauff. - -
RIAKatecholamine
im 24-h-UrinMN im 24-h-Urin Bildgebung
30
pg/ml ermittelt. Der Wert des 3. Quartils von NMN ergab 68,4 pg/ml während der Wert des 3.
Quartils von MN bei 46,2 pg/ml lag.
Insgesamt wurden 12 MN- und 7 NMN- Werte festgestellt, die den zugehörigen oberen
Referenzwert überschritten. Dabei entfielen jeweils 6 der 7 NMN-Werte und 6 der 12 MN-
Werte auf Patienten mit einem Phäochromozytom. Die NMN-Werte dieser Patienten
überschritten den zugehörigen oberen Referenzwert um das 1,2fache bis 15,3fache (s.
Abbildung 10). Die MN-Bestimmungen der Phäochromozytompatienten lagen um das
1,1fache bis 25,5fache oberhalb des Referenzbereiches. Insgesamt wurden sowohl bei der
Messung des Plasma-NMN als auch bei der Messung des Plasma-MN 2 falsch-negative
Werte bestimmt. Die Sensitivität für NMN und MN lag jeweils bei 75,0%. Jedoch waren bei
den falsch-negativen NMN-Messungen die MN im Plasma deutlich erhöht und umgekehrt, so
dass bei der zusammengefassten Betrachtung der Plasmametanephrine keine falsch-negativen
Ergebnisse zu verzeichnen waren und die Sensitivität folglich bei 100% lag.
Bei 6 der insgesamt 105 Patienten ohne nachgewiesenes Phäochromozytom wurde ein falsch-
positives Testergebnis festgestellt, wobei 5 Patienten isoliert erhöhte MN-Werte aufwiesen.
Von diesen 5 Patienten waren 3 stationär wegen rezidivierender hypertensiver Entgleisungen
bei bestehender arterieller Hypertonie und 5-6facher antihypertensiver Therapie. Zusätzlich
litt 1 Patient an einer chronischen Niereninsuffizienz mit einer glomerulären Filtrationsrate
Grenzwert
NMN
Pla
sma
no
rme
tane
ph
rin
(p
g/m
l)
0
500
1000
1500
2000
MN
Pla
smam
eta
nephrin (
pg/m
l)
0
500
1000
1500
2000
2500
3000
Grenzwert
Abbildung 9 Plasmakonzentration von Metanephrin und Normetanephrin mit RIA bei 113 Patienten mit
klinischem Verdacht auf Phäochromozytom
31
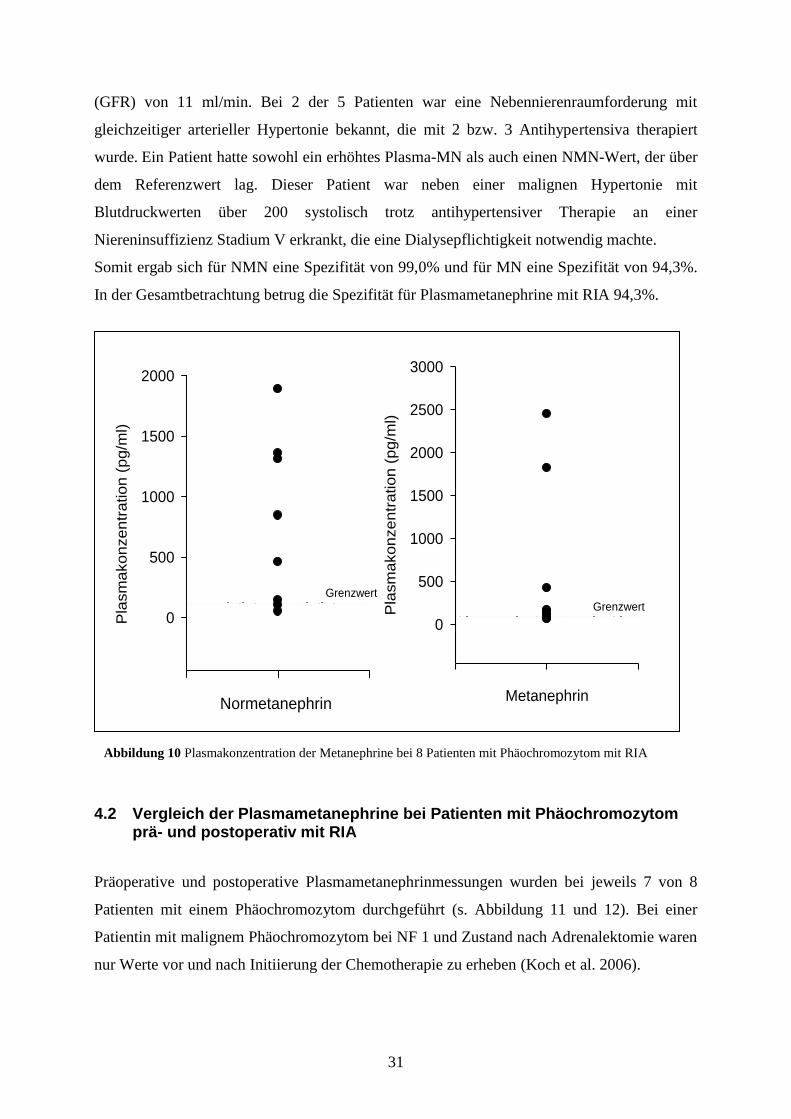
(GFR) von 11 ml/min. Bei 2 der 5 Patienten war eine Nebennierenraumforderung mit
gleichzeitiger arterieller Hypertonie bekannt, die mit 2 bzw. 3 Antihypertensiva therapiert
wurde. Ein Patient hatte sowohl ein erhöhtes Plasma-MN als auch einen NMN-Wert, der über
dem Referenzwert lag. Dieser Patient war neben einer malignen Hypertonie mit
Blutdruckwerten über 200 systolisch trotz antihypertensiver Therapie an einer
Niereninsuffizienz Stadium V erkrankt, die eine Dialysepflichtigkeit notwendig machte.
Somit ergab sich für NMN eine Spezifität von 99,0% und für MN eine Spezifität von 94,3%.
In der Gesamtbetrachtung betrug die Spezifität für Plasmametanephrine mit RIA 94,3%.
4.2 Vergleich der Plasmametanephrine bei Patienten mit Phäochromozytom prä- und postoperativ mit RIA
Präoperative und postoperative Plasmametanephrinmessungen wurden bei jeweils 7 von 8
Patienten mit einem Phäochromozytom durchgeführt (s. Abbildung 11 und 12). Bei einer
Patientin mit malignem Phäochromozytom bei NF 1 und Zustand nach Adrenalektomie waren
nur Werte vor und nach Initiierung der Chemotherapie zu erheben (Koch et al. 2006).
Abbildung 10 Plasmakonzentration der Metanephrine bei 8 Patienten mit Phäochromozytom mit RIA
Normetanephrin
Pla
sm
akon
zen
tration (
pg/m
l)
0
500
1000
1500
2000
Metanephrin
Pla
sm
akon
zen
tration (
pg/m
l)
0
500
1000
1500
2000
2500
3000
GrenzwertGrenzwert
32
a) Normetanephrin
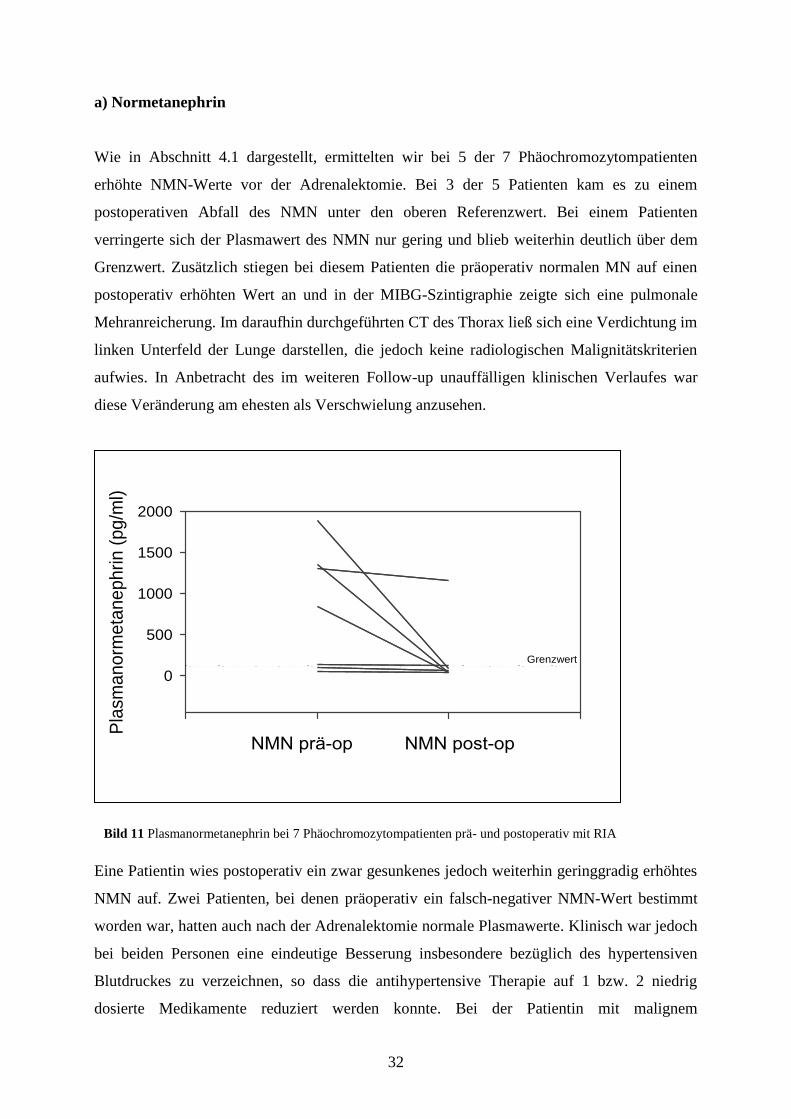
Wie in Abschnitt 4.1 dargestellt, ermittelten wir bei 5 der 7 Phäochromozytompatienten
erhöhte NMN-Werte vor der Adrenalektomie. Bei 3 der 5 Patienten kam es zu einem
postoperativen Abfall des NMN unter den oberen Referenzwert. Bei einem Patienten
verringerte sich der Plasmawert des NMN nur gering und blieb weiterhin deutlich über dem
Grenzwert. Zusätzlich stiegen bei diesem Patienten die präoperativ normalen MN auf einen
postoperativ erhöhten Wert an und in der MIBG-Szintigraphie zeigte sich eine pulmonale
Mehranreicherung. Im daraufhin durchgeführten CT des Thorax ließ sich eine Verdichtung im
linken Unterfeld der Lunge darstellen, die jedoch keine radiologischen Malignitätskriterien
aufwies. In Anbetracht des im weiteren Follow-up unauffälligen klinischen Verlaufes war
diese Veränderung am ehesten als Verschwielung anzusehen.
Eine Patientin wies postoperativ ein zwar gesunkenes jedoch weiterhin geringgradig erhöhtes
NMN auf. Zwei Patienten, bei denen präoperativ ein falsch-negativer NMN-Wert bestimmt
worden war, hatten auch nach der Adrenalektomie normale Plasmawerte. Klinisch war jedoch
bei beiden Personen eine eindeutige Besserung insbesondere bezüglich des hypertensiven
Blutdruckes zu verzeichnen, so dass die antihypertensive Therapie auf 1 bzw. 2 niedrig
dosierte Medikamente reduziert werden konnte. Bei der Patientin mit malignem
Bild 11 Plasmanormetanephrin bei 7 Phäochromozytompatienten prä- und postoperativ mit RIA
Grenzwert
NMN prä-op NMN post-op
Pla
sm
an
orm
eta
ne
ph
rin
(p
g/m
l)
0
500
1000
1500
2000
33
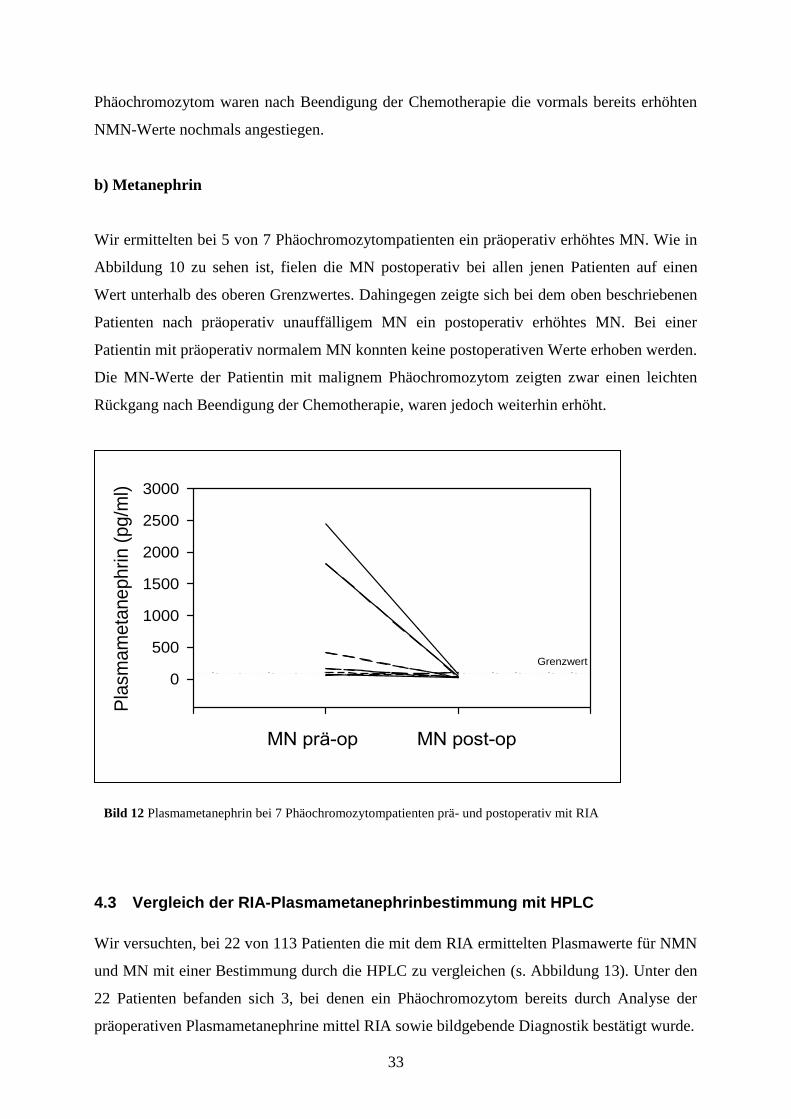
Phäochromozytom waren nach Beendigung der Chemotherapie die vormals bereits erhöhten
NMN-Werte nochmals angestiegen.
b) Metanephrin
Wir ermittelten bei 5 von 7 Phäochromozytompatienten ein präoperativ erhöhtes MN. Wie in
Abbildung 10 zu sehen ist, fielen die MN postoperativ bei allen jenen Patienten auf einen
Wert unterhalb des oberen Grenzwertes. Dahingegen zeigte sich bei dem oben beschriebenen
Patienten nach präoperativ unauffälligem MN ein postoperativ erhöhtes MN. Bei einer
Patientin mit präoperativ normalem MN konnten keine postoperativen Werte erhoben werden.
Die MN-Werte der Patientin mit malignem Phäochromozytom zeigten zwar einen leichten
Rückgang nach Beendigung der Chemotherapie, waren jedoch weiterhin erhöht.
4.3 Vergleich der RIA-Plasmametanephrinbestimmung mit HPLC
Wir versuchten, bei 22 von 113 Patienten die mit dem RIA ermittelten Plasmawerte für NMN
und MN mit einer Bestimmung durch die HPLC zu vergleichen (s. Abbildung 13). Unter den
22 Patienten befanden sich 3, bei denen ein Phäochromozytom bereits durch Analyse der
präoperativen Plasmametanephrine mittel RIA sowie bildgebende Diagnostik bestätigt wurde.
MN prä-op MN post-op
Pla
sm
am
eta
nephrin (
pg/m
l)
0
500
1000
1500
2000
2500
3000
Grenzwert
Bild 12 Plasmametanephrin bei 7 Phäochromozytompatienten prä- und postoperativ mit RIA
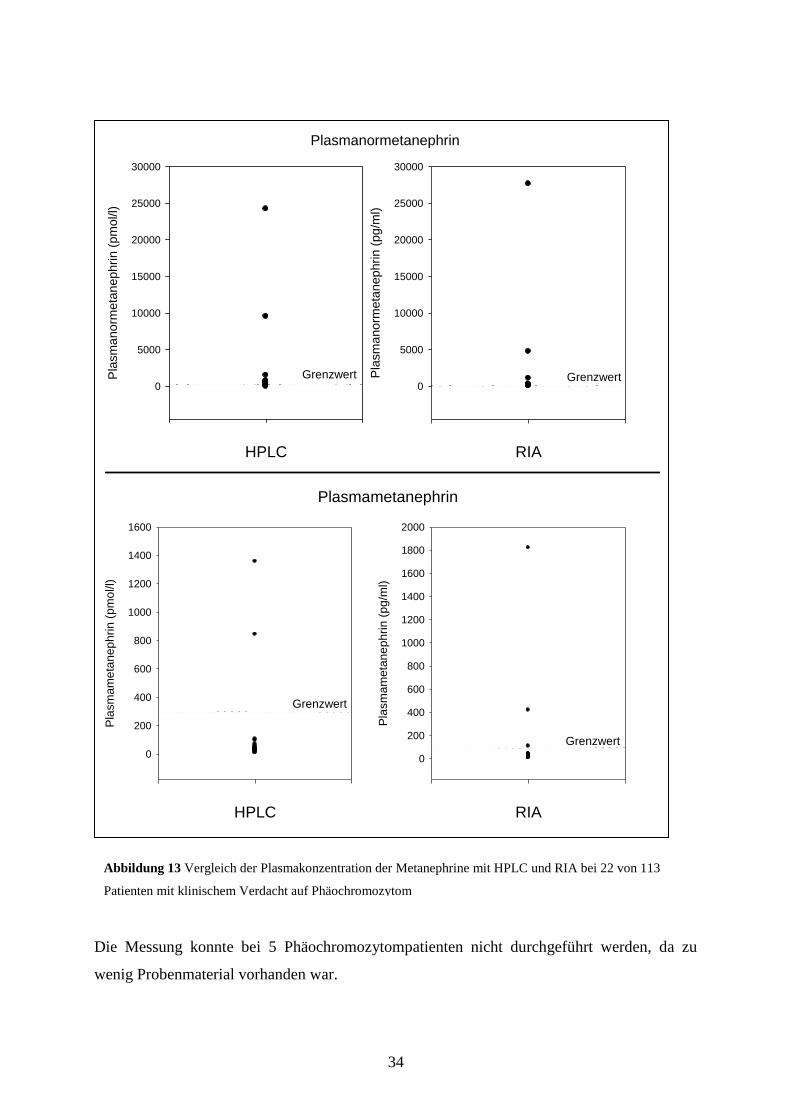
34
Die Messung konnte bei 5 Phäochromozytompatienten nicht durchgeführt werden, da zu
wenig Probenmaterial vorhanden war.
Abbildung 13 Vergleich der Plasmakonzentration der Metanephrine mit HPLC und RIA bei 22 von 113
Patienten mit klinischem Verdacht auf Phäochromozytom
Plasmanormetanephrin
HPLC
Pla
sm
ano
rmeta
nep
hrin (
pm
ol/l)
0
5000
10000
15000
20000
25000
30000
RIA
Pla
sm
ano
rmeta
nep
hrin (
pg
/ml)
0
5000
10000
15000
20000
25000
30000
HPLC
Pla
sm
am
eta
ne
ph
rin
(p
mo
l/l)
0
200
400
600
800
1000
1200
1400
1600
Plasmametanephrin
RIA
Pla
sm
am
eta
ne
ph
rin
(p
g/m
l)
0
200
400
600
800
1000
1200
1400
1600
1800
2000
Grenzwert
Grenzwert
Grenzwert
Grenzwert
35
a) HPLC
Die Untersuchungen wurden freundlicherweise von Prof. Lenders/Nijmwegen durchgeführt.
Durch die Bestimmung der Plasmametanephrine mittels HPLC wurden insgesamt 5 erhöhte
NMN- und 3 erhöhte MN-Werte ermittelt.
In der Gruppe der Patienten ohne nachgewiesenes Phäochromozytom hatten alle Patienten
einen MN-Wert, der unter dem Referenzwert von 300 pmol/l blieb. Bei 2 der 19 Patienten
wurde ein erhöhtes Plasma-NMN festgestellt. Die Charakteristika der Patienten sind in
Tabelle 14 dargestellt. Insgesamt lag der Median in der Gruppe für Patienten ohne
Phäochromozytom für Plasma-NMN bei 346 pmol/l, der Median für Plasma-MN betrug 150
pmol/l. Der Wert des 3. Quartils von NMN ergab 444 pmol/l. Für das 3. Quartil von MN
wurde ein Ergebnis von 179,5 pmol/l ermittelt.
Abbildung 14 Charakteristika von 2 Patienten mit NMN-Erhöhung mittels HPLC. Abkürzungen: NN-RF,
Nebennierenraumforderung; KHK, koronare Herzkrankheit; COPD, Chronic Obstructive Pulmonary Disease
Bei den 3 Phäochromozytompatienten zeigte sich ein erhöhtes NMN und MN. Der Wert des
MN übertraf den zugehörigen Grenzwert um das 3,6-fache bis 92-fache. Ebenso zeigte sich
ein Anstieg des Plasma-NMN um das 2,5fache bis 40fache über dem oberen Grenzwert.
Somit zeigten sich sowohl beim MN als auch beim NMN keine falsch-negativen Ergebnisse.
Die Sensitivität lag somit bei 100%. Eine postoperative Auswertung der Plasmametanephrine
mit HPLC wurde bei 1 Patientin durchgeführt. Hier konnte ein Abfall des präoperativ
erhöhten NMN und MN in den Normbereich beobachtet werden.
Patient 1 676 NN-RF (3x3 cm) negativ Captopril
Nierenzysten bds. Hydrochlorothiazid
KHK ASS
COPD Theophyllin
Diabetes mellitus Typ II Clenubterol
Pravastatin
Metformin
Tolterodin
Patient 2 752 NN-RF (6x6 cm) nicht durch- Estramustin
metastasierendes geführt Clodronsäure
Prostatakarzinom Ramipril
arterielle Hypertonie Torasemid
MIBG-
Szintigraphie
NMN
(< 600 pmol/l)Erkrankungen Medikamente

36
b) RIA
Wir analysierten bei 22 Patienten die Plasmametanephrine mit einem RIA. Es zeigten sich
insgesamt 3 erhöhte NMN- und 2 erhöhte MN-Werte.
In der Gruppe der Patienten ohne Phäochromozytom lagen sowohl beim Plasma-NMN als
auch beim Plasma-MN alle gemessenen Werte innerhalb des Referenzbereiches. Der Median
von NMN lag bei 31,9 pg/ml, der Median für MN bei 25,5 pg/ml. Der Wert des 3. Quartils
ergab 42,9 pg/ml für NMN und 34,5 pg/ml für MN.
Bei den Phäochromozytompatienten wurde bei einem Patienten ein falsch-negativer NMN-
Wert gemessen. Die übrigen NMN- und MN-Werte überschritten den jeweiligen
Referenzwert um das 6,8fache bis 12,3fache bzw. 1,1fache bis 18,9fache und waren somit
positiv.
Im direkten Vergleich der beiden Methoden konnten somit folgende Werte ermittelt werden:
NMN-Messungen mit der HPLC hatten auf Grund von 2 falsch-positiven NMN-Werten eine
Spezifität von 89,47%, MN-Messungen eine Spezifität von 100%. Die Sensitivität lag für
beide Werte jeweils bei 100%. Dadurch, dass in dieser Patientengruppe keine falsch-positiven
Ergebnisse durch den RIA ermittelt wurden, ergab sich eine Spezifität für MN und NMN von
jeweils 100%. Die Sensitivität lag insgesamt bei 100%, in der Einzeldarstellung der
Ergebnisse jedoch bei 66% für NMN und 100% für MN.
4.4 Bestimmung der Urinkatecholamine mit HPLC
Wir analysierten bei 79 der 113 Patienten, die auf das Vorhandensein eines
Phäochromozytoms durch Analyse der Plasmametanephrine mittels RIA untersucht wurden,
zur weiteren Validierung unserer Diagnostik die Urinkatecholamine im 24-h-Sammelurin. Bei
8 der 79 Patienten war ein Phäochromozytom bereits bestätigt worden. Wie in Abbildung 15
zu sehen ist, wurden insgesamt 5 erhöhte Adrenalin- und 6 erhöhte Noradrenalinspiegel
bestimmt. Davon entfielen 3 Adrenalin- bzw. 5 Noradrenalinwerte auf Patienten mit
nachgewiesenem Phäochromozytom. Die Werte überschritten den oberen Referenzbereich um
das 4- bis 34-fache (Adrenalin) bzw. 1,5- bis 11fache (Noradrenalin). Insgesamt waren bei 3
Phäochromozytompatienten sowohl Adrenalin-, als auch Noradrenalinwerte innerhalb des
Normbereiches.
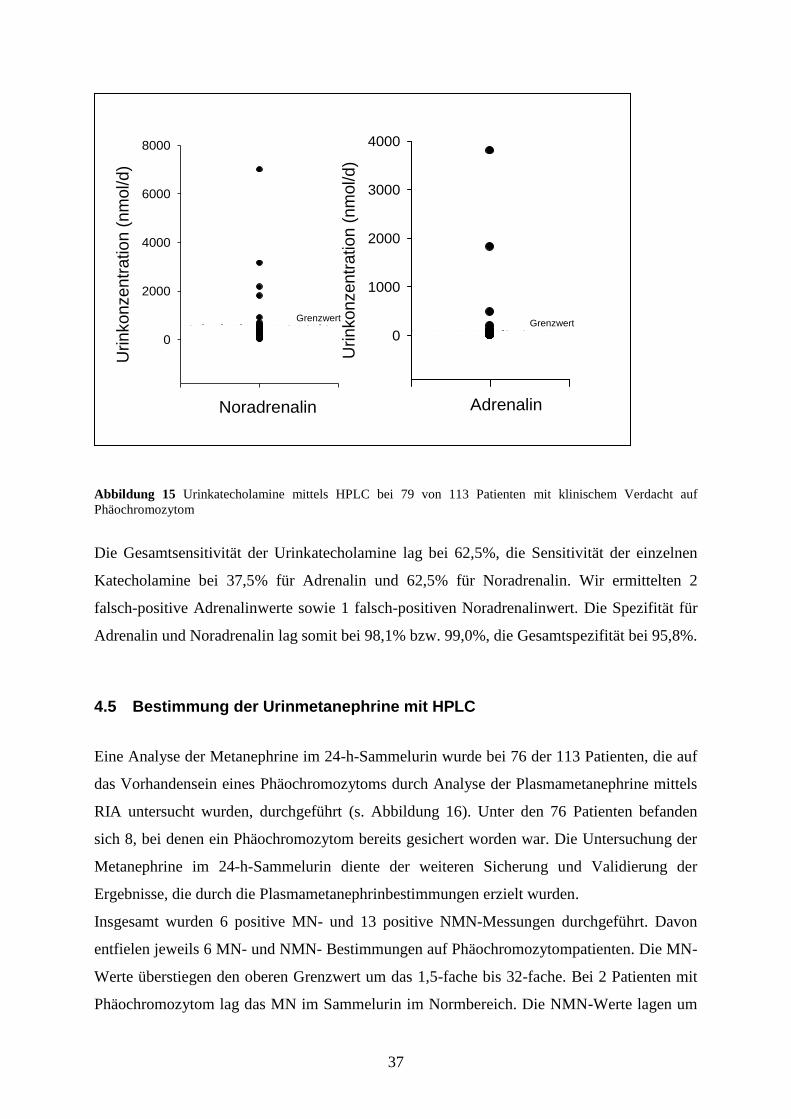
37
Abbildung 15 Urinkatecholamine mittels HPLC bei 79 von 113 Patienten mit klinischem Verdacht auf
Phäochromozytom
Die Gesamtsensitivität der Urinkatecholamine lag bei 62,5%, die Sensitivität der einzelnen
Katecholamine bei 37,5% für Adrenalin und 62,5% für Noradrenalin. Wir ermittelten 2
falsch-positive Adrenalinwerte sowie 1 falsch-positiven Noradrenalinwert. Die Spezifität für
Adrenalin und Noradrenalin lag somit bei 98,1% bzw. 99,0%, die Gesamtspezifität bei 95,8%.
4.5 Bestimmung der Urinmetanephrine mit HPLC
Eine Analyse der Metanephrine im 24-h-Sammelurin wurde bei 76 der 113 Patienten, die auf
das Vorhandensein eines Phäochromozytoms durch Analyse der Plasmametanephrine mittels
RIA untersucht wurden, durchgeführt (s. Abbildung 16). Unter den 76 Patienten befanden
sich 8, bei denen ein Phäochromozytom bereits gesichert worden war. Die Untersuchung der
Metanephrine im 24-h-Sammelurin diente der weiteren Sicherung und Validierung der
Ergebnisse, die durch die Plasmametanephrinbestimmungen erzielt wurden.
Insgesamt wurden 6 positive MN- und 13 positive NMN-Messungen durchgeführt. Davon
entfielen jeweils 6 MN- und NMN- Bestimmungen auf Phäochromozytompatienten. Die MN-
Werte überstiegen den oberen Grenzwert um das 1,5-fache bis 32-fache. Bei 2 Patienten mit
Phäochromozytom lag das MN im Sammelurin im Normbereich. Die NMN-Werte lagen um
Adrenalin
Urinkonzentr
ation (
nm
ol/d)
0
1000
2000
3000
4000
Noradrenalin
Urinkonzentr
ation (
nm
ol/d)
0
2000
4000
6000
8000
GrenzwertGrenzwert
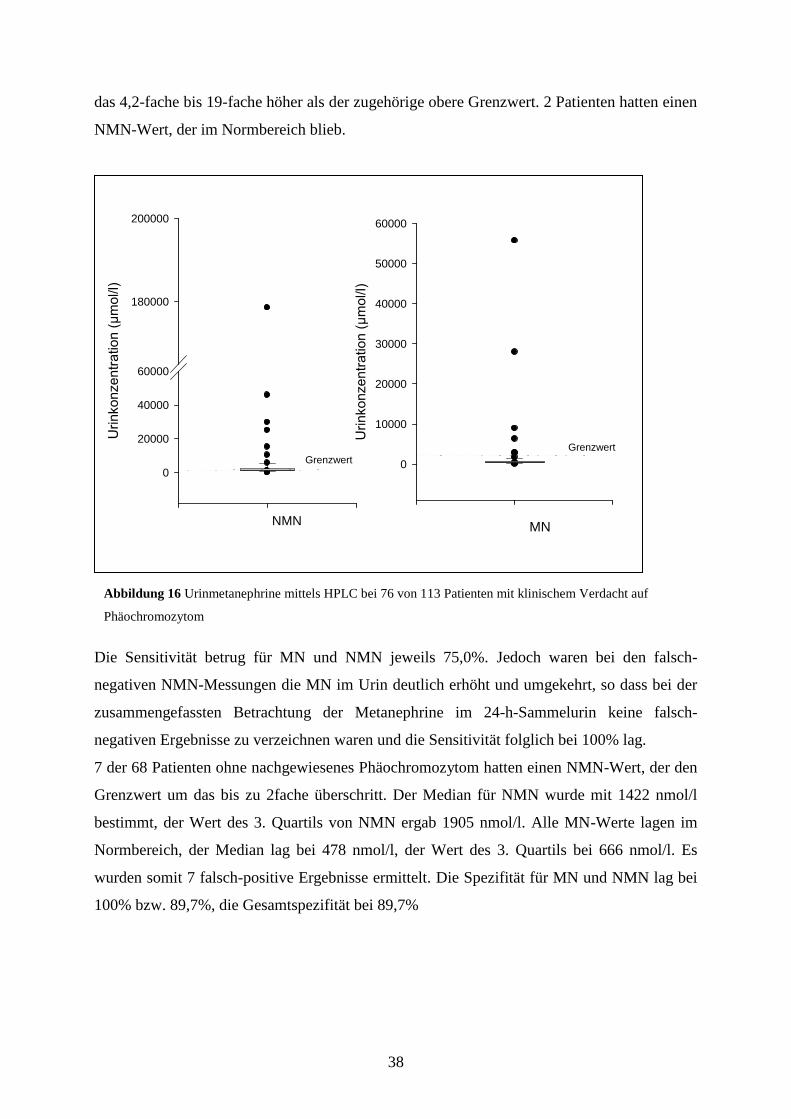
38
das 4,2-fache bis 19-fache höher als der zugehörige obere Grenzwert. 2 Patienten hatten einen
NMN-Wert, der im Normbereich blieb.
Die Sensitivität betrug für MN und NMN jeweils 75,0%. Jedoch waren bei den falsch-
negativen NMN-Messungen die MN im Urin deutlich erhöht und umgekehrt, so dass bei der
zusammengefassten Betrachtung der Metanephrine im 24-h-Sammelurin keine falsch-
negativen Ergebnisse zu verzeichnen waren und die Sensitivität folglich bei 100% lag.
7 der 68 Patienten ohne nachgewiesenes Phäochromozytom hatten einen NMN-Wert, der den
Grenzwert um das bis zu 2fache überschritt. Der Median für NMN wurde mit 1422 nmol/l
bestimmt, der Wert des 3. Quartils von NMN ergab 1905 nmol/l. Alle MN-Werte lagen im
Normbereich, der Median lag bei 478 nmol/l, der Wert des 3. Quartils bei 666 nmol/l. Es
wurden somit 7 falsch-positive Ergebnisse ermittelt. Die Spezifität für MN und NMN lag bei
100% bzw. 89,7%, die Gesamtspezifität bei 89,7%
Abbildung 16 Urinmetanephrine mittels HPLC bei 76 von 113 Patienten mit klinischem Verdacht auf
Phäochromozytom
NMN
Uri
nkonzentr
ation (
µm
ol/l)
0
20000
40000
60000
180000
200000
MN
Uri
nkonzentr
ation (
µm
ol/l)
0
10000
20000
30000
40000
50000
60000
Grenzwert
Grenzwert
39
4.6 Bestimmung des Serum-Chromogranin A mittels RIA
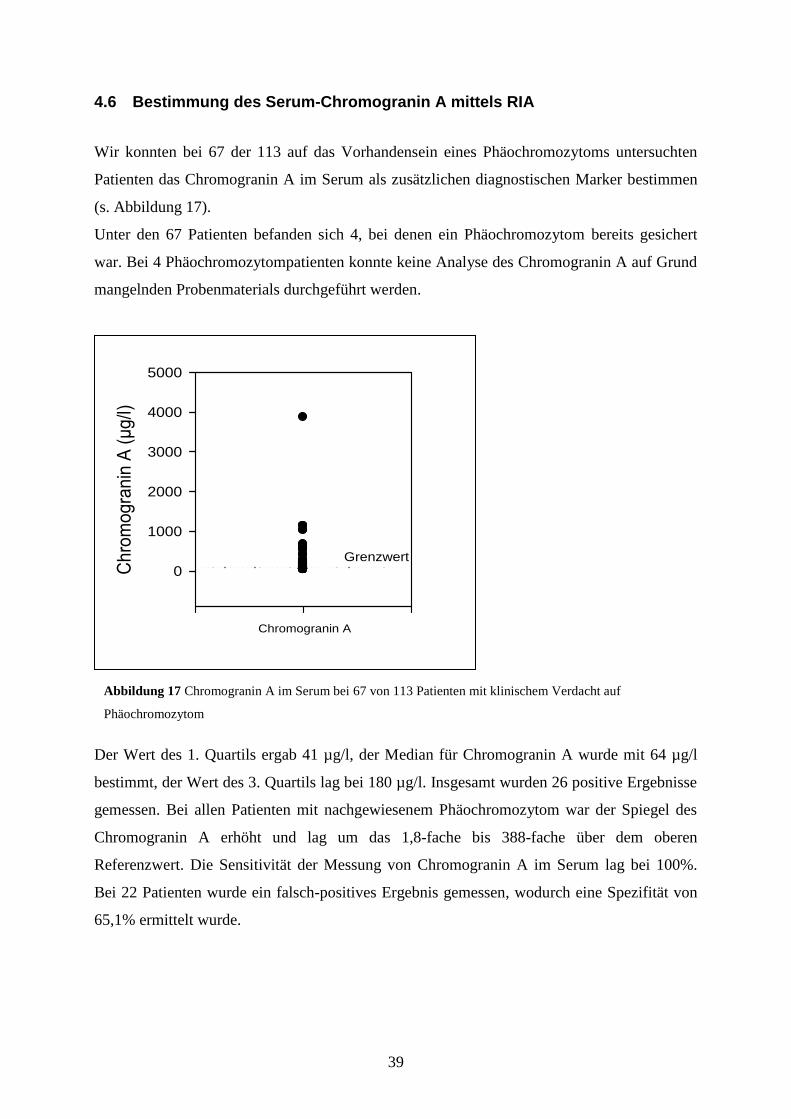
Wir konnten bei 67 der 113 auf das Vorhandensein eines Phäochromozytoms untersuchten
Patienten das Chromogranin A im Serum als zusätzlichen diagnostischen Marker bestimmen
(s. Abbildung 17).
Unter den 67 Patienten befanden sich 4, bei denen ein Phäochromozytom bereits gesichert
war. Bei 4 Phäochromozytompatienten konnte keine Analyse des Chromogranin A auf Grund
mangelnden Probenmaterials durchgeführt werden.
Der Wert des 1. Quartils ergab 41 µg/l, der Median für Chromogranin A wurde mit 64 µg/l
bestimmt, der Wert des 3. Quartils lag bei 180 µg/l. Insgesamt wurden 26 positive Ergebnisse
gemessen. Bei allen Patienten mit nachgewiesenem Phäochromozytom war der Spiegel des
Chromogranin A erhöht und lag um das 1,8-fache bis 388-fache über dem oberen
Referenzwert. Die Sensitivität der Messung von Chromogranin A im Serum lag bei 100%.
Bei 22 Patienten wurde ein falsch-positives Ergebnis gemessen, wodurch eine Spezifität von
65,1% ermittelt wurde.
Abbildung 17 Chromogranin A im Serum bei 67 von 113 Patienten mit klinischem Verdacht auf
Phäochromozytom
Grenzwert
Chromogranin A
Ch
rom
ogra
nin
A (
µg/l)
0
1000
2000
3000
4000
5000
40
4.7 Mehrfachbestimmung der Metanephrine im Plasma mit RIA
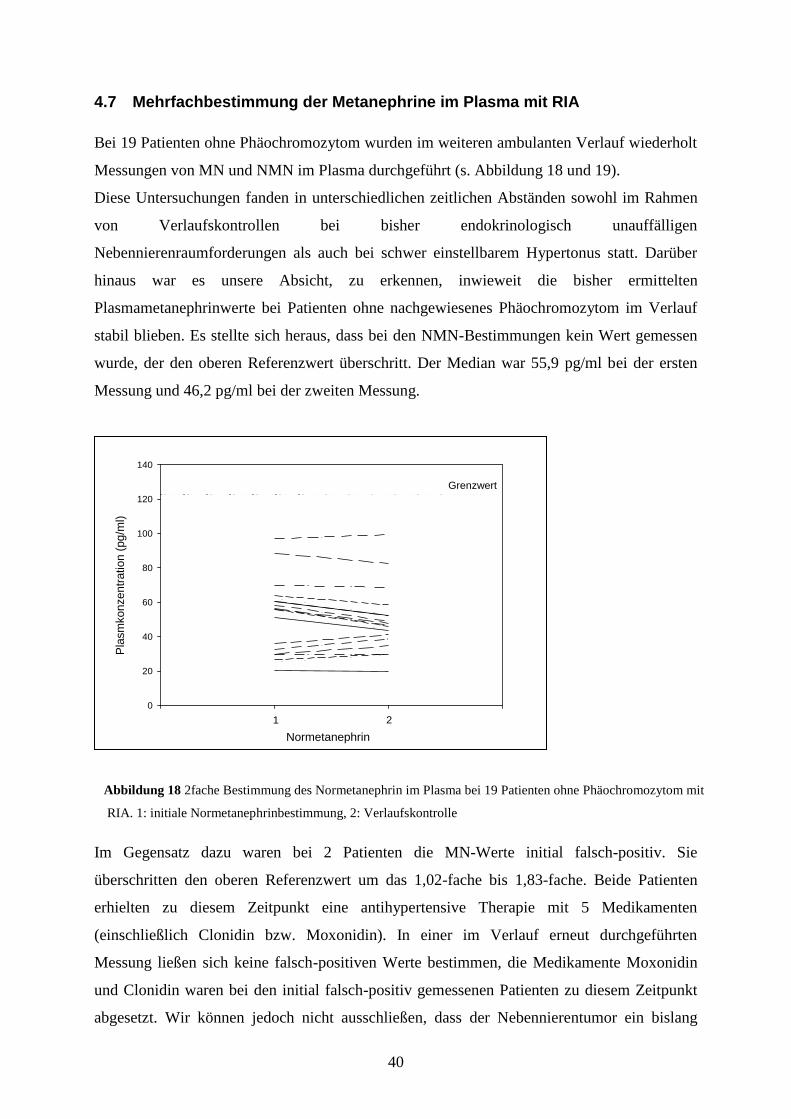
Bei 19 Patienten ohne Phäochromozytom wurden im weiteren ambulanten Verlauf wiederholt
Messungen von MN und NMN im Plasma durchgeführt (s. Abbildung 18 und 19).
Diese Untersuchungen fanden in unterschiedlichen zeitlichen Abständen sowohl im Rahmen
von Verlaufskontrollen bei bisher endokrinologisch unauffälligen
Nebennierenraumforderungen als auch bei schwer einstellbarem Hypertonus statt. Darüber
hinaus war es unsere Absicht, zu erkennen, inwieweit die bisher ermittelten
Plasmametanephrinwerte bei Patienten ohne nachgewiesenes Phäochromozytom im Verlauf
stabil blieben. Es stellte sich heraus, dass bei den NMN-Bestimmungen kein Wert gemessen
wurde, der den oberen Referenzwert überschritt. Der Median war 55,9 pg/ml bei der ersten
Messung und 46,2 pg/ml bei der zweiten Messung.
Im Gegensatz dazu waren bei 2 Patienten die MN-Werte initial falsch-positiv. Sie
überschritten den oberen Referenzwert um das 1,02-fache bis 1,83-fache. Beide Patienten
erhielten zu diesem Zeitpunkt eine antihypertensive Therapie mit 5 Medikamenten
(einschließlich Clonidin bzw. Moxonidin). In einer im Verlauf erneut durchgeführten
Messung ließen sich keine falsch-positiven Werte bestimmen, die Medikamente Moxonidin
und Clonidin waren bei den initial falsch-positiv gemessenen Patienten zu diesem Zeitpunkt
abgesetzt. Wir können jedoch nicht ausschließen, dass der Nebennierentumor ein bislang
Abbildung 18 2fache Bestimmung des Normetanephrin im Plasma bei 19 Patienten ohne Phäochromozytom mit
RIA. 1: initiale Normetanephrinbestimmung, 2: Verlaufskontrolle
Normetanephrin
Pla
sm
konzentr
ation (
pg/m
l)
0
20
40
60
80
100
120
140
Grenzwert
21
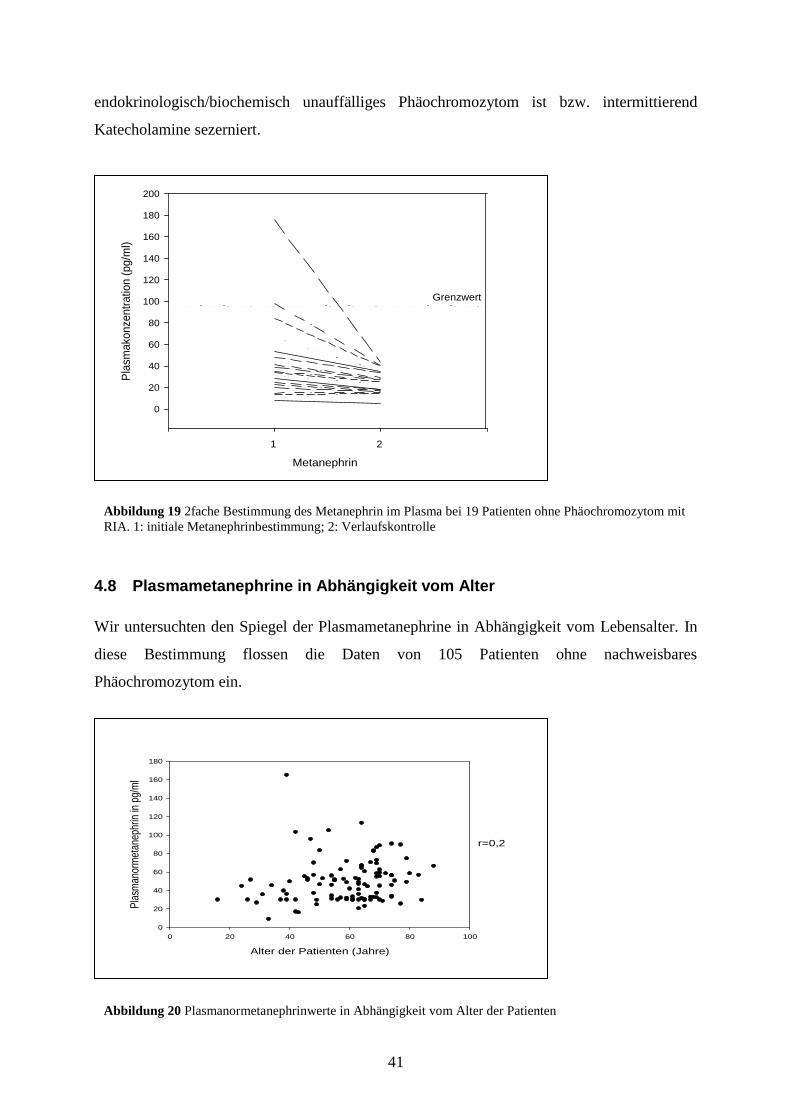
41
endokrinologisch/biochemisch unauffälliges Phäochromozytom ist bzw. intermittierend
Katecholamine sezerniert.
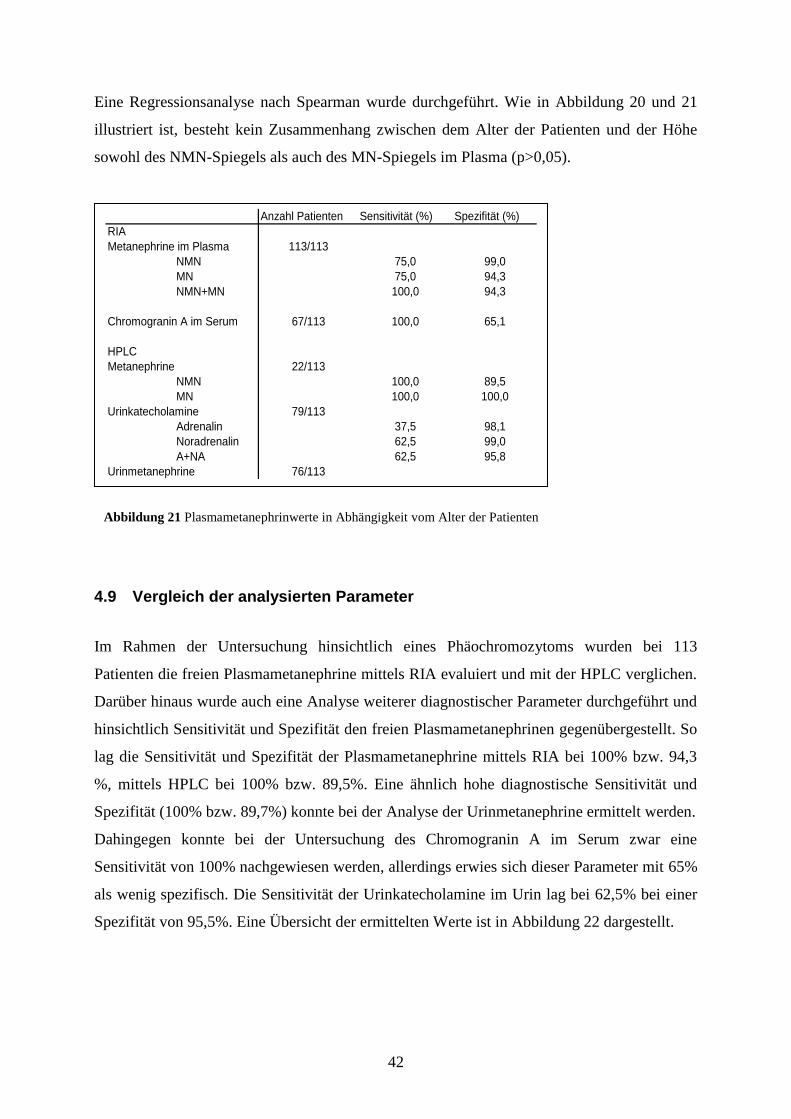
4.8 Plasmametanephrine in Abhängigkeit vom Alter
Wir untersuchten den Spiegel der Plasmametanephrine in Abhängigkeit vom Lebensalter. In
diese Bestimmung flossen die Daten von 105 Patienten ohne nachweisbares
Phäochromozytom ein.
Abbildung 20 Plasmanormetanephrinwerte in Abhängigkeit vom Alter der Patienten
r=0,2
Alter der Patienten (Jahre)
0 20 40 60 80 100
Pla
sman
orm
etan
ephr
in in
pg/
ml
0
20
40
60
80
100
120
140
160
180
Abbildung 19 2fache Bestimmung des Metanephrin im Plasma bei 19 Patienten ohne Phäochromozytom mit
RIA. 1: initiale Metanephrinbestimmung; 2: Verlaufskontrolle
Metanephrin
Pla
smako
nze
ntr
atio
n (
pg/m
l)
0
20
40
60
80
100
120
140
160
180
200
Grenzwert
1 2
42
Eine Regressionsanalyse nach Spearman wurde durchgeführt. Wie in Abbildung 20 und 21
illustriert ist, besteht kein Zusammenhang zwischen dem Alter der Patienten und der Höhe
sowohl des NMN-Spiegels als auch des MN-Spiegels im Plasma (p>0,05).
4.9 Vergleich der analysierten Parameter
Im Rahmen der Untersuchung hinsichtlich eines Phäochromozytoms wurden bei 113
Patienten die freien Plasmametanephrine mittels RIA evaluiert und mit der HPLC verglichen.
Darüber hinaus wurde auch eine Analyse weiterer diagnostischer Parameter durchgeführt und
hinsichtlich Sensitivität und Spezifität den freien Plasmametanephrinen gegenübergestellt. So
lag die Sensitivität und Spezifität der Plasmametanephrine mittels RIA bei 100% bzw. 94,3
%, mittels HPLC bei 100% bzw. 89,5%. Eine ähnlich hohe diagnostische Sensitivität und
Spezifität (100% bzw. 89,7%) konnte bei der Analyse der Urinmetanephrine ermittelt werden.
Dahingegen konnte bei der Untersuchung des Chromogranin A im Serum zwar eine
Sensitivität von 100% nachgewiesen werden, allerdings erwies sich dieser Parameter mit 65%
als wenig spezifisch. Die Sensitivität der Urinkatecholamine im Urin lag bei 62,5% bei einer
Spezifität von 95,5%. Eine Übersicht der ermittelten Werte ist in Abbildung 22 dargestellt.
Anzahl Patienten Sensitivität (%) Spezifität (%)
RIA
Metanephrine im Plasma 113/113
NMN 75,0 99,0
MN 75,0 94,3
NMN+MN 100,0 94,3
Chromogranin A im Serum 67/113 100,0 65,1
HPLC
Metanephrine 22/113
NMN 100,0 89,5
MN 100,0 100,0
Urinkatecholamine 79/113
Adrenalin 37,5 98,1
Noradrenalin 62,5 99,0
A+NA 62,5 95,8
Urinmetanephrine 76/113
Abbildung 21 Plasmametanephrinwerte in Abhängigkeit vom Alter der Patienten
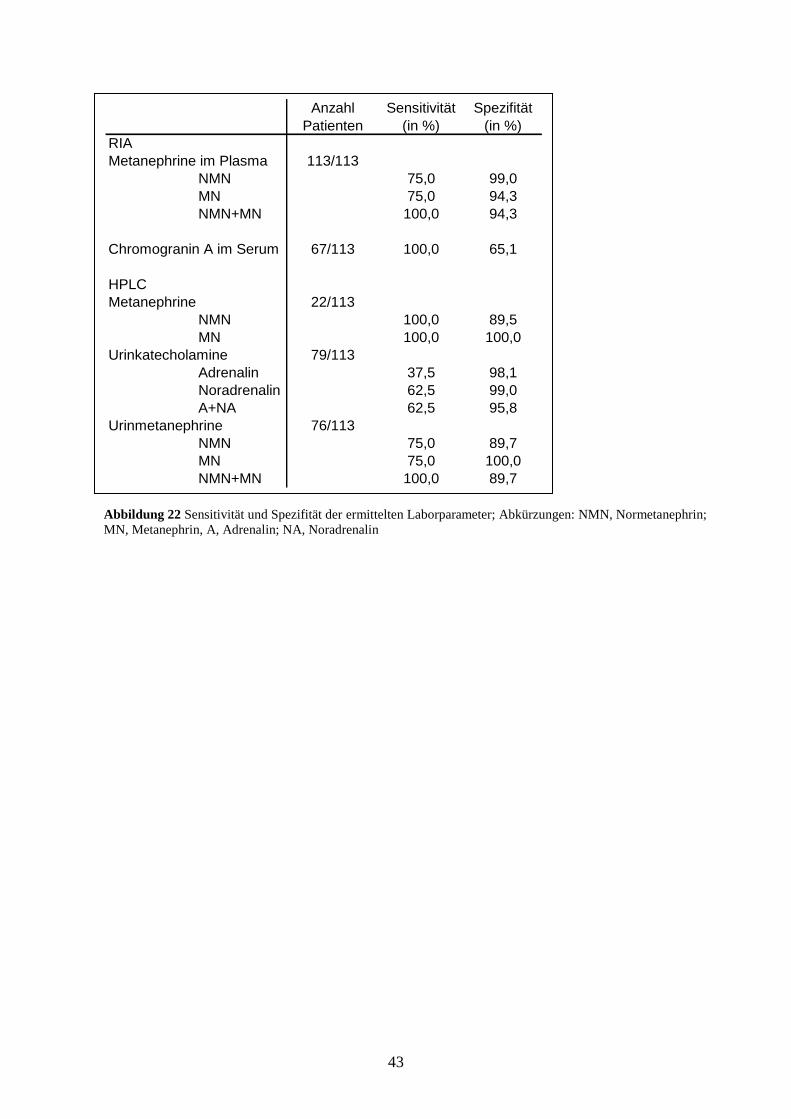
43
Abbildung 22 Sensitivität und Spezifität der ermittelten Laborparameter; Abkürzungen: NMN, Normetanephrin;
MN, Metanephrin, A, Adrenalin; NA, Noradrenalin
RIA
Metanephrine im Plasma 113/113
NMN 75,0 99,0
MN 75,0 94,3
NMN+MN 100,0 94,3
Chromogranin A im Serum 67/113 100,0 65,1
HPLC
Metanephrine 22/113
NMN 100,0 89,5
MN 100,0 100,0
Urinkatecholamine 79/113
Adrenalin 37,5 98,1
Noradrenalin 62,5 99,0
A+NA 62,5 95,8
Urinmetanephrine 76/113
NMN 75,0 89,7
MN 75,0 100,0
NMN+MN 100,0 89,7
Sensitivität
(in %)
Spezifität
(in %)
Anzahl
Patienten

44
5 Diskussion
5.1 Überblick
In den vergangenen Jahren hat sich die Bestimmung der freien Metanephrine im Plasma
mittels HPLC als Mittel der Wahl in der Diagnostik des Phäochromozytoms etabliert
(Eisenhofer 2003c).
Im Rahmen einer Untersuchung verschiedener biochemischer Parameter hinsichtlich der
jeweiligen Sensitivität und Spezifität in der Phäochromozytomdiagnostik konnten Lenders et
al. (1995) erstmals zeigen, dass die freien Plasmametanephrine als diagnostisches Mittel
eingesetzt werden können. In sich hierauf anschließenden Studien wurde dieses Ergebnis
nicht nur bestätigt, sondern es zeigte sich die diagnostische Überlegenheit der freien
Plasmametanephrine gegenüber anderen Parametern (Eisenhofer et al. 1999, Goldstein et al.
2004, Unger et al. 2006, Hickman et al. 2009).
Als Gründe der verbesserten Sensitivität und Spezifität der freien Plasmametanephrine
wurden aufgeführt:
Die Expression von Catechol-O-Methyltransferase (COMT) innerhalb des Tumors. So
erfolgt bereits intratumoral eine kontinuierliche Umwandlung von aus durchlässigen
Speichervesikeln stammendem Adrenalin und Noradrenalin zu MN und NMN. In
einer Studie (Eisenhofer et al. 1995) wurde nachgewiesen, dass dadurch die
Konzentration an freien Metanephrinen innerhalb des Phäochromozytoms auf das
13.000fache bis 23.000fache ansteigen kann.
Im Gegensatz zu den oftmals nur periodisch sezernierten Katecholaminen ist bei den
Phäochromozytompatienten durch den o.g. Mechanismus eine beständige Erhöhung
der freien Metanephrine im Plasma zu verzeichnen.
91% des zirkulierenden Plasma-MN und 23% des Plasma-NMN stammen aus der
Nebenniere selbst. Sie stellt damit die größte natürliche Quelle der Metanephrine im
menschlichen Körper dar.
Mit der HPLC steht ein Labortest zur Verfügung, der eine sensitive und zugleich spezifische
Bestimmung der Metanephrine ermöglicht. Auf Grund dessen, dass dieses Verfahren durch
eine komplizierte Analytik sehr kosten- und arbeitsintensiv ist, steht es derzeit nur wenigen
Zentren zur kommerziellen Verfügung (Sawka et al. 2004a). Dies ist insbesondere
dahingehend ein Nachteil, als durch die stetig verbesserte Bildgebung die Zahl der entdeckten

45
adrenalen Inzidentalome beständig ansteigt und dadurch die Phäochromozytomdiagnostik
auch in kleineren Kliniken an Bedeutung gewinnt (Aron 2001, Thompson und Young. 2003).
Darüber hinaus sind verschiedene Substanzen beschrieben worden- insbesondere
Medikamente wie trizyklische Antidepressiva, Phenoxybenzamin und Sympathomimetika-
die mit der HPLC interferieren und falsch-positive Ergebnisse verursachen können
(Eisenhofer et al. 2003b, Wassell et al. 1999).
Neben der HPLC existieren weitere Verfahren, die in der Diagnostik des Phäochromozytoms
eingesetzt werden und über charakteristische Vor- und Nachteile verfügen.
Die Gaschromatographische Massenspektrometrie (GC-MS) ist sehr zeitaufwändig und durch
die notwendige große Laborausstattung nur in wenigen klinischen Zentren vorhanden.
Darüber hinaus ist es mit dieser Methode zwar möglich, konjugierte Metanephrine im Urin zu
bestimmen, jedoch ist es bisher nicht gelungen, freie Metanephrine im Plasma nachzuweisen
(Crockett et al. 2002).
Im Gegensatz zur GC-MS und HPLC ist ein immunologischer Assay einfach durchzuführen
und bietet dadurch eine breite Anwendbarkeit. Allerdings konnten bis zum heutigen Zeitpunkt
auch mit diesem Verfahren nur die konjugierten Metanephrine im Urin und nicht die freien
Metanephrine im Plasma analysiert werden (Wolthers et al. 1997).
Eine weitere Methode ist die Flüssigchromatographie mit Massenspektrometrie-Kopplung
(LC-MS), mit der die Analyse freier Metanephrine im Plasma möglich ist. Ein Nachteil dieses
Verfahren ist jedoch die Anfälligkeit gegenüber interferierenden Substanzen. So beobachtete
Lagerstedt (2004) eine Beeinflussung der Messergebnisse von Metanephrinen im Plasma
durch Adrenalin.
Es lässt sich konstatieren, dass keines der vorgestellten Verfahren die Anforderungen in der
Diagnostik des Phäochromozytoms vollständig erfüllt. Eine gute Sensitivität und Spezifität
einiger Methoden wird nur durch hohe Kosten oder einen immensen Zeitaufwand erreicht.
Auf der anderen Seite ist es bei schnell und einfach durchzuführenden Methoden bisher nicht
möglich gewesen, freie Metanephrine im Plasma nachzuweisen. Zusätzlich bereiten
interferierende Substanzen immer wieder Probleme in der exakten Bestimmung der
jeweiligen Parameter.
Aus diesen Gesichtspunkten heraus leitet sich die Anwendung eines RIA ab, der auf Grund
seiner Einfachheit und breiten Verfügbarkeit zur Bestimmung von freien Metanephrinen im
Plasma bei der Diagnostik des Phäochromozytoms denkbar ist. Zusätzlich ist die Verwendung
eines RIA an die Erwartung einer der HPLC mindestens gleichwertigen Sensitivität und
Spezifität geknüpft. Vor diesem Hintergrund wurde in der vorliegenden Untersuchung die

46
Bestimmung der Metanephrine im Plasma mittels RIA im Vergleich zu bisher angewandten
Verfahren, insbesondere der HPLC, in der Diagnostik des Phäochromozytoms durchgeführt.
Es wurden 163 Patienten eingeschlossen, von denen lediglich 4 Patienten die klassische
klinische Symptomtrias bestehend aus Palpitationen, Kopfschmerz und Schwitzen angaben.
Dies kontrastiert mit anderen Studien. So fanden Plouin et al. (1981) bei Vorhandensein
dieser Trias und Bluthochdruck bei 2585 Bluthochdruckpatienten solche mit einem
Phäochromozytom mit einer Sensitivität von 91% und Spezifität von 94%.
Die Ergebnisse der Untersuchung sind in Kapitel 4 dargestellt. Es zeigt sich, dass der hier
getestete RIA freie Metanephrine im Plasma sensitiv und spezifisch nachweist und somit eine
gute Alternative zur HPLC ist.
5.2 Plasmametanephrine
Wir untersuchten in dieser Studie 163 Patienten auf das Vorhandensein eines
Phäochromozytoms durch Bestimmung der Plasmametanephrine mit Hilfe eines RIA. In der
Anfangsphase unserer Arbeit standen uns keine validierten Referenzwerte für Metanephrin
und Normetanephrin mittels RIA zur Verfügung. Wir ermittelten daher bei den ersten 50 der
163 Patienten, bei denen ein Phäochromozytom durch bildgebende Verfahren nahezu
ausgeschlossen wurde eigene Grenzwerte (s. 3.2.2). Hierbei orientierten wir uns an den von
Unger et al. (2004) in einer klinischen Studie ermittelten Grenzwerten für Normetanephrin
und Metanephrin. Nachdem wir die Grenzwerte bei den ersten 50 Patienten bestätigen
konnten, wandten wir sie bei der Untersuchung der übrigen 113 Patienten an. Die so
ermittelte Sensitivität und Spezifität der Plasmametanephrine lag bei 100% bzw. 94,3%. Es
zeigte sich, dass die von Unger et al. (2004) ermittelten Grenzwerte auch bei unseren Proben
valide waren und in der Diagnostik des Phäochromozytoms eingesetzt werden können.
Bei 8 der 113 Patienten konnte ein Phäochromozytom histologisch nachgewiesen werden. In
den analysierten Plasmametanephrinen zeigte sich entweder ein erhöhter MN- oder NMN-
Wert. Es wurden sowohl bei den NMN als auch den MN jeweils 2 Werte im Normbereich
ermittelt. Das führte zu einer Sensitivität von 75% bei der Verwendung beider Parameter.
Jedoch waren bei den normwertigen NMN-Bestimmungen die MN deutlich erhöht und
umgekehrt, so dass insgesamt kein falsch-negatives Ergebnis ermittelt wurde und die
Sensitivität der Plasmametanephrine somit bei 100% lag. Diese Ergebnisse stehen im
Einklang mit anderen Studien, in denen eine ähnliche hohe Sensitivität der
Plasmametanephrine ermittelt wurde (Raber et al. 2000, Giovanella et al. 2006, Unger et al.

47
2006, Hickman et al. 2009).
Ferner führten wir eine Analyse der prä- und postoperativen Werte für NMN und MN bei 7
von 8 Phäochromozytompatienten durch. Auf Grund der niedrigen Fallzahl konnten keine
statistisch zu belegenden Aussagen getroffen werden. Es ließen sich jedoch folgende
Vermutungen äußern, die durch weitere Untersuchungen bestätigt werden müssen:
bei den meisten Patienten ist eine rasche Normalisierung bzw. starker Abfall von präoperativ
erhöhten NMN- oder MN-Werten zu verzeichnen. Dies geht gleichzeitig einher mit einem
deutlichen Rückgang bzw. einer kompletten Regredienz der jeweiligen klinischen
Beschwerdesymptomatik der Patienten. Darüber hinaus scheinen nach der Entfernung eines
malignen Phäochromozytoms die Plasmametanephrine auch im weiteren Verlauf erhöht zu
bleiben. In unserer Untersuchung gab die Patientin mit malignem Phäochromozytom auch
nach Adrenalektomie weiterhin Beschwerden (Palpitationen, Kopfschmerzen) an. Sollten bei
initial benignen Phäochromozytomen postoperativ weiterhin erhöhte Spiegel für NMN oder
MN bestehen, sind engmaschige Kontrollen der Werte zu empfehlen, so dass ein eventueller
maligner Verlauf diagnostiziert werden kann. Insgesamt scheint die Bestimmung der
Plasmametanephrine mittels RIA zum postoperativen Follow-up geeignet zu sein.
Bei 6 der 105 Patienten ohne Phäochromozytom wurden falsch-positive Werte mit dem RIA
ermittelt, so dass wir eine Spezifität von 99,0% für NMN und 94,3% für MN ermittelten Dies
sind deutlich höhere Werte als in vorausgegangenen Studien, in denen die Spezifität für
Plasmametanephrine mit 79% bis 89% angegeben wird (Lenders et al. 2002b, Sawka et al.
2003, Unger et al. 2006). Eine mögliche Ursache ist die Verwendung von niedrigeren
Grenzwerten, welche eine höhere Sensitivität ermöglichen, allerdings auf Kosten einer
geringeren Spezifität.
In den letzten Jahren ist immer wieder das diagnostische Dilemma bei Patienten mit
terminaler Niereninsuffizienz oder Dialysepflichtigkeit und erhöhten Plasmakatecholaminen
beschrieben worden (Stumvoll et al. 1995, Box et al. 1997, Saeki et al. 2003). Morioka et al.
(2002) versuchten, dieses Problem durch die Festlegung von separaten Normalwerten zu
lösen. Durch den Einsatz der Metanephrine im Plasma wurde hier ein bedeutender Fortschritt
in der Verbesserung der Phäochromozytomdiagnostik erreicht, da sie unabhängig von der
renalen Funktion und hauptsächlich durch aktive extraneurale Wiederaufnahme aus dem Blut
entfernt werden. So fanden Marini et al. (1994) keinen Unterschied in der Konzentration von
fraktionierten freien Metanephrinen im Plasma zwischen 10 Patienten mit Phäochromozytom
und normaler Nierenfunktion und 11 Patienten ohne Phäochromozytom, die auf Grund einer
terminalen Niereninsuffizienz dialysepflichtig waren. Trotzdem sind auch hier weiterhin

48
Berichte über falsch-positive Metanephrinmessungen bei Patienten mit Niereninsuffizienz
veröffentlicht worden (Eisenhofer et al. 2005b). In unserer Untersuchung waren 6 Patienten
an einer terminalen Niereninsuffizienz erkrankt. Bei keinem von ihnen zeigten sich falsch-
positive Werte für NMN oder MN, so dass unserer Ansicht nach die Plasmametanephrine gut
bei dieser Patientenklientel eingesetzt werden können.
Im Rahmen einer Kontrolluntersuchung wurde bei 19 der 105 Patienten eine zweite Messung
der Plasmametanephrine durchgeführt. Es zeigten sich keine falsch-positiven Ergebnisse. Die
bei der ersten Untersuchung festgestellten MN-Erhöhungen bei 2 Patienten waren regredient.
Beide Patienten waren bei der ersten Untersuchung unter antihypertensiver Therapie mit
Moxonidin bzw. Clonidin. Nach Absetzen der Medikation fielen die Werte der
Plasmametanephrine in den Normbereich. Eine Interferenz der Medikamente mit dem RIA
scheint deswegen möglich zu sein. Dies steht im Gegensatz zu bisherigen Studien, in denen
vor allem trizyklische Antidepressiva, Sympathomimetika und Phenoxybenzamin als
mögliche pharmakologische Ursachen für falsch-positive Messungen der Plasmametanephrine
genannt wurden (Eisenhofer et al. 2003b, Harding et al. 2005). Insgesamt zeigen die
ermittelten Ergebnisse jedoch, dass die Werte der Plasmametanephrine mittels RIA im
Verlauf stabil bleiben und damit nur geringen Schwankungen unterliegen.
Sawka et al. (2005) untersuchten 158 Patienten (inklusive 23 Patienten mit histologisch
nachgewiesenem Phäochromozytom) auf das Vorhandensein eines Phäochromozytoms mit
Hilfe der freien Metanephrine im Plasma. Sie wiesen eine direkte Korrelation der Höhe der
Plasmametanephrinspiegel mit dem Alter der Patienten nach und konnten durch
altersspezifische Referenzwerte eine signifikante Verbesserung der Spezifität beider
Parameter erzielen. In unserer Untersuchung konnte kein Zusammenhang zwischen dem
Lebensalter und der Höhe des Plasmaspiegels der freien Metanephrine gefunden werden
(p>0,05).
Wir versuchten darüber hinaus, die Bestimmung der Plasmametanephrine durch den RIA mit
einer HPLC hinsichtlich Sensitivität und Spezifität zu vergleichen. Die Spezifität lag mit der
HPLC bei 100% für MN bzw. 89,5% für NMN verglichen mit 94,3% für MN und 99,0% für
NMN beim RIA. Es wurde eine Sensitivität sowohl für NMN als auch für MN von 100% mit
der HPLC ermittelt. Die Sensitivität für MN und NMN mit Hilfe des RIA lag jeweils bei
75,0%. Jedoch waren bei den normwertigen NMN-Bestimmungen die MN deutlich erhöht
und umgekehrt, so dass insgesamt kein falsch-negatives Ergebnis ermittelt wurde und die
Sensitivität ebenfalls bei 100% lag. Vergleicht man die Ergebnisse des RIA mit denen der
HPLC, wird jeweils eine Sensitivität von 100% erreicht bei ebenso vergleichbarer Spezifität.

49
Daher erscheint die Anwendung des RIA zur Bestimmung der Plasmametanephrine auch aus
diesem Gesichtspunkt heraus möglich.
Es lassen sich insgesamt folgende Schlussfolgerungen ziehen:
Die Bestimmung der Plasmametanephrine mit Hilfe des RIA zeigte eine hohe
diagnostische Sensitivität. In unserer Untersuchung war ein Anstieg der Metanephrine
um das 1,1fache bis 25,5fache und der Normetanephrine um das 1,2fache bis
15,3fache über den Grenzwert zu verzeichnen.
Darüber hinaus konnte eine hohe diagnostische Spezifität in der Determination der
Plasmametanephrine mittels RIA erzielt werden. In Anbetracht dieser Ergebnisse ist
ein Einsatz des RIA zur Messung der Metanephrine im Plasma in der Diagnostik des
Phäochromozytoms empfehlenswert.
Die Evaluation der Plasmametanephrine scheint beim postoperativen Follow-up
geeignet zu sein. Bei benignen klinischen Verläufen ist ein rascher postoperativer
Abfall der Werte zu verzeichnen, welcher auch mit einer abnehmenden
Beschwerdesymptomatik der Patienten einhergeht. Nach Durchführung einer
Adrenalektomie eines malignen Phäochromozytoms ist die Tendenz nachweisbar, dass
die Plasmametanephrine auch im weiteren Verlauf erhöht bleiben.
Der Ausschluss eines Phäochromozytoms bei multimorbiden Patienten, insbesondere
bei Patienten mit Niereninsuffizienz ist - im Gegensatz zu Chromogranin A im Serum-
durch die Bestimmung der Plasmametanephrine gut möglich.
Die HPLC ist ein sensitives und zugleich spezifisches Verfahren bei der Bestimmung
der freien Plasmametanephrine. Ein Nachteil der Methode liegt darin, dass sie nur an
wenigen Standorten verfügbar ist. Dies ist insbesondere dadurch bedingt, dass das
Verfahren kostenintensiv, zeitaufwändig und auf Grund von präanalytischen
Extraktionsschritten in der Methodik sehr schwierig ist. Des Weiteren sind einige
Nahrungsmittel und Medikamente beschrieben worden, die mit der HPLC
interferieren (Westphal 2005). Deswegen werden dem Patienten vor der Bestimmung
eine 12-stündige Nahrungskarenz sowie das Absetzen bestimmter Medikamente
empfohlen. Durch die Ergebnisse unserer Untersuchung konnten wir die
vorbeschriebene hohe Spezifität der HPLC bestätigen.

50
Der RIA ist im Vergleich zur HPLC wesentlich kostengünstiger und dadurch auch
besser verfügbar. Hinzu kommt, dass er pro Probe weniger zeitintensiv ist und viele
Proben gleichzeitig analysiert werden können. Ein Nachteil ist der Umgang mit
radioaktiven Substanzen, der nur dadurch vermieden werden kann, dass anstelle des
RIA ein Enzyme-linked immunosorbent assay (ELISA) verwandt wird. Eine
Beeinflussung des RIA durch Medikamente ist bisher nicht beschrieben worden. In
der durchgeführten Untersuchung scheint jedoch eine Interferenz durch bestimmte
Antihypertensiva möglich (z.B. Moxonidin, Clonidin). Insgesamt zeichnet sich der
hier getestete RIA durch eine hohe Sensitivität und Spezifität aus.
5.2.1 Katecholamine und Metanephrine im 24-h-Sammelurin
In der Gruppe der Phäochromozytome wurden 3 falsch-negative Werte bei der Bestimmung
der 24-h-Urinkatecholamine gemessen. Die Sensitivität lag bei 62,5%. Bei 71 Patienten ohne
nachgewiesenes Phäochromozytom traten 3 falsch-positive Ergebnisse auf. Somit ergab sich
eine Spezifität von 95,8%.
Es gab keine falsch-negativen Ergebnisse in der Analyse der 24-h-Urinmetanephrine bei den
Patienten mit Phäochromozytom. Jedoch waren 7 falsch-positive Ergebnisse in der
Kontrollgruppe zu verzeichnen. Eine mögliche Ursache könnte bei 2 Patienten eine
symptomatische Hypoglykämie sowie bei einem weiteren Patienten eine intrazerebrale
Blutung bei Hypertension und Therapie mit oralen Antikoagulanzien bei Vorhofflimmern
gewesen sein. Wie bereits in anderen Studien beschrieben, wird durch bestimmte klinische
Situationen (unter anderem Operationen, Hypoglykämie, intrazerebrale Blutungen, Manie) die
sympatho-adrenale Achse aktiviert (Gerlo und Sevens 1994). Dies fördert die Freisetzung der
Katecholamine und ihrer Metabolite, was zu deren falsch-positiver Messung führen kann.
Eine stationäre Behandlung wurde bei 4 Patienten wegen rezidivierender hypertensiver
Entgleisungen notwendig. Die Therapie erfolgte mit einer 2fachen bis 6fachen
antihypertensiven Kombinationsmedikation. Die Sensitivität und Spezifität der 24-h-
Urinmetanephrine lag bei 100% bzw. 89,7%. Insgesamt können folgende Aussagen getroffen
werden:
Betrachtet man die Urinkatecholamine, so sind sie in der Diagnostik des Phäochromozytoms,
insbesondere im Vergleich zur Messung von Plasma- und Urinmetanephrinen nur
eingeschränkt empfehlenswert. So zeigt sich eine hohe Spezifität von Adrenalin und
Noradrenalin bei jedoch geringer Sensitivität. Eine Erklärung für diese Tatsache liegt in der

51
Eigenschaft von Phäochromozytomen, Katecholamine nur periodisch abzugeben. Dadurch
können endokrinologisch aktive Phasen des Tumors verpasst werden, woraus falsch-negative
Messungen resultieren. Daher wird der diagnostischer Einsatz der Urinkatecholamine von
verschiedenen Autoren nur als Ergänzung zu den oben genannten Parametern empfohlen
(Lipsic et al. 2004, Karagiannis et al. 2007).
Wie diese Untersuchung ebenfalls zeigt, ist der Einsatz von Urinmetanephrinen in der
Diagnostik des Phäochromozytoms sinnvoll. Es zeigte sich eine Sensitivität von 100% bei
einer Spezifität von 89,7%. Damit kontrastieren die Ergebnisse mit denen anderer Studien, in
denen vor allem eine niedrigere Sensitivität ermittelt wurde (Lenders el al. 2002b, Unger et al.
2006). Dies wurde mit der fehlenden Organspezifität der Urinmetanephrine begründet,
wodurch die zusätzlich von einem Phäochromozytom gebildeten Metanephrine theoretisch
schlechter zu detektieren sind. In dieser Untersuchung lässt sich dies jedoch nicht nachweisen.
5.2.2 Chromogranin A im Serum
Eine Erhöhung des Chromogranin A im Serum konnte bei allen Phäochromozytompatienten,
bei denen diese Untersuchung durchgeführt wurde, nachgewiesen werden. Somit wurden
keine falsch-negativen Werte bestimmt. Bei 63 Patienten ohne nachgewiesenes
Phäochromozytom wurde das Chromogranin A ebenfalls analysiert. Hier traten 22 falsch-
positive Messungen auf. Die Spezifität lag somit insgesamt bei 65,1%. Die hohe Anzahl
falsch-positiver Messergebnisse in dieser Studie könnte unter anderem aus der Tatsache
resultieren, dass 6 der 22 Patienten mit falschpositivem Chromogranin A eine eingeschränkte
Nierenfunktion (bei diabetischer Nephropathie) aufwiesen sowie 9 Patienten mit einem
Protonenpumpenhemmer behandelt wurden. Es ist bekannt, dass diese Medikamentengruppe
ebenfalls zu einer Erhöhung des Chromogranin A beitragen kann (Blausque et al. 2003,
Wiedmann et al. 2009). Darüber hinaus beschreiben einige Studien einen Zusammenhang
zwischen der Entstehung eines Diabetes mellitus Typ 2 und dem pituitary adenylate cyclase-
activating polypeptide (PACAP) (Filipsson et al. 2001, Vaudry et al. 2002). PACAP hat
neben den Effekten auf das Pankreas auch einen stimulierenden Einfluss auf die Biosynthese
von Katecholaminen und die Sekretion von Chromogranin A, so dass eine Verbindung von
Diabetes mellitus Typ 2 und erhöhten Serumwerten von Chromogranin A bestehen könnte. In
dieser Studie scheint sich dies zu bestätigen, da 14 der 22 Patienten mit falschpositivem
Chromogranin A einen Diabetes mellitus Typ 2 hatten.

52
Anhand der hier vorliegenden Daten können jedoch folgende Feststellungen getroffen
werden:
Chromogranin A hat als Phäochromozytommarker eine hohe Sensitivität, jedoch eine
niedrige Spezifität. Die Ursache hierfür liegt darin, dass Chromogranin A
hauptsächlich renal eliminiert wird. Dadurch steigen die Serumwerte bei
beeinträchtigter Nierenfunktion schnell an. Darüber hinaus ist eine Beeinträchtigung
der Chromograninwerte durch Protonenpumpeninhibitoren und Diabetes mellitus Typ
2 möglich. Insgesamt stützen die Ergebnisse dieser Studie die Empfehlung einiger
Autoren, das Serumchromogranin A als ergänzenden Marker in der
endokrinologischen Diagnostik adrenaler Inzidentalome zu verwenden (Miehle et al.
2005, Algeciras-Schimnich et al. 2007).
5.2.3 Präklinische Einflussfaktoren
Die Bestimmung biochemischer Marker im 24-h-Sammelurin zur Diagnostik des
Phäochromozytoms hat Vor- und Nachteile. So stellt der erforderliche lange Sammelzeitraum
oftmals eine Belastung für den Patienten dar. Darüber hinaus gestaltet sich die Urinsammlung
gerade bei Kindern schwierig oder ist bei ambulanten Patienten nahezu unmöglich
durchzuführen. Auf der anderen Seite gleicht eine 24-h-Sammlung Zeiten des
Phäochromozytoms, in denen der Tumor endokrinologisch inaktiv ist, aus. Ein Problem
stellen jedoch Patienten mit Niereninsuffizienz dar. Bei ihnen ist auf Grund der
beeinträchtigten Nierenfunktion die Interpretation von Urinbestimmungen eingeschränkt. Es
wird jedoch in einigen Studien darüber diskutiert, dieses Problem zum Beispiel durch die
Bestimmung des Metanephrin-Kreatinin-Verhältnisses zu lösen (Heron et al. 1996).
Die Analyse der biochemischen Parameter zur Phäochromozytomdiagnostik im Blut weist
einige Vorteile gegenüber dem 24-h-Sammelurin auf. So entfällt die für den Patienten
aufwändige Urinsammlung und es muss nur einmal venös punktiert werden. Dies
gewährleistet eine gute Anwendbarkeit bei fast allen Patienten, insbesondere bei Kindern. Des
Weiteren wird so auch eine ambulante Diagnostik ermöglicht. Allerdings wird von einigen
Wissenschaftlern empfohlen, vor der Blutentnahme eine nächtliche Diät einzuhalten und die
Blutentnahme nach einer 20-minütigen Ruhezeit beim liegenden Patienten aus einer
Venenverweilkanüle durchzuführen, da dies falsch-positive Messergebnisse durch einen

53
erhöhten Sympathikotonus vermeiden würde (Lenders et al. 2007). In unserer Untersuchung
verzichteten wir auf das Einhalten spezifischer Maßnahmen (z.B. die Anlage einer
Venenverweilkanüle oder das Einhalten einer nächtlichen Diät) im Vorfeld der Blutentnahme.
Wie in der Diagnostik des 24-h-Sammelurins auch, findet eine Beeinträchtigung der
Aussagekraft verschiedener Serummarker bei Niereninsuffizienz statt. Dies betrifft
insbesondere die sulfatierten Metanephrine, die Vanillinmandelsäure und das Chromogranin
A. In einigen Studien wurde auch eine leichte Erhöhung der freien Plasmametanephrine
nachgewiesen (Eisenhofer et al. 2005b). Insgesamt jedoch werden die freien Metanephrine
hinsichtlich ihrer diagnostischen Aussagefähigkeit durch eine Niereninsuffizienz kaum
beeinflusst und stellen hier das Mittel der Wahl dar.
5.3 Einschränkungen dieser Studie
Hauptschwäche dieser Studie ist die geringe Anzahl an Phäochromozytompatienten. Es ist
daher nötig, die bisher ermittelten Daten durch weitere Untersuchungen und eine größere
Anzahl an Patienten mit Phäochromozytom zu bestätigen. Ferner gelang es nicht, bei allen
Patienten ohne nachgewiesenes Phäochromozytom Messungen der Urinkatecholamine,
Urinmetanephrine, Plasmametanephrine mit HPLC und Chromogranin A durchzuführen.
Die Stärke dieser Studie liegt in dem Einschluss aller zur Abklärung einer sekundären
Hypertonie untersuchten Patienten. Hierdurch wurden die freien Metanephrine im Plasma bei
Patienten mit einem breiten Spektrum verschiedener Krankheiten und der
Differentialdiagnose Phäochromozytom analysiert.
5.4 Ökonomische Gesichtspunkte
In einer in den USA durchgeführten Studie wurden verschiedene Screeningalgorithmen zur
Phäochromozytomdiagnostik aufgestellt und hinsichtlich ihrer Kosten-Nutzen-Relation
überprüft (Sawka et al. 2004b). Bei einer angenommenen Prävalenz des Phäochromozytoms
von 0,5% pro 100.000 Personen mit Bluthochdruck wurden je nach Algorithmus zwischen
457 und 489 Phäochromozytompatienten identifiziert. Demgegenüber standen Kosten
zwischen 28,6 und 56,6 Millionen Dollar. Als ursächlich für den hohen finanziellen Aufwand
werden zum einen die bei 80% bis 90% liegende Spezifität verschiedener biochemischer
Tests und zum anderen die niedrige Prävalenz der Erkrankung verantwortlich gemacht. So
errechneten Sawka et al. (2003) eine Post-Test-Wahrscheinlichkeit eines Phäochromozytoms

54
bei einem Patienten mit positiven Plasmametanephrinen und Hypertonie von 3%, bei einem
Patienten mit einem adrenalen Inzidentalom und erhöhten Plasmametanephrinen von 25%.
Auch in Deutschland steht man dem Dilemma gegenüber, ein lebensgefährliches
Krankheitsbild wie das Phäochromozytom einerseits nicht verpassen zu wollen und können,
andererseits jedoch die hohen Kosten und Folgekosten des Screenings einer sekundären
Hypertonie vermeiden zu wollen. Ein mögliches Lösungskonzept muss an vielen Punkten
angreifen. So ist zum einen ein Routinescreening auf ein Phäochromozytom bei Patienten mit
Hypertonie nicht notwendig. Viel wichtiger ist, dass der erfahrene Arzt die oftmals typische
Symptomatik des Phäochromozytompatienten erkennt und die Wahrscheinlichkeit auf das
Vorhandensein eines endokrinologischen Tumors analysiert (Goldstein et al. 1999, Mannelli
et al. 1999, Young WF 2007). Darüber hinaus können biochemische Tests, die besser (vor
allem in der Spezifität), günstiger und damit breiter verfügbar sind, die Diagnostik wesentlich
erleichtern. Der in dieser Studie getestete RIA (bzw. auch HPLC in Speziallaboratorien)
scheint dies zu ermöglichen.
In Abbildung 23 ist ein möglicher diagnostischer Ablauf zur Phäochromozytomdiagnostik
dargestellt. Er betont den Stellenwert der Metanephrine im Plasma, indem diese auf Grund
ihrer hohen diagnostischen Sensitivität und Spezifität als Initialdiagnostik des
Phäochromozytoms vorgeschlagen werden. Sollte keine Erhöhung der Metanephrine im
Plasma detektiert werden, so ist keine weitere Diagnostik hinsichtlich eines
Phäochromozytoms notwendig. Bei einer Erhöhung der Metanephrine um das mindestens 4-
fache gilt die Diagnose Phäochromozytom als gesichert und es sollte eine bildgebende
Diagnostik eingeleitet werden. Bei nur leichter Erhöhung der Plasmametanephrinwerte stellt
der 1981 von Bravo et al. vorgestellte Clonidinsuppressionstest weiterhin ein Mittel dar,
falsch-positive von richtig-positiven Testergebnissen zu unterscheiden (Bravo et al. 1981). In
Abhängigkeit des jeweiligen Befundes schließt sich auch hierauf eine anatomische und
funktionelle Bildgebung zur Lokalisation und Differenzierung des Tumors an. Ein nicht zu
unterschätzender Aspekt ist allerdings die Existenz von so genannten klinisch (Normotension
etc.) und biochemisch ‚silent’ Phäochromozytomen, die in den letzten Jahren insbesondere
durch die verbesserten bildgebenden Maßnahmen in den Fokus der Wissenschaft gelangt sind
(Kopetschke et al. 2009). Hier sind weitere Forschungen notwendig, um eine verbesserte
biochemische Diagnostik und Therapieeinleitung zu gewährleisten.

55
refraktäre Hypertonie, Risikogruppe
freie Plasmametanephrine (RIA)
sehr hoch (>4fach) grenzwertig, 2-3fache Erhöhung
negativ
Clonidin-Test
Phäo
ausgeschlossen
biochemische Bestätigung
CT / MRT des Abdomen
keine Raumforderung
Raumforderung CT / MRT Hals
keine Raumforderung
MIBG-Szinti / PET / Octreotidscan negativ
Phäo ausgeschlossen
Phäo bestätigt
operative Behandlung
Abbildung 23 Diagnostisches Vorgehen bei Verdacht auf Phäochromozytom. Abkürzungen: CT, Computerto-
mographie, MRT, Magnetresonanztomographie; PET, Positronenemissionstomographie, in Anlehnung an:
Karagiannis et al. 2007.

56
6 Zusammenfassung
Der hier verwendete RIA zeigt eine hohe diagnostische Sensitivität und Spezifität bei der
Bestimmung der freien Metanephrine im Plasma.
Der Einsatz des RIA bei Patienten mit dem Verdacht auf ein Phäochromozytom erscheint
nach Datenlage dieser Studie eine sinnvolle diagnostische Option, die gegenüber anderen
Methoden (u. a. HPLC, GC-MS) einige Vorteile aufweist:
Der RIA ist relativ zeitnah durchzuführen, kostengünstig und dadurch breit verfügbar.
Die exakte Analyse der freien Plasmametanephrine ist auch bei Patienten mit
Komorbiditäten (z.B. Niereninsuffizienz) möglich.
Eine genaue Bestimmung der freien Plasmametanephrine im Blut ist durch die schnell
und einfach zu realisierende Probenasservierung auch im ambulanten Bereich
möglich.
Es konnte keine Korrelation zwischen dem Lebensalter der Patienten und der Höhe der
Plasmametanephrinwerte festgestellt werden. Dahingehend ist eine altersspezifische
Anpassung der Grenzwerte der freien Plasmametanephrine nicht notwendig.
Die freien Plasmametanephrine mittels RIA scheinen für das postoperative Follow-up
geeignet zu sein. Ein rascher postoperativer Abfall des Normetanephrin und Metanephrin
einhergehend mit einer deutlichen klinischen Verbesserung der Symptomatik ist bei den
meisten benignen Verläufen zu beobachten. Bei einem malignen Phäochromozytom scheinen
die freien Plasmametanephrine auch im weiteren Verlauf erhöht zu bleiben. Die Daten dieser
Studie sind jedoch auf Grund einer zu geringen Patientenanzahl nur eingeschränkt
aussagekräftig. Weitere Untersuchungen müssen deshalb zur Beantwortung dieser
Fragestellung erfolgen.
Auf Grund der Ergebnisse dieser Studie ist eine Beeinflussung der Plasmametanephrinwerte
durch Erkrankungen wie arterielle Hypertonie, Niereninsuffizienz oder Diabetes mellitus
unwahrscheinlich bzw. nur in geringem Maße vorhanden. Jedoch scheint eine Interferenz des
RIA mit antihypertensiven Medikamenten wie Clonidin oder Moxonidin möglich. Hier sind
weitere Untersuchungen nötig, um diesen Verdacht zu bestätigen oder auszuschließen.

57
In dieser Arbeit zeigte sich weiterhin, dass auch ohne das Befolgen spezifischer
Verhaltensregeln vor der Blutentnahme (z.B. nächtliche Nahrungskarenz, Entnahme des
Blutes aus einer Venenverweilkanüle) keine vermehrten falsch-positiven Messergebnisse
auftreten.
Der getestete RIA bietet sich somit für den Einsatz in der Routinediagnostik des
Phäochromozytoms an, wodurch verbesserte Aussagen zum Ausschluss bzw. zur Bestätigung
der Anwesenheit des Tumors möglich werden.

58
Abkürzungsverzeichnis
Aldosereduktase AR
Alkoholdehydrogenase ADH
Catechol-O-Methyltransferase COMT
Chromogranin A CgA
Enzyme Linked Immunosorbent Assay ELISA
Flüssigchromatographische Massenspektrometrie LC-MS
Gaschromatographische Massenspektrometrie GC-MS
High Pressure Liquid Chromatography HPLC
Metanephrin MN
Monoaminooxidase MAO
Multiple Endokrine Neoplasie MEN
Metajodobenzylguanidin-Szintigraphie MIBG-Szintigraphie
Neurofibromatose 1 NF 1
Normetanephrin NMN
Paragangliomsyndrom 1-4 PGL 1-4
Polyethylenglykol PEG
Radioimmunoassay RIA
Rearranged during Transfection RET
Succinat-Dehydrogenase-Subunit A-D SDHA-D
Vanillinmandelsäure VMS
Von Hippel-Lindau-Syndrom VHL
3-Methoxy-4-hydroxyphenylglycol MHPG
3,4-Dihydroxyphenylglycolaldehyd DHPGA
3,4-Dihydroxyphenylglycol DGPG

59
Abbildungsverzeichnis
Abbildung 1 Differentialdiagnose der hereditären Phäochromozytome und
Paragangliome
Abbildung 2 Abbauwege der Katecholamine
Abbildung 3 Möglichkeiten der Katecholamin- und Metanephrinbestimmung
Abbildung 4 Charakteristika von 163 untersuchten Patienten
Abbildung 5 Charakteristika der Patienten zur Validierung der Grenzwerte
Abbildung 6 Plasmametanephrine bei 50 Patienten mit klinischem Verdacht
jedoch ohne Nachweis eines Phäochromozytoms zur Validierung
der Grenzwerte
Abbildung 7 Diagnostisches Prozedere bei 163 Patienten mit Verdacht auf
bzw. zum Ausschluss eines Phäochromozytoms
Abbildung 8 Charakteristika von 14 Patienten mit Erhöhung der
Plasmametanephrine
Abbildung 9 Plasmakonzentration von Metanephrin und Normetanephrin mit
RIA bei 113 Patienten mit klinischem Verdacht auf
Phäochromozytom
Abbildung 10 Plasmakonzentration der Metanephrine bei 8 Patienten mit
Phäochromozytom mit RIA
Abbildung 11 Plasmanormetanephrin bei 7 Phäochromozytompatienten prä- und
postoperativ mit RIA
Abbildung 12 Plasmametanephrin bei 7 Phäochromozytompatienten prä- und
postoperativ mit RIA

60
Abbildung 13 Vergleich der Plasmakonzentration der Metanephrine mit HPLC
und RIA bei 22 von 113 Patienten mit klinischem Verdacht auf
Phäochromozytom
Abbildung 14 Charakteristika von 2 Patienten mit NMN-Erhöhung mittels
HPLC
Abbildung 15 Urinkatecholamine mittels HPLC bei 79 von 113 Patienten mit
klinischem Verdacht auf Phäochromozytom
Abbildung 16 Urinmetanephrine mittels HPLC bei 76 von 113 Patienten mit
klinischem Verdacht auf Phäochromozytom
Abbildung 17 Chromogranin A im Serum bei 67 von 113 Patienten mit
klinischem Verdacht auf Phäochromozytom
Abbildung 18 2fache Bestimmung des Normetanephrin im Plasma bei 19
Patienten ohne Phäochromozytom mit RIA
Abbildung 19 2fache Bestimmung des Metanephrin im Plasma bei 19 Patienten
ohne Phäochromozytom mit RIA
Abbildung 20 Plasmanormetanephrinwerte in Abhängigkeit vom Alter der
Patienten
Abbildung 21 Plasmametanephrinwerte in Abhängigkeit vom Alter der
Patienten
Abbildung 22 Sensitivität und Spezifität der ermittelten Laborparameter
Abbildung 23 Diagnostisches Vorgehen bei Verdacht auf Phäochromozytom

61
Literaturverzeichnis
Alezais, MM und Peyron A. (1908). Un groupe nouveau de tumeurs epithéliales: les
paraganglions C.R. Seances Soc. Biol. Paris 65, 745-747.
Algeciras-Schimnich, A., Preissner, C. M., Young, W. F. Jr., Singh, R. J. und Grebe, S.K.
(2007): Plasma Chromogranin A or urine fractionated metanephrines follow-up testing
improves the diagnostic accuracy of plasma fractionated metanephrines for
pheochromocytoma. J Clin Endocrinol Metab. 93(1), 91-95.
Amar, L., Bertherat, J., Baudin, E., Ajzenberg, C., Bressac-de Paillerets, B., Chabre, O.,
Chamontin, B., Delemer, B., Giraud, S., Murat, A., Niccoli-Sire, P., Richard, S., Rohmer, V.,
Sadoul, J. L., Strompf, L., Schlumberger, M., Bertagna, X., Plouin, P. F., Jeunemaitre, X. und
Gimenez-Roqueplo, A. P. (2005): Genetic testing in pheochromocytoma or functional
paraganglioma. J. Clin. Oncol. 23, 8812-8818.
Andersen, G. S., Toftdahl, D. B., Lund, J. O., Strandgaard, S. und Nielsen, P. E. (1988): The
incidence rate of phaeochromocytoma and Conn's syndrome in Denmark, 1977-1981. J. Hum.
Hypertens. 2, 187-189.
Arighi, E., Borrello, M. G. und Sariola, H. (2005): RET tyrosine kinase signaling in
development and cancer. Cytokine Growth Factor Rev. 16, 441-467.
Aron, D. C. (2001): The adrenal incidentaloma: disease of modern technology and public
health problem. Rev. Endocr. Metab Disord. 2, 335-342.
Astuti, D., Latif, F., Dallol, A., Dahia, P. L., Douglas, F., George, E., Skoldberg, F., Husebye,
E. S., Eng, C. und Maher, E. R. (2001): Gene mutations in the succinate dehydrogenase
subunit SDHB cause susceptibility to familial pheochromocytoma and to familial
paraganglioma. Am. J. Hum. Genet. 69, 49-54.
Baguet, J. P., Hammer, L., Mazzuco, T. L., Chabre, O., Mallion, J. M., Sturm, N. und
Chaffanjon, P. (2004): Circumstances of discovery of phaeochromocytoma: a retrospective
study of 41 consecutive patients. Eur. J. Endocrinol. 150, 681-686.

62
Balogh, K., Hunyady, L., Patocs, A., Gergics, P., Valkusz, Z., Toth, M. und Racz, K. (2007):
MEN1 gene mutations in Hungarian patients with multiple endocrine neoplasia type 1. Clin.
Endocrinol. (Oxf) 67, 727-734.
Bargsten, G. und Grube, D. (1992): Serotonin storage and chromogranins: an experimental
study in rat gastric endocrine cells. J. Histochem. Cytochem. 40, 1147-1155.
Basu, T. N., Gutmann, D. H., Fletcher, J. A., Glover, T. W., Collins, F. S. und Downward, J.
(1992): Aberrant regulation of ras proteins in malignant tumour cells from type 1
neurofibromatosis patients. Nature 356, 713-715.
Bausch, B., Borozdin, W. und Neumann, H. P. (2006): Clinical and genetic characteristics of
patients with neurofibromatosis type 1 and pheochromocytoma. N. Engl. J. Med. 354, 2729-
2731.
Baysal, B. E., Ferrell, R. E., Willett-Brozick, J. E., Lawrence, E. C., Myssiorek, D., Bosch,
A., van der, M. A., Taschner, P. E., Rubinstein, W. S., Myers, E. N., Richard, C. W., III,
Cornelisse, C. J., Devilee, P. und Devlin, B. (2000): Mutations in SDHD, a mitochondrial
complex II gene, in hereditary paraganglioma. Science 287, 848-851.
Baysal, B. E., Willett-Brozick, J. E., Filho, P. A., Lawrence, E. C., Myers, E. N. und Ferrell,
R. E. (2004): An Alu-mediated partial SDHC deletion causes familial and sporadic
paraganglioma. J. Med. Genet. 41, 703-709.
Beard, C. M., Sheps, S. G., Kurland, L. T., Carney, J. A. und Lie, J. T. (1983): Occurrence of
pheochromocytoma in Rochester, Minnesota, 1950 through 1979. Mayo Clin. Proc. 58, 802-
804.
Benn, D. E., Gimenez-Roqueplo, A. P., Reilly, J. R., Bertherat, J., Burgess, J., Byth, K.,
Croxson, M., Dahia, P. L., Elston, M., Gimm, O., Henley, D., Herman, P., Murday, V.,
Niccoli-Sire, P., Pasieka, J. L., Rohmer, V., Tucker, K., Jeunemaitre, X., Marsh, D. J., Plouin,
P. F. und Robinson, B. G. (2006): Clinical presentation and penetrance of
pheochromocytoma/paraganglioma syndromes. J. Clin. Endocrinol. Metab 91, 827-836.

63
Bernini, G., Moretti, A., Fontana, V., Orlandini, C., Miccoli, P., Berti, P., Basolo, F., Faviana,
P., Bardini, M. und Salvetti, A. (2004): Plasma chromogranin A in incidental non-
functioning, benign, solid adrenocortical tumors. Eur. J. Endocrinol. 151, 215-222.
Biausque, F., Jaboureck, O., Devos, P., d´Herbomez, M., Hainaut, P., Carre, A. und Mounier-
Vehier, C. (2003): Clinical significant of serum chromogranin A levels for diagnosing
pheochromocytoma in hypertensive patients. Arch Mal Coeur Vaiss. 96 (7-8), 780-783.
Bílek, R, Safarík, L, Ciprová, V, Vlcek, P und Lisá L. (2008): Chromogranin A, a member of
neuroendocrine secretory proteins as a selective marker for laboratory diagnosis of pheo-
chromocytoma. Physiol Res. 57 Suppl 1, S171-9.
Birch-Machin, M. A., Taylor, R. W., Cochran, B., Ackrell, B. A. und Turnbull, D. M. (2000):
Late-onset optic atrophy, ataxia, and myopathy associated with a mutation of a complex II
gene. Ann. Neurol. 48, 330-335.
Bollag, G., Clapp, D. W., Shih, S., Adler, F., Zhang, Y. Y., Thompson, P., Lange, B. J.,
Freedman, M. H., McCormick, F., Jacks, T. und Shannon, K. (1996): Loss of NF1 results in
activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells.
Nat. Genet. 12, 144-148.
Boltze, C., Mundschenk, J., Unger, N., Schneider-Stock, R., Peters, B., Mawrin, C., Hoang-
Vu, C., Roessner, A. und Lehnert, H. (2003): Expression profile of the telomeric complex
discriminates between benign and malignant pheochromocytoma. J. Clin. Endocrinol. Metab
88, 4280-4286.
Bourgeron, T., Rustin, P., Chretien, D., Birch-Machin, M., Bourgeois, M., Viegas-Pequignot,
E., Munnich, A. und Rotig, A. (1995): Mutation of a nuclear succinate dehydrogenase gene
results in mitochondrial respiratory chain deficiency. Nat. Genet. 11, 144-149.
Box, J. C., Braithwaite, M. D., Duncan, T. und Lucas, G. (1997): Pheochromocytoma, chronic
renal insufficiency, and hemodialysis: a combination leading to a diagnostic and therapeutic
dilemma. Am. Surg. 63, 314-316.

64
Bravo, E. L., Tarazi R. C., Fouad F. M., Vidt D. G. und Gifford R. W. jr. (1981): Clonidine
suppression: a useful aid in the diagnosis of pheochromocytoma. N Engl J Med. 305, 623-
626.
Bravo, E. L. und Tagle, R. (2003): Pheochromocytoma: state-of-the-art and future prospects.
Endocr. Rev. 24, 539-553.
Bravo, E. L. (2004): Pheochromocytoma: current perspectives in the pathogenesis, diagnosis,
and management. Arq Bras. Endocrinol. Metabol. 48, 746-750.
Buchberg, A. M., Cleveland, L. S., Jenkins, N. A. und Copeland, N. G. (1990): Sequence
homology shared by neurofibromatosis type-1 gene and IRA-1 and IRA-2 negative regulators
of the RAS cyclic AMP pathway. Nature 347, 291-294.
Canale, M. P. und Bravo, E. L. (1994): Diagnostic specificity of serum chromogranin-A for
pheochromocytoma in patients with renal dysfunction. J. Clin. Endocrinol. Metab 78, 1139-
1144.
Cawthon, R. M., O'Connell, P., Buchberg, A. M., Viskochil, D., Weiss, R. B., Culver, M.,
Stevens, J., Jenkins, N. A., Copeland, N. G. und White, R. (1990): Identification and
characterization of transcripts from the neurofibromatosis 1 region: the sequence and genomic
structure of EVI2 and mapping of other transcripts. Genomics 7, 555-565.
Chandrasekharappa, S. C., Guru, S. C., Manickam, P., Olufemi, S. E., Collins, F. S., Emmert-
Buck, M. R., Debelenko, L. V., Zhuang, Z., Lubensky, I. A., Liotta, L. A., Crabtree, J. S.,
Wang, Y., Roe, B. A., Weisemann, J., Boguski, M. S., Agarwal, S. K., Kester, M. B., Kim, Y.
S., Heppner, C., Dong, Q., Spiegel, A. M., Burns, A. L. und Marx, S. J. (1997): Positional
cloning of the gene for multiple endocrine neoplasia-type 1. Science 276, 404-407.
Cotesta, D., Caliumi, C., Alo, P., Petramala, L., Reale, M. G., Masciangelo, R., Signore, A.,
Cianci, R., D'Erasmo, E. und Letizia, C. (2005): High plasma levels of human chromogranin
A and adrenomedullin in patients with pheochromocytoma. Tumori 91, 53-58.

65
Crockett, D. K., Frank, E. L. und Roberts, W. L. (2002): Rapid analysis of metanephrine and
normetanephrine in urine by gas chromatography-mass spectrometry. Clin. Chem. 48, 332-
337.
d'Herbomez, M., Gouze, V., Huglo, D., Nocaudie, M., Pattou, F., Proye, C., Wemeau, J. L.
und Marchandise, X. (2001): Chromogranin A assay and (131)I-MIBG scintigraphy for
diagnosis and follow-up of pheochromocytoma. J. Nucl. Med. 42, 993-997.
Eisenhofer, G., Rundquist, B., Aneman, A., Friberg, P., Dakak, N., Kopin, I. J., Jacobs, M. C.
und Lenders, J. W. (1995): Regional release and removal of catecholamines and extraneuronal
metabolism to metanephrines. J. Clin. Endocrinol. Metab 80, 3009-3017.
Eisenhofer, G., Aneman, A., Hooper, D., Rundqvist, B. und Friberg, P. (1996): Mesenteric
organ production, hepatic metabolism, and renal elimination of norepinephrine and its
metabolites in humans. J. Neurochem. 66, 1565-1573.
Eisenhofer, G., Keiser, H., Friberg, P., Mezey, E., Huynh, T. T., Hiremagalur, B., Ellingson,
T., Duddempudi, S., Eijsbouts, A. und Lenders, J. W. (1998): Plasma metanephrines are
markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J.
Clin. Endocrinol. Metab 83, 2175-2185.
Eisenhofer, G., Lenders, J. W., Linehan, W. M., Walther, M. M., Goldstein, D. S. und Keiser,
H. R. (1999): Plasma normetanephrine and metanephrine for detecting pheochromocytoma in
von Hippel-Lindau disease and multiple endocrine neoplasia type 2. N. Engl. J. Med. 340,
1872-1879.
Eisenhofer, G., Walther, M., Keiser, H. R., Lenders, J. W., Friberg, P. und Pacak, K. (2000):
Plasma metanephrines: a novel and cost-effective test for pheochromocytoma. Braz. J. Med.
Biol. Res. 33, 1157-1169.
Eisenhofer, G., Goldstein, D. S., Kopin, I. J. und Crout, J. R. (2003a): Pheochromocytoma:
rediscovery as a catecholamine-metabolizing tumor. Endocr. Pathol. 14, 193-212.

66
Eisenhofer, G., Goldstein, D. S., Walther, M. M., Friberg, P., Lenders, J. W., Keiser, H. R.
und Pacak, K. (2003b): Biochemical diagnosis of pheochromocytoma: how to distinguish
true- from false-positive test results. J. Clin. Endocrinol. Metab 88, 2656-2666.
Eisenhofer, G. (2003c): Biochemical Diagnosis of Pheochromocytoma – is it Time to Switch
to Plasma-Free Metanephrines? J Clin Endocrinol Metab. 88 (2), 550-552.
Eisenhofer, G., Lenders, J. W. und Pacak, K. (2004): Biochemical diagnosis of
pheochromocytoma. Front Horm Res. 31, 76-106.
Eisenhofer, G., Huysmans, F., Pacak, K., Walther, M. M., Sweep, F. C. und Lenders, J. W.
(2005): Plasma metanephrines in renal failure. Kidney Int. 67, 668-677.
Elsayes, K. M., Narra, V. R., Leyendecker, J. R., Francis, I. R., Lewis, J. S., Jr. und Brown, J.
J. (2005): MRI of adrenal and extraadrenal pheochromocytoma. AJR Am. J. Roentgenol. 184,
860-867.
Emlet, JR, Grimson, KS, Bell, DM und Orgain, ES (1951): use of piperoxan and regitine as
routine test in patients with hypertension. JAMA 146, 1383-1386.
Eng, C., Clayton, D., Schuffenecker, I., Lenoir, G., Cote, G., Gagel, R. F., van Amstel, H. K.,
Lips, C. J., Nishisho, I., Takai, S. I., Marsh, D. J., Robinson, B. G., Frank-Raue, K., Raue, F.,
Xue, F., Noll, W. W., Romei, C., Pacini, F., Fink, M., Niederle, B., Zedenius, J.,
Nordenskjold, M., Komminoth, P., Hendy, G. N. und Mulligan, L. M. (1996): The
relationship between specific RET proto-oncogene mutations and disease phenotype in
multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA
276, 1575-1579.
Eras, M., Yenigun, M., Acar, C., Kumbasar, B., Sar, F. und Bilge, T. (2004): Pancreatic
involvement in Von Hippel-Lindau disease. Indian J. Cancer 41, 159-161.
Erem, C., Onder, E. H., Ukinc, K., Hacihasanoglu, A., Alhan, E., Cobanoglu, U., Kocak, M.
und Erdol, H. (2007): Neurofibromatosis type 1 associated with pheochromocytoma: a case
report and a review of the literature. J. Endocrinol. Invest 30, 59-64.

67
Erlic, Z. und Neumann, H. P. (2009): Familial pheochromocytoma. Hormones (Athens). 8 (1),
29-38.
Fernandez-Calvet, L. und Garcia-Mayor, R. V. (1994): Incidence of pheochromocytoma in
South Galicia, Spain. J. Intern. Med. 236, 675-677.
Ferner, R. E. (2007): Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century
perspective. Lancet Neurol. 6, 340-351.
Filipsson, K., Kvist-Reimer, M. und Ahren, B. (2001): The neuropeptide pituitary adenylate
cyclase-activating polypeptide and islet function. Diabetes 50, 1959-1969.
Flamme, I., Krieg, M. und Plate, K. H. (1998): Up-regulation of vascular endothelial growth
factor in stromal cells of hemangioblastomas is correlated with up-regulation of the
transcription factor HRF/HIF-2alpha. Am. J. Pathol. 153, 25-29.
Fränkel Felix (1886): Ein Fall von doppelseitigem, völlig latent verlaufenem
Nebennierentumor und gleichzeitiger Nephritis mit Veränderungen am Circulationsapparat
und Retinitis. Arch Pathol. Anat Physiol Klin Med 103, 244-263.
Gerlo, E. A. und Sevens, C. (1994): Urinary and plasma catecholamines and urinary
catecholamine metabolites in pheochromocytoma: diagnostic value in 19 cases. Clin. Chem.
40, 250-256.
Gimenez-Roqueplo, A. P., Favier, J., Rustin, P., Mourad, J. J., Plouin, P. F., Corvol, P., Rotig,
A. und Jeunemaitre, X. (2001): The R22X mutation of the SDHD gene in hereditary
paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory
chain and activates the hypoxia pathway. Am. J. Hum. Genet. 69, 1186-1197.
Gimenez-Roqueplo, A. P., Favier, J., Rustin, P., Rieubland, C., Kerlan, V., Plouin, P. F.,
Rotig, A. und Jeunemaitre, X. (2002): Functional consequences of a SDHB gene mutation in
an apparently sporadic pheochromocytoma. J. Clin. Endocrinol. Metab 87, 4771-4774.

68
Gimm, O. (2001): Multiple endocrine neoplasia type 2: clinical aspects. Front Horm. Res. 28,
103-130.
Giovanella, L., Squin, N., Ghelfo, A. und Ceriani, L. (2006): Chromogranin A
immunoradiometric assay in diagnosis of pheochromocytoma: comparison with plasma
metanephrines and 123I-MIBG scan. Q. J. Nucl. Med. Mol. Imaging 50, 344-347.
Glasker, S. (2005): Central nervous system manifestations in VHL: genetics, pathology and
clinical phenotypic features. Fam. Cancer 4, 37-42.
Gnarra, J. R., Zhou, S., Merrill, M. J., Wagner, J. R., Krumm, A., Papavassiliou, E., Oldfield,
E. H., Klausner, R. D. und Linehan, W. M. (1996): Post-transcriptional regulation of vascular
endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc.
Natl. Acad. Sci. U. S. A 93, 10589-10594.
Goldstein, D. S., Eisenhofer, G. und Kopin, I. J. (2003): Sources and significance of plasma
levels of catechols and their metabolites in humans. J. Pharmacol. Exp. Ther. 305, 800-811.
Goldstein, D. S., Eisenhofer, G., Flynn, J. A., Wand, G. und Pacak, K. (2004): Diagnosis and
localization of pheochromocytoma. Hypertension 43, 907-910.
Goldstein RE, O'Neill JA Jr, Holcomb GW 3rd, Morgan WM 3rd, Neblett WW 3rd, Oates JA,
Brown N, Nadeau J, Smith B, Page DL, Abumrad NN, Scott HW (1999): Clinical experience
over 48 years with pheochromocytoma. Jr Ann Surg. 229(6):755-64.
Gorr, S. U., Shioi, J. und Cohn, D. V. (1989): Interaction of calcium with porcine adrenal
chromogranin A (secretory protein-I) and chromogranin B (secretogranin I). Am. J. Physiol
257, E247-E254.
Gortz, B., Roth, J., Speel, E. J., Krahenmann, A., de Krijger, R. R., Matias-Guiu, X., Muletta-
Feurer, S., Rutmann, K., Saremaslani, P., Heitz, P. U. und Komminoth, P. (1999): MEN1
gene mutation analysis of sporadic adrenocortical lesions. Int. J. Cancer 80, 373-379.

69
Grossrubatscher, E., Dalino, P., Vignati, F., Gambacorta, M., Pugliese, R., Boniardi, M.,
Rossetti, O., Marocchi, A., Bertuzzi, M. und Loli, P. (2006): The role of chromogranin A in
the management of patients with phaeochromocytoma. Clin. Endocrinol. (Oxf) 65, 287-293.
Guillemot, J., Barbier, L., Thouennon, E., Vallet-Erdtmann, V., Montero-Hadjadje, M.,
Lefebvre, H., Klein, M., Muresan, M., Plouin, P. F., Seidah, N., Vaudry, H., Anouar, Y. und
Yon, L. (2006): Expression and processing of the neuroendocrine protein secretogranin II in
benign and malignant pheochromocytomas. Ann. N. Y. Acad. Sci. 1073, 527-532.
Gujral, T. S. und Mulligan, L. M. (2006): Molecular implications of RET mutations for
pheochromocytoma risk in multiple endocrine neoplasia 2. Ann. N. Y. Acad. Sci. 1073, 234-
240.
Harding, J. L., Yeh, M. W., Robinson, B. G., Delbridge, L. W. und Sidhu, S. B. (2005):
Potential pitfalls in the diagnosis of phaeochromocytoma. Med. J. Aust. 182, 637-640.
Hartley, L. und Perry-Keene, D. (1985): Phaeochromocytoma in Queensland--1970-83. Aust.
N. Z. J. Surg. 55, 471-475.
Heppner, C., Reincke, M., Agarwal, S. K., Mora, P., Allolio, B., Burns, A. L., Spiegel, A. M.
und Marx, S. J. (1999): MEN1 gene analysis in sporadic adrenocortical neoplasms. J. Clin.
Endocrinol. Metab 84, 216-219.
Hernandez, F. C., Sanchez, M., Alvarez, A., Diaz, J., Pascual, R., Perez, M., Tovar, I. und
Martinez, P. (2000): A five-year report on experience in the detection of pheochromocytoma.
Clin. Biochem. 33, 649-655.
Heron, E., Chatellier, G., Billaud, E., Foos, E. und Plouin, P. F. (1996): The urinary
metanephrine-to-creatinine ratio for the diagnosis of pheochromocytoma. Ann. Intern. Med.
125, 300-303.
Heutink, P., van der Mey, A. G., Sandkuijl, L. A., van Gils, A. P., Bardoel, A., Breedveld, G.
J., van Vliet, M., van Ommen, G. J., Cornelisse, C. J., Oostra, B. A. und . (1992): A gene
subject to genomic imprinting and responsible for hereditary paragangliomas maps to
chromosome 11q23-qter. Hum. Mol. Genet. 1, 7-10.

70
Hickman; P. E., Leong; M., Chang; J., Wilson; S. R. und McWhinney, B. (2009): Plasma free
metanephrines are superior to urine and plasma catecholamines and urine catecholamine
metabolites for the investigation of phaeochromocytoma. Pathology 41 (2), 173-177.
Huang, H., Abraham, J., Hung, E., Aberbuch, S., Merino, M., Steinberg, S. M., Pacak, K. und
Fojo, T. (2008): Treatment of malignant pheochromocytoma/paraganglioma with
cyclophosphamide, vincristine and dacarbazine: recommendation from a 22-Year Follow-up
of 18 Patients. Cancer 113 (8), 2020-2028.
Huang, S. C., Koch, C. A., Vortmeyer, A. O., Pack, S. D., Lichtenauer, U. D., Mannan, P.,
Lubensky, I. A., Chrousos, G. P., Gagel, R. F., Pacak, K. und Zhuang, Z. (2000): Duplication
of the mutant RET allele in trisomy 10 or loss of the wild-type allele in multiple endocrine
neoplasia type 2-associated pheochromocytomas. Cancer Res. 60, 6223-6226.
Iwai, K., Yamanaka, K., Kamura, T., Minato, N., Conaway, R. C., Conaway, J. W., Klausner,
R. D. und Pause, A. (1999): Identification of the von Hippel-lindau tumor-suppressor protein
as part of an active E3 ubiquitin ligase complex. Proc. Natl. Acad. Sci. U. S. A 96, 12436-
12441.
Jimenez, C. und Gagel, R. F. (2004): Genetic testing in endocrinology: lessons learned from
experience with multiple endocrine neoplasia type 2 (MEN2). Growth Horm. IGF. Res. 14
Suppl A, S150-S157.
John, A. M., Ruggieri, M., Ferner, R. und Upadhyaya, M. (2000): A search for evidence of
somatic mutations in the NF1 gene. J. Med. Genet. 37, 44-49.
Kamura, T., Sato, S., Iwai, K., Czyzyk-Krzeska, M., Conaway, R. C. und Conaway, J. W.
(2000): Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL)
tumor suppressor complex. Proc. Natl. Acad. Sci. U. S. A 97, 10430-10435.
Karagiannis, A., Mikhailidis, D. P., Athyros, V. G. und Harsoulis, F. (2007):
Pheochromocytoma: an update on genetics and management. Endocr. Relat Cancer 14, 935-
956.

71
Katai, M., Sakurai, A., Itakura, Y., Ikeo, Y., Nakajima, K., Hara, M., Iijima, S., Kaneko, T.,
Kobayashi, M., Ichikawa, K., Aizawa, T. und Hashizume, K. (1997): Multiple endocrine
neoplasia type 1 is not rare in Japan. Endocr. J. 44, 841-845.
Knudson, A. G., Jr. und Strong, L. C. (1972): Mutation and cancer: neuroblastoma and
pheochromocytoma. Am. J. Hum. Genet. 24, 514-532.
Knudson, A. G., Jr. (1986): Genetics of human cancer. Annu. Rev. Genet. 20, 231-251.
Koch, C. A., Vortmeyer, A. O., Huang, S. C., Alesci, S., Zhuang, Z. und Pacak, K. (2001):
Genetic aspects of pheochromocytoma. Endocr. Regul. 35, 43-52.
Koch, C. A., Pacak, K. und Chrousos, G. P. (2002): The molecular pathogenesis of hereditary
and sporadic adrenocortical and adrenomedullary tumors. J. Clin. Endocrinol. Metab 87,
5367-5384.
Koch, C. A. (2005): Molecular pathogenesis of MEN2-associated tumors. Fam. Cancer 4, 3-7.
Koch, C. A., Gimm, O., Vortmeyer A. O., Al-Ali, H. K., Lamesch, P., Ott, R., Kluge, R.,
Bierbach, U. und Tannapfel, A. (2006): Does the expression of c-kit (CD117) in
neuroendocrine tumors represent a target for therapy? Ann N Y Acad Sci 1073, 517-526.
Kopetschke, R., Slisko, M., Kilisli, A., Tuschy, U., Wallaschofski, H., Fassnacht, M., Ventz,
M., Beuschlein, F., Reincke, M., Reisch, N. und Quinkler, M. (2009): Frequent incidental
discovery of phaeochromocytoma: data from a German cohort of 201 phaeochromocytoma.
Eur J Endocrinol. 161 (2), 355-361
Krieg, M., Haas, R., Brauch, H., Acker, T., Flamme, I. und Plate, K. H. (2000): Up-regulation
of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal
carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19,
5435-5443.

72
Kudva, Y. C., Sawka, A. M. und Young, W. F., Jr. (2003): Clinical review 164: The
laboratory diagnosis of adrenal pheochromocytoma: the Mayo Clinic experience. J. Clin.
Endocrinol. Metab 88, 4533-4539.
Lagerstedt, S. A., O'Kane, D. J. und Singh, R. J. (2004): Measurement of plasma free
metanephrine and normetanephrine by liquid chromatography-tandem mass spectrometry for
diagnosis of pheochromocytoma. Clin. Chem. 50, 603-611.
Lakhani, V. T., You, Y. N. und Wells, S. A. (2007): The multiple endocrine neoplasia
syndromes. Annu. Rev. Med. 58, 253-265.
Lammert, M., Friedman, J. M., Kluwe, L. und Mautner, V. F. (2005): Prevalence of
neurofibromatosis 1 in German children at elementary school enrollment. Arch. Dermatol.
141, 71-74.
Latif, F., Tory, K., Gnarra, J., Yao, M., Duh, F. M., Orcutt, M. L., Stackhouse, T., Kuzmin, I.,
Modi, W., Geil, L. und . (1993): Identification of the von Hippel-Lindau disease tumor
suppressor gene. Science 260, 1317-1320.
Lee, N. C. und Norton, J. A. (2000): Multiple endocrine neoplasia type 2B--genetic basis and
clinical expression. Surg. Oncol. 9, 111-118.
Lenders, J. W., Eisenhofer, G., Armando, I., Keiser, H. R., Goldstein, D. S. und Kopin, I. J.
(1993): Determination of metanephrines in plasma by liquid chromatography with
electrochemical detection. Clin. Chem. 39, 97-103.
Lenders, J. W., Keiser, H. R., Goldstein, D. S., Willemsen, J. J., Friberg, P., Jacobs, M. C.,
Kloppenborg, P. W., Thien, T. und Eisenhofer, G. (1995): Plasma metanephrines in the
diagnosis of pheochromocytoma. Ann. Intern. Med. 123, 101-109.
Lenders, J. W., Pacak, K. und Eisenhofer, G. (2002a): New advances in the biochemical
diagnosis of pheochromocytoma: moving beyond catecholamines. Ann. N. Y. Acad. Sci. 970,
29-40.

73
Lenders, J. W., Pacak, K., Walther, M. M., Linehan, W. M., Mannelli, M., Friberg, P., Keiser,
H. R., Goldstein, D. S. und Eisenhofer, G. (2002b): Biochemical diagnosis of
pheochromocytoma: which test is best? JAMA 287, 1427-1434.
Lenders, J. W., Willemsen, J. J., Eisenhofer, G., Ross, H. A., Pacak, K., Timmers, H. J. und
Sweep, C. G. (2007): Is supine rest necessary before blood sampling for plasma
metanephrines? Clin. Chem. 53, 352-354.
Lipsic, E., Balazovjech, I., Kosmalova, V., Makaiova, I., Dekret, J. und Zanou, D. F. (2004):
Epinephrine producing pheochromocytoma. Is the secretory pattern decisive for the clinical
manifestation? J. Endocrinol. Invest 27, 691-694.
Lisztwan, J., Imbert, G., Wirbelauer, C., Gstaiger, M. und Krek, W. (1999): The von Hippel-
Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity.
Genes Dev. 13, 1822-1833.
Lonser, R. R., Glenn, G. M., Walther, M., Chew, E. Y., Libutti, S. K., Linehan, W. M. und
Oldfield, E. H. (2003): von Hippel-Lindau disease. Lancet 361, 2059-2067.
Machens, A., Gimm, O., Hinze, R., Hoppner, W., Boehm, B. O. und Dralle, H. (2001):
Genotype-phenotype correlations in hereditary medullary thyroid carcinoma: oncological
features and biochemical properties. J. Clin. Endocrinol. Metab 86, 1104-1109.
Maher, E. R., Iselius, L., Yates, J. R., Littler, M., Benjamin, C., Harris, R., Sampson, J.,
Williams, A., Ferguson-Smith, M. A. und Morton, N. (1991): Von Hippel-Lindau disease: a
genetic study. J. Med. Genet. 28, 443-447.
Mannelli, M., Ianni, L., Cilotti, A. und Conti, A. (1999): Pheochromocytoma in Italy: a
multicentric retrospective study. Eur. J. Endocrinol. 141, 619-624.
Mariman, E. C., van Beersum, S. E., Cremers, C. W., Struycken, P. M. und Ropers, H. H.
(1995): Fine mapping of a putatively imprinted gene for familial non-chromaffin
paragangliomas to chromosome 11q13.1: evidence for genetic heterogeneity. Hum. Genet. 95,
56-62.

74
Marini, F., Falchetti, A., Del Monte, F., Carbonell, S. S., Tognarini, I., Luzi, E. und Brandi,
M. L. (2006): Multiple endocrine neoplasia type 2. Orphanet. J. Rare. Dis. 1, 45
Marini, M., Fathi, M. und Vallotton, M. (1994): [Determination of serum metanephrines in
the diagnosis of pheochromocytoma]. Ann. Endocrinol. (Paris) 54, 337-342.
Marx, S. J. (2005): Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat.
Rev. Cancer 5, 367-375.
Mayo C. H. (1924): Paroxysmal hypertension with tumor of retroperitoneal nerve: report of a
case. JAMA 89, 1047-1050.
Miehle, K., Kratzsch, J., Lenders, J. W., Kluge, R., Paschke, R. und Koch, C. A. (2005):
Adrenal incidentaloma diagnosed as pheochromocytoma by plasma chromogranin A and
plasma metanephrines. J. Endocrinol. Invest 28, 1040-1042.
Morioka, M., Yuihama, S., Nakajima, T., Kobayashi, T., Sone, A., Furukawa, Y., Tanaka, H.
und Saiki, T. (2002): Incidentally discovered pheochromocytoma in long-term hemodialysis
patients. Int. J. Urol. 9, 700-703.
Mulligan, L. M., Kwok, J. B., Healey, C. S., Elsdon, M. J., Eng, C., Gardner, E., Love, D. R.,
Mole, S. E., Moore, J. K., Papi, L. und . (1993): Germ-line mutations of the RET proto-
oncogene in multiple endocrine neoplasia type 2A. Nature 363, 458-460.
Neumann, H. P. und Wiestler, O. D. (1991): Clustering of features and genetics of von
Hippel-Lindau syndrome. Lancet 338, 258
Neumann, H. P., Berger, D. P., Sigmund, G., Blum, U., Schmidt, D., Parmer, R. J., Volk, B.
und Kirste, G. (1993): Pheochromocytomas, multiple endocrine neoplasia type 2, and von
Hippel-Lindau disease. N. Engl. J. Med. 329, 1531-1538.
Neumann, H. P., Bausch, B., McWhinney, S. R., Bender, B. U., Gimm, O., Franke, G.,
Schipper, J., Klisch, J., Altehoefer, C., Zerres, K., Januszewicz, A., Eng, C., Smith, W. M.,

75
Munk, R., Manz, T., Glaesker, S., Apel, T. W., Treier, M., Reineke, M., Walz, M. K., Hoang-
Vu, C., Brauckhoff, M., Klein-Franke, A., Klose, P., Schmidt, H., Maier-Woelfle, M.,
Peczkowska, M., Szmigielski, C. und Eng, C. (2002): Germ-line mutations in nonsyndromic
pheochromocytoma. N. Engl. J. Med. 346, 1459-1466.
Neumann, H. P., Vortmeyer, A., Schmidt, D., Werner, M., Erlic, Z., Cascon, A., Bausch, B.,
Januszewicz, A. und Eng, C. (2007): Evidence of MEN-2 in the original description of classic
pheochromocytoma. N. Engl. J. Med. 357, 1311-1315.
Nguyen, L., Niccoli-Sire, P., Caron, P., Bastie, D., Maes, B., Chabrier, G., Chabre, O.,
Rohmer, V., Lecomte, P., Henry, J. F. und Conte-Devolx, B. (2001): Pheochromocytoma in
multiple endocrine neoplasia type 2: a prospective study. Eur. J. Endocrinol. 144, 37-44.
Niemann, S., Steinberger, D. und Muller, U. (1999): PGL3, a third, not maternally imprinted
locus in autosomal dominant paraganglioma. Neurogenetics. 2, 167-170.
Nobels, F. R., Kwekkeboom, D. J., Coopmans, W., Schoenmakers, C. H., Lindemans, J., De
Herder, W. W., Krenning, E. P., Bouillon, R. und Lamberts, S. W. (1997): Chromogranin A
as serum marker for neuroendocrine neoplasia: comparison with neuron-specific enolase and
the alpha-subunit of glycoprotein hormones. J. Clin. Endocrinol. Metab 82, 2622-2628.
Opocher, G., Conton, P., Schiavi, F., Macino, B. und Mantero, F. (2005): Pheochromocytoma
in von Hippel-Lindau disease and neurofibromatosis type 1. Fam. Cancer 4, 13-16.
Peczkowska, M. und Januszewicz, A. (2005): Multiple endocrine neoplasia type 2. Fam.
Cancer 4, 25-36.
Pick L. (1912): Das Ganglioma embryonale sympathicum (Sympathoma embryonale), eine
typische bösartige Geschwulstform des sympathischen Nervensystems. Berl. Klin.
Wochenschr. 49, 16-22.
Pisano J. J. (1960): A simple analysis for normetanephrine and metanephrine in urine. Clin
Chim Acta 5, 406-414.

76
Pisano, J. J., Crout, J. R. and Abraham, D. (1962): Determination of 3-methoxy-4-
hydroxymandelic acid in urine. Clin Chim Acta 7, 285-291.
Plouin, P. F., Duclos, J. M., Menard, J., Comoy, E., Bohuon, C. und Alexandre, J. M. (1981):
Biochemical tests for diagnosis of phaeochromocytoma: urinary versus plasma
determinations. Br. Med. J. (Clin. Res. Ed) 282, 853-854.
Pollard, P. J., Briere, J. J., Alam, N. A., Barwell, J., Barclay, E., Wortham, N. C., Hunt, T.,
Mitchell, M., Olpin, S., Moat, S. J., Hargreaves, I. P., Heales, S. J., Chung, Y. L., Griffiths, J.
R., Dalgleish, A., McGrath, J. A., Gleeson, M. J., Hodgson, S. V., Poulsom, R., Rustin, P. und
Tomlinson, I. P. (2005): Accumulation of Krebs cycle intermediates and over-expression of
HIF1alpha in tumours which result from germline FH and SDH mutations. Hum. Mol. Genet.
14, 2231-2239.
Poyhonen, M. (2000): A clinical assessment of neurofibromatosis type 1 (NF1) and segmental
NF in Northern Finland. J. Med. Genet. 37, E43
Raber, W., Raffesberg, W., Bischof, M., Scheuba, C., Niederle, B., Gasic, S., Waldhausl, W.
und Roden, M. (2000): Diagnostic efficacy of unconjugated plasma metanephrines for the
detection of pheochromocytoma. Arch. Intern. Med. 160, 2957-2963.
Reisch, N., Peczkowska, M., Januszewicz, A. und Neumann, H. P. (2006):
Pheochromocytoma: presentation, diagnosis and treatment. J. Hypertens. 24, 2331-2339.
Roth, G.M. und Kvale W.F. (1945): “A tentative Test for Pheochromocytoma,” Am J Med
Sci 210, 653:
Rubin, G. L., Sharp, S., Jones, A. L., Glatt, H., Mills, J. A. und Coughtrie, M. W. (1996):
Design, production and characterization of antibodies discriminating between the phenol- and
monoamine-sulphating forms of human phenol sulphotransferase. Xenobiotica 26, 1113-1119.
Saegesser, F. (1984): [Cesar Roux (1857-1934) and his times]. Rev. Med. Suisse Romande
104, 403-464.

77
Saeki, T., Suzuki, K., Yamazaki, H., Miyamura, S., Koike, H., Morishita, H. und Gejyo, F.
(2003): Four cases of pheochromocytoma in patients with end-stage renal disease. Intern.
Med. 42, 1011-1015.
Sawka, A. M., Jaeschke, R., Singh, R. J. und Young, W. F., Jr. (2003): A comparison of
biochemical tests for pheochromocytoma: measurement of fractionated plasma metanephrines
compared with the combination of 24-hour urinary metanephrines and catecholamines. J.
Clin. Endocrinol. Metab 88, 553-558.
Sawka, A. M., Prebtani, A. P., Thabane, L., Gafni, A., Levine, M. und Young, W. F., Jr.
(2004a): A systematic review of the literature examining the diagnostic efficacy of
measurement of fractionated plasma free metanephrines in the biochemical diagnosis of
pheochromocytoma. BMC. Endocr. Disord. 4, 2
Sawka, A. M., Gafni, A., Thabane, L. und Young, W. F., Jr. (2004b): The economic
implications of three biochemical screening algorithms for pheochromocytoma. J. Clin.
Endocrinol. Metab 89, 2859-2866.
Sawka, A. M., Thabane, L., Gafni, A., Levine, M. und Young, W. F., Jr. (2005):
Measurement of fractionated plasma metanephrines for exclusion of pheochromocytoma: Can
specificity be improved by adjustment for age? BMC. Endocr. Disord. 5, 1
Scholz, T., Eisenhofer, G., Pacak, K., Dralle, H. und Lehnert, H. (2007): Clinical review:
Current treatment of malignant pheochromocytoma. J. Clin. Endocrinol. Metab 92, 1217-
1225.
Schussheim, D. H., Skarulis, M. C., Agarwal, S. K., Simonds, W. F., Burns, A. L., Spiegel, A.
M. und Marx, S. J. (2001): Multiple endocrine neoplasia type 1: new clinical and basic
findings. Trends Endocrinol. Metab 12, 173-178.
Selak, M. A., Armour, S. M., MacKenzie, E. D., Boulahbel, H., Watson, D. G., Mansfield, K.
D., Pan, Y., Simon, M. C., Thompson, C. B. und Gottlieb, E. (2005): Succinate links TCA
cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 7,
77-85.

78
Serra, E., Ars, E., Ravella, A., Sanchez, A., Puig, S., Rosenbaum, T., Estivill, X. und Lazaro,
C. (2001): Somatic NF1 mutational spectrum in benign neurofibromas: mRNA splice defects
are common among point mutations. Hum. Genet. 108, 416-429.
Siemeister, G., Weindel, K., Mohrs, K., Barleon, B., Martiny-Baron, G. und Marme, D.
(1996): Reversion of deregulated expression of vascular endothelial growth factor in human
renal carcinoma cells by von Hippel-Lindau tumor suppressor protein. Cancer Res. 56, 2299-
2301.
Solis, D. C., Burnichon, N., Timmers, H. J., Raygada, M. J., Kozupa, A., Merino, M. J.,
Makey, D., Adams, K. T., Venisse, A., Gimenez-Roqueplo, A. P. und Pacak, K. (2009):
Penetrance and clinical consequences of a gross SDHB deletion in a large family. Clin Genet.
75, 354-363
Stenstrom, G. und Svardsudd, K. (1986): Pheochromocytoma in Sweden 1958-1981. An
analysis of the National Cancer Registry Data. Acta Med. Scand. 220, 225-232.
Stumvoll, M., Radjaipour, M. und Seif, F. (1995): Diagnostic considerations in
pheochromocytoma and chronic hemodialysis: case report and review of the literature. Am. J.
Nephrol. 15, 147-151.
Tanimoto, K., Makino, Y., Pereira, T. und Poellinger, L. (2000): Mechanism of regulation of
the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein.
EMBO J. 19, 4298-4309.
Taupenot, L., Harper, K. L. und O'Connor, D. T. (2003): The chromogranin-secretogranin
family. N. Engl. J. Med. 348, 1134-1149.
Thompson, G. B. und Young, W. F., Jr. (2003): Adrenal incidentaloma. Curr. Opin. Oncol.
15, 84-90.
Timmers, H. J., Kozupa, A., Eisenhofer, G., Raygada, M., Adams, K. T., Solis, D., Lenders, J.
W. und Pacak, K. (2007): Clinical presentations, biochemical phenotypes, and genotype-

79
phenotype correlations in patients with succinate dehydrogenase subunit B-associated
pheochromocytomas and paragangliomas. J. Clin. Endocrinol. Metab 92, 779-786.
Tonelli, F., Fratini, G., Falchetti, A., Nesi, G. und Brandi, M. L. (2005): Surgery for
gastroenteropancreatic tumours in multiple endocrine neoplasia type 1: review and personal
experience. J. Intern. Med. 257, 38-49.
Unger, N., Pitt, C., Schmitt, I. L:, Walz, M. K., Philipp T., Mann, K. und Petersenn S. (2004):
Diagnostic value of various biochemical parameters for the diagnosis of pheochromocytoma.
Exp Clin Endocrinol Diabetes 112, v042
Unger, N., Pitt, C., Schmidt, I. L., Walz, M. K., Schmid, K. W., Philipp, T., Mann, K. und
Petersenn, S. (2006): Diagnostic value of various biochemical parameters for the diagnosis of
pheochromocytoma in patients with adrenal mass. Eur. J. Endocrinol. 154, 409-417.
van Houtum, W. H., Corssmit, E. P., Douwes Dekker, P. B., Jansen, J. C., van der Mey, A.
G., Brocker-Vriends, A. H., Taschner, P. E., Losekoot, M., Frolich, M., Stokkel, M. P.,
Cornelisse, C. J. und Romijn, J. A. (2005): Increased prevalence of catecholamine excess and
phaeochromocytomas in a well-defined Dutch population with SDHD-linked head and neck
paragangliomas. Eur. J. Endocrinol. 152, 87-94.
Vaudry, D. und Taupenot, L. (2002): Fast-breaking results on the PACAP/VIP/secretin
peptide family in chromaffin cells. Ann. N. Y. Acad. Sci. 971, 460-466.
Viskochil, D., Buchberg, A. M., Xu, G., Cawthon, R. M., Stevens, J., Wolff, R. K., Culver,
M., Carey, J. C., Copeland, N. G., Jenkins, N. A. und . (1990): Deletions and a translocation
interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 62, 187-192.
Von Euler, U. und Lishaiko, F. (1961): Improved technique for the fluorimetric estimation of
catecholamines. Acta Physiol. Scand. 51, 348-355.
Wallace, M. R., Marchuk, D. A., Andersen, L. B., Letcher, R., Odeh, H. M., Saulino, A. M.,
Fountain, J. W., Brereton, A., Nicholson, J., Mitchell, A. L. und . (1990): Type 1

80
neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients.
Science 249, 181-186.
Walther, M. M., Reiter, R., Keiser, H. R., Choyke, P. L., Venzon, D., Hurley, K., Gnarra, J.
R., Reynolds, J. C., Glenn, G. M., Zbar, B. und Linehan, W. M. (1999): Clinical and genetic
characterization of pheochromocytoma in von Hippel-Lindau families: comparison with
sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J. Urol.
162, 659-664.
Wassell, J., Reed, P., Kane, J. und Weinkove, C. (1999): Freedom from drug interference in
new immunoassays for urinary catecholamines and metanephrines. Clin. Chem. 45, 2216-
2223.
Watkinson, A. und Dockray, G. J. (1992): Functional control of chromogranin A and B
concentrations in the body of the rat stomach. Regul. Pept. 40, 51-61.
Westphal, S. A. (2005): Diagnosis of Pheochromocytoma. Am J Med Sci 329 (1), 18-21.
Widimsky, J. Jr. (2006): Recent Advances in the Diagnosis and Treamtent of
Pheochromocytoma. Kidney Blood Press Res. 29, 321-326.
Wiedmann, W. M., Mössner, J., Stumvoll, M. und Führer, D. (2009): Case report: extreme
CgA elevation under PPI confounding assessment of adrenal mass. Z Gastroenterol. 47 (7),
671-673.
Wiesener, M. S., Munchenhagen, P. M., Berger, I., Morgan, N. V., Roigas, J., Schwiertz, A.,
Jurgensen, J. S., Gruber, G., Maxwell, P. H., Loning, S. A., Frei, U., Maher, E. R., Grone, H.
J. und Eckardt, K. U. (2001): Constitutive activation of hypoxia-inducible genes related to
overexpression of hypoxia-inducible factor-1alpha in clear cell renal carcinomas. Cancer Res.
61, 5215-5222.
Willenberg, H. S., Schott, M., Saeger, W., Tries, A., Scherbaum, W. A. und Bornstein, S. R.
(2006): Expression of connexins in chromaffin cells of normal human adrenals and in benign
and malignant pheochromocytomas. Ann. N. Y. Acad. Sci. 1073, 578-583.

81
Wolthers, B. G., Kema, I. P., Volmer, M., Wesemann, R., Westermann, J. und Manz, B.
(1997): Evaluation of urinary metanephrine and normetanephrine enzyme immunoassay
(ELISA) kits by comparison with isotope dilution mass spectrometry. Clin. Chem. 43, 114-
120.
World Health Organization 2004 WHO classification of tumors 2004. In Pathology and
Genetics of Tumors of Endocrine Organs, Eds RA De Lellis, RV Lloyd, PV Heitz and Eng.
France: IARC Press
Xu, G. F., O'Connell, P., Viskochil, D., Cawthon, R., Robertson, M., Culver, M., Dunn, D.,
Stevens, J., Gesteland, R., White, R. und . (1990): The neurofibromatosis type 1 gene encodes
a protein related to GAP. Cell 62, 599-608.
Yohay, K. (2006): Neurofibromatosis types 1 and 2. Neurologist. 12, 86-93.
Young WF Jr. (2007). Adrenal causes of hypertension: pheochromocytoma and primary
hyperaldosteronism. Rev Endocr Metab Disord. 8 (4):309-320.
Zhou, G.W., Wei, Y., Chen, X., Jiang, X., Ning, G. und Li H. (2009): Diagnosis and surgical
treatment of multiple endocrine neoplasia. Chin Med J. 122 (13):1495-1500.

82
Erklärung über die eigenständige Abfassung der Arbeit
Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne unzulässige Hilfe
oder Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Ich versichere, dass
Dritte von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten
haben, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen, und dass
die vorgelegte Arbeit weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer
anderen Prüfungsbehörde zum Zweck einer Promotion oder eines anderen Prüfungsverfahrens
vorgelegt wurde. Alles aus anderen Quellen und von anderen Personen übernommene
Material, das in der Arbeit verwendet wurde oder auf das direkt Bezug genommen wird,
wurde als solches kenntlich gemacht. Insbesondere wurden alle Personen genannt, die direkt
an der Entstehung der vorliegenden Arbeit beteiligt waren.
15.03.2010............................... ....................................
Datum Unterschrift

83
Lebenslauf
Persönliche Daten
Name: Ulrich Miehe
Anschrift: Brockhausstr. 20
04229 Leipzig
Geburtsdatum: 06.06.1981
Geburtsort: Magdeburg
Nationalität: deutsch
Familienstand: ledig, keine Kinder
Schullaufbahn
08/1988-07/2000 Grundschule Magdeburg
Gymnasium Magdeburg
Gymnasium: Oschersleben
Abschluss: Allgemeine Hochschulreife, Note 1,2
Grundwehrdienst
09/2000-07/2001 Panzergrenadierlehrbataillon 92 in
Munster/Niedersachsen
Studium
10/2001 Studium der Humanmedizin, Universität Leipzig
08/2003 Ärztliche Vorprüfung, Note 2,6
08/2006 Beginn des Praktischen Jahres
Famulaturen
02/2004 Kardiologie, Klinikum St. Georg/Leipzig
02/2005 Gastroenterologie, University of Sydney/Australia
02/2006 Notfallambulanz, Universitätsklinikum Leipzig
03/2006 Neurologie, Universitätsklinikum Leipzig
Praktisches Jahr
08/2006-12/2006 Chirurgie am Spital Bern Tiefenau/Schweiz
12/2006-04/2007 Innere Medizin: 8 Wochen Universitätsklinikum
Leipzig (Rheumatologie), 8 Wochen Herzzentrum
Leipzig (Kardiologie)
04/2007-08/2007 Pädiatrie: Universitätsklinikum Leipzig

84
Berufliche Erfahrungen Seit 02/2008 Assistenzarzt in Weiterbildung, Universitätsklinikum
Leipzig, Abteilung für Pädiatrie
Promotion
Evaluation eines Radioimmunoassays zur Bestimmung der freien Metanephrine im Plasma
im Vergleich zu bisher angewandten biochemischen Verfahren in der Diagnostik des
Phäochromozytoms. Beginn: 06/2004
Betreuer:
Prof. Dr. med. C. A. Koch, Division of Endocrinology, University of Mississippi Medical
Center, Jackson, Mississippi/USA
Prof. Dr. rer. nat. J. Kratzsch, Institut für Laboratoriumsmedizin, Klinische Chemie und
Molekulare Diagnostik, Universitätsklinkum Leipzig
Zusatzqualifikationen
Sprachen: Sehr gute Kenntnisse in Englisch und Latein
Grundkenntnisse in Französisch
EDV-Kenntnisse: Microsoft Office, SPSS
Fortbildungskurse 15th
Budapest Nephrology School 26.8-31.8.2008
Hochschullehrertraining Leipzig 3.10.-06.10.2009
Vorträge: Schuster, V., Falkenberg, C., Miehe, U. 8. Interdisziplinäres
Leipziger Allergiegespräch, 27.5.2009
Prenzel, F., Miehe, U., Hammerschmidt, S. „Kinderzentrum-
Kinderpneumologie“, Leipzig, 30.09.2009
Interessen: Leichtathletik und Handball (jeweils 7 Jahre Vereinsspieler)
Leipzig, 15.03.2010





























