Embed Size (px)
Citation preview

Hämorrhagische Diathesen
Modul 3.4 Erkrankungen des Blutes

Hämostase – StörungenEinteilungen
• Blutungs - Neigungen• Thrombose - Neigungen• Leicht• Schwer• Angeboren• Erworben • Vasculär• Thrombozytär• Plasmatisch
Primäre Hämostase
Sekundäre Hämostase
Tertiäre Hämostase

Vorlesung Hämostaseologie
Angeborene hämorrhagische Diathesen– Hämophilie– Von Willebrand Syndrom– Morbus Osler
Klinik und Differentialdiagnostik

Geschichte der Hämophilie
Babylonischer Talmud (ca. 500 n. Chr.)Bericht über 4 Schwestern zu Sephoris
„ Die erste ließ ihren Sohn beschneiden, und er starb; die zweite, und er starb; die dritte, und er starb; die vierte trat dann vor Rabban Simeon ben Gamaliel, der zu ihr sprach: Seht ab von der Beschneidung.., denn es gibt Familien, deren Blut locker ist, andere, bei denen es gerinnt."

Geschichte der Hämophilie
Begriff “Hämophilie“ (=Vorliebe zu Blut) geprägt1828 durch Schüler von Johann Lukas Schoenlein(Dissertation des Friedrich Hopf: „Über die Hämophilie oder die erbliche Neigung zu tödlichen Blutungen“)Vorheriger Terminus: Hämorrhaphilia = Neigung zu Blutungen
Verlängerung der Gerinnungszeit bei HämophilieWright 1893
Identifizierung des Faktor VIIIBrinkhaus & Quick 1947
Identifizierung des F IX (Christmas Factor) Aggeler 1952

Geschichte der Hämophilie
Ab 1957 Behandlung mit F VIII angereichertem Plasmasowie den ersten PPSB-Präparaten
Ab 1970 Bereitstellung lagerfähiger F VIII und PPSB-Konzentrate -> Voraussetzung für Selbstbehandlung
Um 1983 Anstieg der Mortalität der HämophilienDurch Virusinfektionen (Hepatitis B und C, AIDS)
Ab 1986 ausschließlicher Einsatz virusinaktivierter Präparate
1988 Entwicklung von gentechnisch hergestellten Präparaten
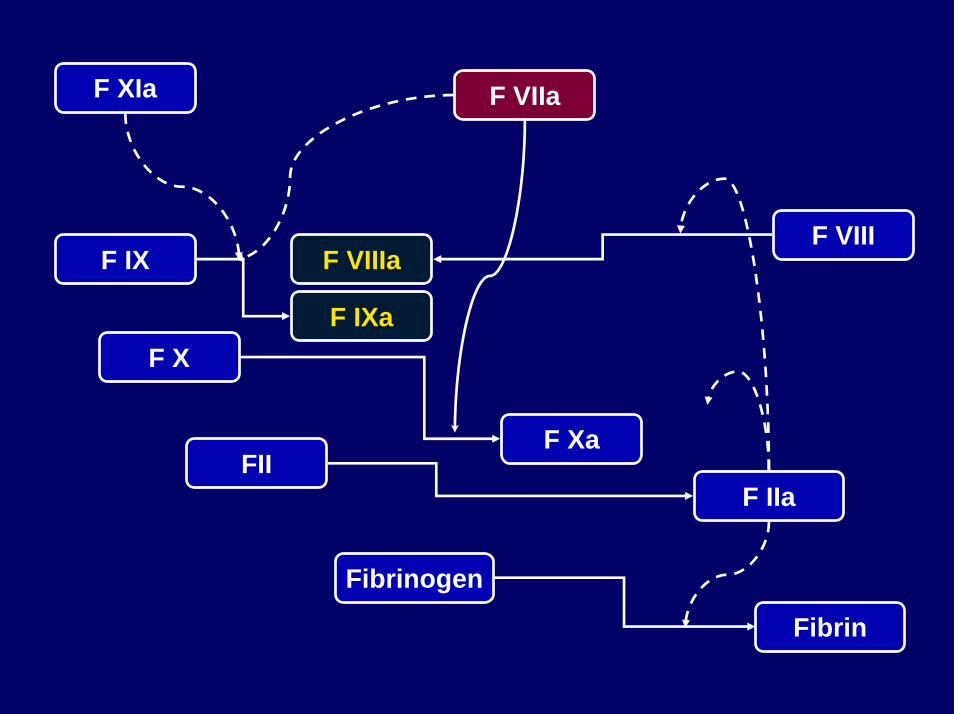
F IX
F IXaF X
F Xa
F VIIIaF VIII
FIIF IIa
Fibrinogen
Fibrin
F XIa F VIIa

Funktion des Faktor VIII
• Katalysiert (keine Proteolyse) als Kofaktor die Aktivierung des Faktors X durch aktivierten Faktor IX (IXa)
• Weitere für diesen Prozess notwendige Kofaktoren sindCalzium und Phospholipide
• Durch die Interaktion mit Faktor IXa wird die Aktivierungvon Faktor X (zu Xa) um das 300.000-fache beschleunigt

Faktor VIII
• Spurenprotein mit ca. 280.000 D• Wird durch Thrombin zu FVIIIa aktiviert• Wird nach Synthese in der Leber ins Plasma
sezerniert• Halbwertszeit ca. 8 bis 12 Stunden• Zirkuliert gebunden an Willebrand Faktor

Genomdefekte der Hämophilie
Kein einheitliche Mutationsvariante, sondern Vielzahl an quantitativ und qualitativ unterschiedlichen Veränderungen(z.B. 597 unterschiedliche Mutationen bei 1535 untersuchten Patienten mit Hämophilie B lt. Gianelli et al., 1997)
Mutationsformen im Faktor VIII oder Faktor IX Gen
- Punktmutationen (> 80) Meist leichtere Hämophilieformen - Nonsensmutationen hohes Risiko für schwere Hämophilie- Deletionsmutationen hohes Risiko für schwere Hämophilie
Bei Hämophilie A > 60 bekannt, Länge 1 Basenpaar bis 200 kBVor allem bei den längerem Deletionenstypen Neigung zur Hemmkörperbildung
- Insertionsmutatationen (selten)- Stopmutationen

Klassifizierung der Hämophilie A / B
F VIIIc / F IX
• Schwere Hämophilie A < 1 % (<0,01E/ml)
• Mittelschwere Hämophilie A 1-5 %
• Leichte Hämophilie A 5-15 %
• Subhämophilie A 15-40 %

Klinik der Hämophilien• Schwere Formen
( < 3% Restaktivität, Neigung zu spontanen Blutungen)
und leichte Formen ( > 3 % Restaktivität, Blutung meist erst nach Traumata)
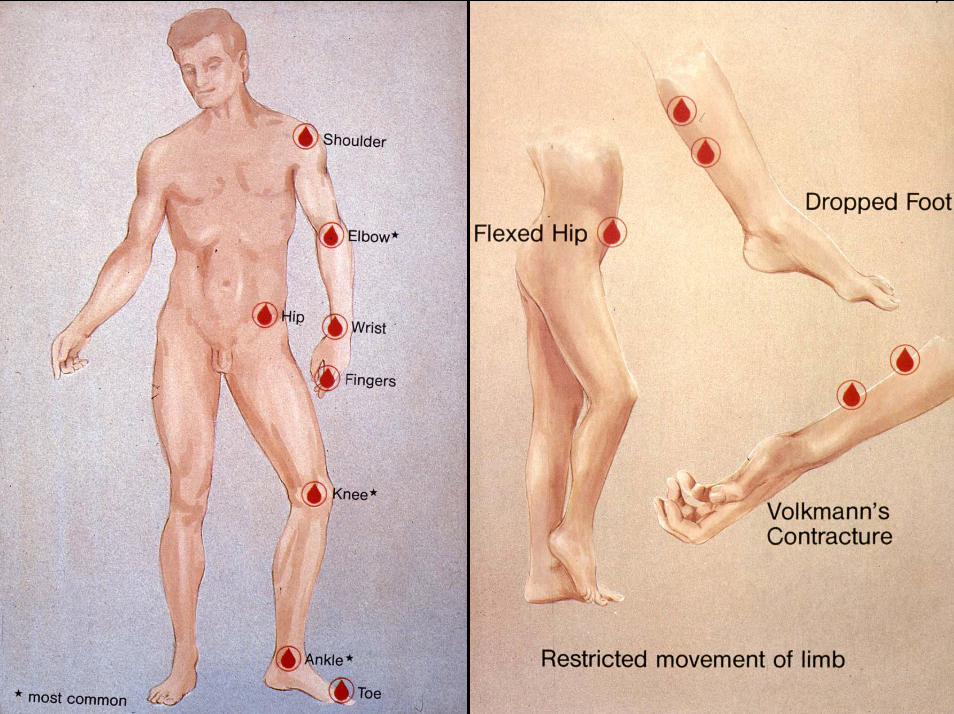
• X-chromosomal vererbt, ca. 1: 10.000 (A) / 1:30.000 (B)• Gelenkblutungen (Knie, Sprunggelenk)• Hämophile Arthropathie• Muskelblutungen• Hirnblutungen• Blutungen nach Operationen & Wundheilungsstörungen• Bei einigen Patienten Aura vor spontanen Blutungen

Erstmanifestationen der schweren Hämophilie A und BLechner K, Handbuch der Inneren Medizin
• Spontanblutungen 36 (67%)– Hämatome 18 (33%)– Gelenksblutungen 20 (37%)– Muskelblutungen 2– Epistaxis 2– Hämaturie 1
• Posttraumatische /perioperative Blutungen 18 (33%)– Lippen-Zungen-Biß 7– Traumatische Hämatome 7– Postoperative Blutungen 4
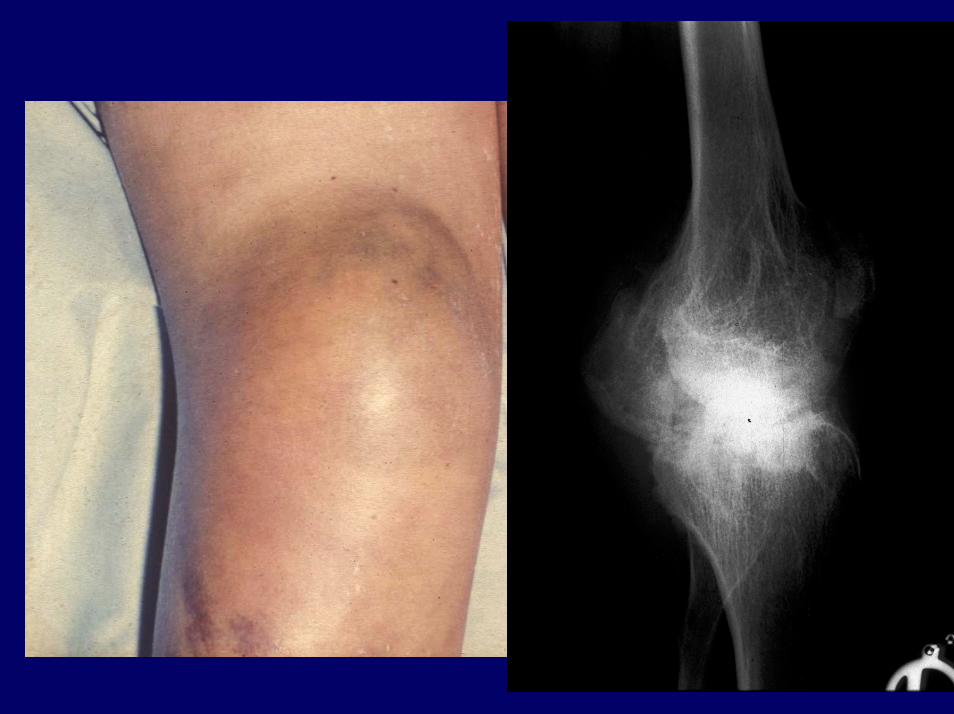

Stadien der hämophilen ArthropathieP. Hoffmann et al., 1982
• Stadium I- Akute hämophile Arthropathie– Intraartikuläre Blutung– akute Synovitis– Schonhaltung
• Stadium II- Chronische hämophile Arthropathie– chronische Synovitis– muskulo-ligamentäre Instabilität– reflektorische Kontraktur
• Stadium III- Synovialhyperplasie– Muskelatrophie– Bandinsuffizienz– myogene Kontraktur
• Stadium IV- Degenerative hämophile Arthropathie– Fibrosierung der Synovia– weitgehender Muskelschwund– ligamentär fixierte Kontraktur
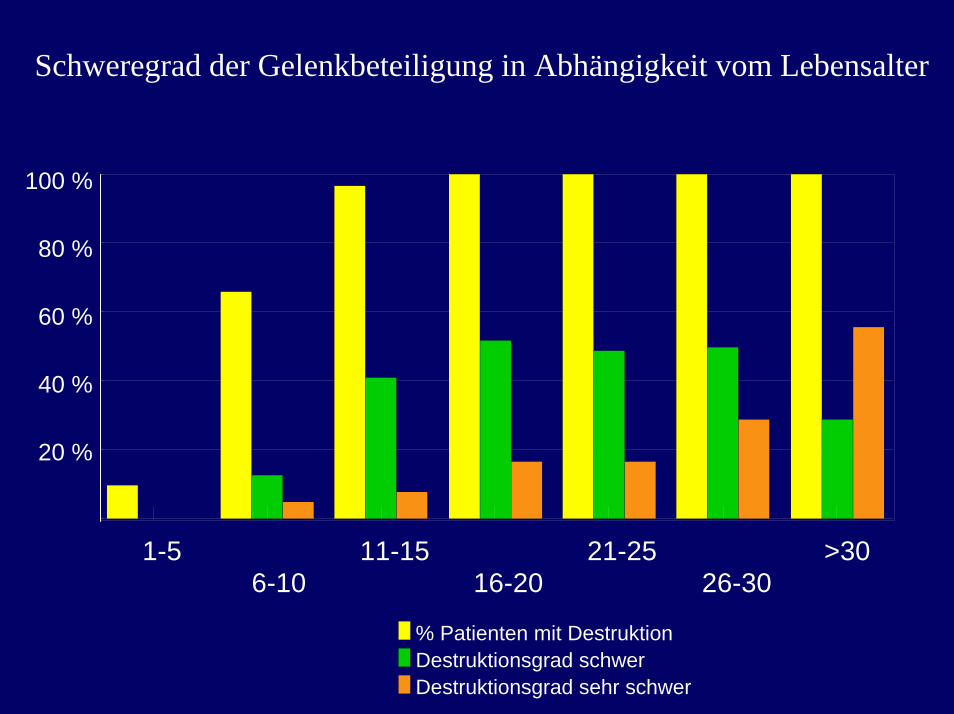
Schweregrad der Gelenkbeteiligung in Abhängigkeit vom Lebensalter
1-56-10
11-1516-20
21-2526-30
>300
20
40
60
80
100
% Patienten mit DestruktionDestruktionsgrad schwerDestruktionsgrad sehr schwer
100 %
80 %
60 %
40 %
20 %

Labordiagnostik der Hämophilie
• aPTT – Verlängerung • Thrombelastogramm
(Reaktionszeit bis zu 60-100 Minuten)• Einzelfaktoranalyse (F VIII:c / F IX)
Cave: Mehrfachanalyse, für zukünftige Planung der Substitution auch nach Gabe vonFaktorenkonzentraten oder DDAVP
• Molekulargenetische Untersuchung zur Bestimmung der zu Grunde liegenden Mutation

Behandlung der Hämophilie
• Bei den schweren Hämophilien muss der fehlende Gerinnungsfaktor ersetzt werden– Hämophilie A: Faktor VIII-Konzentrat– Hämophilie B: Faktor IX-Konzentrat
Faustregel:
1 E/kg führt zu einem Anstieg des jeweiligen Faktors um 1-2 %
Halbwertszeiten: Faktor VIII:c 8-12 Std. Faktor IX 18-24 Std.

Behandlung der HämophilieProphylaktische Therapie- 20-30 E/kg 3x/Woche bei Hämophilie A- Wegen längerer Halbwertszeit seltener bei Hämophilie B
Therapie bei BedarfSubstitution nur - nach eingetretenen Blutungsereignissen
- vor besonderen Belastungen
Initialdosis [E/kg] Initialdosis [E/kg]
Gelenkblutungen 20-40 Epistaxis, Hämaturie 20-40Weichteilblutungen Operationen- Bedrohlich / ausgedehnt 40-60 - Große Wundfläche bzw.- Kleinere Haut-/Muskelblutung 15-30 Blutungsgefahr 50-80Gastrointestinal 30-60 - Kleine Eingriffe 25-40

Behandlung der Hämophilie
• Bei den schweren Hämophilien muss der fehlende Gerinnungsfaktor ersetzt werden– Hämophilie A: Faktor VIII-Konzentrat– Hämophilie B: Faktor IX-Konzentrat
• Behandlung von Blutungen• Blutungsverhütende Therapie
– HeimselbstbehandlungPatient in eine weniger schwere Hämophilie überführen
– Klinik im Rahmen von Operationen bis zum Abschluss der Wundheilung

Alternative bzw. ergänzende Therapeutika
• Desmopressin- DDAVP, 1-Desamino-8-D-arginin-vasopressin, Analogon des Anti-diuretischen Hormons ADH, Minirin, 0,4 µg/kg
- führt zur Ausschüttung von FVIII, variables Ansprechen- nur bei leichterer Hämophilie nach individueller Prüfung,
bei kleinerem Eingriff
• Fibrinkleber• zahnärztlichen Eingriffen unterstützend• Antifibrinolytika
- -Aminocapronsäure (50 mg/kg, mehrmals täglich)- Tranexamsäure (Ugorol, 25 mg/kg)
• Glukokortikoide (Hämaturie)

Komplikationen der Hämophilie-Behandlung
• Inhibitorbildung– Kein Ansprechen auf Therapie
• Allergische Reaktionen
• Infektiöse Komplikationen– Heutzutage selten– Früher HIV, HCV, HBV
FVIII

HemmkörperhämophilieFVIIIInduzierte Antikörperbildung gegen allogen
substituierte Faktoren, fast nur bei schweren Verlaufsformen
Bei ausgeprägten molekularen Gendefekten (“große Deletion“)6-10 x häufiger als bei leichten Gendefekten (“Punktmutation“)Genetische Prädisposition zur Hemmkörperbildung wirdvermutet (HLA-Klasse II-Allele DQB0602 & DR15)
Häufigkeit: 10-15 % bei Hämophilie Aca. 3 % bei Hämophilie B
Sonderfall: Erworbene Hemmkörper (1:1.000.000 Personen/Jahr)führen in Form von Auto-Antikörpern bei Gesundenzu schweren Blutungen (nach Schwangerschaften oder bei Autoimmunerkrankungen)

Hemmkörperhämophilie - EinteilungFVIII
Bestimmung der Stärke des Hemmkörpers Erfolgt nach Methode von Bethesta:
Eine Bethesta Einheit (BE) entspricht der Menge an Antikörper, welche die Faktor VIII/IX-Aktivitätin Normalplasma um 50 % verringert
- Permanent hochtitrige Hemmkörper (> 5 BE, “high responder“)- Permenant niedrigtitrige Hemmkörper (< 5 BE, “low responder“)- Transiente Hemmkörper (meist niedrigtitrig, passageres Auftreten)

HemmkörperhämophilieFVIIITherapie
akut zur Blutstillung– extrem hohe FVIII-Dosen, porciner FVIII– Plasmaaustausch, Immunadsorption– FEIBA (factor VIII bypassing activity)– Rekombinanter Faktor VIIa
• zur Behandlung des Inhibitors– Immuntoleranz durch hohe FVIII-Dosen
(150 E/kg zweimal täglich über mehrere Monate !)
– Immunsuppression, high-dose IgG

Konsensus-Empfehlungen zur Hämophiliebehandlung in Deutschland
W. Schramm, Hämostaseologie 14:81, 1994
• Ziele der Hämophilie-Therapie– Die Verhütung von Blutungen.– Die Behandlung von Blutungen, deren
Komplikationen und Folgeschäden.– Die Erhaltung und/oder Wiederherstellung der
Gelenkfunktionen.– Die Integration des Hämophilen in ein normales
soziales Leben.

Zusammenfassung Hämophilie A
• Inzidenz Hämophilie A ca. 1:10.000, Hämophilie B 5x seltener• x-chromosomal vererbt- ca. 25% Neumutationen• klinisch schwer oder mittelschwer/ (ca. 50%) oder leicht• bei schwerer Hämophilie Spontan- und posttraumatische
Blutungen, v.a. Gelenks-, Muskel-, aber auch gastrointestinaleund cerebrale Blutungen, unbehandelte postoperative Blutungen führen oft zu tage- bis wochenlangen Nachblutungen
• bei leichteren Hämophilien stehen postoperative/traumatische Blutungen im Vordergrund, sie werden oft spät erkannt (leere Anamnese bei Neuauftreten!)
• Therapie der 1. Wahl sind Faktorenkonzentrate• bei kleineren Eingriffen und leichteren Hämophilien können
alternativ Desmopressin, Antifibrinolytika, lokale Hämostyptikaeingesetzt werden (individuell, erfordert klinische Erfahrung)

Willebrand Syndrom(von Willebrand- Jürgensche Erkrankung)
• Störung der Primärhämostase, die durch einen Defekt des Willebrand- Faktors hervorgerufen wird.
• Der Willebrand Faktor ist für die Interaktion von Thrombozyten mit dem Subendothel essentiell.

Willebrand Faktor
• Genort: Kurzer Arm Chromosom 12, 178 kB• Endothelzellen und Megakaryozyten• Monomer 2050 AS, 260.000 D• Multimere mit MG 10.000.000 D• Bildung in Endothelzellen und Megakaryozyten• 7µg/ml; Halbwertszeit ca. 12 h

Klassifikation der Willebrand´schen Erkrankung
Typ I- vWF vermindert, dominant, variable Expression
Typ II- Qualitativer Defekt des vWF,
meist dominant
Typ III- Kein vWF, rezessiv
Der Faktor VIII kann bei der von Willebrand´schenErkrankung ebenfalls vermindert sein, da derWillebrand Faktor Trägerprotein für Faktor VIII ist.

Klinik der Willebrand´schen Erkrankung
• Nasenbluten und Hämatomneigung
• Nach chirurgischen Eingriffen Nachblutungen,v. a. Zahnextraktionen und Tonsillektomie
• Verstärkte Regelblutung bei Frauen
• Bei schwerer Erkrankung (Typ III) kann eine hämophilieartige Symptomatik mit Gelenk-und Muskelblutungen auftreten
• Häufigkeit ca. 1. 5000 - 1: 10.000, erbliche Erkrankung

Diagnostik & Laboranalysen des vWF
• vWF- Antigen• Ristocetin-induzierte Aggregation (RIPA)• Ristocetin-Cofactor• vWF: Collagen- Bindung• Multimeren-Analyse• Molekularbiologische Diagnostik• Faktor VIII häufig vermindert• Blutungszeit

(In-Vivo-) Blutungszeit nach Ivy
Durchführung- 40 mmHg Stau (Manschette)- Definierter Hautschnitt- Repetetives Abtupfen des
sich bildenen Bluttropfens, bis die Blutung steht
InterpretationBis 10 Minuten: normal10-15 Minuten: leicht verlängert> 15 Minuten: pathologisch
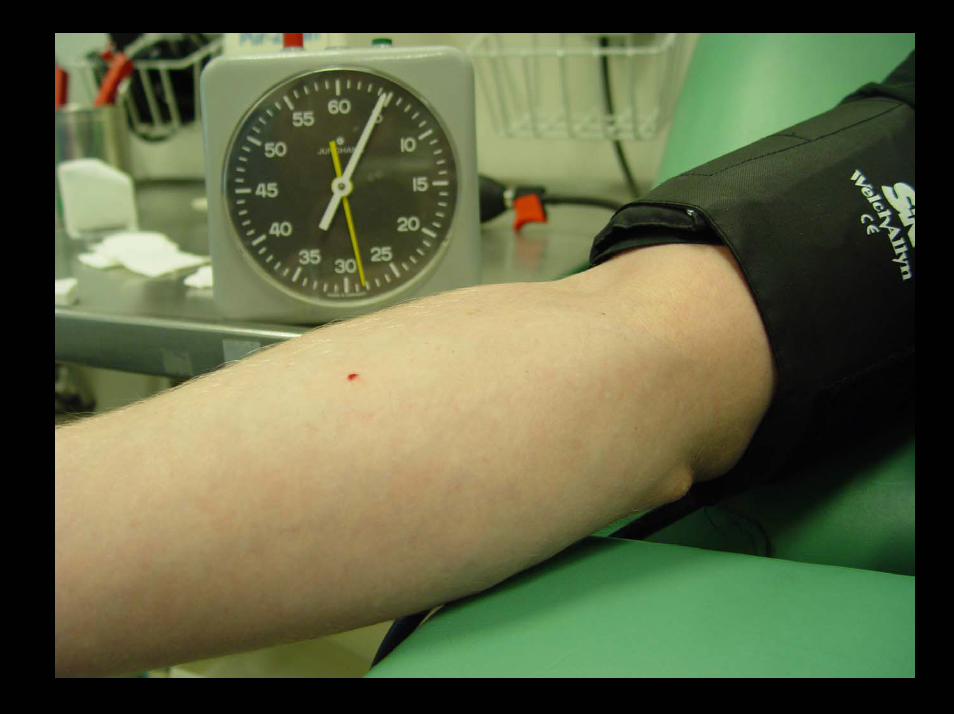

(In-Vivo-) Blutungszeit nach Ivy
Bewertung
- Pathologisch verlängerte Blutungszeit spricht fürthrombozytäre Funktionsstörung inklusive Störung der thrombozytären Adhäsion (vWF !) oder
- Vaskuläre Blutungsneigung
- Plasmatisch bedingte Hämorrhagien führen nicht zueiner Verlängerung der Blutungszeit

Therapie der Willebrand´schen Erkrankung
• Bei leichten Formen Therapieversuch mit Desmopressin (Minirin), variables Ansprechen, führt zur Freisetzung des vWF aus den Weibel-Palade Körperchen
• Bei schweren Formen Substitution mit vWF-reichem FVIII- Konzentrat (Haemate HS®)bzw. vWF-Konzentrat (30-50 E/kg)
• Lokale Blutstillung• Antifibrinolytika

Zusammenfassung von Willebrand Syndrom
• Inzidenz ca. 1:10.000• Leichtere Formen dominant, schwere rezessiv vererbt• klinisch schwere Form (vor allem Typ III) selten• bei leichten Formen Epistaxis, Hämatomneigung, verstärkte
Regelblutung bei Frauen sowie Nachblutung bei chirurgischen Eingriffen
• bei schwerer Form z.T. hämophilieartige Symptomatik• Diagnostisch Verlängerung der Blutungszeit• Therapie der 1. Wahl sind Faktorenkonzentrate• bei kleineren Eingriffen und leichteren Formen können
alternativ Desmopressin, Antifibrinolytika, lokale Hämostyptika eingesetzt werden

Erworbene Hämostasestörungen
• Thrombozytopenie• Medikamentös induzierte Hämostasestörungen• Verdünnungskoagulopathie• Vitamin K- Mangel• Lebererkrankungen• Urämie• Disseminierte Intravasale Gerinnung (DIG)

Erkrankungen der Gefässe
• Primäre Purpura• Sekundäre Purpura
– Purpura fulminans– Allergische purpura– Dysproteinämisch
• Bindegewebserkrankungen– Fabry´sche Erkrankung– Ehlers-Danlos Syndrom– Hereditäre haemorrhagische Teleangiektasie
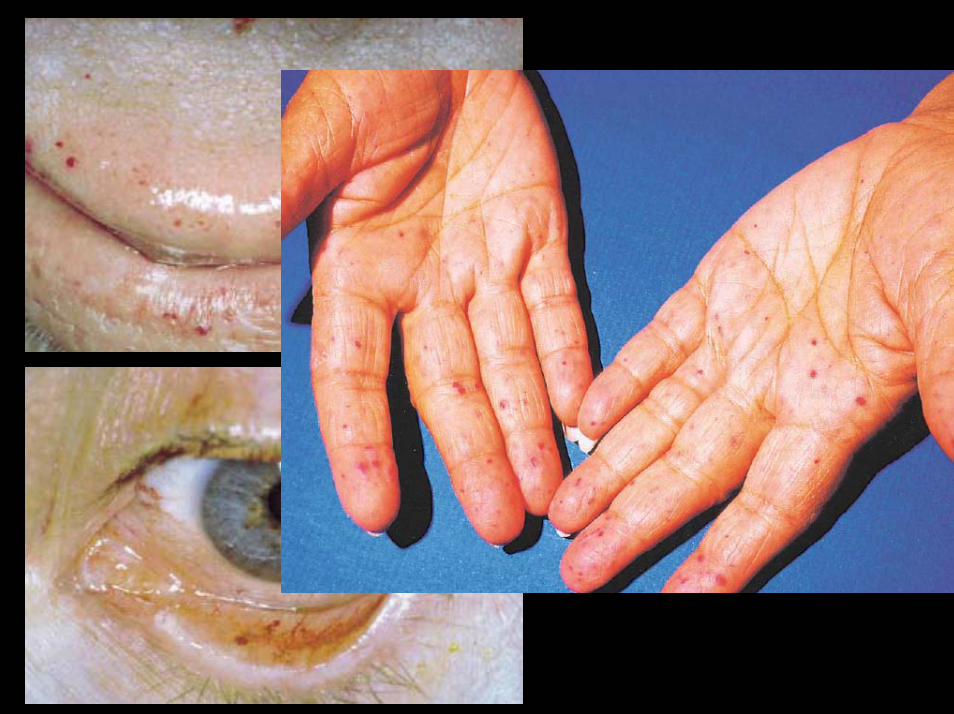

Hereditäre hämorrhagische TeleangiektasieRendu-Osler-Weber Syndrom
• Angeborene, autosomal dominante (variable Penetranz) Erkrankung
• Häufigkeit zwischen 1: 2.000 und 1: 40.000• Blickdiagnose: Teleangieektasien von Haut
und Schleimhäuten• Teleangieektasien auch in Darm, Lunge,
Gehirn, etc.• Klinik: Nasenbluten, Hautblutungen,
Darmblutungen (Blutungsanämie)

Anamnese bei hämorrhagischen Diathesen
• Jetzige Beschwerden, warum kommt der Patient• Eigenanamnese
– Spontanblutungen, oder Blutungen bei Belastung– Hämatomneigung, Nasenbluten, verstärkte Regelblutung,
Blutung bei Zahnextraktionen, Tonsillektomie oder nah anderen operativen Eingriffen
– Weitere Erkrankungen, v.a. Leber• Familienanamnese
– Familiäre Häufung, Erbgang?• Medikamentenanamnese

Untersuchung bei hämorrhagischen Diathesen
• Inspektion– Blutungen, petechial oder flächig– Narbenbildung– Gefässanomalien– Bindegewebserkrankungen
• Hinweise für Lebererkrankungen• Blutungsanämie• Blutungszeit


















