Embed Size (px)
Citation preview

1
Kapitel 6
Grundtypen organisch-
chemischer Reaktionen

2
Arten von organisch- chemischen Reaktionen
Reaktion Reaktionstyp Reaktions-
geschehen
A + B-C → A-B + C Substitution Ersatz eines
Teils des Moleküls
durch ein anderes
Teilchen
A + B-C → A-B-C Addition aus zwei oder mehr
Teilchen entsteht ein
Molekül
A-B-C → A-B + C Eliminierung von einem Molekül
werden ein oder
mehrere Teilchen
abgespalten
A → B Umlagerung Umwandlung in ein
isomeres Molekül

3
Teilchen, die an Reaktionen teilnehmen
• Nucleophile
„kernliebende“ Teilchen,
selbst elektronenreich (negativ polarisiert, oft mit negativer Ladung);
reagieren mit elektronenarmen Teilchen
• Elektrophile
„elektronenliebende“ Teilchen,
selbst elektronenarm (positiv polarisiert, oft mit positiver Ladung);
reagieren mit elektronenreichen Teilchen
• Radikale
Teilchen mit ungepaarten Elektronen (meist 1 e-);
reagieren mit anderen Radikalen oder mit anderen Teilchen
(können polar oder unpolar sein)
4
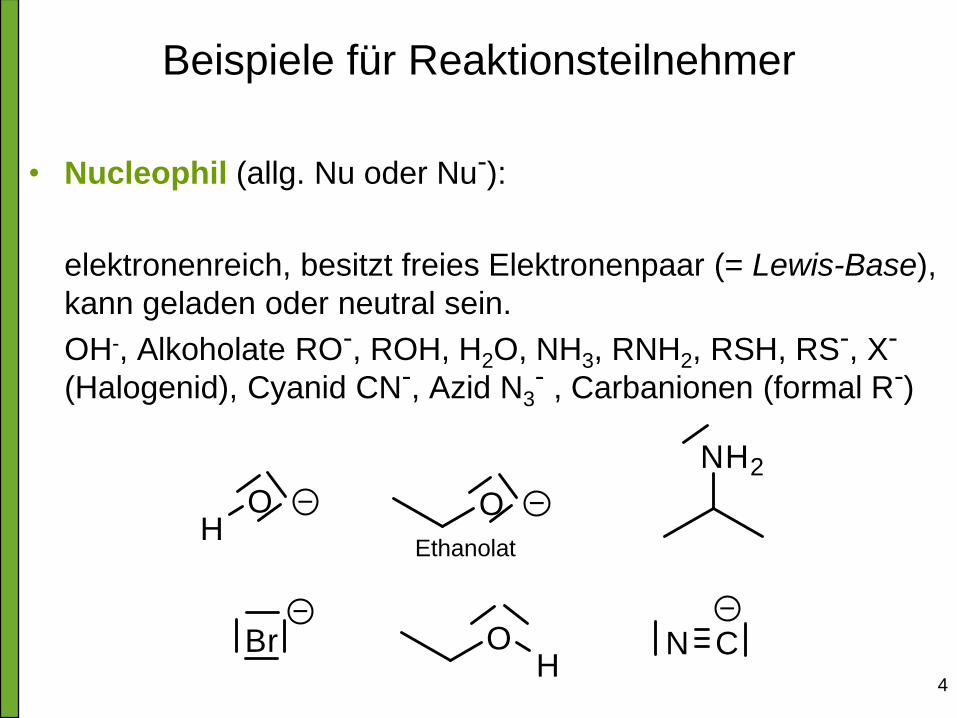
Beispiele für Reaktionsteilnehmer
• Nucleophil (allg. Nu oder Nu-):
elektronenreich, besitzt freies Elektronenpaar (= Lewis-Base),
kann geladen oder neutral sein.
OH-, Alkoholate RO-, ROH, H2O, NH3, RNH2, RSH, RS-, X-
(Halogenid), Cyanid CN-, Azid N3- , Carbanionen (formal R-)
HO O
NH2
OH
Br N C
Ethanolat
5
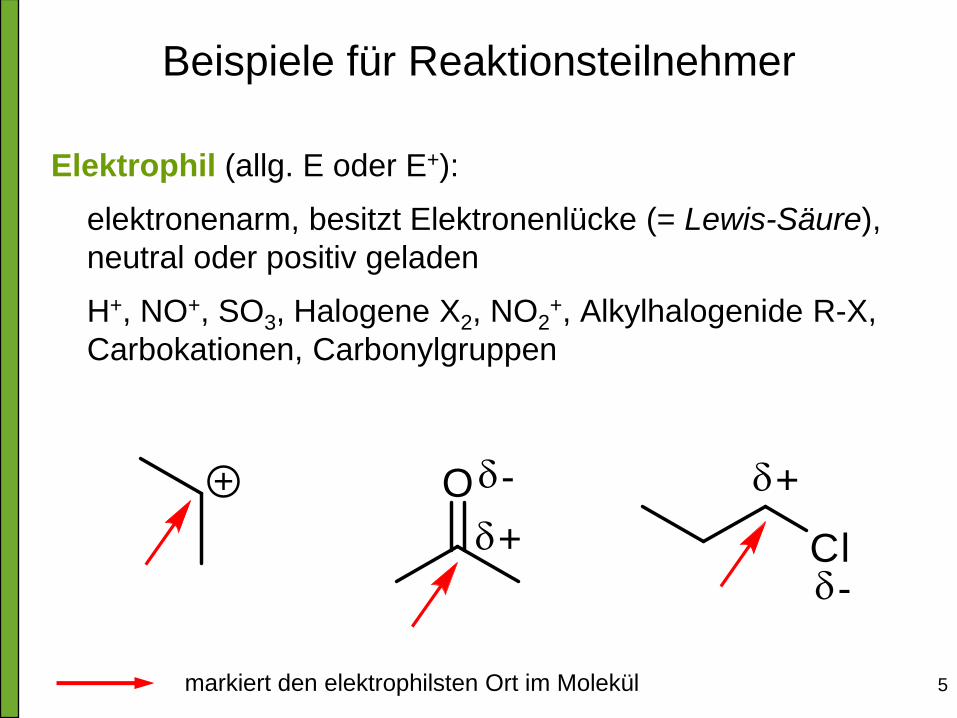
Beispiele für Reaktionsteilnehmer
Elektrophil (allg. E oder E+):
elektronenarm, besitzt Elektronenlücke (= Lewis-Säure),
neutral oder positiv geladen
H+, NO+, SO3, Halogene X2, NO2+, Alkylhalogenide R-X,
Carbokationen, Carbonylgruppen
O
+
-
Cl-
+
markiert den elektrophilsten Ort im Molekül
6
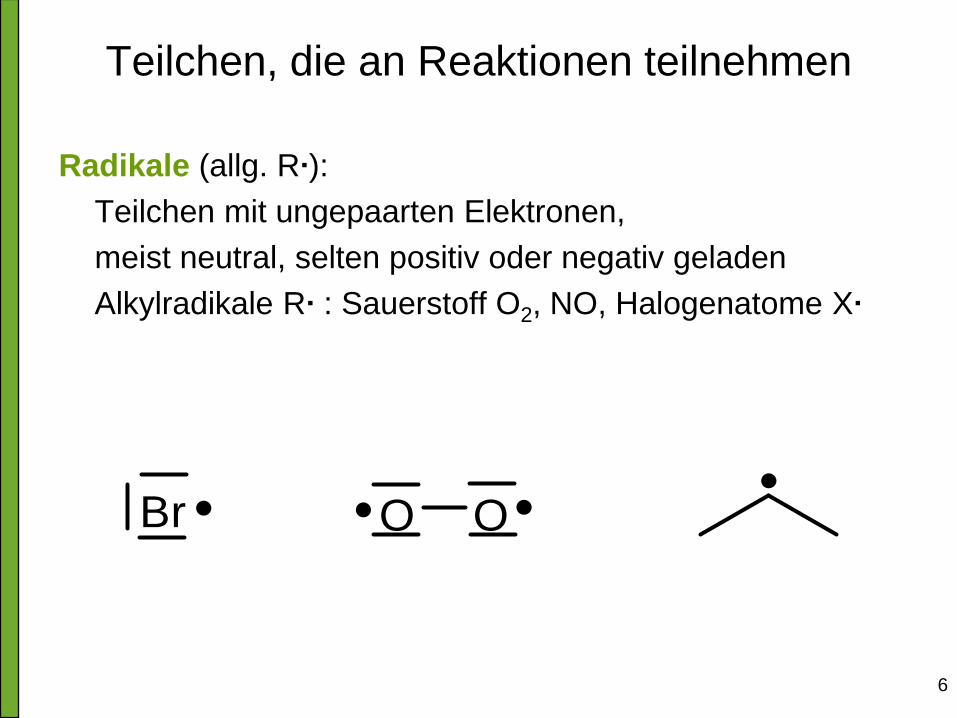
Teilchen, die an Reaktionen teilnehmen
Radikale (allg. R∙):
Teilchen mit ungepaarten Elektronen,
meist neutral, selten positiv oder negativ geladen
Alkylradikale R∙ : Sauerstoff O2, NO, Halogenatome X∙
Br O O

7
Regeln der Organische Chemie
• 1. Kohlenstoff ist NIE fünfbindig!
wegen der Oktettregel kann Kohlenstoff maximal vier Einfachbindungen
ausbilden
• 2. Elektrophil reagiert mit Nucleophil
also:
entgegen gesetzte Ladungen (oder Ladungsschwerpunkte) ziehen sich an
ein Nucleophil reagiert NICHT mit einem anderen Nucleophil
ein Elektrophil reagiert NICHT mit einem anderen Elektrophil

8
Eigenschaften organischer Reaktionen
• organische Reaktionen sind meist polare Reaktionen
• Nucleophil reagiert mit Elektrophil
• Anion reagiert mit Kation
• Partialladung + reagiert mit -
• Lewis-Base reagiert mit Lewis-Säure
Es kommt zur Verschiebung von Elektronenpaaren vom
Nucleophil auf das Elektrophil
Nu- + E+ → Nu-E
(koordinative) Bindung

9
Wo sind die Reaktionszentren in einem Molekül?
• Wichtigste Frage:
Wo sind in den reagierenden Molekülen elektrophile
(elektronenarme) und nucleophile (elektronenreiche)
Stellen?
• Feststellung dieser Orte unter Berücksichtigung der:
Polarität von Bindungen
Ladungen an bestimmten Atomen
Elektronegativität von Elementen
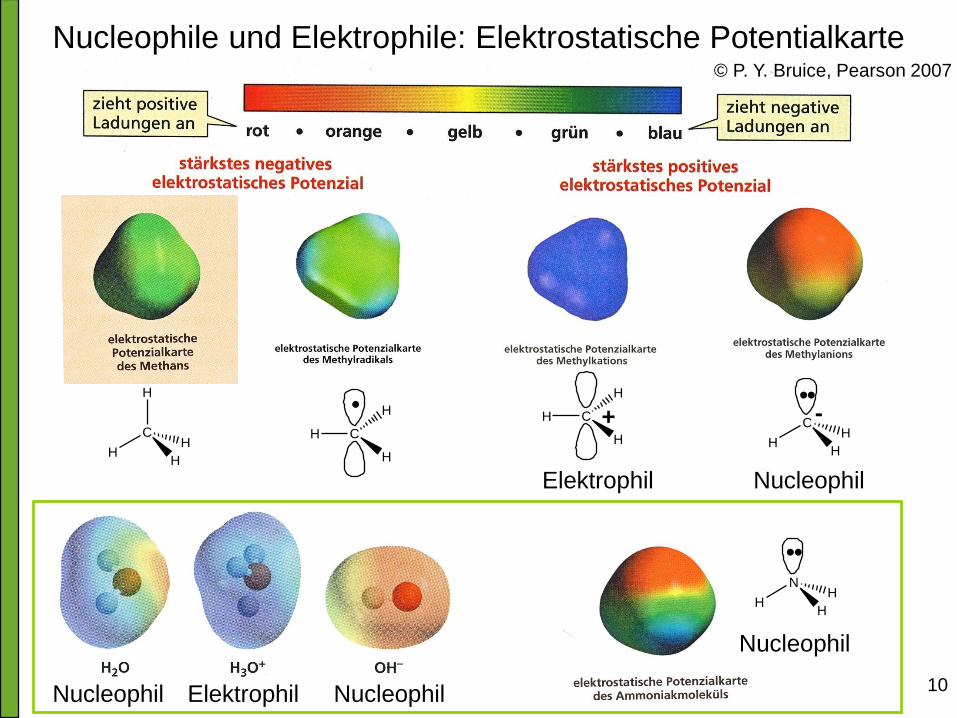
Nucleophile und Elektrophile: Elektrostatische Potentialkarte
10 Nucleophil Nucleophil Elektrophil
Nucleophil
H
C
HH
H
C
HH
H
• • -
Nucleophil Elektrophil
H
C
H
H +
N
HH
H
• •
H
C
H
H
•
© P. Y. Bruice, Pearson 2007

Substitutionsreaktionen
A + B-C → A-B + C
Edukt1 (Nu) + Edukt2 (R-X) → Produkt (R-Nu) + Abgangsgruppe
11
12
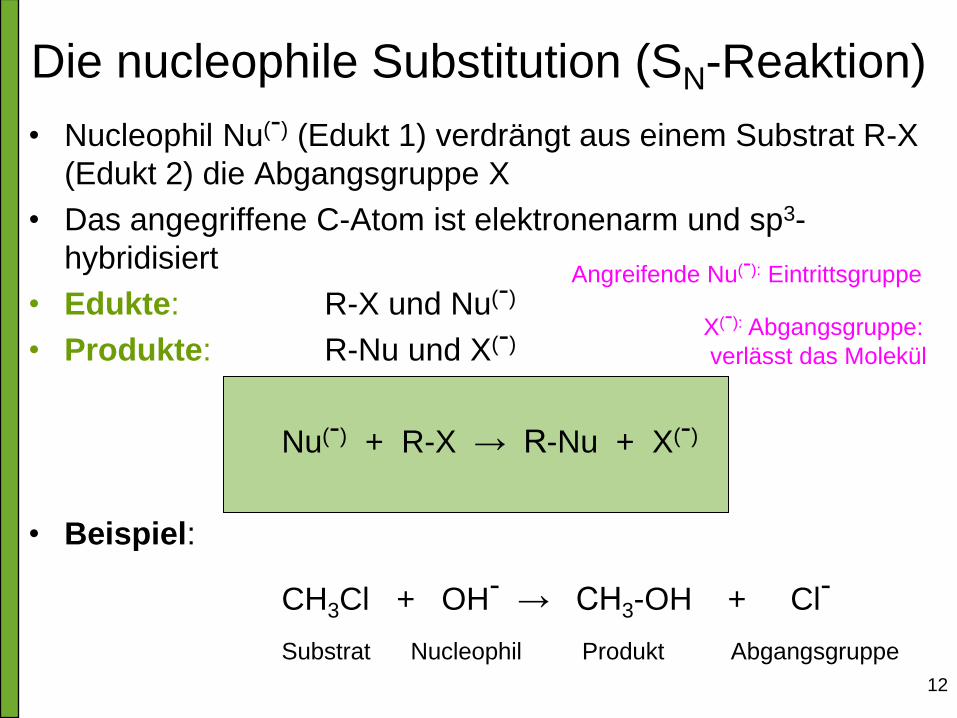
Die nucleophile Substitution (SN-Reaktion)
• Nucleophil Nu(-) (Edukt 1) verdrängt aus einem Substrat R-X
(Edukt 2) die Abgangsgruppe X
• Das angegriffene C-Atom ist elektronenarm und sp3-
hybridisiert
• Edukte: R-X und Nu(-)
• Produkte: R-Nu und X(-)
Nu(-) + R-X → R-Nu + X(-)
• Beispiel:
CH3Cl + OH- → CH3-OH + Cl- Substrat Nucleophil Produkt Abgangsgruppe
Angreifende Nu(-): Eintrittsgruppe
X(-): Abgangsgruppe:
verlässt das Molekül
13
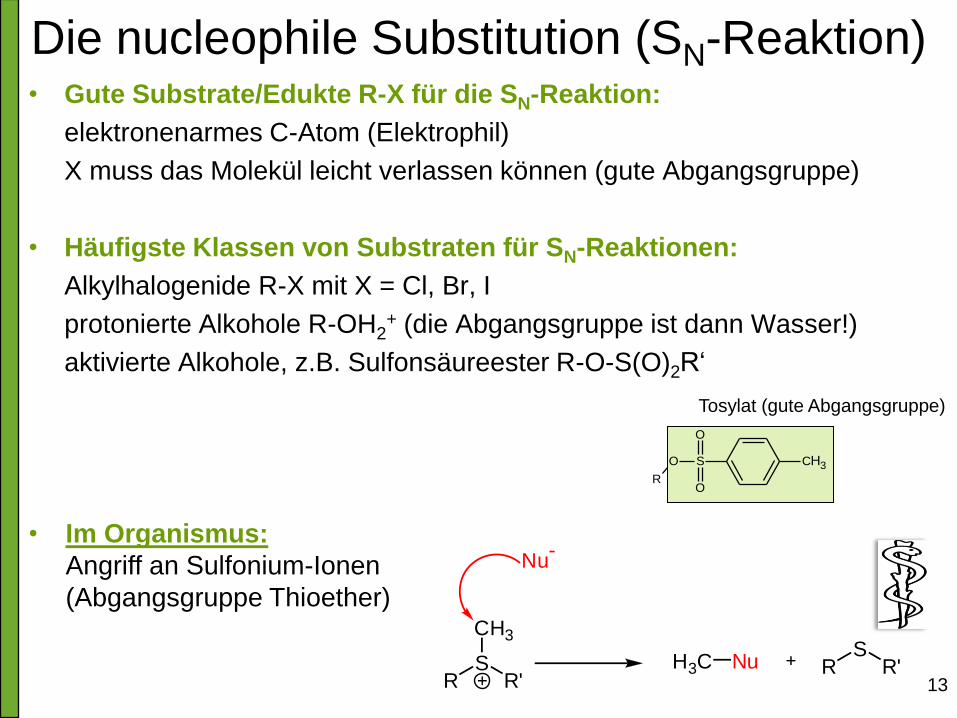
Die nucleophile Substitution (SN-Reaktion) • Gute Substrate/Edukte R-X für die SN-Reaktion:
elektronenarmes C-Atom (Elektrophil)
X muss das Molekül leicht verlassen können (gute Abgangsgruppe)
• Häufigste Klassen von Substraten für SN-Reaktionen:
Alkylhalogenide R-X mit X = Cl, Br, I
protonierte Alkohole R-OH2+ (die Abgangsgruppe ist dann Wasser!)
aktivierte Alkohole, z.B. Sulfonsäureester R-O-S(O)2R‘
RS
R'
CH3
Nu-
H3C Nu RS
R'
• Im Organismus:
Angriff an Sulfonium-Ionen
(Abgangsgruppe Thioether)
CH3S
O
O
O
R
Tosylat (gute Abgangsgruppe)
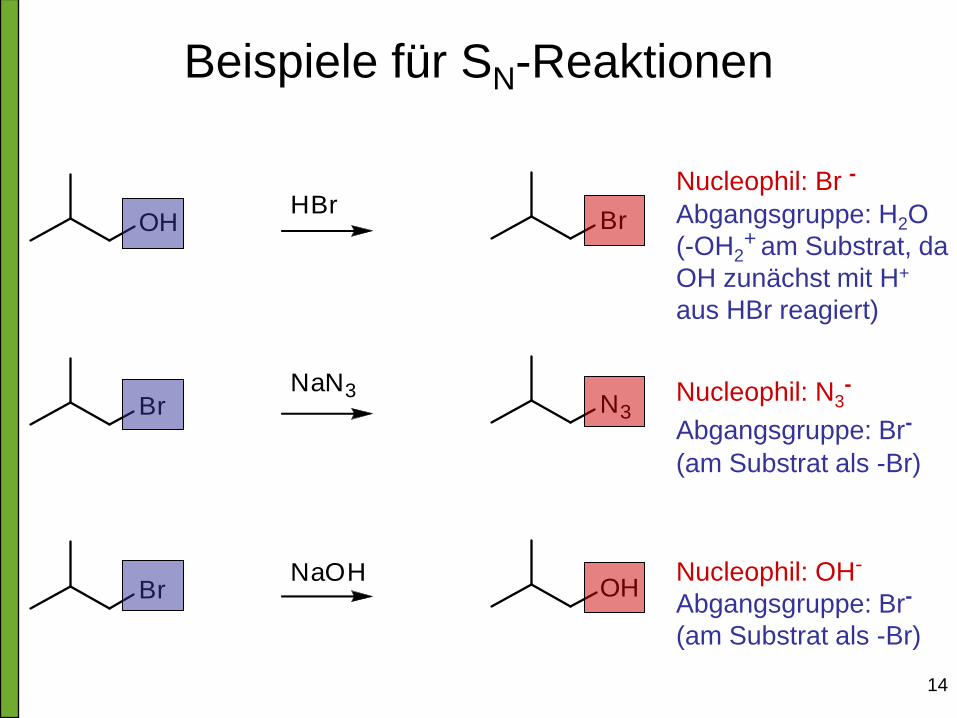
14
Br
Br
OH
N3
OH
BrHBr
NaN3
NaOH
Beispiele für SN-Reaktionen
Nucleophil: Br - Abgangsgruppe: H2O
(-OH2+
am Substrat, da
OH zunächst mit H+
aus HBr reagiert)
Nucleophil: N3
-
Abgangsgruppe: Br- (am Substrat als -Br)
Nucleophil: OH-
Abgangsgruppe: Br-
(am Substrat als -Br)

15
Die nucleophile Substitution (SN-Reaktion) • Gute Nucleophile:
Anionen oder neutrale Moleküle. Nucleophilie hängt von verschiedenen
Faktoren ab: Größe, Ladung, Lösemittel.
• häufig Halogenide, O-, N- und S-Nucleophile
außerdem: CN-, NH3, N3
-, Carbanionen usw.
• O-Nucleophile: H2O, ROH (z.B. in Serin, Tyrosin und in Kohlehydraten),
oder ihre geladenen Analoga (OH-, RO
-), Carboxylate RC(=O)O
- (z.B. in
Aspartat, Glutamat)
• N-Nucleophile: RNH2 (z.B. in Lysin, Guanin etc.), Amidgruppe RCONH2
(z.B. in Asparagin, Glutamin)
• S-Nucleophile: Thiolgruppe RSH (z.B. in Cystein, Coenzym-A)

16
Warum ist das Substrat polarisiert? • Situation an der C-H-Bindung
kaum Unterschied der Elektronegativität zwischen C (2,55) und H (2,20)
die Bindung ist fast unpolar und es gibt keine Partialladungen
• Situation an der R-X-Bindung
je nach X großer Unterschied der Elektronegativität zwischen C und X
(O: 3,44, Cl: 3,16, Br: 2,96)
X zieht die Elektronen zu sich herüber → die Bindung wird polarisiert und
am C befindet sich positive Partialladung
polarisierte Bindungen können heterolytisch gespalten werden
heterolytisch: die beiden Elektronen der Bindung werden nicht
gleichmäßig verteilt, sondern einem der Bindungspartner zugeschlagen, in
diesem Fall nimmt die Abgangsgruppe die Elektronen mit
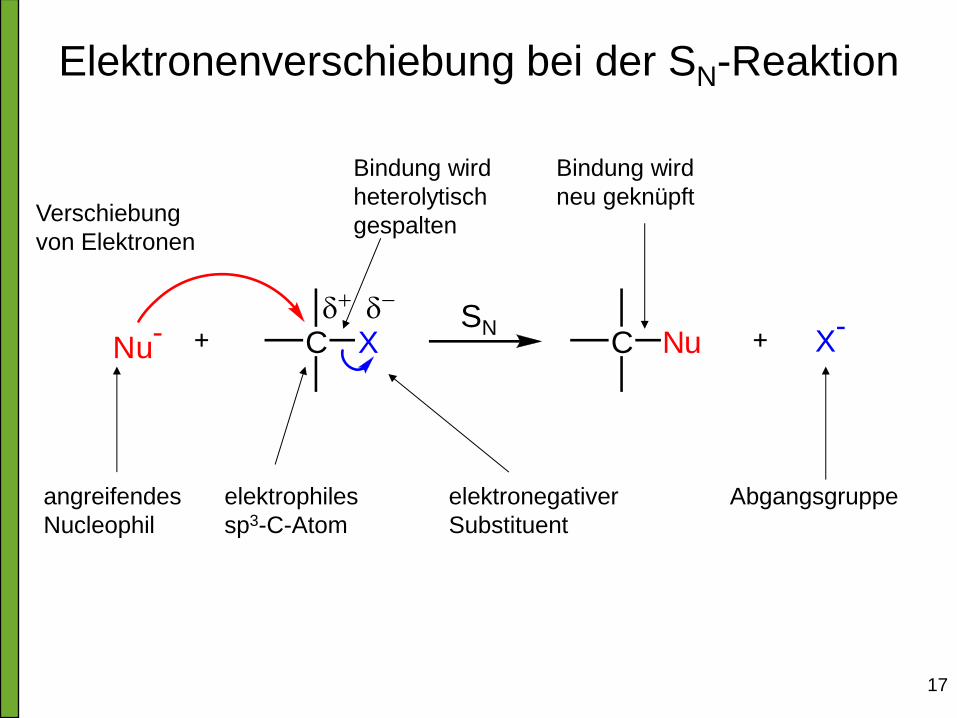
17
Elektronenverschiebung bei der SN-Reaktion
Nu- C X C Nu X
-
elektrophiles
sp3-C-Atom
elektronegativer
Substituent
angreifendes
Nucleophil
Abgangsgruppe
Verschiebung
von Elektronen
Bindung wird
neu geknüpft
Bindung wird
heterolytisch
gespalten
SN

18
Die SN2-Reaktion
• Bindungsbruch (Abspaltung von X) und Bindungsknüpfung (Anbindung
des Nucleophils) finden gleichzeitig (simultan) statt.
• Das Nucleophil bildet mit einem freien Elektronenpaar eine Bindung zum
entsprechenden C-Atom aus
• Wegen der Oktettregel muss gleichzeitig X aus dem Molekül
heraustreten, damit nie mehr als acht Elektronen am C vorhanden sind!
• Teilchen, die stabile Anionen X- bilden, sind gute Abgangsgruppen.
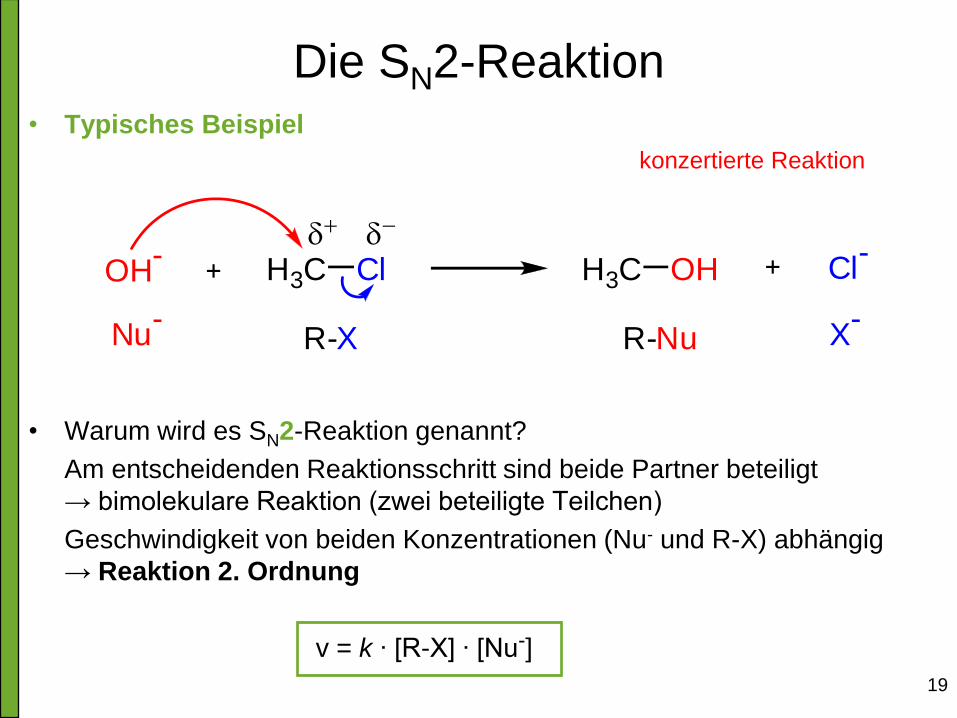
19
Die SN2-Reaktion • Typisches Beispiel
• Warum wird es SN2-Reaktion genannt?
Am entscheidenden Reaktionsschritt sind beide Partner beteiligt
→ bimolekulare Reaktion (zwei beteiligte Teilchen)
Geschwindigkeit von beiden Konzentrationen (Nu- und R-X) abhängig
→ Reaktion 2. Ordnung
v = k ∙ [R-X] ∙ [Nu-]
H3C Cl H3C OH Cl-
OH-
Nu-
R-X R-Nu X-
konzertierte Reaktion
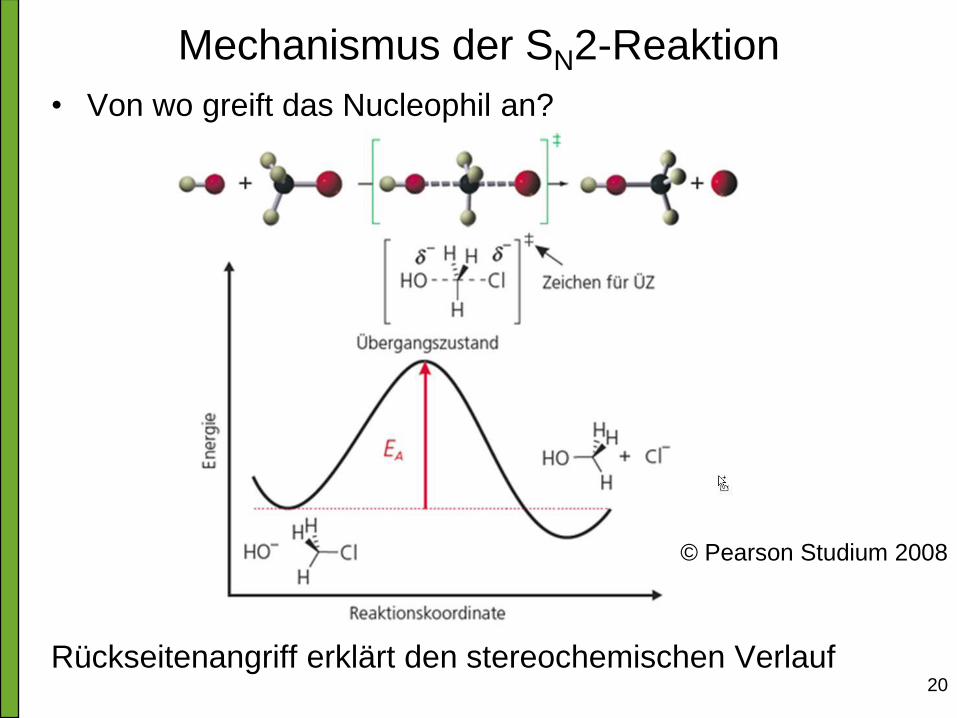
20
Mechanismus der SN2-Reaktion
• Von wo greift das Nucleophil an?
Rückseitenangriff erklärt den stereochemischen Verlauf
© Pearson Studium 2008
21
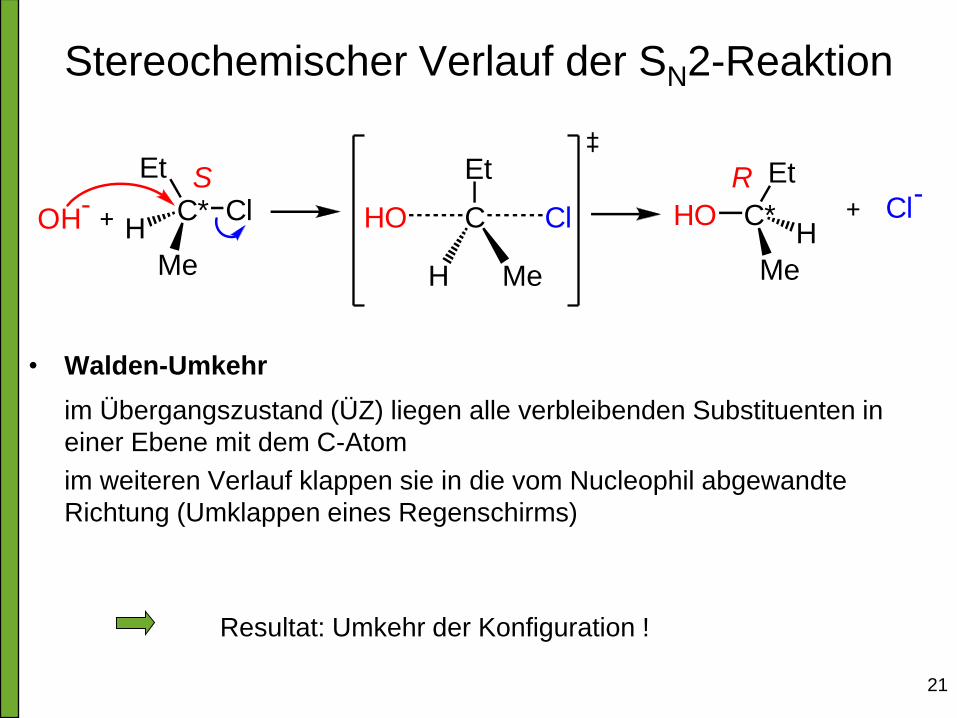
Stereochemischer Verlauf der SN2-Reaktion
• Walden-Umkehr
im Übergangszustand (ÜZ) liegen alle verbleibenden Substituenten in
einer Ebene mit dem C-Atom
im weiteren Verlauf klappen sie in die vom Nucleophil abgewandte
Richtung (Umklappen eines Regenschirms)
Resultat: Umkehr der Konfiguration !
C* Cl
Me
Et
H
SCl
-OH
- C*HO
Me
Et
HHO C Cl
Et
MeH
R
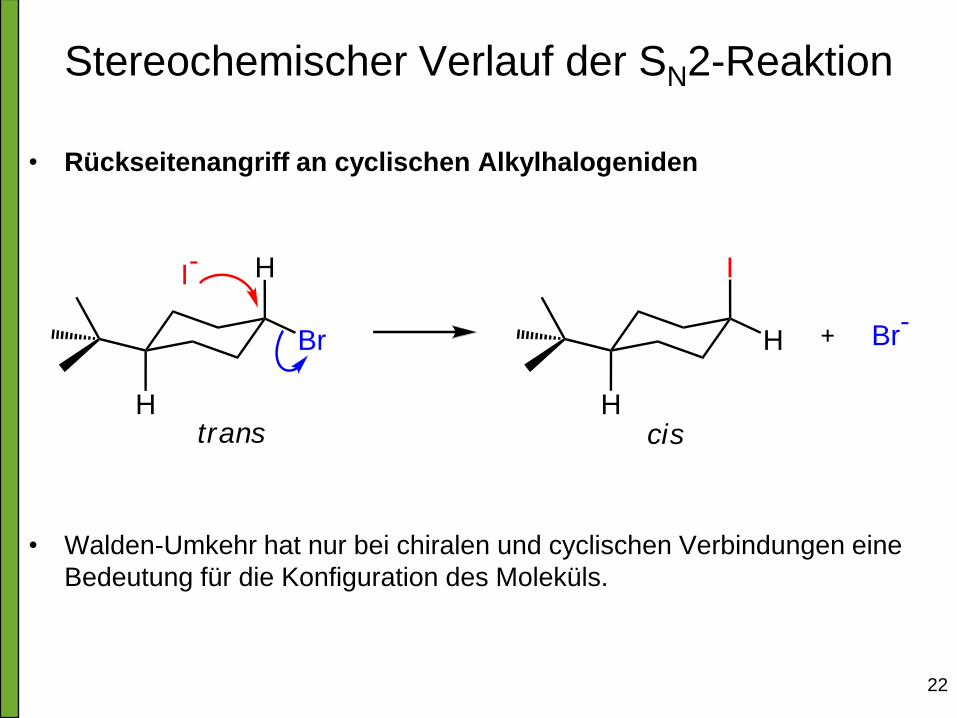
22
Stereochemischer Verlauf der SN2-Reaktion
• Rückseitenangriff an cyclischen Alkylhalogeniden
• Walden-Umkehr hat nur bei chiralen und cyclischen Verbindungen eine
Bedeutung für die Konfiguration des Moleküls.
H
Br
HI-
H
H
I
Br-
trans cis

23
Die SN1-Reaktion
• Bindungsbruch (Abspaltung von X) und Bindungsknüpfung (Anbindung
von Nu) finden nicht gleichzeitig statt.
• Wegen Oktettregel muss zuerst X aus dem Molekül heraustreten, damit
nie mehr als 8 Elektronen am C vorhanden sind!
• Es bildet sich ein Carbokation (Carbenium-Ion).
• Anschließend erfolgt Angriff des Nucleophils
• Die Reaktion ist also zweistufig!
Es finden also zwei Elementarreaktionen statt:
1. Bildung des Carbeniumions; 2. Angriff des Nucleophils
• Der langsamste Schritt bestimmt die Reaktionsgeschwindigkeit:
geschwindigkeitsbestimmender Schritt
• Bei SN1-Reaktion ist die Bildung des Carbenium-Ions limitierend
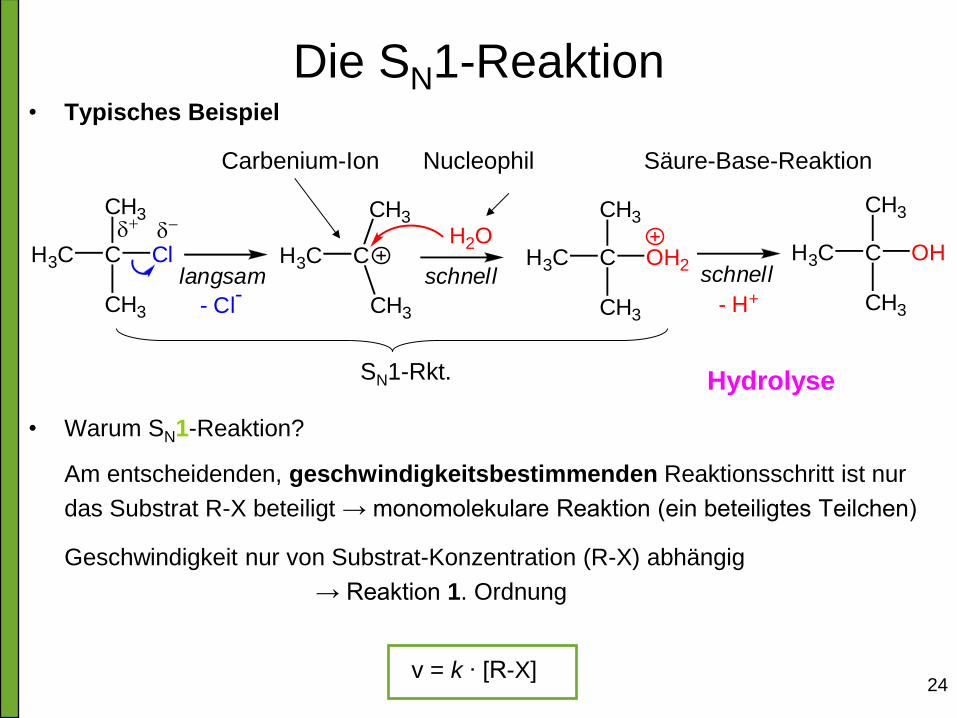
24
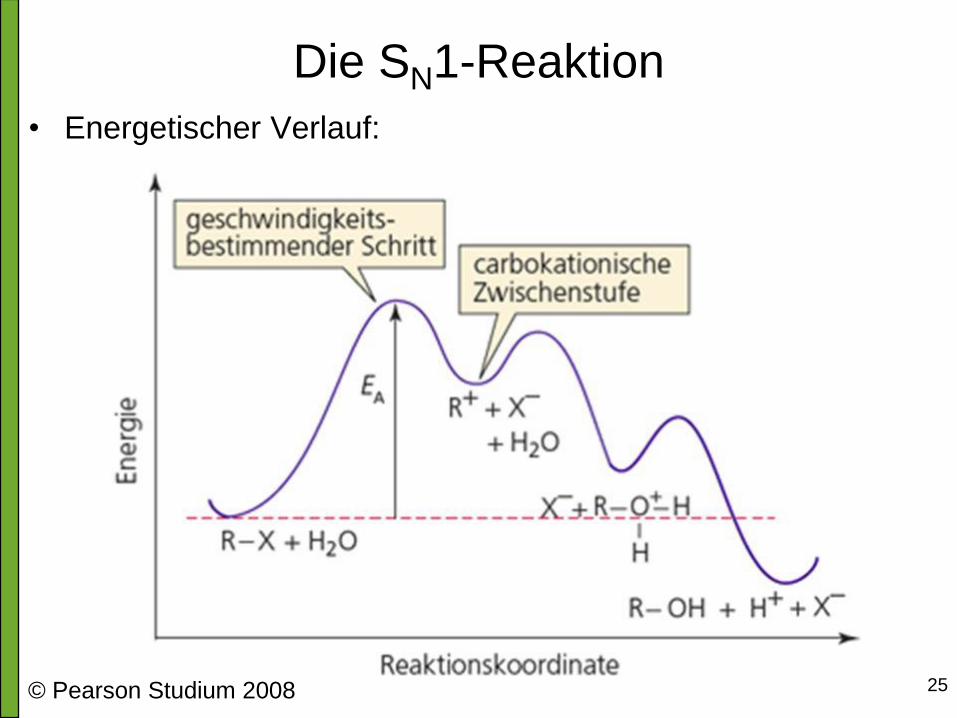
Die SN1-Reaktion • Typisches Beispiel
• Warum SN1-Reaktion?
Am entscheidenden, geschwindigkeitsbestimmenden Reaktionsschritt ist nur
das Substrat R-X beteiligt → monomolekulare Reaktion (ein beteiligtes Teilchen)
Geschwindigkeit nur von Substrat-Konzentration (R-X) abhängig
→ Reaktion 1. Ordnung
v = k ∙ [R-X]
H3C C Cl
CH3
CH3 - Cl-
H3C C
CH3
CH3
H2O
langsam schnellH3C C OH2
CH3
CH3
H3C C OH
CH3
CH3
schnell
- H+
Nucleophil Carbenium-Ion Säure-Base-Reaktion
SN1-Rkt. Hydrolyse
25
Die SN1-Reaktion
• Energetischer Verlauf:
© Pearson Studium 2008
26
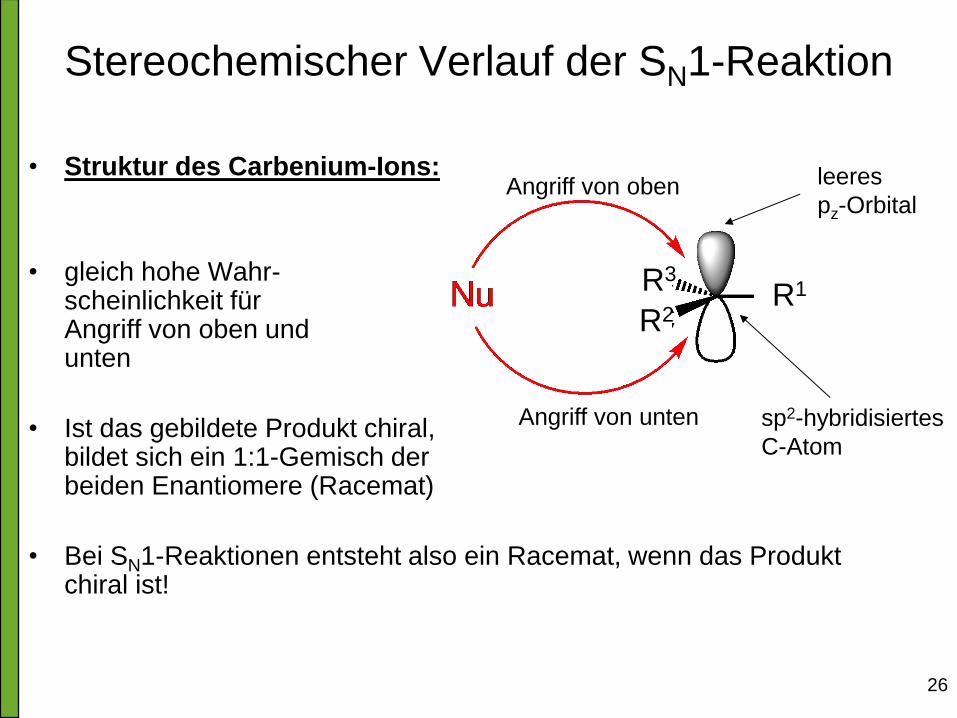
• Struktur des Carbenium-Ions:
• gleich hohe Wahr- scheinlichkeit für Angriff von oben und unten
• Ist das gebildete Produkt chiral, bildet sich ein 1:1-Gemisch der beiden Enantiomere (Racemat)
• Bei SN1-Reaktionen entsteht also ein Racemat, wenn das Produkt chiral ist!
Stereochemischer Verlauf der SN1-Reaktion
sp2-hybridisiertes
C-Atom
leeres
pz-Orbital Angriff von oben
Angriff von unten
R3 R1 R2
27
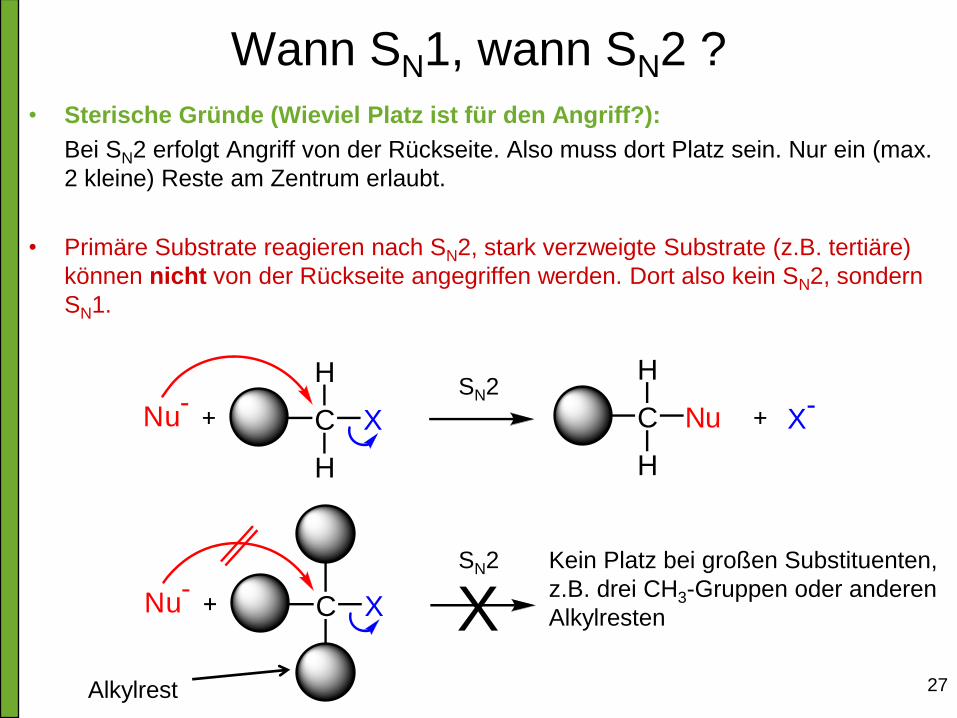
Wann SN1, wann SN2 ?
• Sterische Gründe (Wieviel Platz ist für den Angriff?):
Bei SN2 erfolgt Angriff von der Rückseite. Also muss dort Platz sein. Nur ein (max.
2 kleine) Reste am Zentrum erlaubt.
• Primäre Substrate reagieren nach SN2, stark verzweigte Substrate (z.B. tertiäre)
können nicht von der Rückseite angegriffen werden. Dort also kein SN2, sondern
SN1.
Nu-
C X
H
H
C Nu
H
H
X-
Nu-
C X
Kein Platz bei großen Substituenten,
z.B. drei CH3-Gruppen oder anderen
Alkylresten
SN2
SN2
X Alkylrest
28
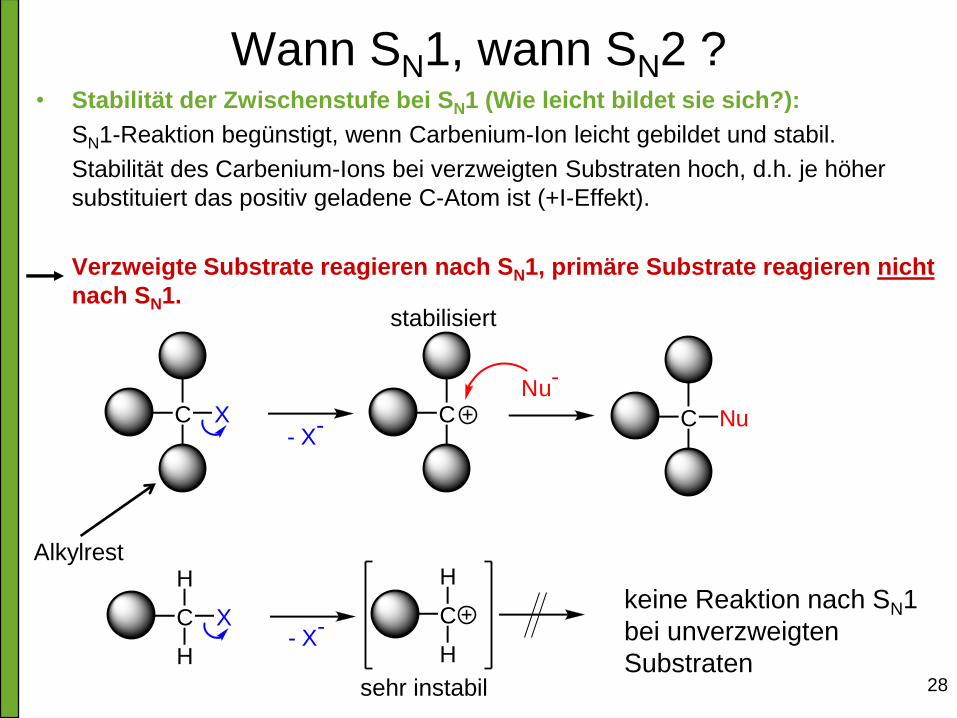
Wann SN1, wann SN2 ? • Stabilität der Zwischenstufe bei SN1 (Wie leicht bildet sie sich?):
SN1-Reaktion begünstigt, wenn Carbenium-Ion leicht gebildet und stabil.
Stabilität des Carbenium-Ions bei verzweigten Substraten hoch, d.h. je höher
substituiert das positiv geladene C-Atom ist (+I-Effekt).
Verzweigte Substrate reagieren nach SN1, primäre Substrate reagieren nicht
nach SN1.
Nu-
C X
H
H
C
H
H- X
-
C X C- X
- C Nu
stabilisiert
sehr instabil
keine Reaktion nach SN1
bei unverzweigten
Substraten
Alkylrest

29
Vergleich SN1 und SN2
Eigenschaft SN1 SN2
Reaktionsverlauf zweistufig einstufig
langsamster Schritt monomolekular bimolekular
zeitliche Abfolge erst Bindungs- Bindungsbruch
bruch, dann und -bildung
-bildung simultan
Zwischenstufe Carbenium-ion keine
Kinetik 1. Ordnung 2. Ordnung
Stereochemie Racemisierung Inversion
Entscheidender Faktor Stabilität des Sterische Zu-
Carbenium-Ions gänglichkeit des
C-Atoms
Mögliche Substrate tertiär, sekundär Methyl-, primär,
sekundär

Eliminierungsreaktionen
A-B-C → A-B + C
30
31
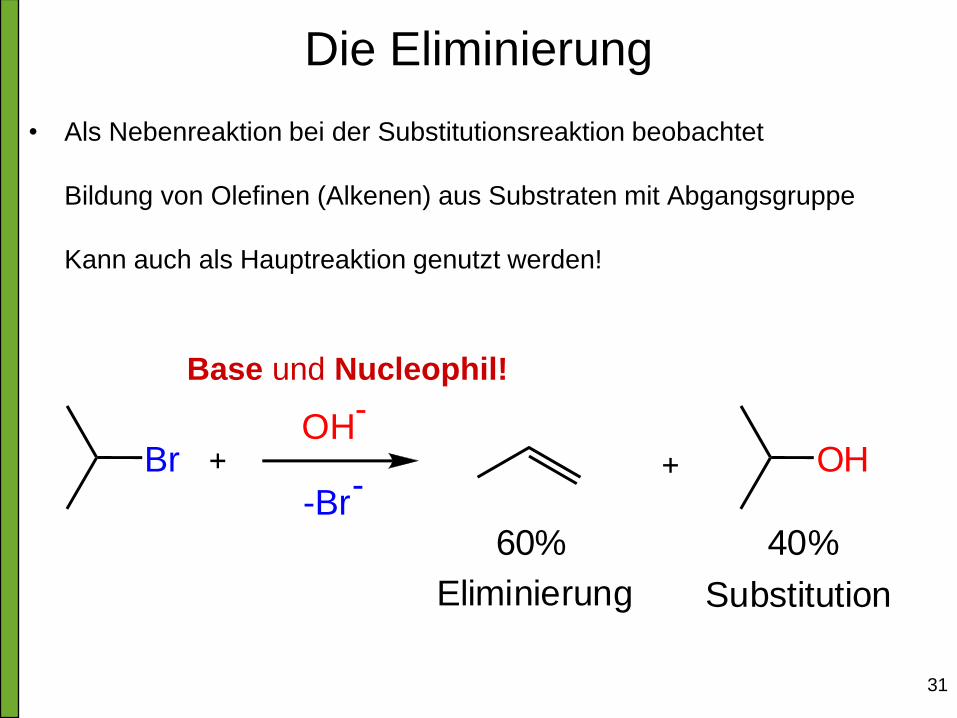
Die Eliminierung
• Als Nebenreaktion bei der Substitutionsreaktion beobachtet
Bildung von Olefinen (Alkenen) aus Substraten mit Abgangsgruppe
Kann auch als Hauptreaktion genutzt werden!
BrOH
-
-Br-
OH
60% 40%
Eliminierung Substitution
Base und Nucleophil!
32
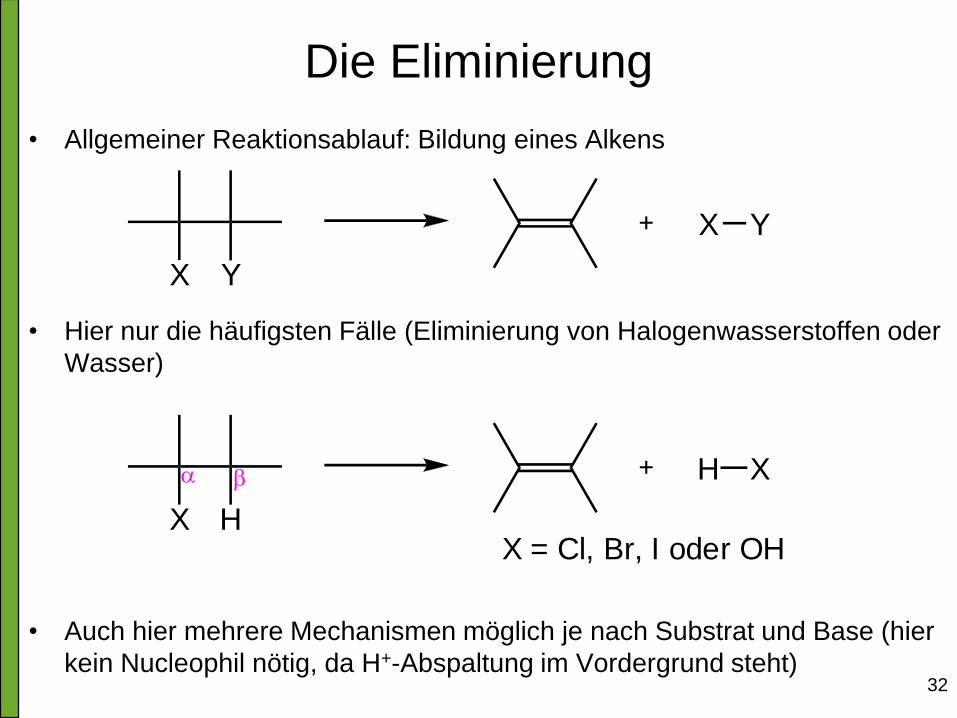
Die Eliminierung
• Allgemeiner Reaktionsablauf: Bildung eines Alkens
• Hier nur die häufigsten Fälle (Eliminierung von Halogenwasserstoffen oder
Wasser)
• Auch hier mehrere Mechanismen möglich je nach Substrat und Base (hier
kein Nucleophil nötig, da H+-Abspaltung im Vordergrund steht)
YX
X Y
HX
H X
X = Cl, Br, I oder OH
a b
33
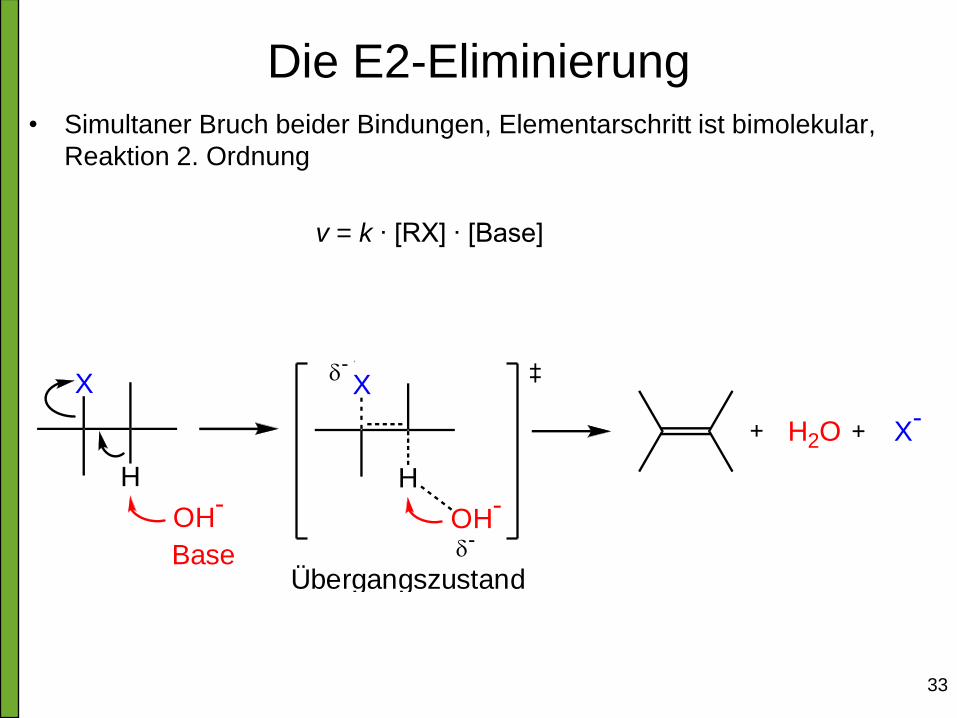
Die E2-Eliminierung • Simultaner Bruch beider Bindungen, Elementarschritt ist bimolekular,
Reaktion 2. Ordnung
v = k ∙ [RX] ∙ [Base]
H
X
OH-
Base
H2O X-
H
X
OH-
+
-
Übergangszustand
-
-
34
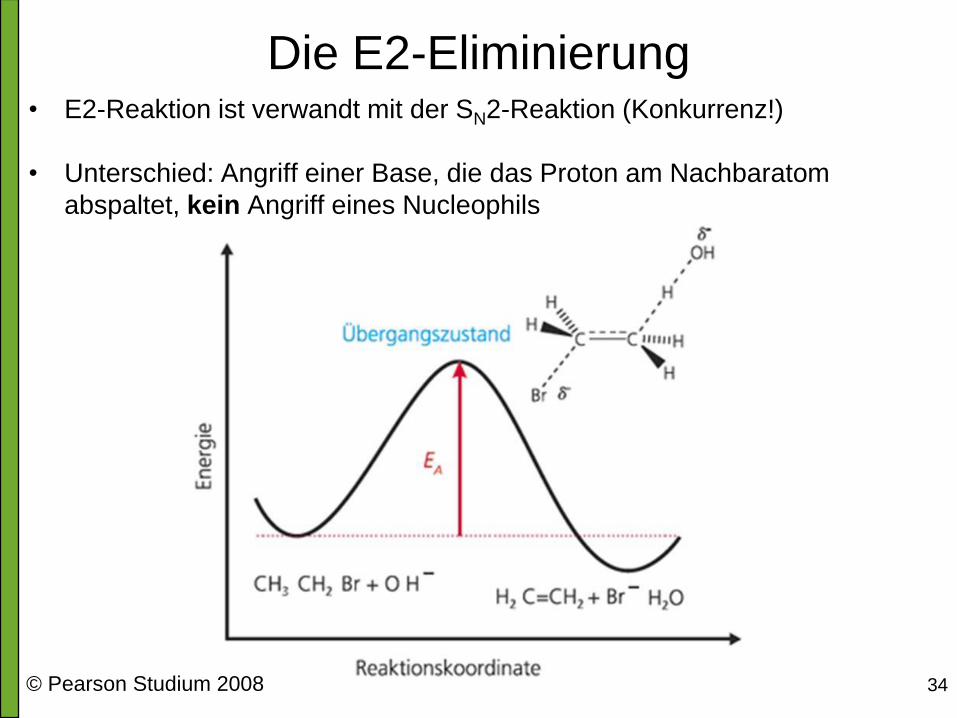
Die E2-Eliminierung • E2-Reaktion ist verwandt mit der SN2-Reaktion (Konkurrenz!)
• Unterschied: Angriff einer Base, die das Proton am Nachbaratom
abspaltet, kein Angriff eines Nucleophils
© Pearson Studium 2008
35
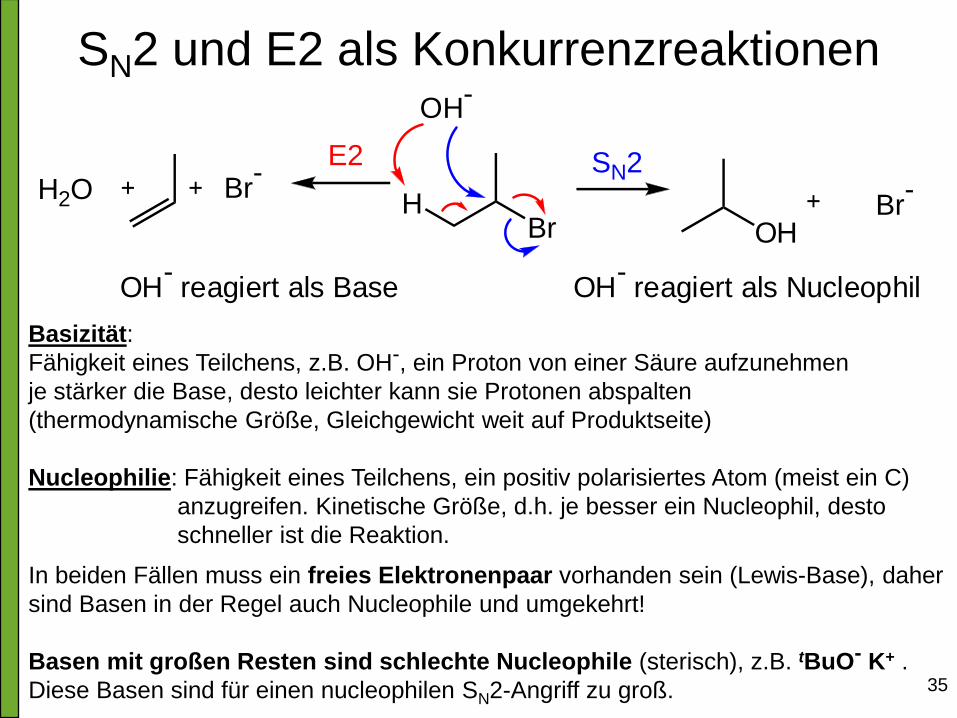
SN2 und E2 als Konkurrenzreaktionen
BrH
OH-
H2O Br-
OHBr
-E2 SN2
OH-
reagiert als Base OH-
reagiert als Nucleophil
Basizität:
Fähigkeit eines Teilchens, z.B. OH-, ein Proton von einer Säure aufzunehmen
je stärker die Base, desto leichter kann sie Protonen abspalten
(thermodynamische Größe, Gleichgewicht weit auf Produktseite)
Nucleophilie: Fähigkeit eines Teilchens, ein positiv polarisiertes Atom (meist ein C)
anzugreifen. Kinetische Größe, d.h. je besser ein Nucleophil, desto
schneller ist die Reaktion.
In beiden Fällen muss ein freies Elektronenpaar vorhanden sein (Lewis-Base), daher
sind Basen in der Regel auch Nucleophile und umgekehrt!
Basen mit großen Resten sind schlechte Nucleophile (sterisch), z.B. tBuO- K+ .
Diese Basen sind für einen nucleophilen SN2-Angriff zu groß.
36
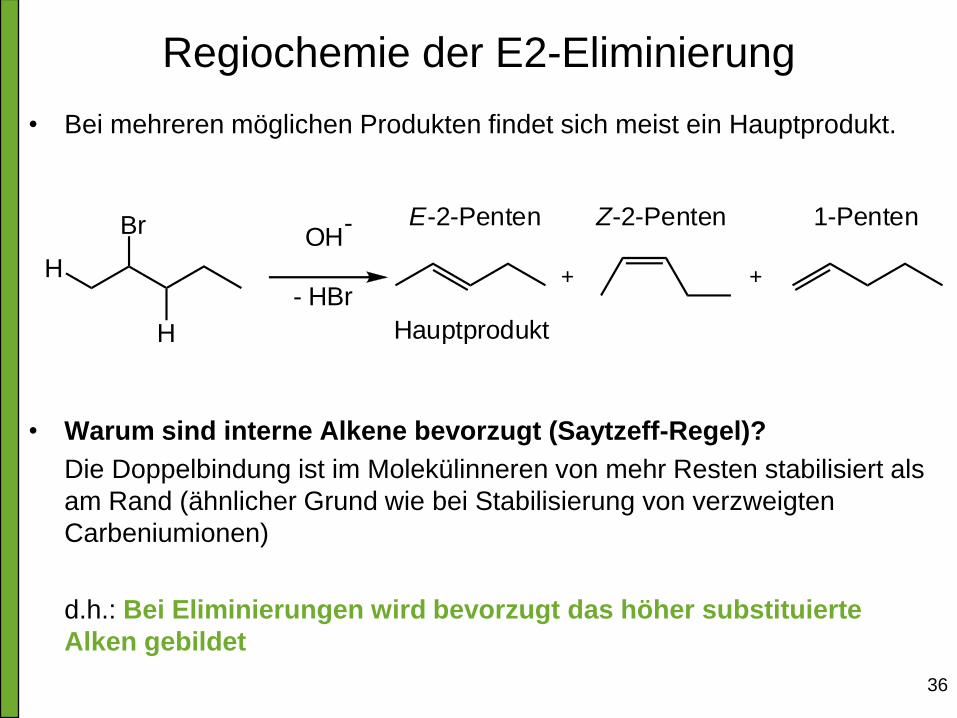
Regiochemie der E2-Eliminierung
• Bei mehreren möglichen Produkten findet sich meist ein Hauptprodukt.
• Warum sind interne Alkene bevorzugt (Saytzeff-Regel)?
Die Doppelbindung ist im Molekülinneren von mehr Resten stabilisiert als
am Rand (ähnlicher Grund wie bei Stabilisierung von verzweigten
Carbeniumionen)
d.h.: Bei Eliminierungen wird bevorzugt das höher substituierte
Alken gebildet
Br
H
H
OH-
- HBr
Hauptprodukt
E-2-Penten Z-2-Penten 1-Penten
37
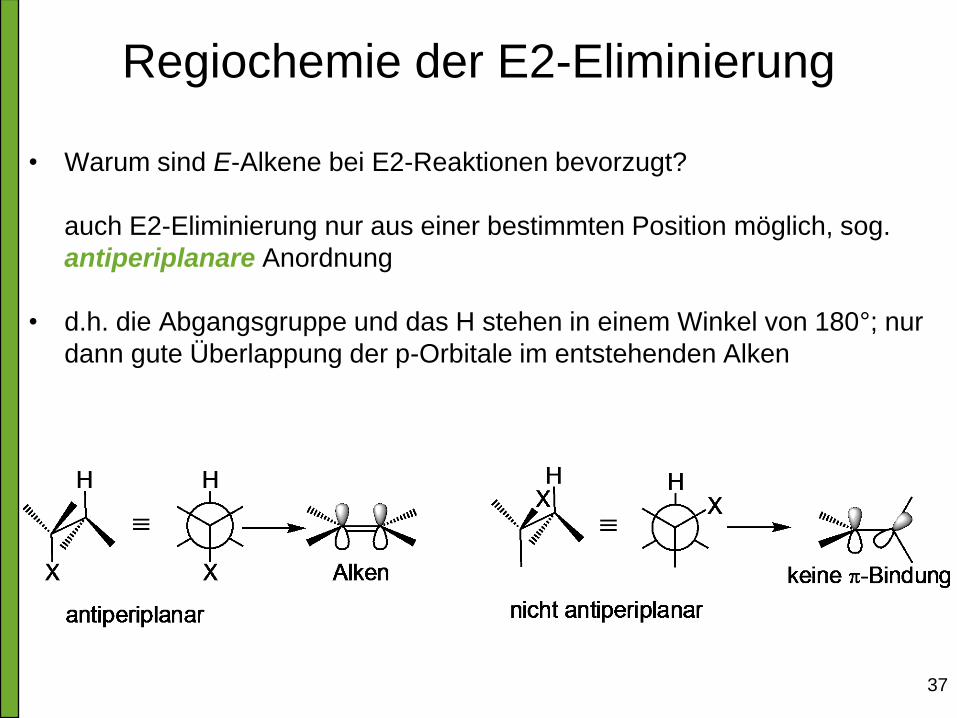
Regiochemie der E2-Eliminierung
• Warum sind E-Alkene bei E2-Reaktionen bevorzugt?
auch E2-Eliminierung nur aus einer bestimmten Position möglich, sog.
antiperiplanare Anordnung
• d.h. die Abgangsgruppe und das H stehen in einem Winkel von 180°; nur
dann gute Überlappung der p-Orbitale im entstehenden Alken
38
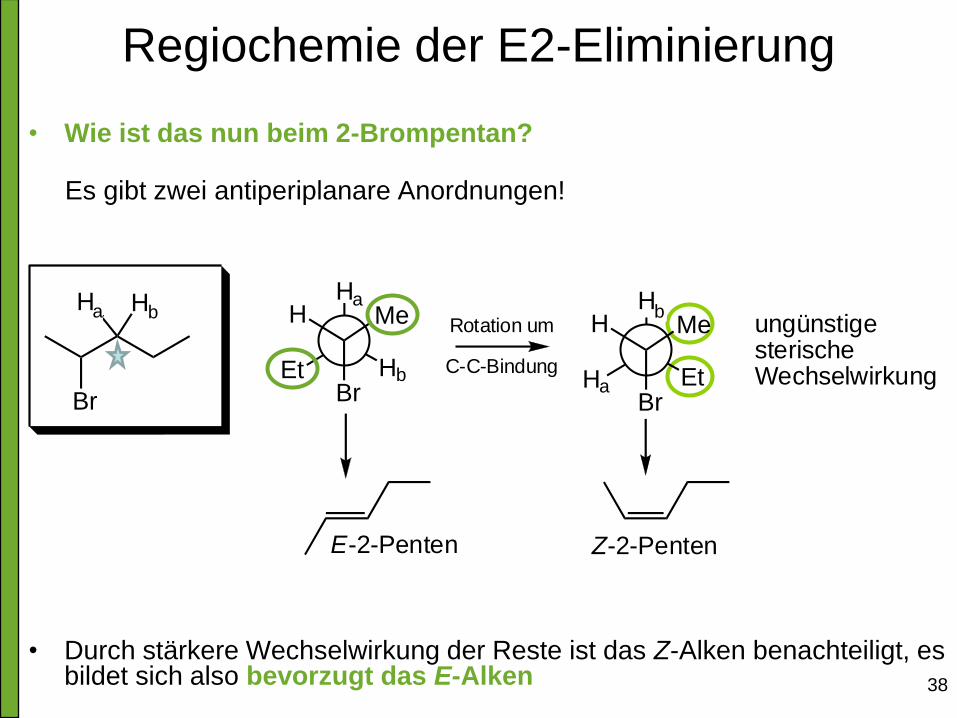
Regiochemie der E2-Eliminierung
• Wie ist das nun beim 2-Brompentan?
Es gibt zwei antiperiplanare Anordnungen!
• Durch stärkere Wechselwirkung der Reste ist das Z-Alken benachteiligt, es bildet sich also bevorzugt das E-Alken
H1
Et H2
H
Br
Me
Br
H1 H2 H2
H1Et
H
Br
Me ungünstigesterischeWechselwirkung
E-2-Penten Z-2-Penten
Rotation um
C-C-Bindung
a a
a
b
b
b
39
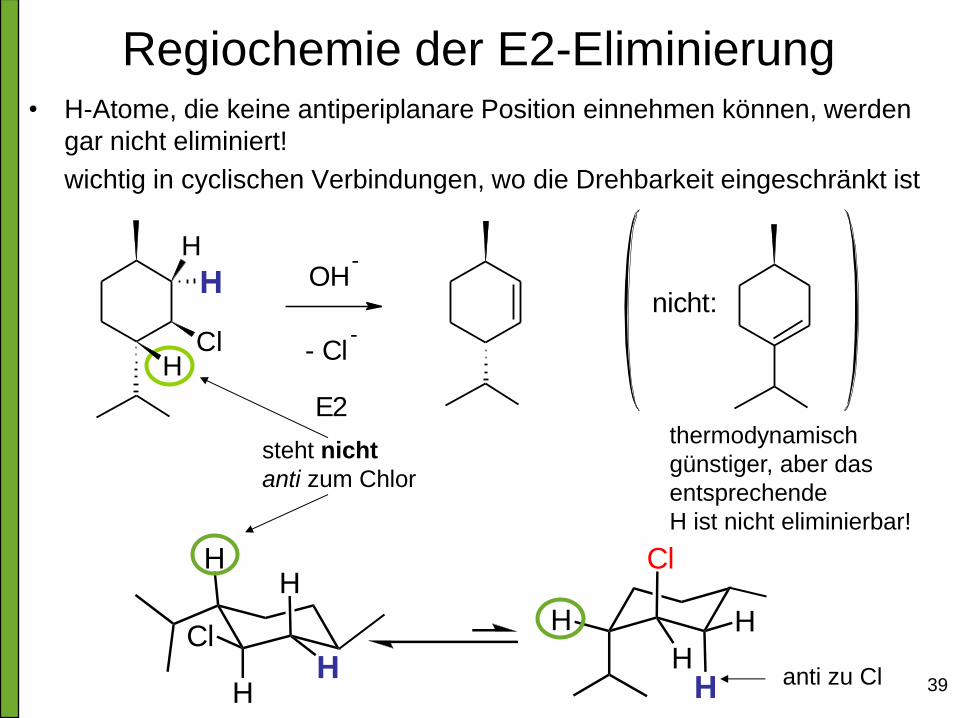
Regiochemie der E2-Eliminierung • H-Atome, die keine antiperiplanare Position einnehmen können, werden
gar nicht eliminiert!
wichtig in cyclischen Verbindungen, wo die Drehbarkeit eingeschränkt ist
thermodynamisch
günstiger, aber das
entsprechende
H ist nicht eliminierbar!
steht nicht
anti zum Chlor
H
Cl
H
H
H
H
Cl
HH
H
anti zu Cl
H
H
ClH
OH-
- Cl-
E2
nicht: H
H H
40
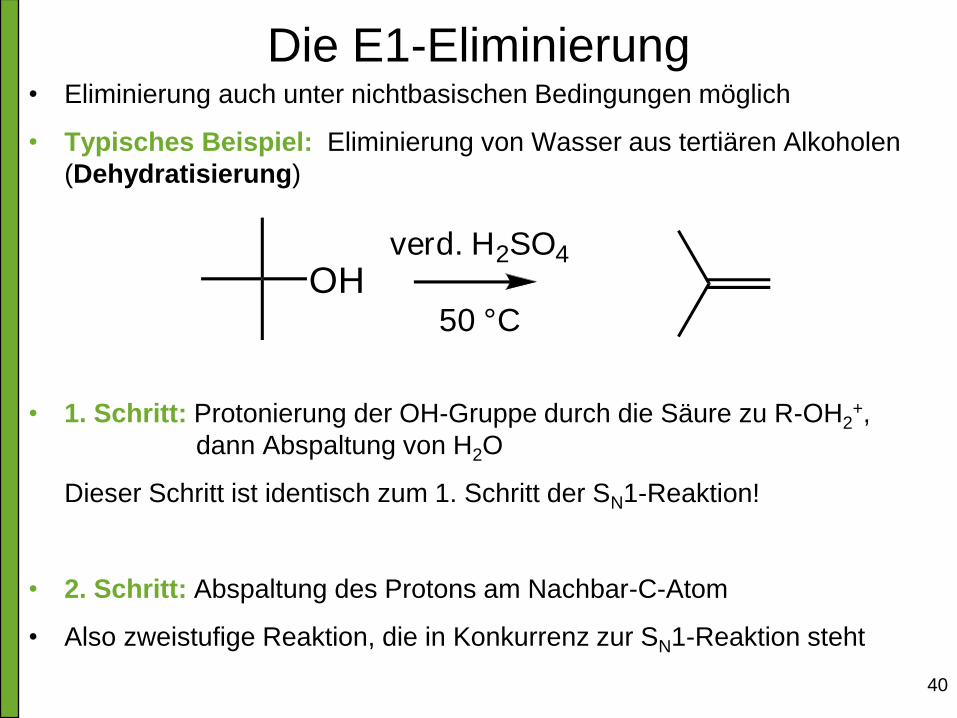
Die E1-Eliminierung • Eliminierung auch unter nichtbasischen Bedingungen möglich
• Typisches Beispiel: Eliminierung von Wasser aus tertiären Alkoholen
(Dehydratisierung)
• 1. Schritt: Protonierung der OH-Gruppe durch die Säure zu R-OH2+,
dann Abspaltung von H2O
Dieser Schritt ist identisch zum 1. Schritt der SN1-Reaktion!
• 2. Schritt: Abspaltung des Protons am Nachbar-C-Atom
• Also zweistufige Reaktion, die in Konkurrenz zur SN1-Reaktion steht
OHverd. H2SO4
50 °C
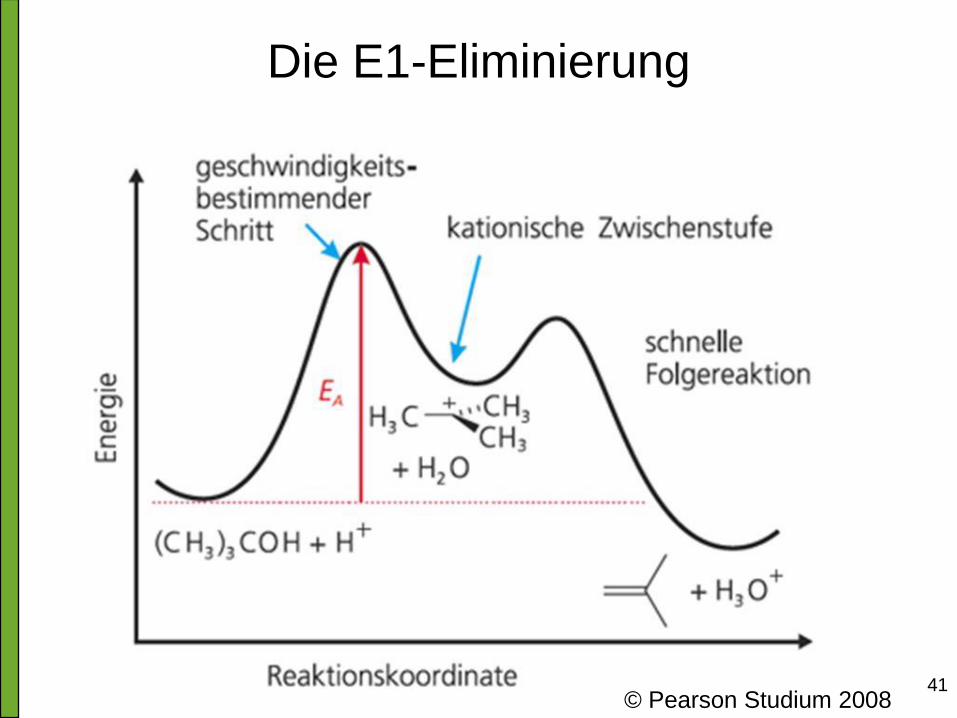
41 © Pearson Studium 2008
Die E1-Eliminierung
42
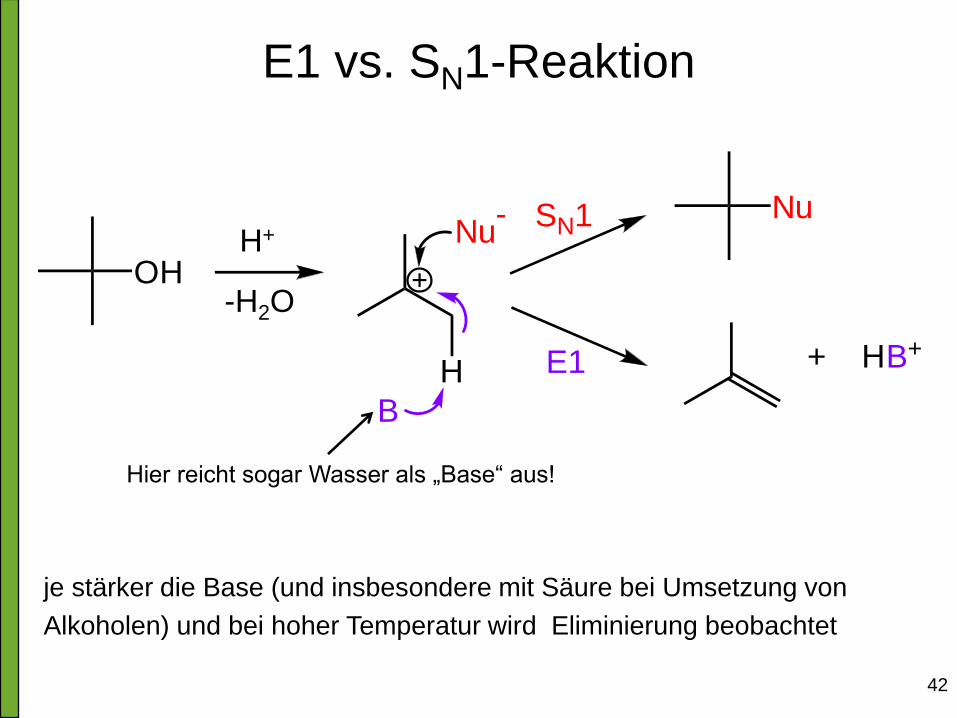
E1 vs. SN1-Reaktion
OH
H
Nu-
B
E1
SN1 Nu
+ HB+
je stärker die Base (und insbesondere mit Säure bei Umsetzung von
Alkoholen) und bei hoher Temperatur wird Eliminierung beobachtet
-H2O
H+
Hier reicht sogar Wasser als „Base“ aus!

43
Vergleich von E1 und E2-Reaktion
Eigenschaft E1 E2
Reaktionsverlauf zweistufig einstufig
langsamster Schritt monomolekular bimolekular
zeitliche Abfolge erst C-X-Bruch, beide
dann C-H-Bruch Bindungsbrüche
simultan
Zwischenstufe Carbenium-Ion keine
Kinetik 1. Ordnung 2. Ordnung
Bedingungen oft sauer oder starke Base
neutral nötig
Einschränkungen nur wenn antiperiplanare
Carbenium-Ion Anordnung von
gebildet H und X
Mögliche Substrate tertiär, sekundär Methyl-, primär,
sekundär, tertiär
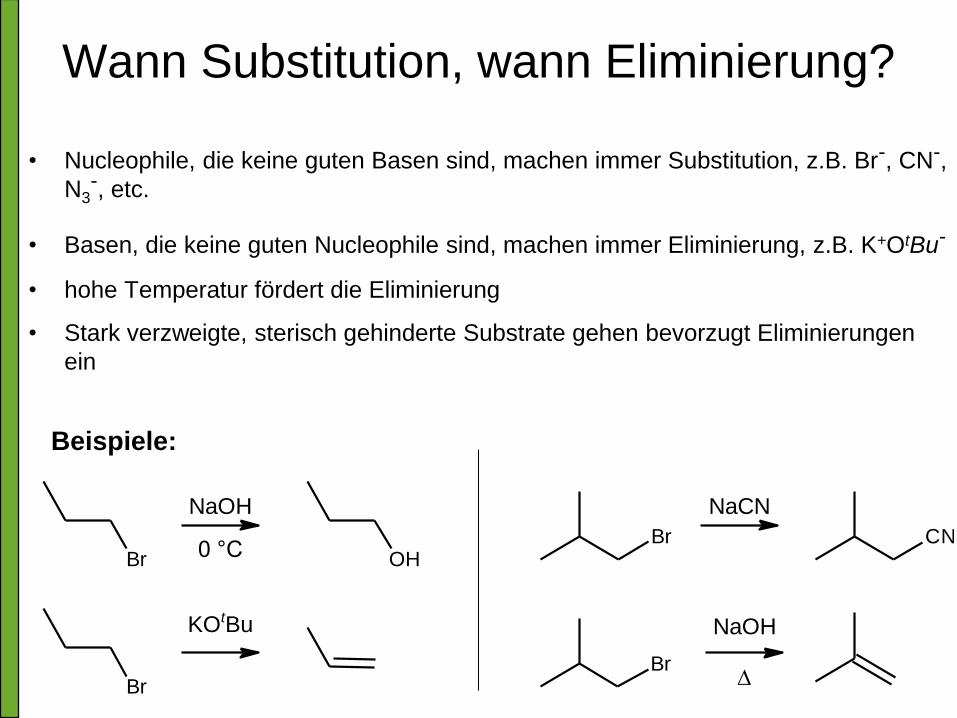
Wann Substitution, wann Eliminierung?
• Nucleophile, die keine guten Basen sind, machen immer Substitution, z.B. Br-, CN-,
N3-, etc.
• Basen, die keine guten Nucleophile sind, machen immer Eliminierung, z.B. K+OtBu-
• hohe Temperatur fördert die Eliminierung
• Stark verzweigte, sterisch gehinderte Substrate gehen bevorzugt Eliminierungen
ein
Beispiele:
Br
BrBr
Br
KOtBu
NaCNNaOH
0 °C OHCN
NaOH

Additionsreaktionen
A + B-C → A-B-C
45
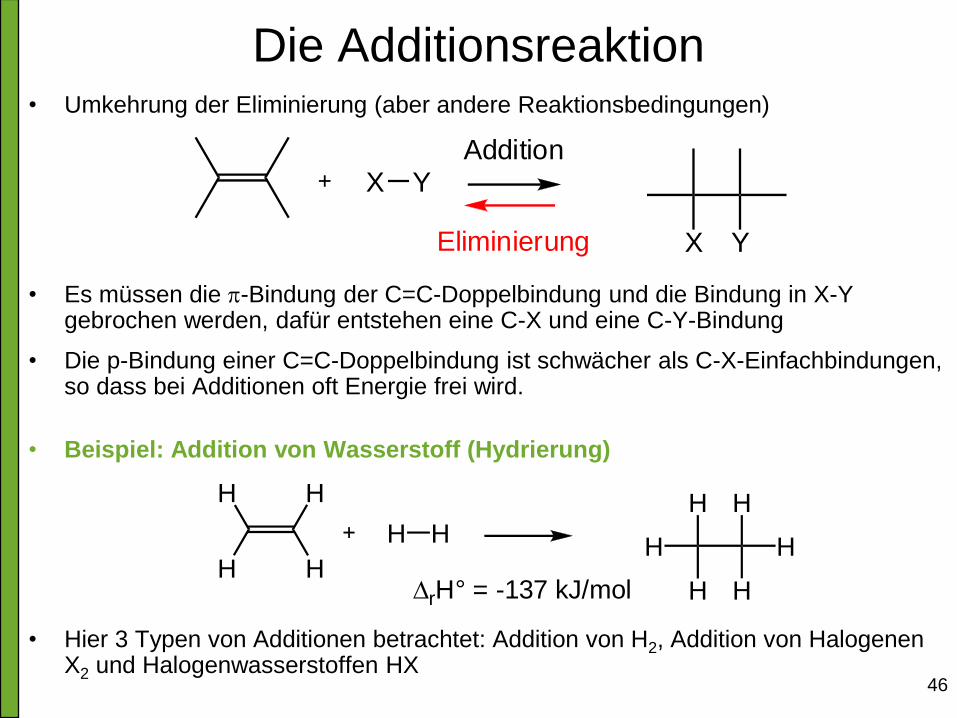
46
Die Additionsreaktion • Umkehrung der Eliminierung (aber andere Reaktionsbedingungen)
• Es müssen die p-Bindung der C=C-Doppelbindung und die Bindung in X-Y gebrochen werden, dafür entstehen eine C-X und eine C-Y-Bindung
• Die p-Bindung einer C=C-Doppelbindung ist schwächer als C-X-Einfachbindungen, so dass bei Additionen oft Energie frei wird.
• Beispiel: Addition von Wasserstoff (Hydrierung)
• Hier 3 Typen von Additionen betrachtet: Addition von H2, Addition von Halogenen X2 und Halogenwasserstoffen HX
X Y
YX
Addition
Eliminierung
H
H H
H
H HH
H
H
H
H
HrH° = -137 kJ/mol

47
Additionsreaktionen: Katalytische Hydrierung
• Hydrierung: Addition von Wasserstoff
• Obwohl Addition von H2 an C=C-Bindung exergon ist, läuft die Reaktion so
langsam ab, das nichts passiert.
Grund: Aktivierungsbarriere zu hoch!
Lösung: Absenken der Aktivierungsenergie durch Katalysator
• Mögliche Hydrierkatalysatoren: fein verteilte Metalle wie Nickel, Platin und
Palladium (also heterogene Katalyse an einer festen Oberfläche)
• Wasserstoff adsorbiert auf der Metalloberfläche und die H-H-Bindung wird
gebrochen
• Die Einzel-H-Atome sind sehr viel reaktiver als molekularer Wasserstoff H2 und auf
der Oberfläche beweglich
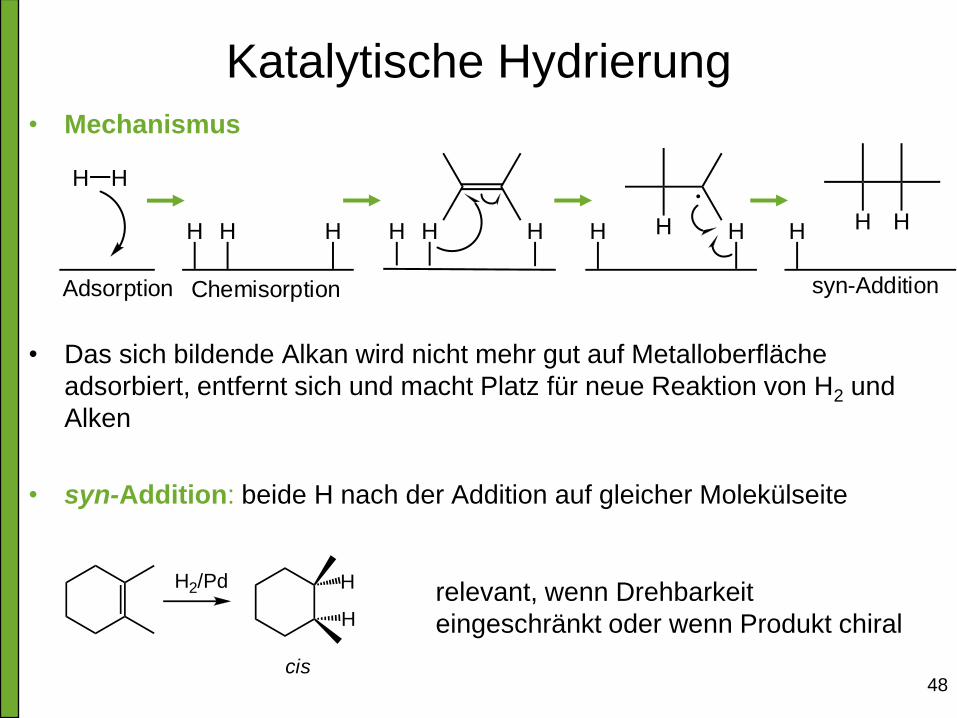
48
Katalytische Hydrierung • Mechanismus
• Das sich bildende Alkan wird nicht mehr gut auf Metalloberfläche
adsorbiert, entfernt sich und macht Platz für neue Reaktion von H2 und
Alken
• syn-Addition: beide H nach der Addition auf gleicher Molekülseite
H H H
H H
H H H HH HH HH
Adsorption Chemisorption syn-Addition
H
H
H2/Pd
cis
relevant, wenn Drehbarkeit
eingeschränkt oder wenn Produkt chiral
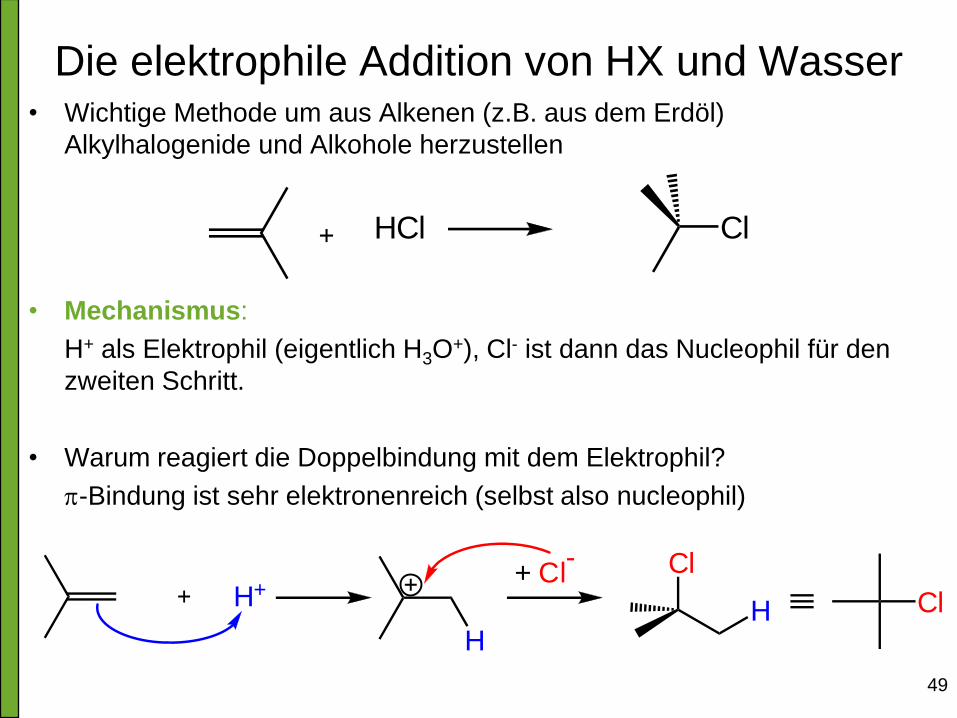
49
Die elektrophile Addition von HX und Wasser
• Wichtige Methode um aus Alkenen (z.B. aus dem Erdöl)
Alkylhalogenide und Alkohole herzustellen
• Mechanismus:
H+ als Elektrophil (eigentlich H3O+), Cl- ist dann das Nucleophil für den
zweiten Schritt.
• Warum reagiert die Doppelbindung mit dem Elektrophil?
p-Bindung ist sehr elektronenreich (selbst also nucleophil)
HCl Cl
H+
H
+ Cl- Cl
H Cl
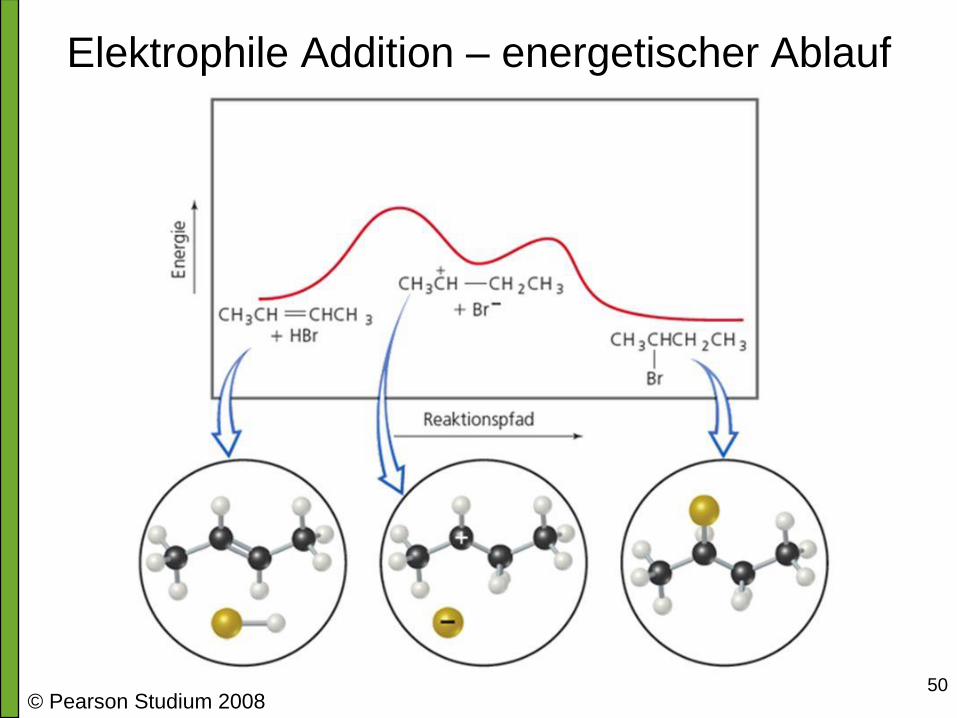
50
Elektrophile Addition – energetischer Ablauf
© Pearson Studium 2008
51
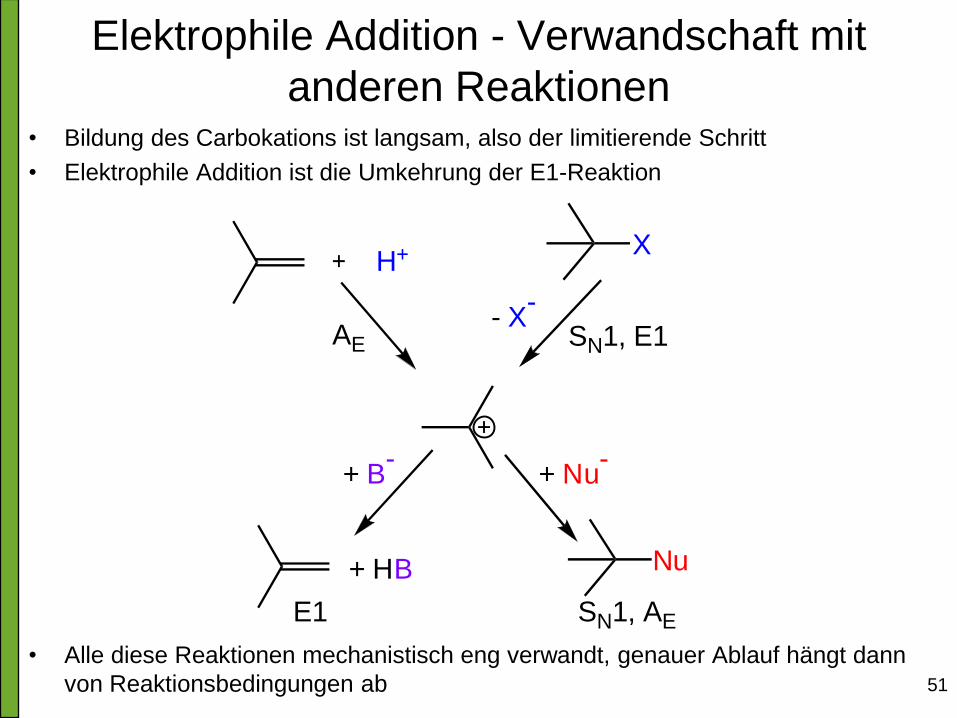
Elektrophile Addition - Verwandschaft mit
anderen Reaktionen • Bildung des Carbokations ist langsam, also der limitierende Schritt
• Elektrophile Addition ist die Umkehrung der E1-Reaktion
• Alle diese Reaktionen mechanistisch eng verwandt, genauer Ablauf hängt dann
von Reaktionsbedingungen ab
H+ X
+ HB
+ B-
+ Nu-
Nu
SN1, E1
SN1, AEE1
AE- X
-
52
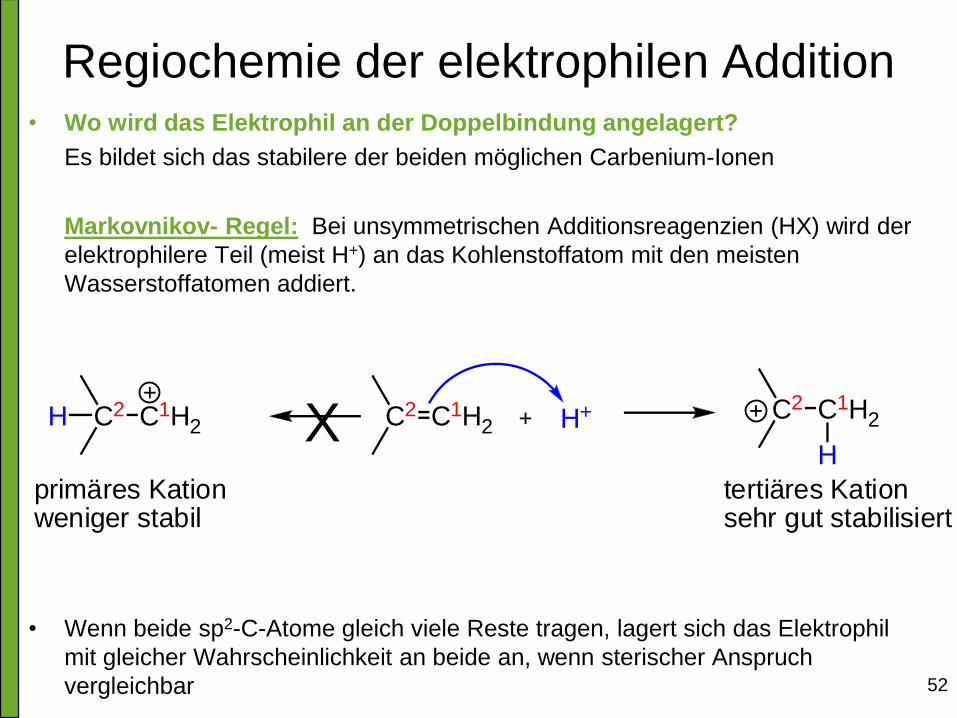
Regiochemie der elektrophilen Addition • Wo wird das Elektrophil an der Doppelbindung angelagert?
Es bildet sich das stabilere der beiden möglichen Carbenium-Ionen
Markovnikov- Regel: Bei unsymmetrischen Additionsreagenzien (HX) wird der
elektrophilere Teil (meist H+) an das Kohlenstoffatom mit den meisten
Wasserstoffatomen addiert.
• Wenn beide sp2-C-Atome gleich viele Reste tragen, lagert sich das Elektrophil
mit gleicher Wahrscheinlichkeit an beide an, wenn sterischer Anspruch
vergleichbar
C2 C1H2 H+XC2 C1H2H C2 C1H2
H
primäres Kationweniger stabil
tertiäres Kationsehr gut stabilisiert
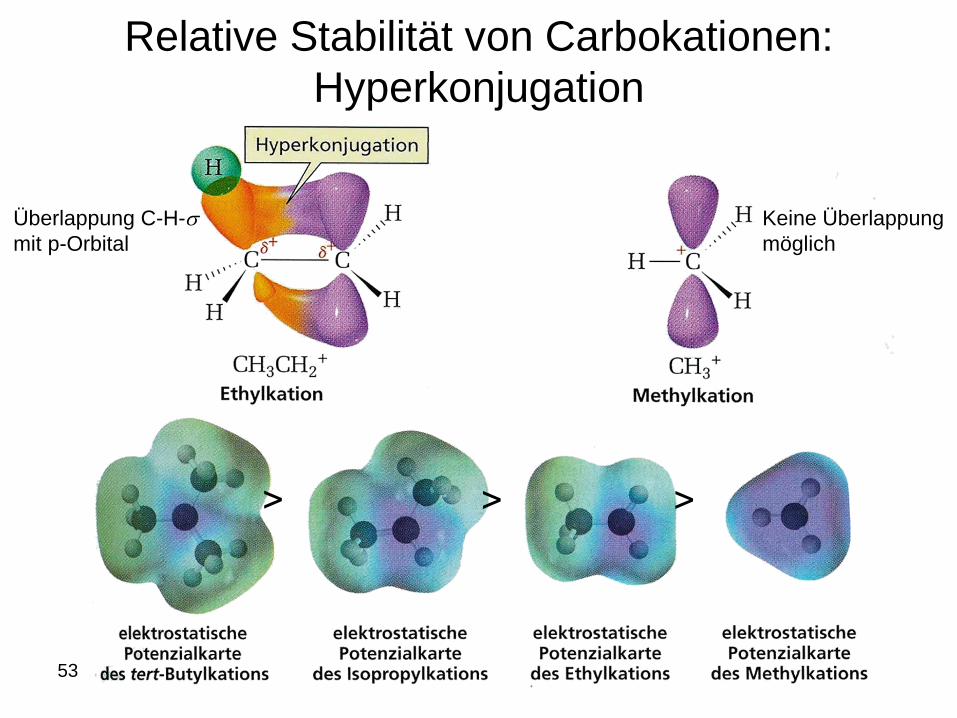
Relative Stabilität von Carbokationen:
Hyperkonjugation
53
Überlappung C-H-s
mit p-Orbital
Keine Überlappung
möglich
˃ ˃ ˃
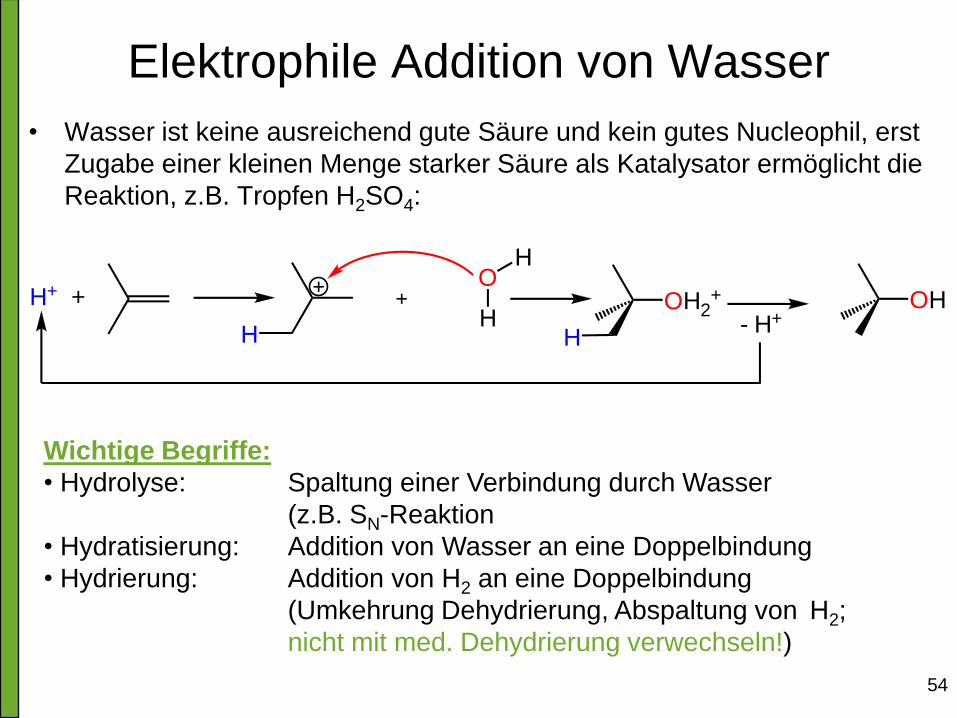
54
Elektrophile Addition von Wasser
• Wasser ist keine ausreichend gute Säure und kein gutes Nucleophil, erst
Zugabe einer kleinen Menge starker Säure als Katalysator ermöglicht die
Reaktion, z.B. Tropfen H2SO4:
Wichtige Begriffe:
• Hydrolyse: Spaltung einer Verbindung durch Wasser
(z.B. SN-Reaktion
• Hydratisierung: Addition von Wasser an eine Doppelbindung
• Hydrierung: Addition von H2 an eine Doppelbindung
(Umkehrung Dehydrierung, Abspaltung von H2;
nicht mit med. Dehydrierung verwechseln!)
H+ +O
H
HOH2
+
H H- H+
OH
55
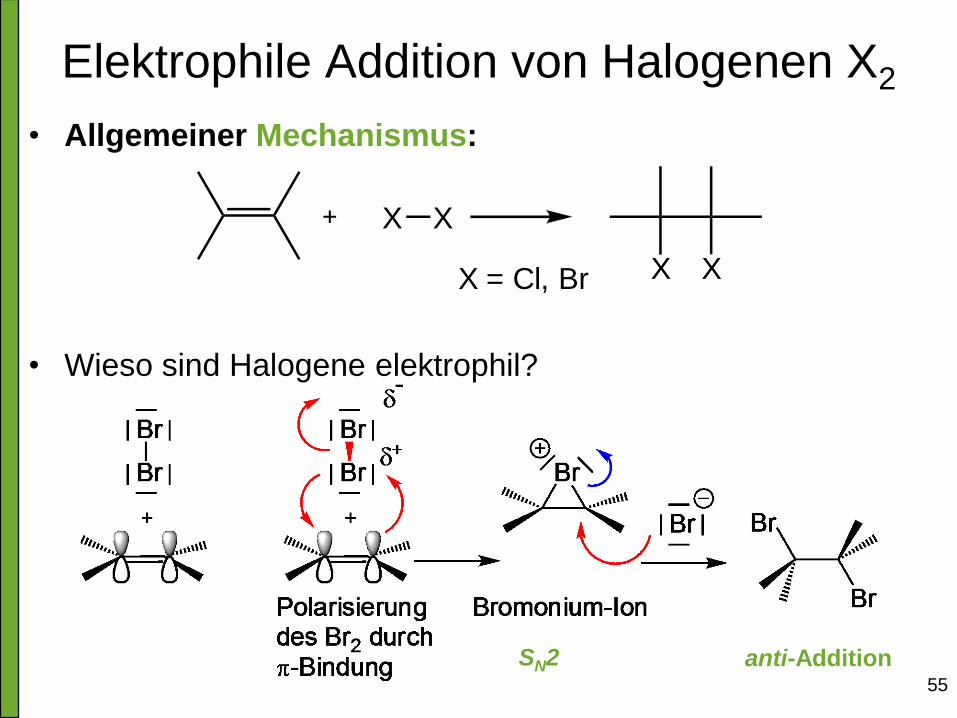
Elektrophile Addition von Halogenen X2
• Allgemeiner Mechanismus:
• Wieso sind Halogene elektrophil?
X X
XXX = Cl, Br
anti-Addition SN2
56
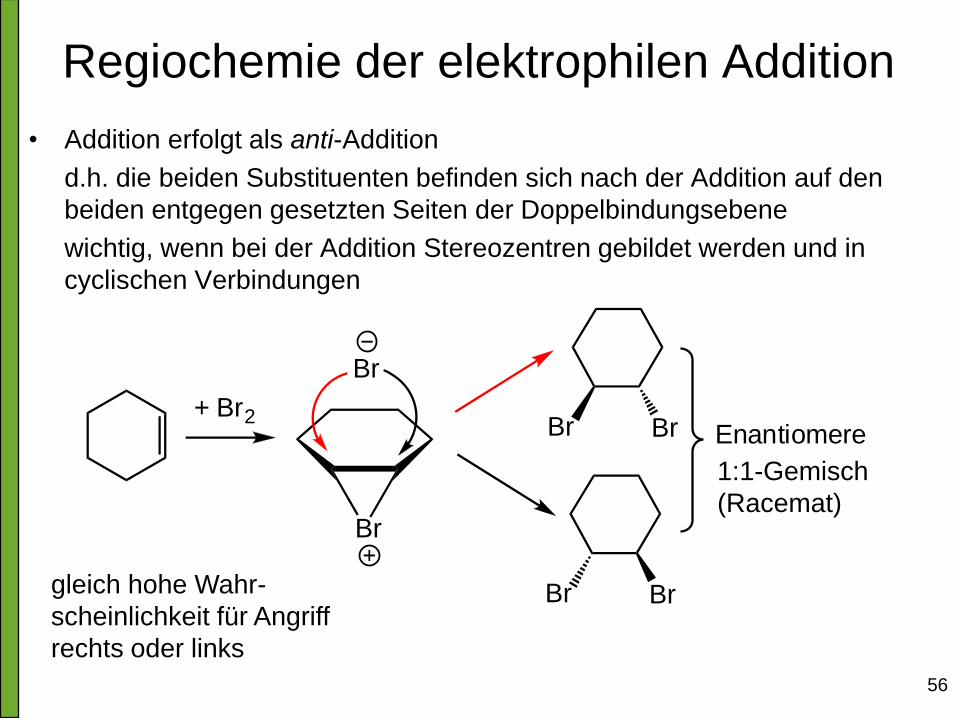
Regiochemie der elektrophilen Addition
• Addition erfolgt als anti-Addition
d.h. die beiden Substituenten befinden sich nach der Addition auf den
beiden entgegen gesetzten Seiten der Doppelbindungsebene
wichtig, wenn bei der Addition Stereozentren gebildet werden und in
cyclischen Verbindungen
Br
Br
Br
Br
Br
Br
Enantiomere+ Br2
gleich hohe Wahr-
scheinlichkeit für Angriff
rechts oder links
1:1-Gemisch
(Racemat)

57
Kapitel 7
Aromaten
und ihre Reaktionen

Acetylsalicylsäure – ein Wirkstoff und seine Geschichte
• Fieber senkende und schmerzstillende Wirkung
von Extrakten der Weidenrinde seit Hippokrates
(460-370 v. Chr.) bekannt
• Extraktion der Salicylsäure (von Salicaceae) gelang Mitte des 19. Jh.,
erste Synthese der Verbindung durch H. Kolbe 1859
• Starke Nebenwirkungen, z.B. Magenblutungen
• Verbesserung der Wirksamkeit und der Verträglichkeit durch Herstellung
eines Derivates mit einer Acetylgruppe
• Handelsname des Präparates bei der entwickelnden Firma Bayer:
Aspirin (andere Namen: ASS, Acesal, Alka-Seltzer usw.)
58
Welche Struktur hat Aspirin und wie
wird es hergestellt? O
OH
O
O
59
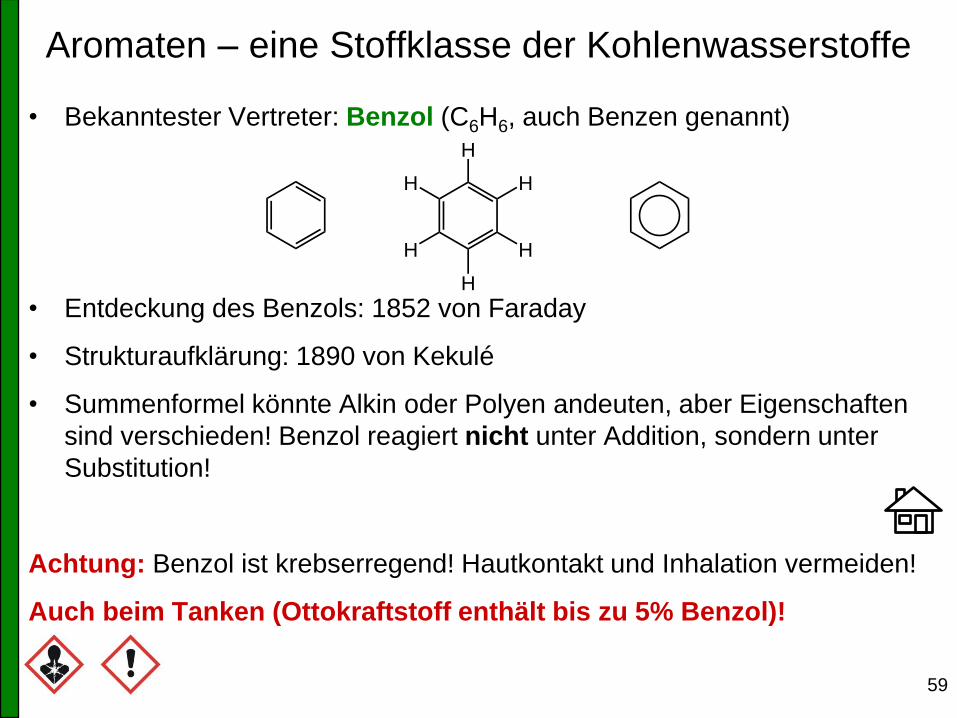
Aromaten – eine Stoffklasse der Kohlenwasserstoffe
• Bekanntester Vertreter: Benzol (C6H6, auch Benzen genannt)
• Entdeckung des Benzols: 1852 von Faraday
• Strukturaufklärung: 1890 von Kekulé
• Summenformel könnte Alkin oder Polyen andeuten, aber Eigenschaften
sind verschieden! Benzol reagiert nicht unter Addition, sondern unter
Substitution!
Achtung: Benzol ist krebserregend! Hautkontakt und Inhalation vermeiden!
Auch beim Tanken (Ottokraftstoff enthält bis zu 5% Benzol)!
H
H
H
H
H
H
60
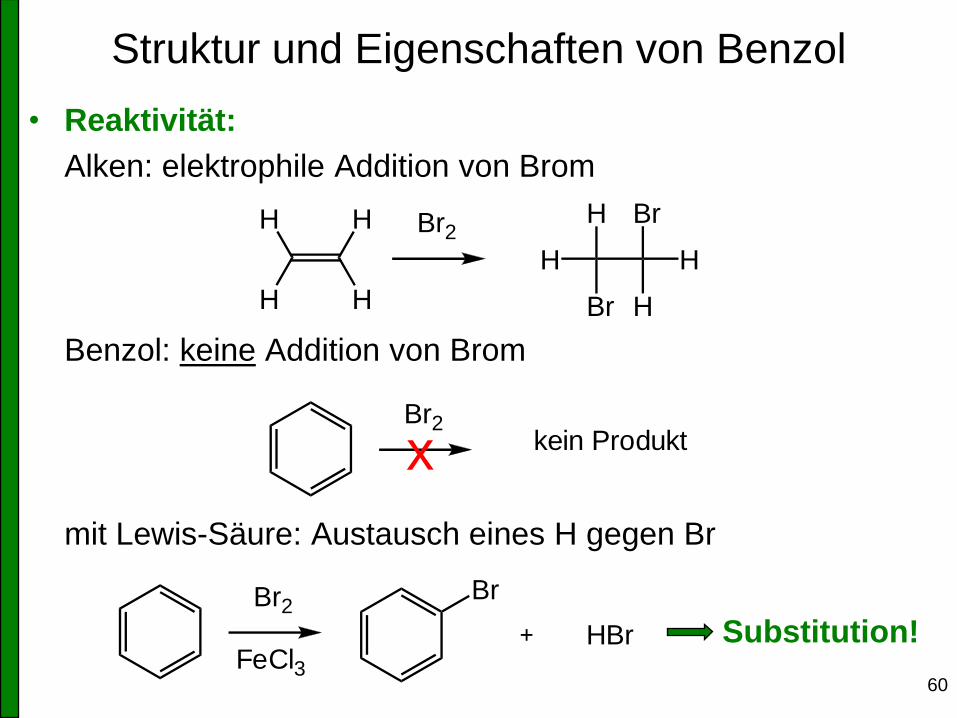
Struktur und Eigenschaften von Benzol
• Reaktivität:
Alken: elektrophile Addition von Brom
Benzol: keine Addition von Brom
mit Lewis-Säure: Austausch eines H gegen Br
Br2H
H
H
H
H
Br
H
H
H
Br
Br2kein Produktx
Br2
FeCl3
Br
HBr Substitution!
61
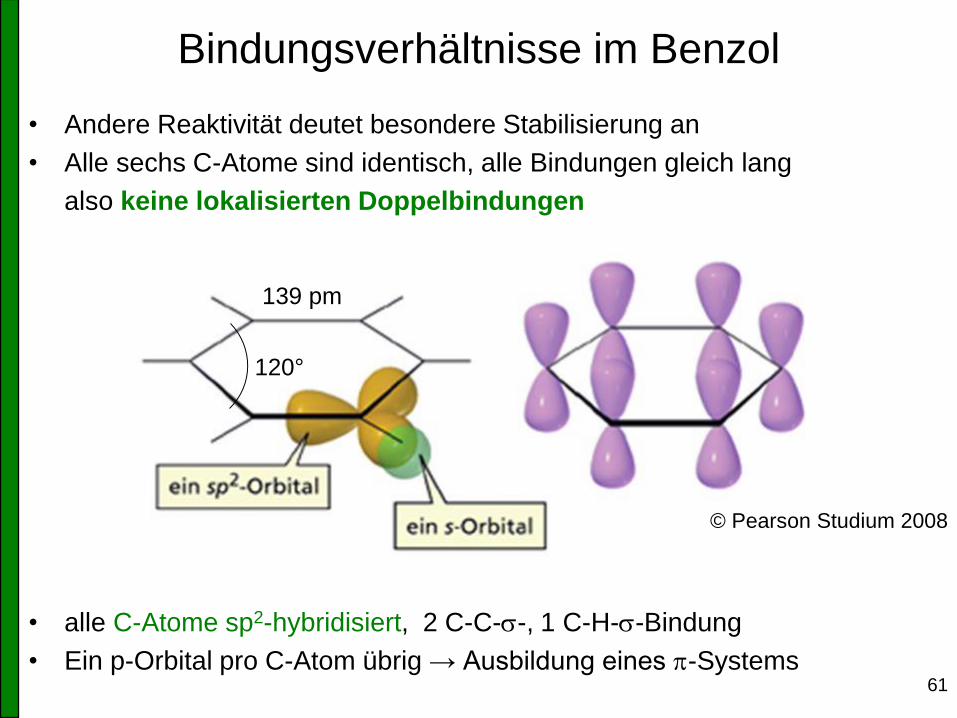
• Andere Reaktivität deutet besondere Stabilisierung an
• Alle sechs C-Atome sind identisch, alle Bindungen gleich lang
also keine lokalisierten Doppelbindungen
• alle C-Atome sp2-hybridisiert, 2 C-C-s-, 1 C-H-s-Bindung
• Ein p-Orbital pro C-Atom übrig → Ausbildung eines p-Systems
Bindungsverhältnisse im Benzol
© Pearson Studium 2008
120°
139 pm

62
Struktur des p-System im Benzol
• Es kommt nicht zur Ausbildung von 3 Doppelbindungen!
sonst wären die Bindungslängen alternierend (Einfachbindung ~154 pm,
Doppelbindung ~134 pm, beobachtet: alle gleich, 139 pm)
• Alle 6 p-Orbitale überlappen ringförmig miteinander
es bildet sich ein cyclisch delokalisiertes p-System
• Die 6 p-Elektronen sind als „Wolke“ gleichmäßig oberhalb und unterhalb
der Molekülebene verteilt
• Elektronendelokalisation (auch: Resonanz, Mesomerie) ist der Grund für
die besonderen Eigenschaften des Benzols
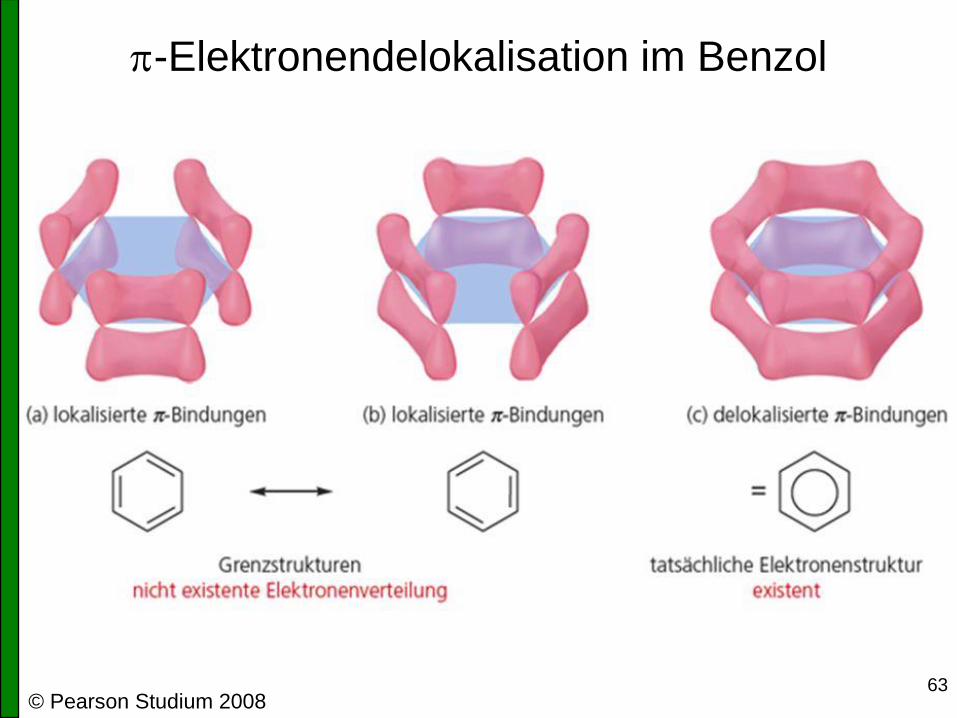
63
p-Elektronendelokalisation im Benzol
© Pearson Studium 2008
64
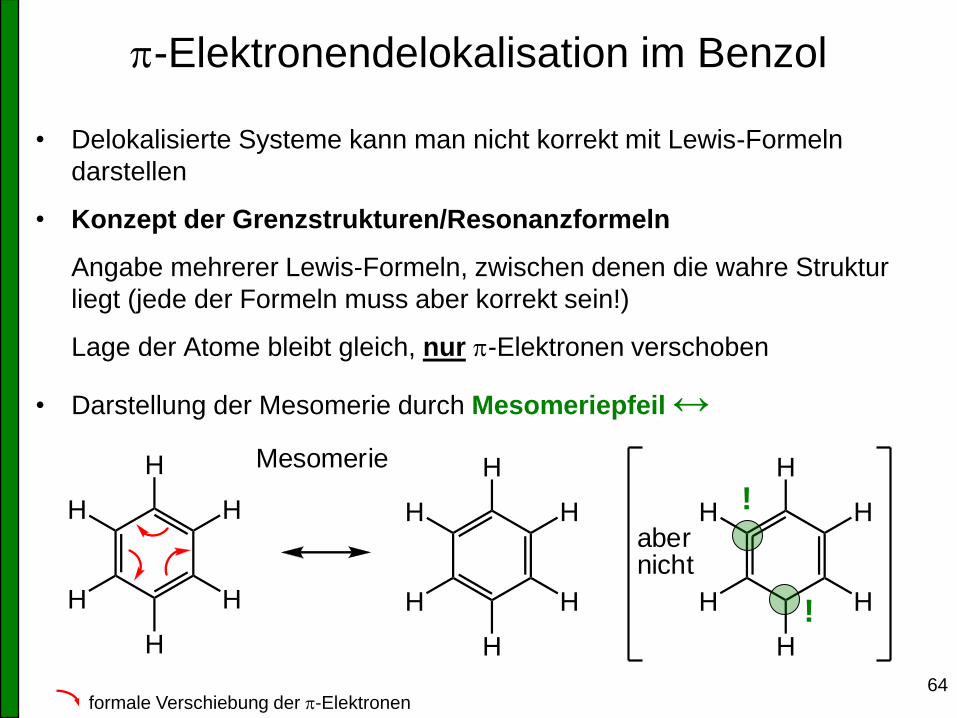
p-Elektronendelokalisation im Benzol
• Delokalisierte Systeme kann man nicht korrekt mit Lewis-Formeln
darstellen
• Konzept der Grenzstrukturen/Resonanzformeln
Angabe mehrerer Lewis-Formeln, zwischen denen die wahre Struktur
liegt (jede der Formeln muss aber korrekt sein!)
Lage der Atome bleibt gleich, nur p-Elektronen verschoben
• Darstellung der Mesomerie durch Mesomeriepfeil ↔
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
H
Habernicht
Mesomerie
formale Verschiebung der p-Elektronen
!
!

65
Das Konzept der Mesomerie
• Mesomerie ist kein Gleichgewicht!
• Die Grenzstrukturen sind nur Gedankenkonstrukte und existieren in der Realität nicht!
• Es existiert nur die delokalisierte Struktur, die zwischen den Resonanzformeln liegt
• Wenn mehrere, nicht-äquivalente Resonanzformeln möglich, ähnelt die wahre Struktur eher der stabileren Formel
• Faustregel: Je mehr sinnvolle und gut stabilisierte Resonanzformeln aufgestellt werden können, desto stabiler ist das Molekül (Benzol: 2 mögliche, sehr stabile Resonanzformeln)
• Wann tritt Resonanz/Mesomerie auf?
wenn mehr als zwei sp2-oder sp-Atome mit parallelen p-Orbitalen direkt benachbart sind, so dass es zu seitlicher Überlappung von mindestens drei p-Orbitalen kommt (im Benzol 6), s.a. Konjugation
66
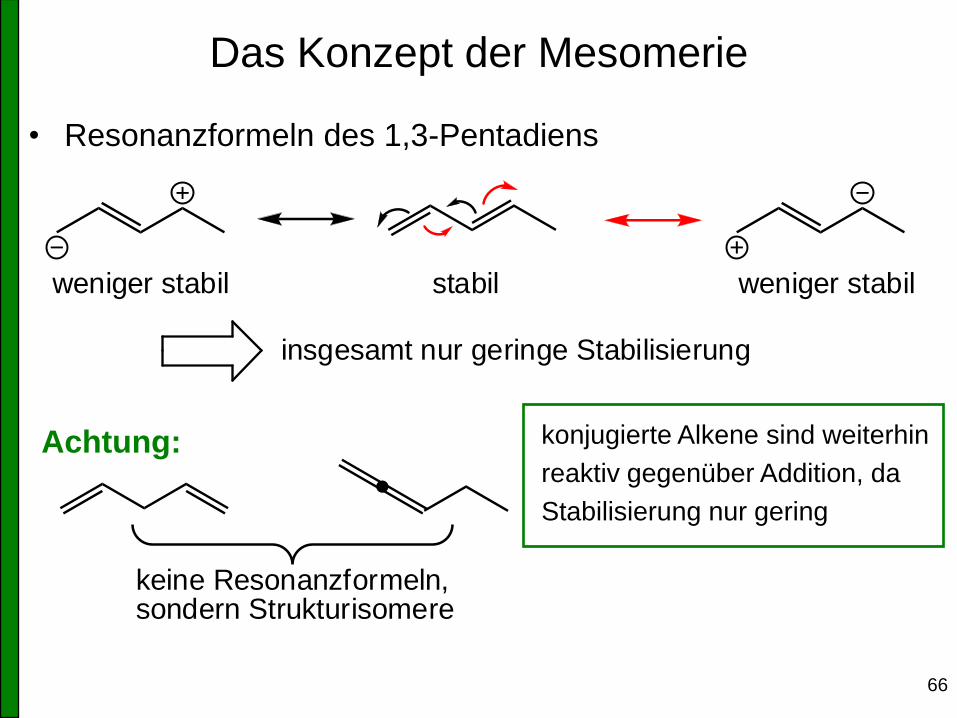
Das Konzept der Mesomerie
• Resonanzformeln des 1,3-Pentadiens
weniger stabilstabilweniger stabil
insgesamt nur geringe Stabilisierung
keine Resonanzformeln,sondern Strukturisomere
konjugierte Alkene sind weiterhin
reaktiv gegenüber Addition, da
Stabilisierung nur gering
Achtung:
67
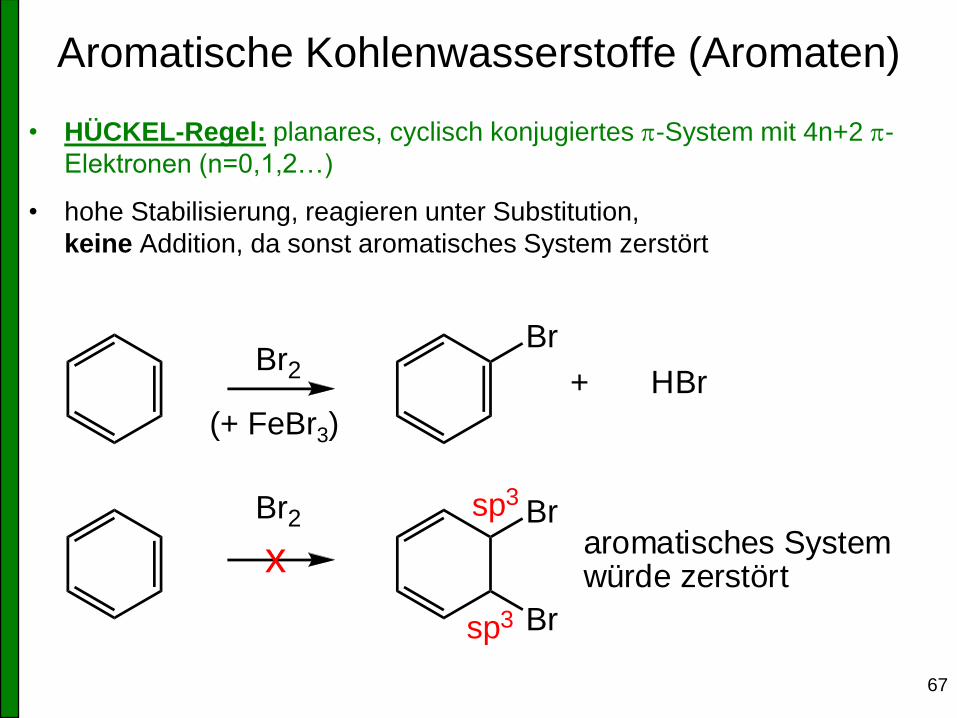
Aromatische Kohlenwasserstoffe (Aromaten)
• HÜCKEL-Regel: planares, cyclisch konjugiertes p-System mit 4n+2 p-
Elektronen (n=0,1,2…)
• hohe Stabilisierung, reagieren unter Substitution,
keine Addition, da sonst aromatisches System zerstört
BrBr2 + HBr
Br
Br
Br2
x
sp3
sp3
aromatisches Systemwürde zerstört
(+ FeBr3)
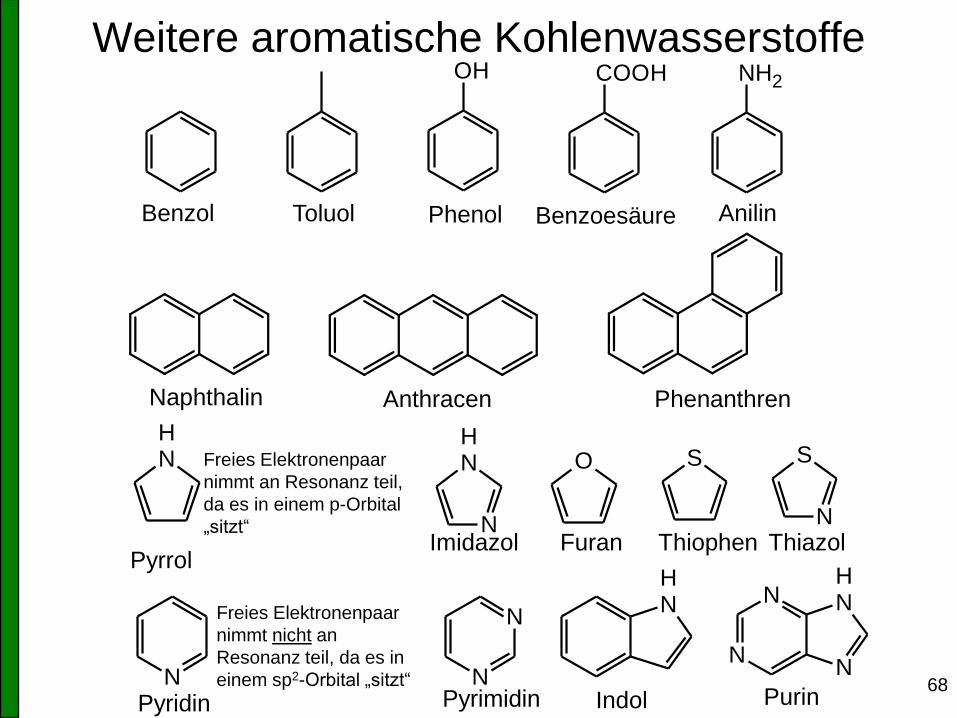
68
Weitere aromatische Kohlenwasserstoffe OH COOH NH2
Benzol Toluol Phenol Benzoesäure Anilin
Naphthalin Anthracen Phenanthren
NH
NH
N
O S S
N
N
N
N
NH
N
N
NH
N
Pyrrol
Pyridin
Imidazol Furan Thiophen Thiazol
Pyrimidin Indol Purin
Freies Elektronenpaar
nimmt an Resonanz teil,
da es in einem p-Orbital
„sitzt“
Freies Elektronenpaar
nimmt nicht an
Resonanz teil, da es in
einem sp2-Orbital „sitzt“
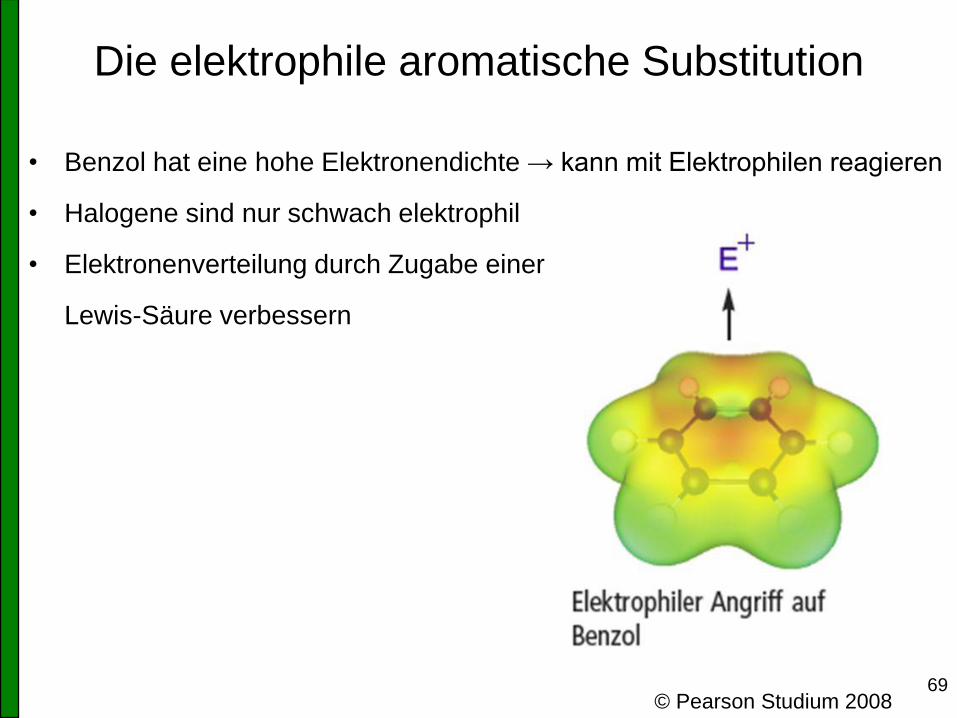
69
Die elektrophile aromatische Substitution
• Benzol hat eine hohe Elektronendichte → kann mit Elektrophilen reagieren
• Halogene sind nur schwach elektrophil
• Elektronenverteilung durch Zugabe einer
Lewis-Säure verbessern
© Pearson Studium 2008
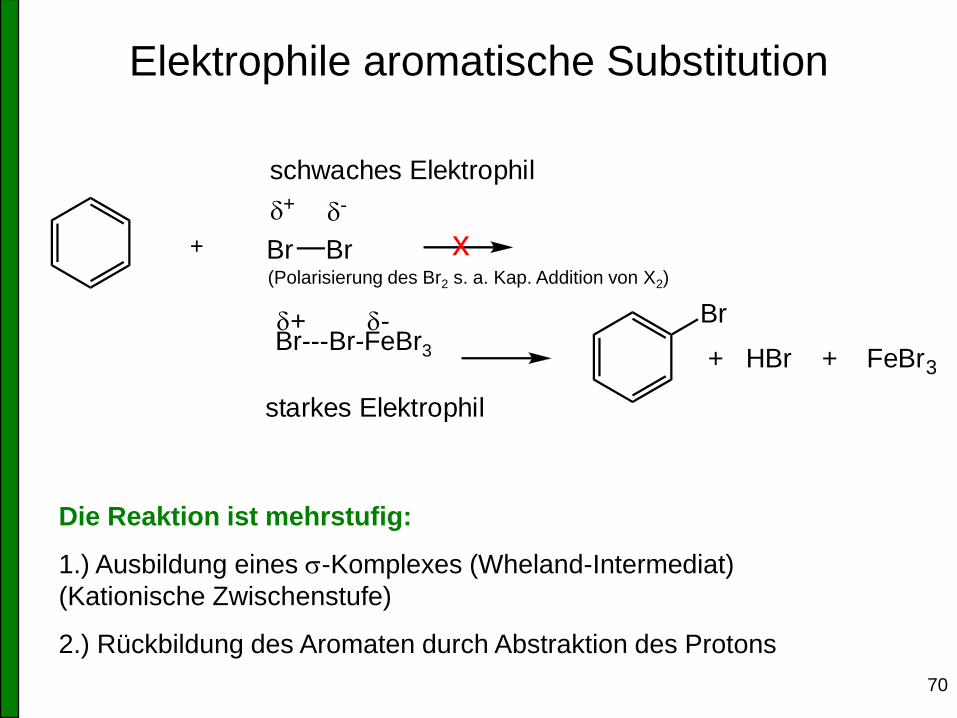
70
Elektrophile aromatische Substitution
Br Br
+ -
Br Br
+
FeBr3
schwaches Elektrophil
starkes Elektrophil
x
+ HBr + FeBr3
Br
Die Reaktion ist mehrstufig:
1.) Ausbildung eines s-Komplexes (Wheland-Intermediat)
(Kationische Zwischenstufe)
2.) Rückbildung des Aromaten durch Abstraktion des Protons
(Polarisierung des Br2 s. a. Kap. Addition von X2)
Br---Br-FeBr3 + -
71
Elektrophile aromatische Substitution
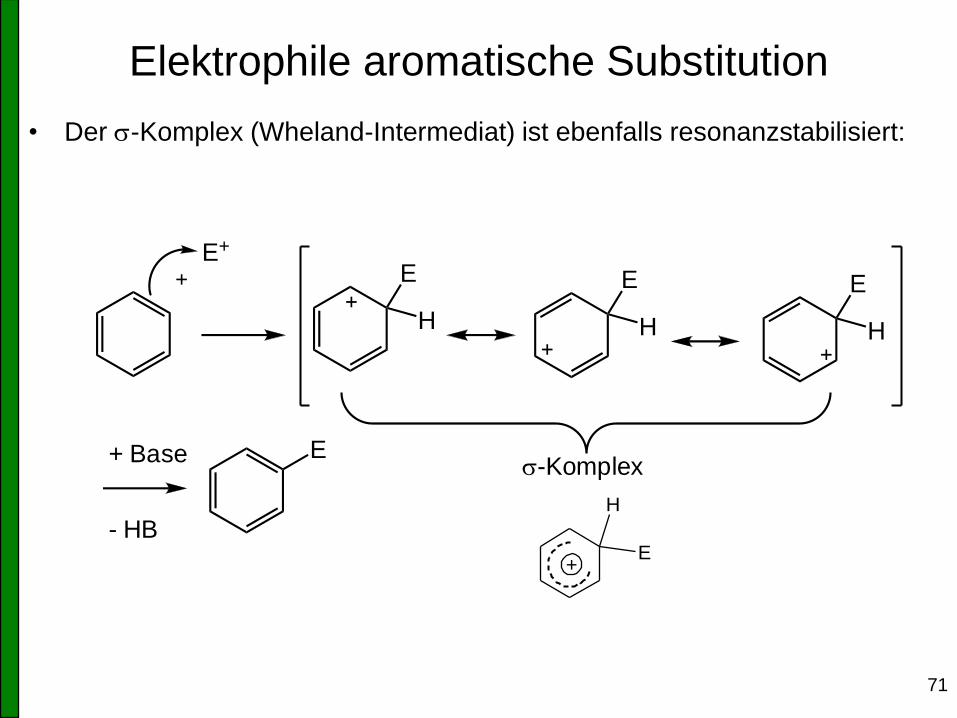
• Der s-Komplex (Wheland-Intermediat) ist ebenfalls resonanzstabilisiert:
E
H
E
H
E
H
s-Komplex
E+
+ Base
- HB
E
H
E+
72
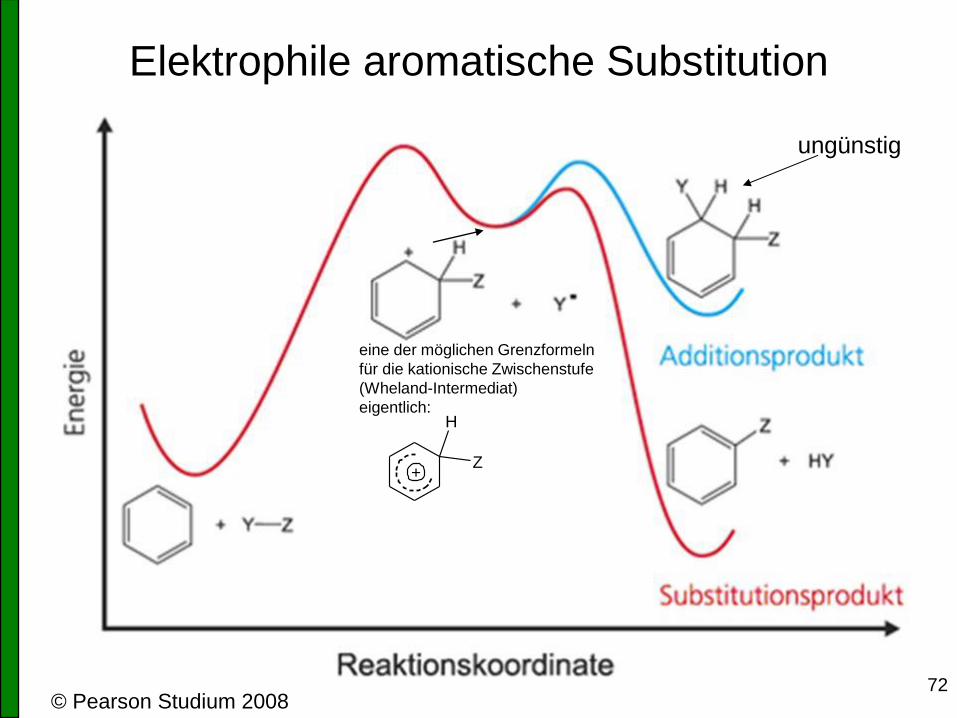
Elektrophile aromatische Substitution
© Pearson Studium 2008
eine der möglichen Grenzformeln
für die kationische Zwischenstufe
(Wheland-Intermediat)
eigentlich:
ungünstig
H
Z+
73
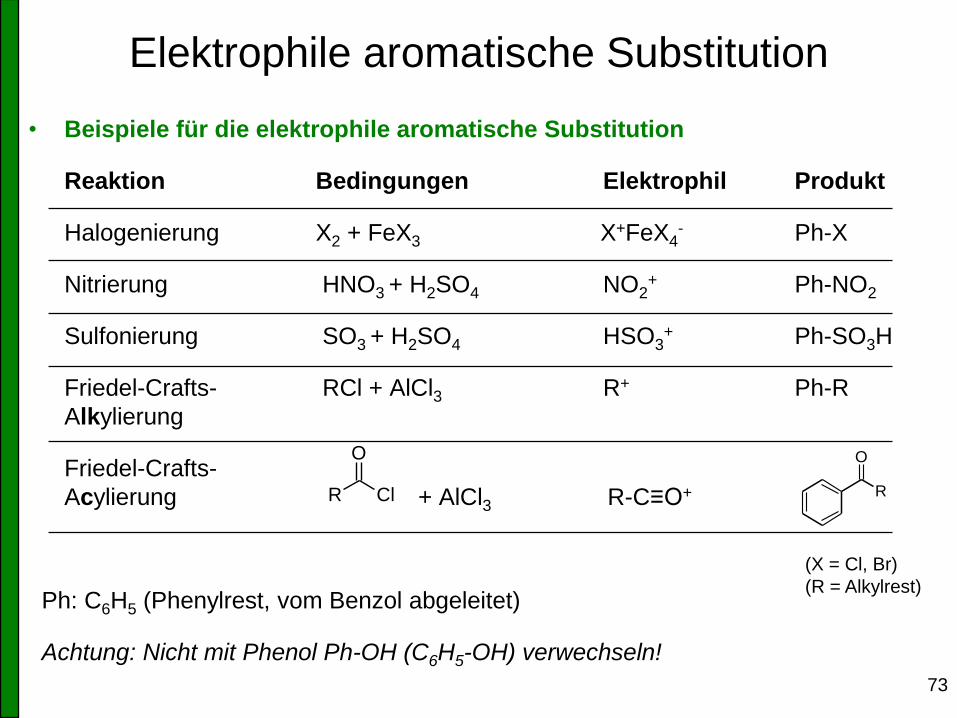
Elektrophile aromatische Substitution
• Beispiele für die elektrophile aromatische Substitution
Reaktion Bedingungen Elektrophil Produkt
Halogenierung X2 + FeX3 X+FeX4- Ph-X
Nitrierung HNO3 + H2SO4 NO2+ Ph-NO2
Sulfonierung SO3 + H2SO4 HSO3+ Ph-SO3H
Friedel-Crafts- RCl + AlCl3 R+ Ph-R
Alkylierung
Friedel-Crafts-
Acylierung + AlCl3 R-C≡O+
Ph: C6H5 (Phenylrest, vom Benzol abgeleitet)
Achtung: Nicht mit Phenol Ph-OH (C6H5-OH) verwechseln!
(X = Cl, Br)
(R = Alkylrest)
R Cl
O
R
O
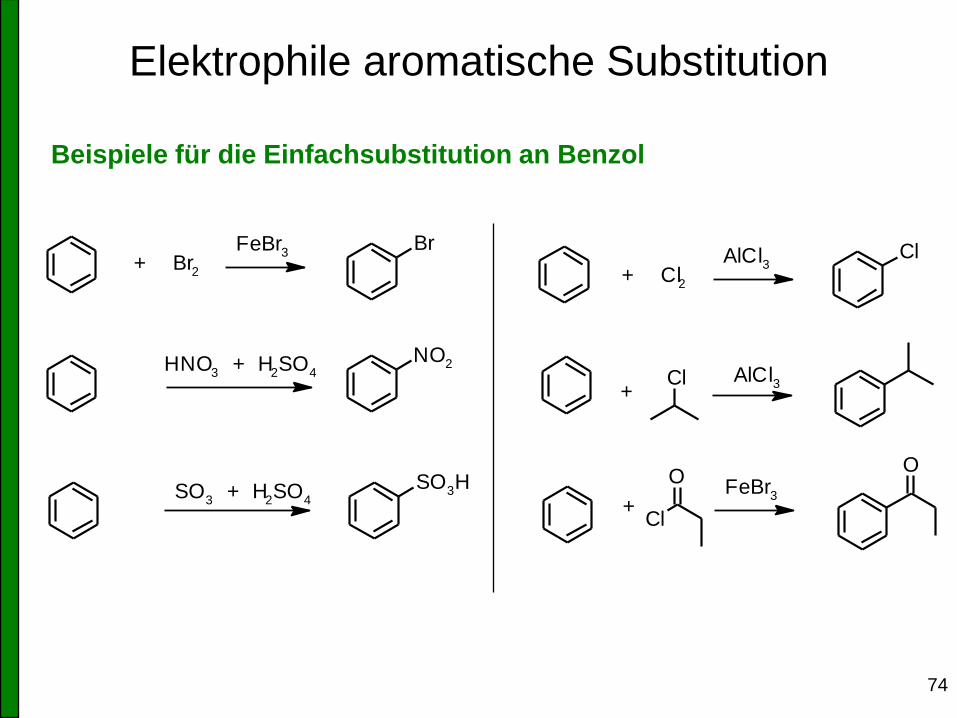
Elektrophile aromatische Substitution
74
Beispiele für die Einfachsubstitution an Benzol
+ Br2
FeBr3
Br
NO2
SO3H
O
Cl
HNO3 + H
2SO
4
+AlCl
3
+ FeBr
3
+ Cl2
AlCl3
SO3 + H
2SO
4
Cl
Cl
O
75
Elektrophile aromatische Substitution
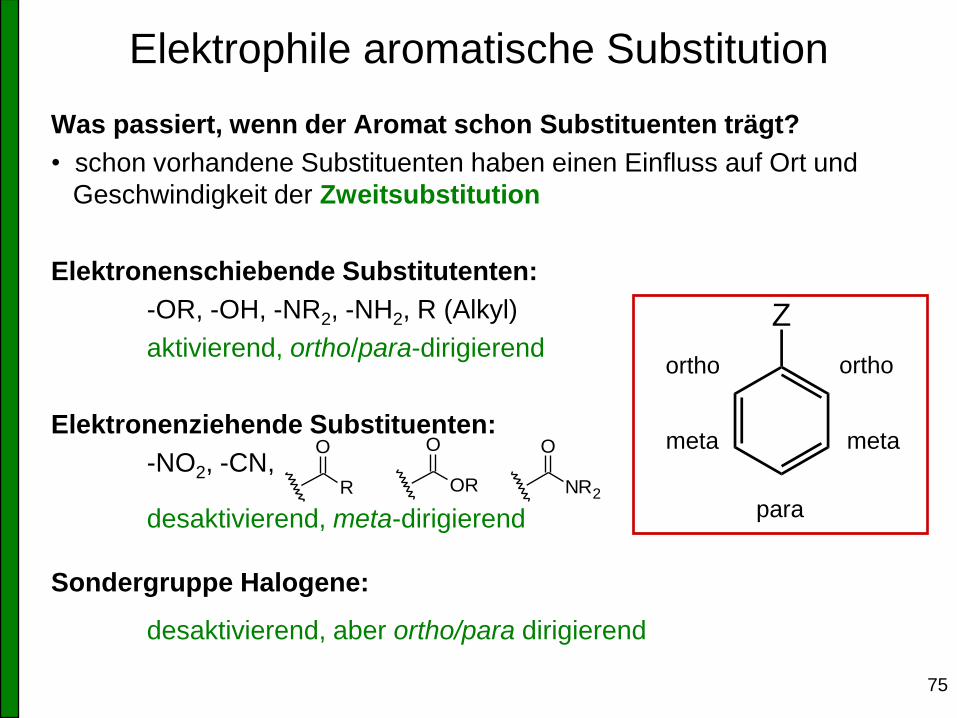
Was passiert, wenn der Aromat schon Substituenten trägt?
• schon vorhandene Substituenten haben einen Einfluss auf Ort und
Geschwindigkeit der Zweitsubstitution
Elektronenschiebende Substitutenten:
-OR, -OH, -NR2, -NH2, R (Alkyl)
aktivierend, ortho/para-dirigierend
Elektronenziehende Substituenten:
-NO2, -CN,
desaktivierend, meta-dirigierend
Sondergruppe Halogene:
desaktivierend, aber ortho/para dirigierend
Z
ortho ortho
meta meta
para R
O
OR
O
NR2
O
76
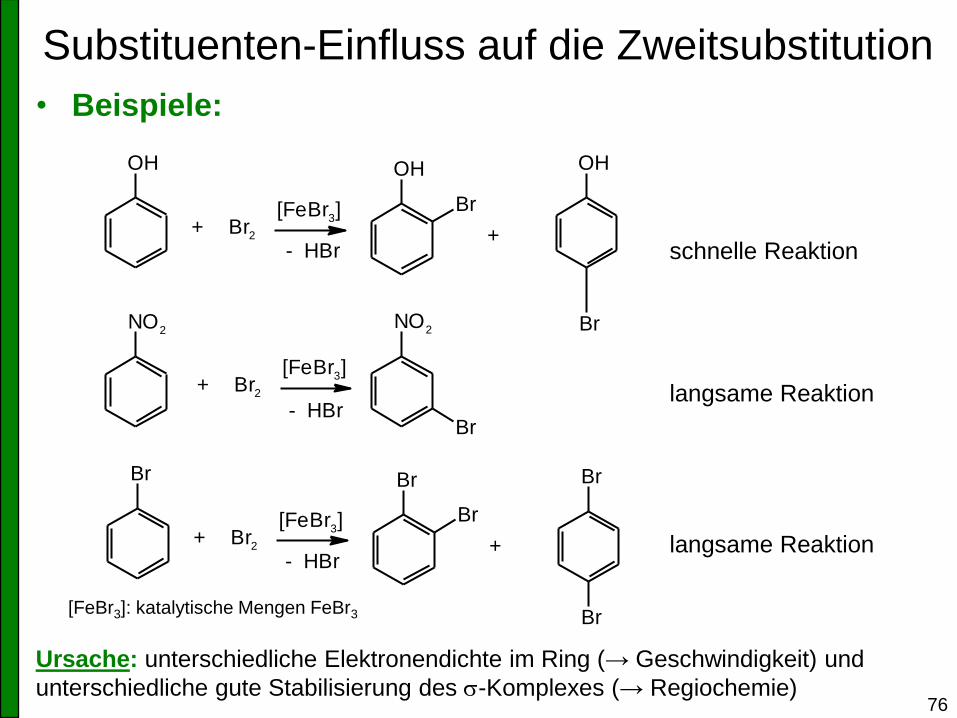
Substituenten-Einfluss auf die Zweitsubstitution
• Beispiele:
Ursache: unterschiedliche Elektronendichte im Ring (→ Geschwindigkeit) und
unterschiedliche gute Stabilisierung des s-Komplexes (→ Regiochemie)
schnelle Reaktion
langsame Reaktion
langsame Reaktion
[FeBr3]: katalytische Mengen FeBr3
OH
+ Br2
[FeBr3] Br
OH
NO2
NO2
Br
+
OH
Br
Br
+ Br2
[FeBr3] Br
Br
+
Br
Br
+ Br2
[FeBr3]
- HBr
- HBr
- HBr
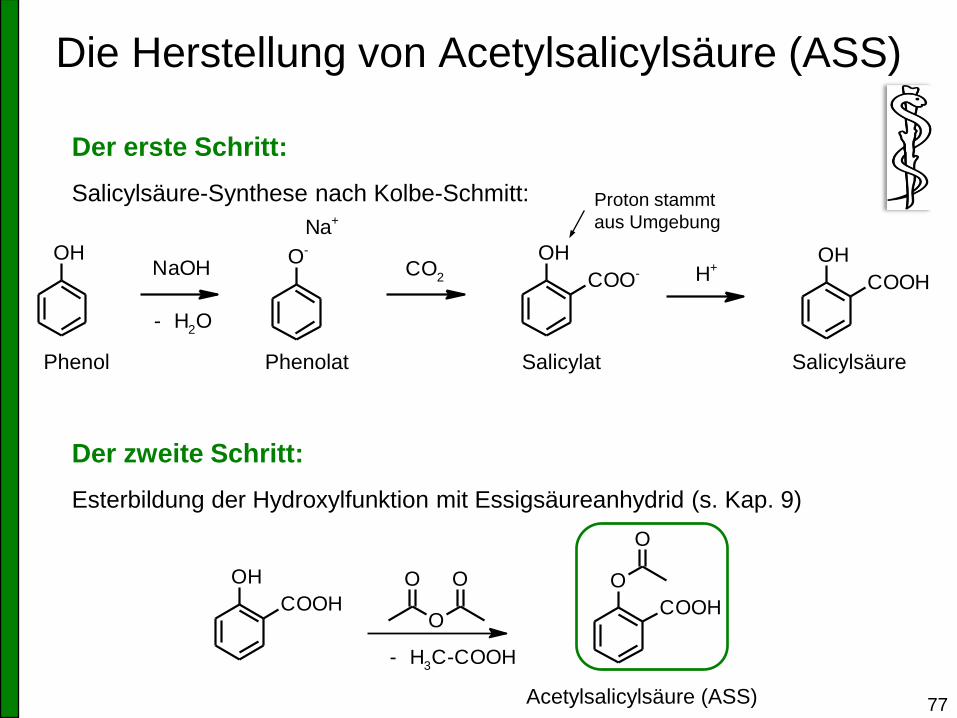
Die Herstellung von Acetylsalicylsäure (ASS)
77
Der erste Schritt:
Salicylsäure-Synthese nach Kolbe-Schmitt:
Der zweite Schritt:
Esterbildung der Hydroxylfunktion mit Essigsäureanhydrid (s. Kap. 9)
OH O- OH
COO-
Na+
NaOH
- H2O
CO2
OH
COOHH+
Phenol Phenolat Salicylat Salicylsäure
OH
COOHO
COOH
O
O
OO
- H3C-COOH
Acetylsalicylsäure (ASS)
Proton stammt
aus Umgebung

78
Kapitel 8
Radikalreaktionen

Luftsauerstoff – ein aggressives Reagenz
• Viele Lebensmittel, z.B. Butter oder Margarine verderben leichter, wenn sie
an der Sonne oder aber in Metallgefäßen gelagert werden.
• Austausch der Luft in Verpackungen gegen Schutzgas (Stickstoff oder
CO2) sowie Lagerung im Dunklen und Kühlen verzögert die Reaktion
• Der Zusatz von Vitamin C (Ascorbinsäure, E300) macht Lebensmittel
länger haltbar (sog. Antioxidationsmittel )
Welche Art von Reaktionen geht Luftsauerstoff ein?
Welche Verbindungen werden von Luftsauerstoff angegriffen?
Wie wirkt Vitamin C? 79
80
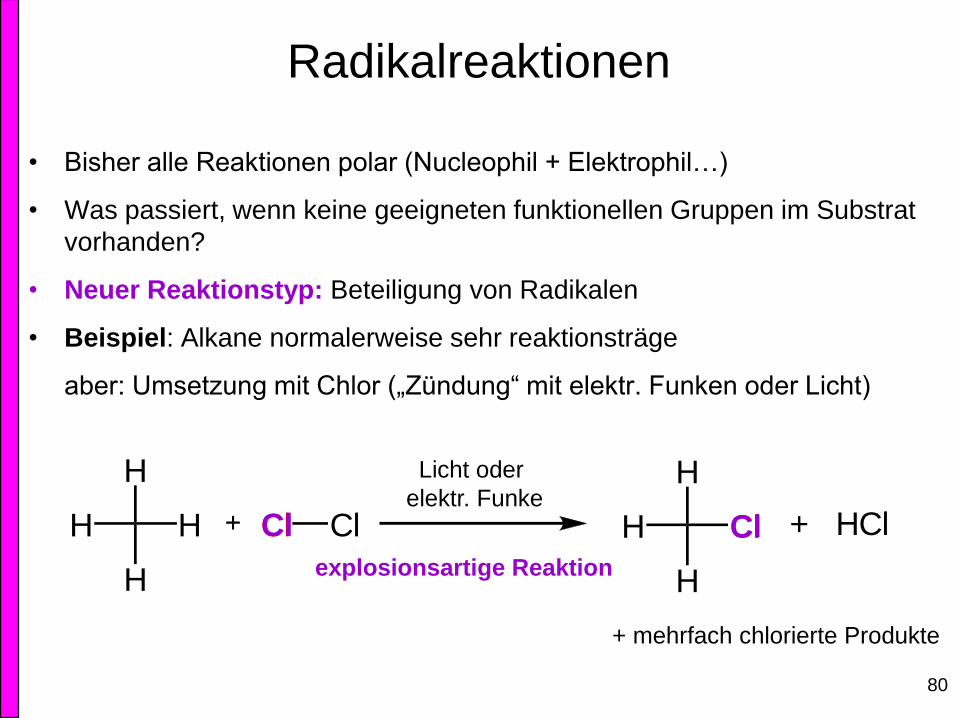
Radikalreaktionen
• Bisher alle Reaktionen polar (Nucleophil + Elektrophil…)
• Was passiert, wenn keine geeigneten funktionellen Gruppen im Substrat
vorhanden?
• Neuer Reaktionstyp: Beteiligung von Radikalen
• Beispiel: Alkane normalerweise sehr reaktionsträge
aber: Umsetzung mit Chlor („Zündung“ mit elektr. Funken oder Licht)
+ mehrfach chlorierte Produkte
H
H H
H
H
H Cl
H
Cl Cl + HCl
Licht oderelektr. Funke
explosionsartige Reaktion
Licht oder
elektr. Funke
Cl Cl

81
Radikalreaktionen
• Woher stammt das Radikal?
homolytische Spaltung der Cl-Cl-Bindung (Bindungsenergie 240 kJ/mol),
die C-H-Bindung ist zu stark (440 kJ/mol)
• Kettenstart-Reaktion:
• Chlor-Radikale (und andere Radikale) sind sehr reaktiv, da sie ein
ungepaartes Elektron und Elektronenmangel (zum Oktett fehlt ein
Elektron) besitzen
• Radikale reagieren mit sehr vielen Substraten, z.B. auch Alkanen
Cl Clh
2 Cl Chloratome, 7 Valenz-Elektronen
(bedeutet Einwirkung von Licht )
82
Radikalreaktionen
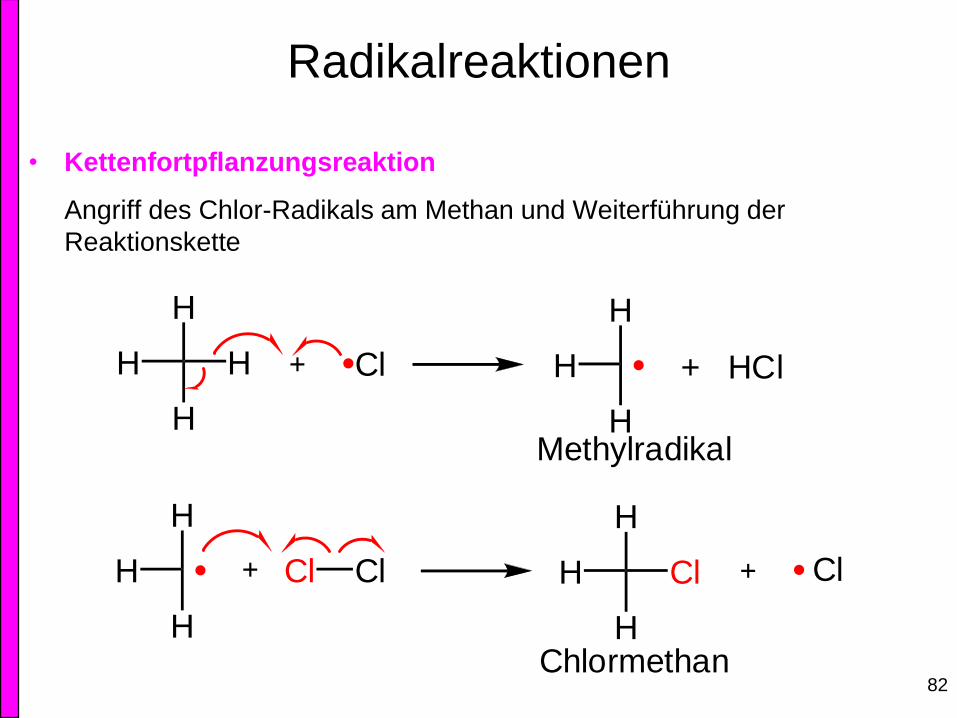
• Kettenfortpflanzungsreaktion
Angriff des Chlor-Radikals am Methan und Weiterführung der
Reaktionskette
H
H H
H
Cl
H
H
H
+ HCl
H
H
H
Cl Cl
H
H Cl
H
Cl
Methylradikal
Chlormethan
83
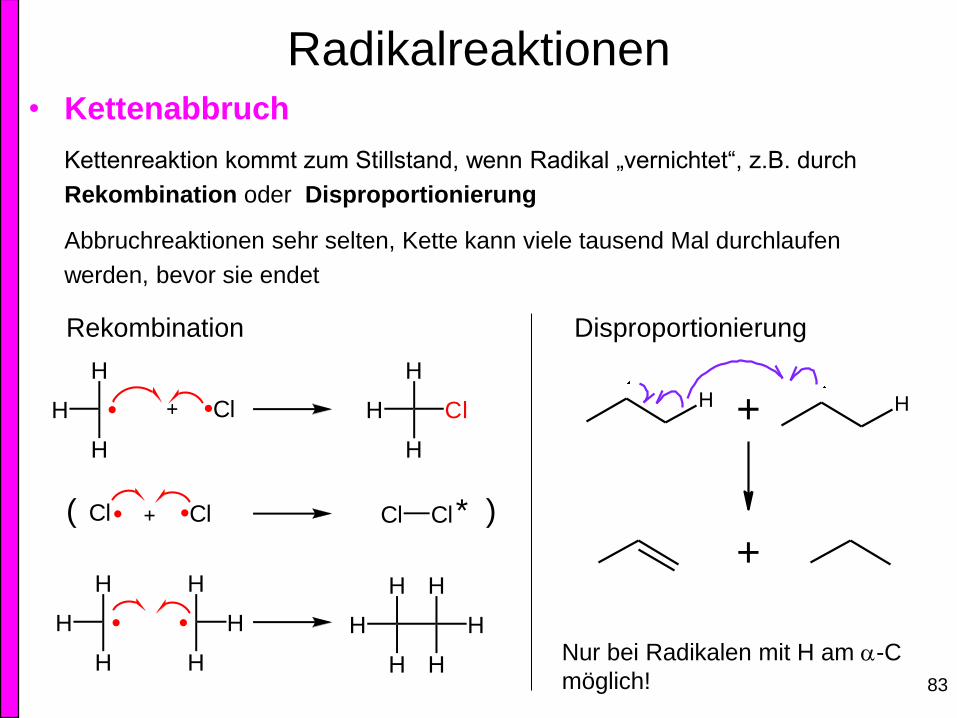
Radikalreaktionen • Kettenabbruch
Kettenreaktion kommt zum Stillstand, wenn Radikal „vernichtet“, z.B. durch
Rekombination oder Disproportionierung
Abbruchreaktionen sehr selten, Kette kann viele tausend Mal durchlaufen
werden, bevor sie endet
Cl
H
H
H
H
H
H
Cl Cl
H
H Cl
H
Cl Cl
H
H
H
H
H
H
H
H
H
Rekombination Disproportionierung
( * )
H H+
+
Nur bei Radikalen mit H am a-C
möglich!
84
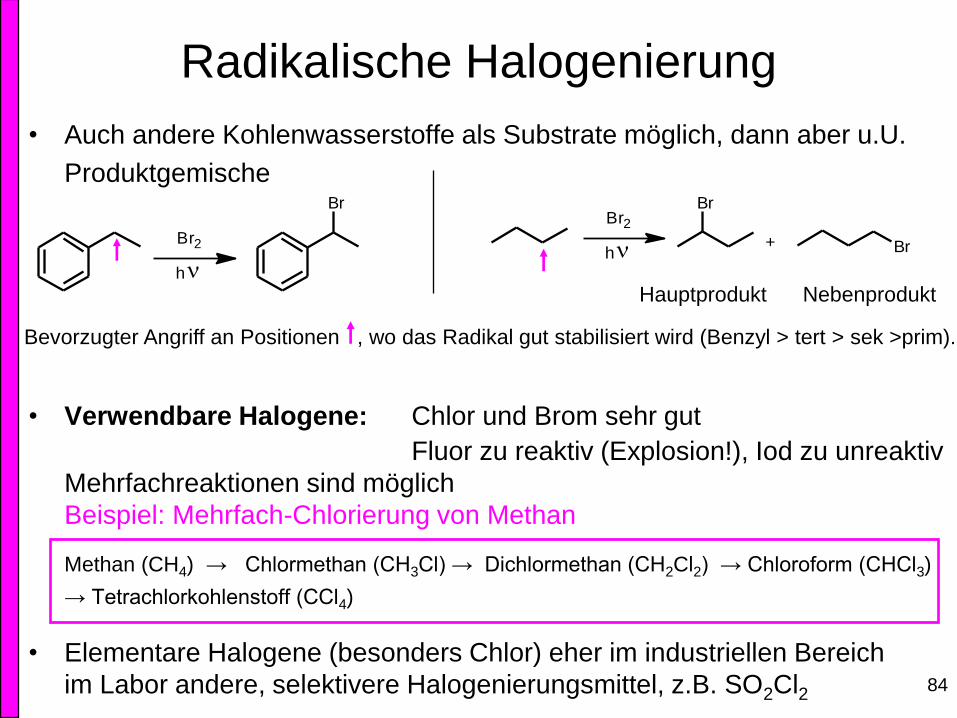
Radikalische Halogenierung
• Auch andere Kohlenwasserstoffe als Substrate möglich, dann aber u.U.
Produktgemische
• Verwendbare Halogene: Chlor und Brom sehr gut
Fluor zu reaktiv (Explosion!), Iod zu unreaktiv
Mehrfachreaktionen sind möglich
Beispiel: Mehrfach-Chlorierung von Methan
Methan (CH4) → Chlormethan (CH3Cl) → Dichlormethan (CH2Cl2) → Chloroform (CHCl3)
→ Tetrachlorkohlenstoff (CCl4)
• Elementare Halogene (besonders Chlor) eher im industriellen Bereich
im Labor andere, selektivere Halogenierungsmittel, z.B. SO2Cl2
Br2
h
Br
Hauptprodukt Nebenprodukt
Br2
h
Br
Br+
Bevorzugter Angriff an Positionen , wo das Radikal gut stabilisiert wird (Benzyl > tert > sek >prim).
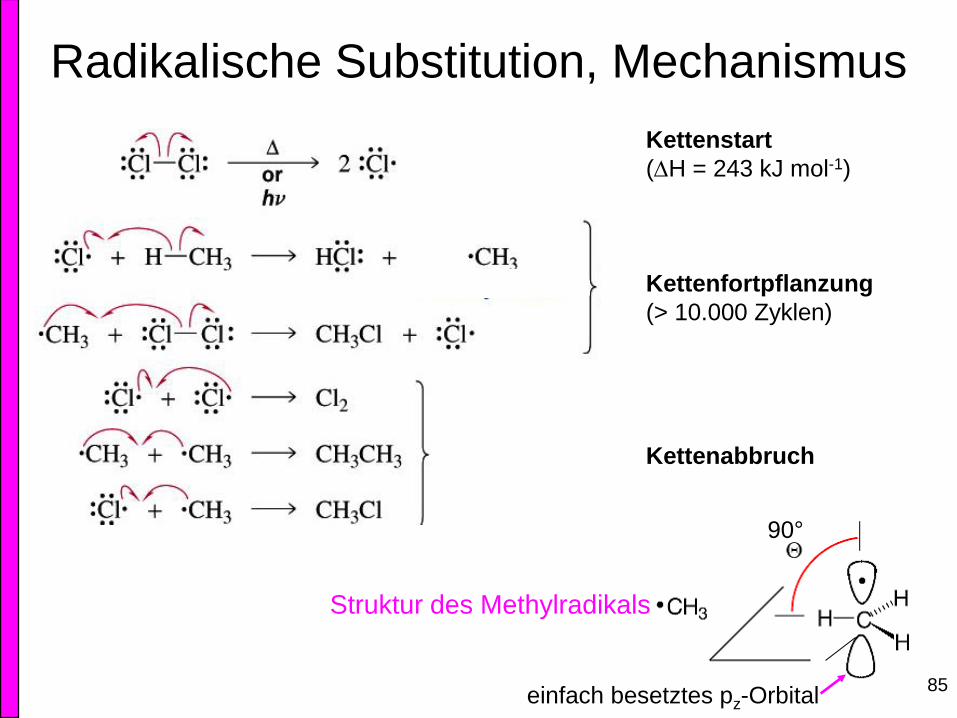
Radikalische Substitution, Mechanismus
Kettenstart
(H = 243 kJ mol-1)
Kettenfortpflanzung
(> 10.000 Zyklen)
Kettenabbruch
85
Struktur des Methylradikals
90°
einfach besetztes pz-Orbital
86
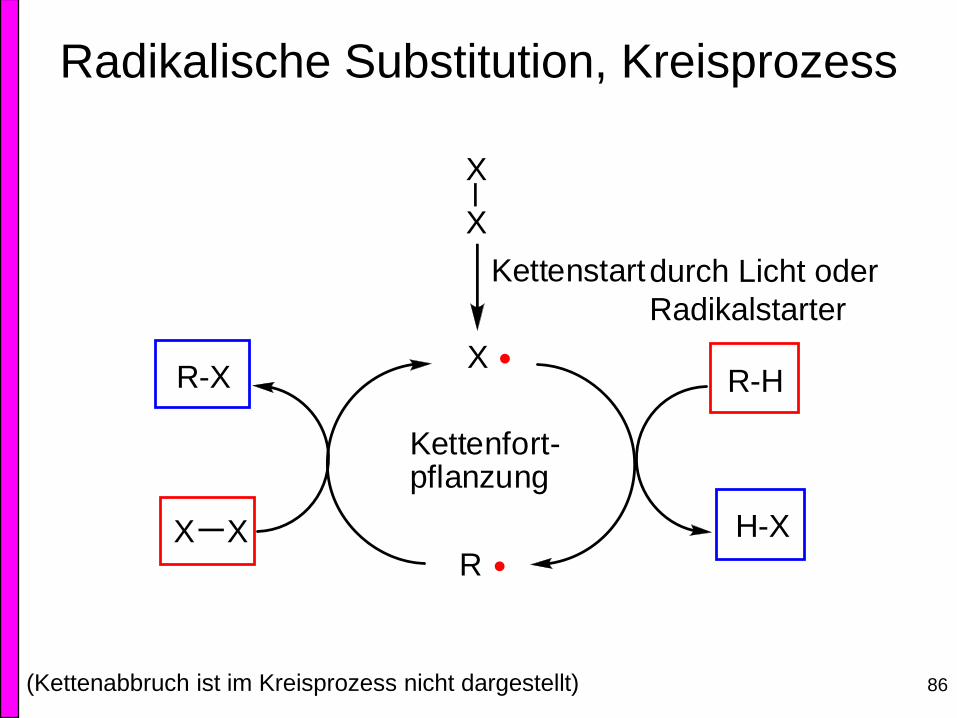
Radikalische Substitution, Kreisprozess
X
X
X
R-H
H-X
RXX
R-X
Kettenstart
Kettenfort-pflanzung
durch Licht oder
Radikalstarter
(Kettenabbruch ist im Kreisprozess nicht dargestellt)
87
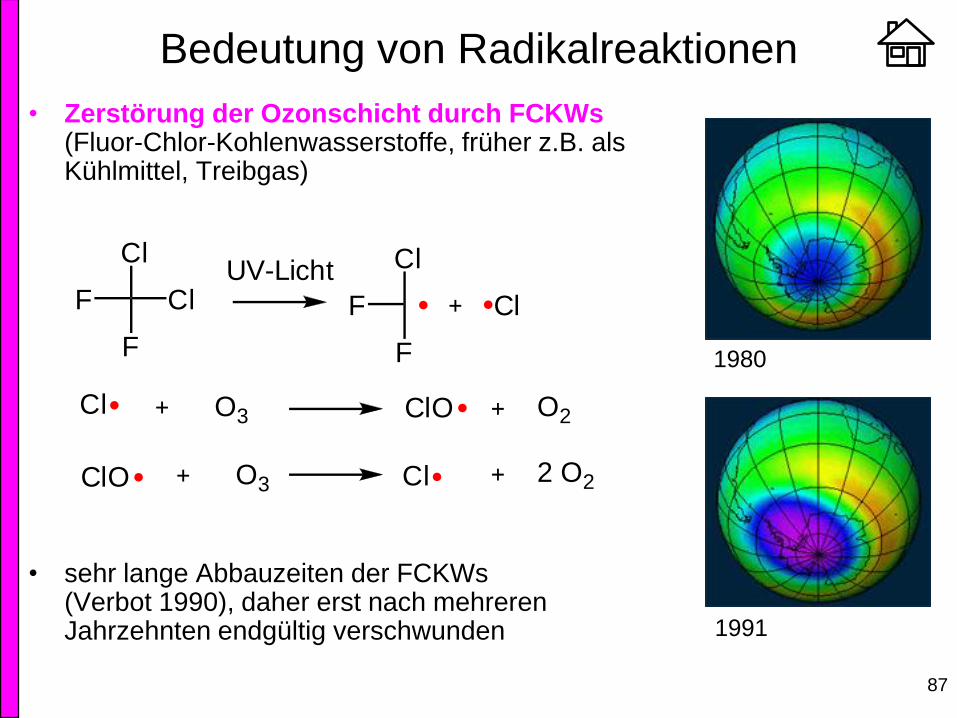
Bedeutung von Radikalreaktionen
• Zerstörung der Ozonschicht durch FCKWs (Fluor-Chlor-Kohlenwasserstoffe, früher z.B. als Kühlmittel, Treibgas)
• sehr lange Abbauzeiten der FCKWs (Verbot 1990), daher erst nach mehreren Jahrzehnten endgültig verschwunden
Cl
F Cl
F
UV-Licht Cl
F
F
Cl
Cl O3 ClO O2
ClO O3 Cl 2 O2
1991
1980
88
Bedeutung von Radikalreaktionen
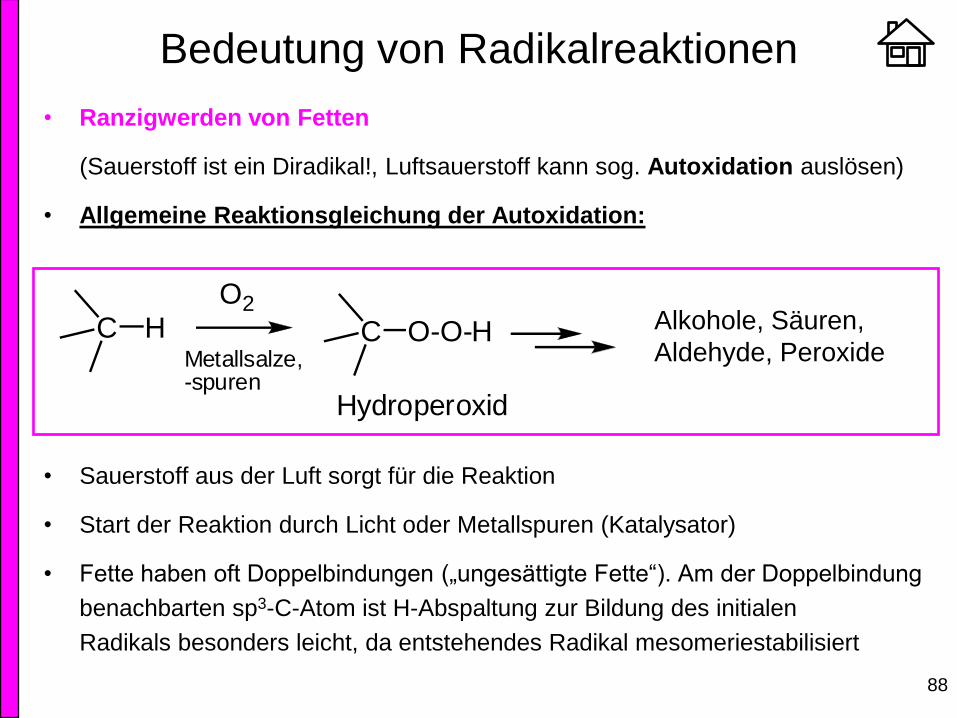
• Ranzigwerden von Fetten
(Sauerstoff ist ein Diradikal!, Luftsauerstoff kann sog. Autoxidation auslösen)
• Allgemeine Reaktionsgleichung der Autoxidation:
• Sauerstoff aus der Luft sorgt für die Reaktion
• Start der Reaktion durch Licht oder Metallspuren (Katalysator)
• Fette haben oft Doppelbindungen („ungesättigte Fette“). Am der Doppelbindung
benachbarten sp3-C-Atom ist H-Abspaltung zur Bildung des initialen
Radikals besonders leicht, da entstehendes Radikal mesomeriestabilisiert
C HO2
Metallsalze,-spuren
C O-O-H
Hydroperoxid
Alkohole, Säuren,
Aldehyde, Peroxide
89
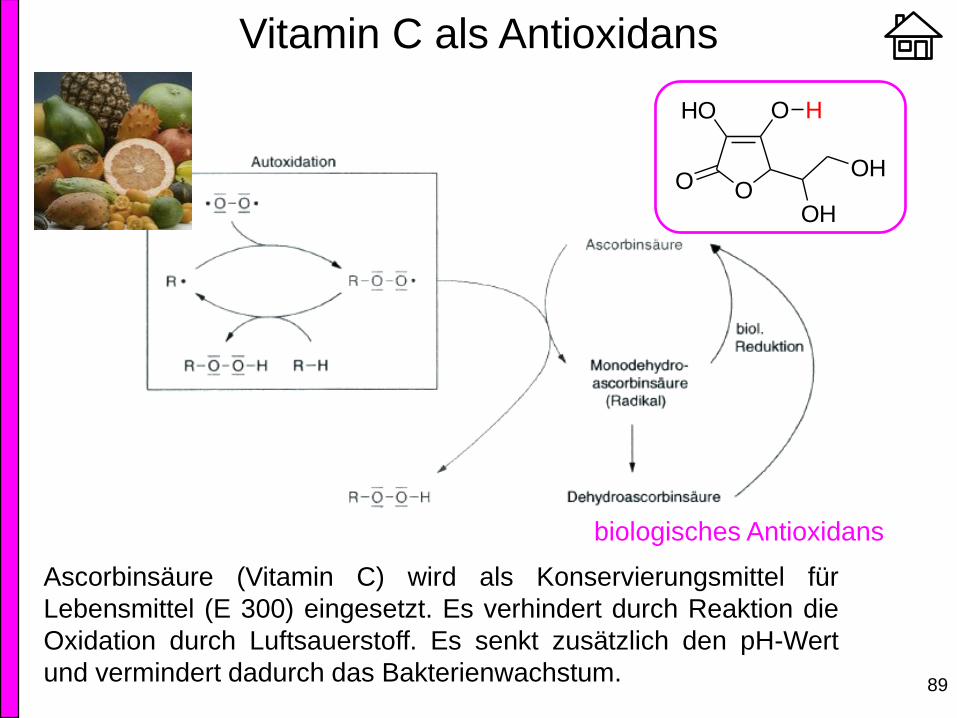
Ascorbinsäure (Vitamin C) wird als Konservierungsmittel für
Lebensmittel (E 300) eingesetzt. Es verhindert durch Reaktion die
Oxidation durch Luftsauerstoff. Es senkt zusätzlich den pH-Wert
und vermindert dadurch das Bakterienwachstum.
biologisches Antioxidans
OO
HO O
OH
OH
H
Vitamin C als Antioxidans
90
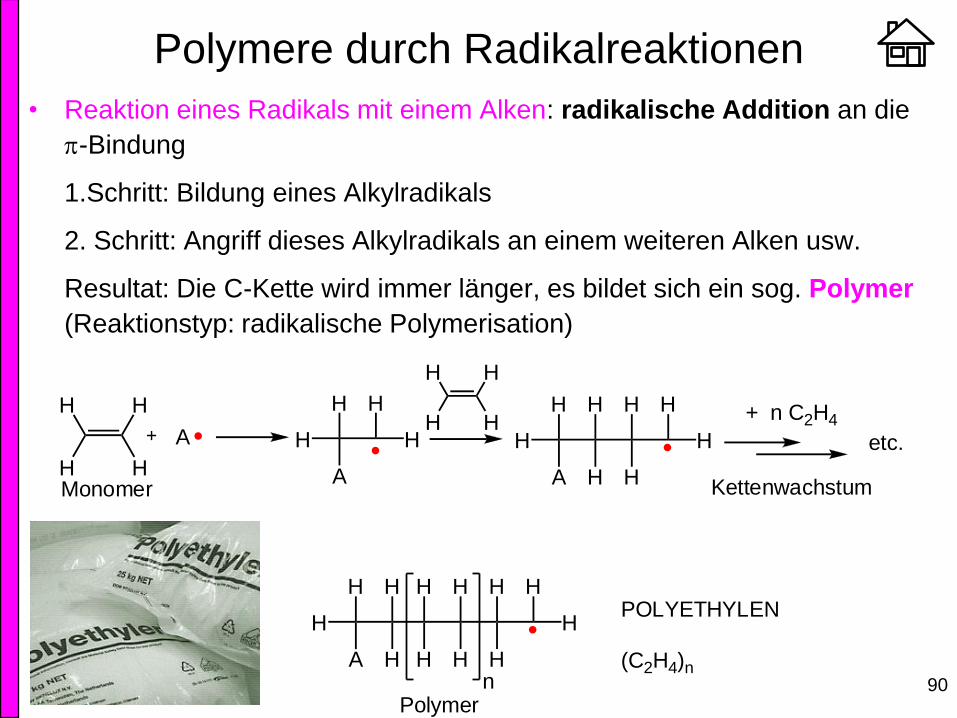
Polymere durch Radikalreaktionen
• Reaktion eines Radikals mit einem Alken: radikalische Addition an die
p-Bindung
1.Schritt: Bildung eines Alkylradikals
2. Schritt: Angriff dieses Alkylradikals an einem weiteren Alken usw.
Resultat: Die C-Kette wird immer länger, es bildet sich ein sog. Polymer
(Reaktionstyp: radikalische Polymerisation)
H
H H
H
A H
H
H
H
A
H
H H
H
H
HH
H
H
H
H
A
H etc.
Monomer Kettenwachstum
+ n C2H4
H
H
H
A
H
HH
H
H
H
H
HHn
Polymer
POLYETHYLEN
(C2H4)n

91
Beispiele für Polymere
H
H H
H
Monomer
H
H
H
H n
F
F F
F
H
H CH3
H
H
H Ph
H
H
H Cl
H
Polymer
F
F
F
F n
H
Me
H
H n
H
Ph
H
H n
H
Cl
H
H n
Polyethylen
Teflon (Polytetrafluorethylen, PTFE)
Ethen (Ethylen)
Propen (Propylen)
Vinylbenzol (Styrol)
Chlorethen(Vinylchlorid)
Tetrafluorethen
Polypropylen (PP)
Polystyrol (PS)
Polyvinylchlorid (PVC)© Andrevan © Ralf Roletschek © Pearson
PE PP PTFE

92
H
H H
H
Monomer
H
H
H
H n
F
F F
F
H
H CH3
H
H
H Ph
H
H
H Cl
H
Polymer
F
F
F
F n
H
Me
H
H n
H
Ph
H
H n
H
Cl
H
H n
Polyethylen
Teflon (Polytetrafluorethylen, PTFE)
Ethen (Ethylen)
Propen (Propylen)
Vinylbenzol (Styrol)
Chlorethen(Vinylchlorid)
Tetrafluorethen
Polypropylen (PP)
Polystyrol (PS)
Polyvinylchlorid (PVC)
Beispiele für Polymere
© Dubaj
Polystyrol-Schaum-Verpackung PVC-Foliengewächshaus PVC-Rohrleitungen
© Was a bee © Takuya Murata
93
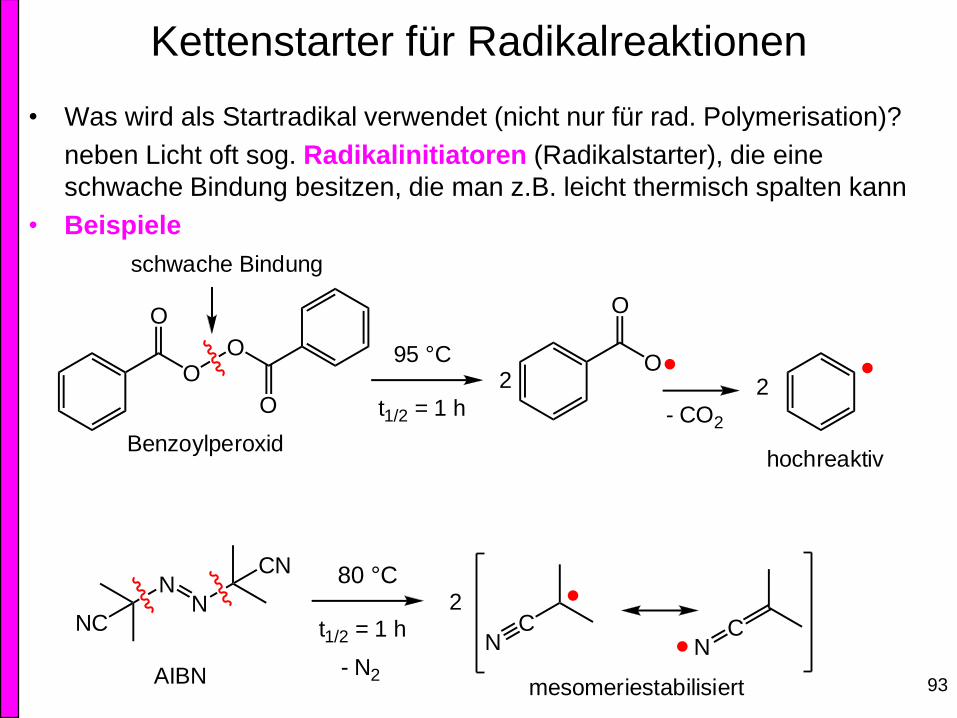
Kettenstarter für Radikalreaktionen
• Was wird als Startradikal verwendet (nicht nur für rad. Polymerisation)?
neben Licht oft sog. Radikalinitiatoren (Radikalstarter), die eine
schwache Bindung besitzen, die man z.B. leicht thermisch spalten kann
• Beispiele
O
OO
O
Benzoylperoxid
schwache Bindung
95 °C
t1/2 = 1 h
O
O
- CO2
2 2
NN
CN
NC
(= °C
t1/2 = 1 h
- N2
C2
NC
N
hochreaktiv
AIBNmesomeriestabilisiert
80 °C
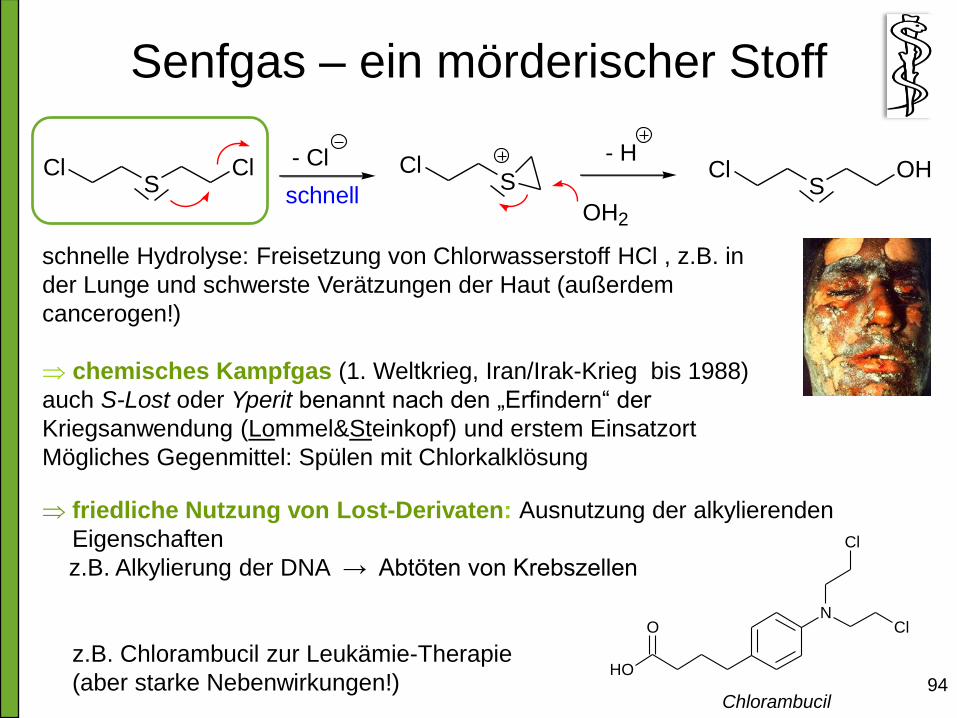
Senfgas – ein mörderischer Stoff
94
SCl Cl - Cl
schnellS
Cl
OH2
SCl OH
- H
schnelle Hydrolyse: Freisetzung von Chlorwasserstoff HCl , z.B. in
der Lunge und schwerste Verätzungen der Haut (außerdem
cancerogen!)
chemisches Kampfgas (1. Weltkrieg, Iran/Irak-Krieg bis 1988)
auch S-Lost oder Yperit benannt nach den „Erfindern“ der
Kriegsanwendung (Lommel&Steinkopf) und erstem Einsatzort
Mögliches Gegenmittel: Spülen mit Chlorkalklösung
friedliche Nutzung von Lost-Derivaten: Ausnutzung der alkylierenden
Eigenschaften
z.B. Alkylierung der DNA → Abtöten von Krebszellen
z.B. Chlorambucil zur Leukämie-Therapie
(aber starke Nebenwirkungen!)
N
Cl
OH
O Cl
Chlorambucil