Embed Size (px)
Citation preview

180 L. EI~DEY und L. K~PL~I~:
Ansehliegend wird mi t K M n 0 4 zum Vanadin(V) oxydier t , der K M n 0 4- ~be r schug mit Ni t r i t entfernt , das nieht umgesetzte Nitr i t mit Harns tof f zerst6rt u n d der Vanadingehal t durch erneute Ti t ra t ion mi t F e S 0 a- L6sung best immt.
Literatur 1 ]~RENNEOKE, E. , u. E. BLASIUS in G. JANDEIr Neuere mal~anMytische Me-
thoden, 4. Auflage, Stuttgart: Enke, 1956. - - e KNoP, J., u. 0. KUBELKOV-~- KNoPovX: diese Z. 122, 183 (1941). - - 3 LANG, 1~., U. F. KtTRTZ: diese Z. 86, 288 (1931). - - t REUTER, B., U. J. SIEWERT: Natttrwissensch~ften 4:4, 440 (1957). - - 5 I~i~DORFF, W., G. WALTER 11. H. BECKER: Z. anorg, allg. Chem. 285, 287 (1956).
:Prof. Dr.-Ing. BE~TOLI)I~EV~E~, BerlimCharlottenburg 2, ttardenbergstraBe 34
Aus 4em Institut ftir Allgemeine Chemie der Technischen Universiti~t Budapest (Ungarn)
Mal~analytische Aseorbins~iurebestimmungen mit Variaminblau als Indicator
Von L.:ERDEY und L. KX~LiR
(Eiugegangen am 17. Februar 1958)
Das ~lteste Verfahren zur maBanalyt ischen Ascorbins~urebest immung ist das Verfahren yon TILLMA~S i9, bei welchem mi t 2,6-Dichlorphenol- indophenollSsung bis zur bleibenden Blauf~rbung der Fliissigkeit t i t r iert wird. Diese 1Y~ethode ist das heute vorzugsweise benutz te Verfahren zur Bes t immung des Vi tamin C-Gehaltes yon Bflanzen und biologischem Material. Obzwar m a n im Laufe der Zeit viele Modifikationen und Ver- besserungen des Verfahrens empfohlen hat , liefert es doch nicht in alien F~llen die erwiinschte Genauigkeit.
Das Ti t ra t ionsverfahren mi t JodlSsung und St~rke als Indicator~,9,15 wird in den PharmacopOen der meisten Lgnder zur Ascorbinsi~ure- bes t immung vorgeschrieben. Der verh~ltnism~Big groge Indicatorfehler und die Ver langsamung der l~eaktion Ascorbins~ure-Jod dutch die Stgrke bedeuten aber erhebliche Nachteile der Methode. Diese Nachteile liegen auch bei dem Joda tver fuhren yon BALLENTI~E i vor, bei welchem unter Zusutz yon KMinmjodid bis zur Stgrkeblaufs t i t r ier t wird. D~s eigentliche Oxydat ionsmit te l ist ja auch hier das J o d .
Eisen(III) -salze (Chlorid oder Alaun) werden als Oxydat ionsmit te l un ter Verwendnng yon Ammoninmrhodan id n,l~,ia,i7 oder Kal inmjodid- Sti~rke 7 Ms Indica toren empfohlen. E~DnY und BoDo~ s haben abet bereits auf die vortei lhafte Anwendbarkei t der Var iaminblauindikat ion bei der Ascorbins~uret i t rat ion mi t Eisen(III)-chloridl6sung hingewiesen.

Mal~analy~ische Bestimmung yon Ascorbinsiure 181
Weitere zur Ascorbins~urebestimmung empfohlene oxydierende Ma~- flfissigkeiten sind Chloramin-T-L6sung s, Permanganafl6sung i~ (beide mit Jodid-St~rkeindikation), Bromatl6sung (mit p-~thoxychrysoidin als Indicator) 14, Kaliumhexacyanoferrat(III)-16sung is und essigsaure Brom- 16sung ~0.
Schwaehe Oxydationsmittel (2,6-Diehlorphenolindophenol, Kupfer(II)- sulfat, Eisen(III)-sulfati% Jod, Jodat, Chloramin T usw.) oxydieren Ascorbins~ure quantitativ zu Dehydroaseorbins~ure. Mit starken Oxy- d~tionsmitteln (W~sserstoffperoxyd, Chroms~ure , Salpetersiure, I-Iypo- ehlorit) kann die 0xydat ion weiter gehen i6. Aus den in Tab. 1 zusammen- gestellten Werten der Re-
Tabelle 1 doxpotentiale versehiede- ner Systeme sieht man, dal~ System E0 Volt
alle dort angegebenen Mal~- C6Hs06 ~ C6H60G d- 2 H + d- 2 e ~- 0,36 16sungen theoretiseh zur 2 J - ~ J~- aqu. H- 2 e d- 0,62 quantitativen Oxydation Fe 2+ ~ Fe 3+ § e + 0,75 der Ascorbinsi~ure geeignet C1- § 2 OH- ~- CIO- + H~O -4- 2 e § 0,95
erscheinen. Man mul~ je- doeh beachten, dab die Versuchsumstande Bestimmungsfehler verursaehen k6nnen. So sind wiH~rige Ascorbinsaurel6sungen besonders in Gegenwart yon Schwermetallspuren sehr unbest~ndig und werden auch dureh die Luft mehr oder weniger oxydiert. Im Falle yon Bflanzenstoffextrakten k6nnen Begleitsubstanzen die Oxydation bescMeunigen oder verz5gern. ~hnliche Wirkungen kSnnen aueh die Temperatur, der pR-Wert und das Lieht hervorrufen.
Die Untersuehungen, fiber die im folgenden beriehtet wird, sind zu dem Zweek ausgeffihrt worden, um die Anwendbarkeit der Variamin- blauindikation bei oxydimetrischen Aseorbinsiurebestimmungen zu prfifen. Als 0xydationsmittel haben wir Chloramin T, Jod, Jodat, Bi- jodat, Eisen(III)-ehlorid, und Eisen(III)-ammoniumsulfat angewendet. Die Priifung erstreekt sieh auf reine Asc0rbinsaurel6sungen sowie auf die Vitamin C-Bestimmung in Arzneimitteln und Pflanzenextrakten.
Experimenteller Teil Liisungen
0,1 n AscorbinsiiurdSsunff. Man 16st ungefihr 10 g Ascorbins/~ure 1o. a. in 1 1 dest. W~sser, 4as in einer Glasapparatur destflliert worden ist. Die Titerstellung erfolgt jodometrisch: Zu 20 ml 0,1 n Kaliumjodatl6sung fiigt m~n 2 ml 2 ~ Sa.]zs/~ure und 0,5 g Kaliumjodid, 1/~Bt die Ascorbins/~urel6sung bis zur he]lgelben Farbe zufiieBen, setzt 3 Tropfen l~oige Variaminblaul6sung un4 so l~nge eine 20~ l~atrium- acetatl6sung hinzu, bis die blaue Farbe des Indicators erscheint, fiigt noch 1 ml Natriumacetatl6sung zu und titriert tropfenweise weiter, bis die L6sung eben farb- los geworden ist 5.
Indicatorl6sung. Man verreibt 0,l g Variaminblau griindlich mit wenig ~r verdiinnt d~nn mit Wasser ~uf 10 ml und filtriert die L6sung durch ein F~ltenfilter.

182 L. EtCDEY und L. KXrr,X~:
0,1 n Chloramin T-L6sung wird wie iiblich bereitet; die Titerstellung erfolgt mit 0,1 n Arsenigs~urelSsung.
0,1 n Jod-, Kalium]odat- und Kaliumbi]odatl6sungen. Zur Bereitung und Titer- stellung verf/~hrt man wie iiblich.
0,1 n Eisen(III)-ammoniumsul/atl6sung. Man 15st 48,221 g ~'eNHa(S04) e �9 12 H20 13. a. in 1 I Wasser.
0,1 n Eisen (III)-chloridl6sung. Man 15st 5,6 g Eisenpulver unter gelinder Erw~r- mtmg in 40 ml konz. Salzs/~ure, ver4iinnt nach AufhSren der Gasentwicklung mit Wasser auf 4as dreifache Volumen, filtriert, oxydiert mit Wasserstoffperoxyd, ver- kocht 4essen UberschuB uncl erg/inzt die LSsung auf 1 1.
0,01 n L68ungen bereitet man stets durch Verdiinnung 4er entsprechenden 0,1 n LSsungen.
2 n EssigsSure. 0,2 n Essigsi~ure-Natrlumacetatpu/]erl6sung yon pE 4,55.
Untersuehungssubstanzen Vitamin C-Tabletten, mit ungef~hr 0,1 g Ascorbins~uregehalt. In#ktionsprdiparate. Die verwendeten Pri~parate enthalten etw~ 10% Vitamin C. P/lanzenextrakte. Man zerkleinert die je nach dem zu erwartenden Vitamin C-
Gehalt gew/ihlte Menge der Pflanzen, Gemiise, Obst usw. mit einem rostfreien Messer und_ verreibt auf einem Glasreibebrett. Die so vorbereitete Substanz wird in einem Glasger&t mit 4 • ml Eisessig extrahiert. Man verdiinnt den Extrakt in einem MeSkolben zur Marke, schiittelt urn, filtriert un4 fiihrt die Bestimmung in eiaem aliquoten Teil des Filtrats durch. Im Falle der Auwendung einer Eisen(III)- malMSsung bew~hrt sich auch eine Extraktion mit Metaphosphorsaure bzw. mit Metaphosphors~ure-Eisessig.
Bestimmung mit Chloramin T-Mal~lSsung
Die T i t r a t i o n mug bei e inem b e s t i m m t e n p m W e r t ansgef i ihr t werden, de r so auszuw/~hlen ist , dag einersei ts die O x y d a t i o n der Ascorbins/ ture m i t Chloramin T q u a n t i t a t i v und ohne s tSrende Neben reak t i onen ver- l~uft , andererse i t s der F a r b u m s c h l a g des Va r i aminb lau ind i ca to r s ge- n t igend schar f ist . Das R e d o x p o t e n t i a l yon Chloramin T gnder t sich mi t dem px~-Wert. I ra a l lgemeinen b e n u t z t m a n eine Chloramin T-LSsung in 5 ~oiger Salzs/ture, m a n k a n n jedoch auch noeh LSsungen m i t den pg - W e r t e n 1 - -3 gu t anwenden. Der V a r i a m i n b l a u i n d i c a t o r a rbe i t e t a m bes ten im pg- Gebie t 3 - -4 . Daraus e rg ib t sich, dal~ ftir d ie Aseorbins~ture- be s t immung m i t Chloramin T der pH-Wer t 3 am bes ten geeignet sein wird. W i t s tel len d iesen pr f -Wer t durch Zusa tz yon Essigs/~ure her, d ie E r k e n n u n g des E n d p u n k t e s wi rd du tch Zugabe yon K a l i u m b r o m i d verbesser t . Bei Verschiebung des p g - W e r t e s in a lka l i scher i~ichtung kann infolge der VerzSgerung des E n d p u n k t e s ein U b e r v e r b r a u c h an ?r yon 0 ,5% e in t re ten , der I n d i c a t o r sehl/~gt d a n n s t a t t nach Blau in die sieh sehnel l zerse tzende r o t e F a r b e urn. Bei p ~ - W e r t e n un te r 2 wird der I n d i c a t o r zers tSr t , die F a r b e des E n d p u n k t e s erscheint versp~te t . Zur Zers tSrung des Var i aminb laus tr/~gt das a u f Zugabe yon Chloramin T i m E n d p u n k t e en t s t ehende Brom bei. Der Vorte i l der I n d i k a t i o n m i t

Maganalytische Bestimmung yon Ascorbinsiiure 183
Ka l i umbromid -Var i aminb l au i s t der, daB die St/irke, die meis t in pharma-
zeut ischen Vi tamin C-Pr/ iparaten vo rhanden ist, n icht stSrt. Den
Aseorbins/~uregehalt dieser Pr/~parate kann man also ohne vorhergehende
En t f e rnung der Sts genau bes t immen.
Arbeitsweise
Zu 4er 50--500 mg Ascorbins/~ure enthaltenden w/iBrigen L6sung fiigt man 20 ml 2 n Essigs/iure, 1 g Kaliumbromicl, 3 Tropfen l~oige Va~iaminblaul6sung und so vie1 Wasser, daB 4as zu erwartende Endvolumen 100 ml betr~gt. Dann ~itriert man langsam unter kr/~ftigem Umschiitteln mit 0,1n Chloramin T-LOsung bis zur bleibenden Blau- f/irbung. Bei Titrationen mit 0,01 n MaB16sung ver- f/~hrt man ebenso, nur Einwaage nnterbleibt die Volumen- c0~o, erg/inzung mitWasser, um mg eine etwaige Oxydation zu vermei4en. 277,48
Zur Ascorbinsiiurebe- 185,28 stimmung in Tabletten 92,46 pulvert man 5 Tabletten in 46,50 einemMOrser und w/~gt 4ar - 37,28 aus eine geeignete Menge 22,42 ein. Man 16st in Wasser, 15,80 s/~uert mit 20 ml 2 n Essig- 7,88 s/~ure an und verf~hrt wei- 3,96 ter wie oben angegeben.
Tabelle 2
~rerbrauch an 0,1 n (bzw. 0,0~ n)
Chloramin T- LOsung
ml
31,51 21,02 10,51 5,27
42,40* 25,51" 17,98" 8,97* 4,52*
Geflmden C6HsOr
mg
277,48 185,10 92,55 46,41 37,34 22,46 15,83 7,90 3,98
Abweichung
~:0 - -0 ,1 +0 ,1 --0,18 + 0,16 + 0,18 + 0 , 2 + 0,25 -- 0,5
Im Falle yon In]ektionen spiilt man den Inhalt der Ampulle mit Wasser in einen 100 ml-MeBkolben un4 s/~uert vor 4em Aufftillen mit 20 ml 2 n Essigs/~ure an. Die Titration erfolgt in eiaem aliquoten Tell der LSsung.
Obst- und Gemi~seextrakte sin4 bereits essigsauer (siehe oben). Zur Titration ver- wendet man aliquote Teile yon 20 bzw. 50 ml.
I n Tab. 2 sind Ergebnisse yon Ascorbins/~urebest immungen in reinen
L6sungen zusammenges te l l t ; die mi t * bezeichneten T i t r a t ionen sind mi t
0,01 n MaB16sung durchgefi ihr t .
Aus 12 Para l l e lbes t immungen ergibt sich der re la t ive Fehler im 0,1 n
MaBstabe zu • 0,1%, im 0,01 n MaB zu • 0 ,2%; die S tandards t reuung
bet rs im 0,1 n MaB :L 0,054 ml, im 0,01 n MaB • 0,14 ml.
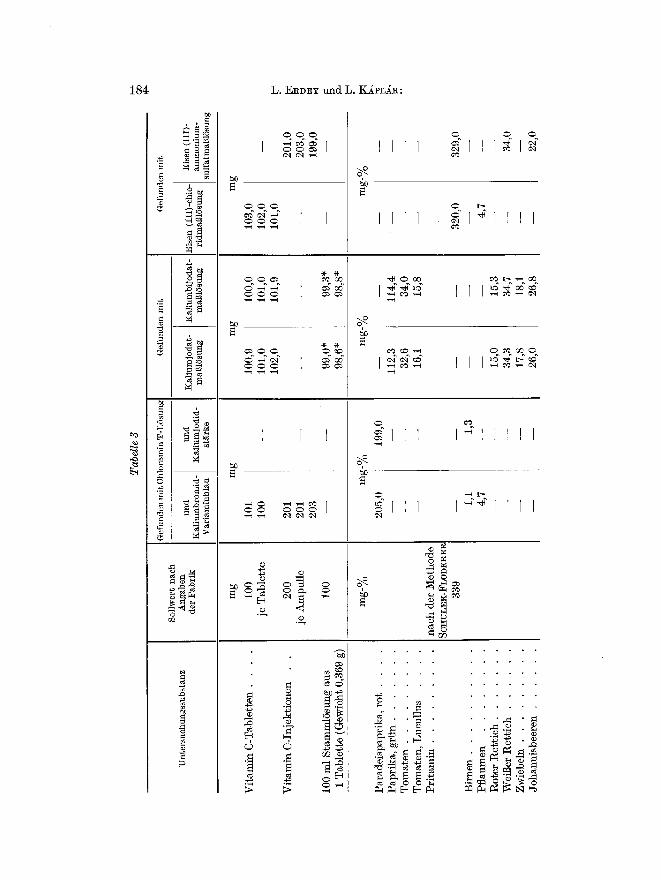
Tab. 3 enth/~lt die Ergebnisse von Ascorbins/~urebest immungen in
Table t ten , In j ek t ionen und einigen der un te r such ten Pf lanzenext rakte .
In der Tabelle sired bei den Arzneimitteln die yon der Fabrik angegebenen Werte, bei Pflanzenextrakten die mittels Kaliumjcdid-St~rkeindikation erhalte~en Werte Ms Sollwerte angegeben. Letztere Bestimmungen wurdea wie iiblich in 5~oiger salzsaurer L6sung ausgefiihrt. Vc'aren die Extrakte gef/~rbt, so konnte man den Endpunkt mit Variaminblau nicht wahrnehmen. In diesem Falle versetzte man die LSsung mit 2 ml Tetrachlorkohlenstoff und titrierte, his sich die organische Schicht durch das im J~quivalenzpunkt frei werdende Brom gelb f~rbte.
Tab
elle
3
Un~
ersu
ctm
ngss
ubs~
anz
7it
~m
in C
-Tab
lett
en
7it
amin
C-I
nje
kti
on
en
. .
Sol
lwer
t na
ch
An
gab
en
der
Fab
rik
mg
100
je T
able
tte
100
ml
St~
mm
l6su
ng
~u
s 1
T~
ble
tto
(G
ewic
ht
0,36
9 g)
P~
rad
eisp
apri
k~
, ro
t ..
..
Pap
rik
a, g
riin
..
..
..
.
To
m.t
en
.
..
..
..
..
T
om
aten
, L
ucu
llu
s ..
..
Pri
t~m
in
..
..
..
..
.
Bir
nen
..
..
..
..
..
P
flau
men
.
..
..
..
.
t~o
ter
Re,
rich
..
..
..
.
Wei
l3cr
Ret
tich
.
..
..
.
Zw
ieb
eln
.
..
..
..
..
Jo
h~
nn
isb
eere
n
..
..
..
200
je A
mp
ull
e
100
rag-
%
Gef
unde
n m
it G
hlor
~m
in T
-L6s
un~
und
Kal
ium
brom
id-
Var
iam
inbl
~u
101
100
201
201
203
n~
ch 4
erM
eth
od
e
SCtIU
LE
K-F
LO
DE
RE
~ 33
9
205,
0
1,1
4,7
und
Kal
ium
jod
id-
stii
rke
Gef
unde
n m
it
Gef
unde
n m
it
Kal
ium
joda
t -
maF
316s
ung
Kal
ium
bijo
da~
- E
isen
(I
II)-
chlo
- m
ai]1
6sun
g ri
dma0
16su
ng
Eis
en (
III)
- am
mon
ium
- su
lfa~
maB
l6su
ng
mg
rag
-%
199,
0
1,3
mg
100,
9 10
1,0
102,
0
99,0
* 98
,6* ra
g-%
112,
3 32
,6
16,1
m 15
,0
34,3
17
,8
26,0
100,
0 10
1,0
101,
9
99,3
* 98
,8*
114,
4 34
,0
15,8
15,3
34
,7
18,1
26
,8
103,
0 10
2,0
101,
0
320,
0
4,7
mg
rag-
% 20
1,0
203,
0 19
9,0
329,
0
34,0
22,0
t2
~176

Mal]analytische Bestimmung yon Ascorbinsi~ure 185
Bestimmung mit Jodliisung Der Endpunkt der Ascorbins/~uretitration mit Jodlbsung ist bei An-
wendung des Variaminblauindicators in mit Essigs/iure-Natriumacetat gepufferter LSsung ausgezeichnet wahrzunehmen. Der erste iiberschiissige Tropfen MaBl6sung ruft sofort die blauviolette Farbe der L6sung hervor. Die Titrationen werden auf die bei der Titerstellung beschriebene Weise ausgeffihrt. Der Vorteil des Variaminblauindicators gegenfiber der Sti~rke besteht darin, daft der Indicatorfehler auch in verdfinnten L6sungen sehr gering ist und dab die Oxydation der Ascorbins/iure rasch und unverzSgert vor sich geht.
Wir bestimmten auf diese Art den Ascorbins/~uregehalt von Vitamin C- Tabletten. Der Gehalt betrug laut Fabriksangabe 0,10 g, wit fanden 0,101 g.
Bestimmung mit Kaliumjodat- und Kaliumbijodatmallliisung E~DEY, Bol)o~ und B u z g s ~ ~ beschrieben eine direkte ascorbinome-
trische Methode der Jodatbestimmung. Zur Bindung des entstehenden Jodids ffigten sie Quecksilber(II)-chlorid zur L6sung. Ascorbins~ture kann umgekehrt auch direkt mit Kaliumjodat titriert werden. Setzt man auch noch Kaliumjodid zur LSsung oder binder man das entstehende Jodidion nicht, so oxydiert das im Laufe der Reaktion
JO 3- q- 5 J - q- 6 H + ~ 3J2 q- 3H20
entstehende Jod die Ascorbinsi~ure. Die KaliumjodatlSsung ist in diesem Falle eigentlich nur eine stabilisierte Jodl6sung.
Man arbeitet am besten folgendermal]en: Man s/~uert die 50--500 mg Ascorbin- s/~ure enthaltende neutrale w/~l~rige LOsung mit 5 ml 2 n Essigs~ure an, fiigt 3 Tropfen 1%ige VariaminblaulSsung hinzu und titriert bis zur schwachblauen F/~rbung. In 0,01 n Mal~bereich ist es zweckm/~l~ig der LSsung noch 0,1 g Kalium- jodi4 zuzusetzem
Unter denselben Bedingungen kann man auch KaliumbijodatmaB15sung anwenden.
Tabelle 4
Einwaage C6HsO~
mg
440,21 266,11 177,70 88,68 44,65 29,71" 19,94 9,95 5,01
Verbrauch au Gln
Kalium- jodatl6sung
ml
Gefunden Abwei- C~Its06 chung
mg %
49,99 440,21 30,21 266,03 20,29 177,79 10,07 88,68 5,08 44,73
33,72* 29,70 22,67* 19,96 11,32" 9,97 5,71" 5,03
i• - - 0,03 I + 0,05
- - 0,04 +0,1 +0,2 + 0,4
Einwaage C6ttaO6
mg
430,79 259,07 173,04 86,30 43,41 30,21 20,25 10,12 5,09
Verbrauch an 0,i n
Kaliumbi- jodatl6sung
ml
49,00 29,44 19,65 9,79 4,94
34,34* 23,02* 11,51" 5,77*
Gefnnden C~Its06
mg
431,49 259,25 173,04 86,21 43,50 30,24 20,27 10,14 5,08
Abweichung
%
+ 0,16 + 0,07 • --0,1 + 0,18 + 0,09 +O,lO +0,2 + 0,2
186 L. ERDEY und L. KAPL~_R: 3/Iaganalytische Bestimmung yon Aseorbinsaure
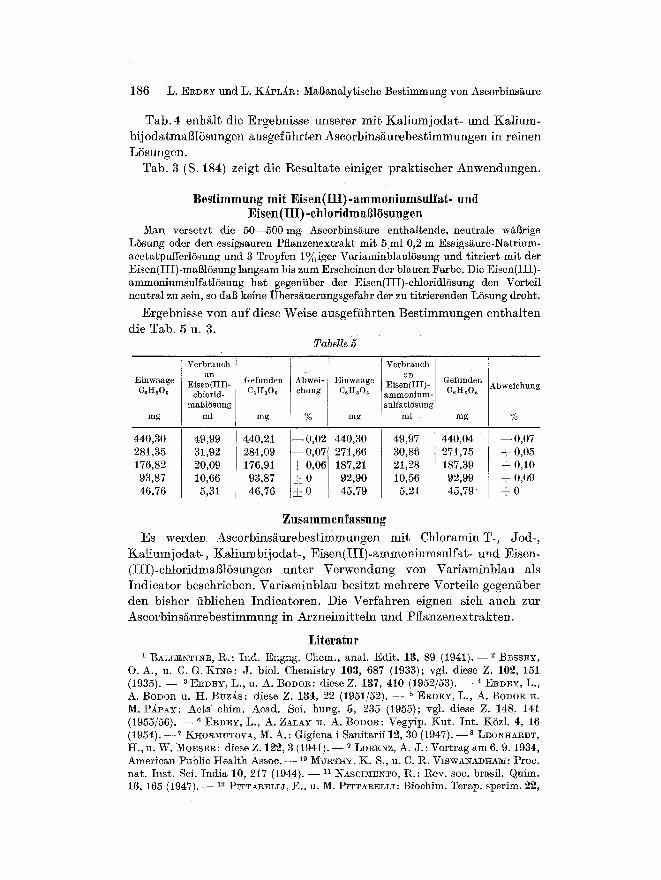
T a b . 4 enh / i l t d ie E r g e b n i s s e u n s e r e r m i t K a l i u m j o d a t - u n d K a l i u m -
bij o d a t m a S 1 5 s u n g e n a u s g e f i i h r t e n A s c o r b i n s / ~ u r e b e s t i m m u n g e n in r e inen
LSsungen .
Tab . 3 (S. 184) ze ig t d i e l~esu l t a t e e in iger p r a k t i s c h e r A n w e n d u n g e n .
Bestimmung mit Eisen(III)-ammoniumsulfat- und E i s e n ( I I I ) - ehlor idmal~l i i sungen
Man versetzt die 50 500 mg Ascorbins~iure enthaltende, neutrale w~l~rige LSsung oder den essigsauren Pflanzenextrakt mit 5 ml 0,2 m Essigs~ure-Natrium- acetatpufferl6sung nnd 3 Tropfen 1%iger Va~iaminblaulSsnng und titriert mit der Eisen(III)-mal315sung langsam bis zum Erscheinen der blauen Farbe. Die Eisen(III)- am'moniumsulfatl6sung ha~ gegeniiber tier Eisen(III)-ehloridl6sung den Vorteil neutral zu sein, so dab keine ~lbers~uerungsgefahr der zu titrierenden LSsung droht.
E r g e b n i s s e y o n a u f diese We i se a u s g e f i i h r t e n B e s t i m m u n g e n e n t h a l t e n
d ie T a b . 5 u. 3.
Einwaage C~HsO~
mg
440,30 281,35 176,82 93,87 46,76
Verbrauch an
Eisen(III)- ehlorid-
mal315sung ml
49,99 31,92 20,09 10,66 5,31
Gefunden
mg
Tabelle 5
Verbrauch ]
Abwei-Einwaage Eise:~III)- Gefunden chung CetIs06 ~,mmonium- C~HsO~
, mlfatl6sung % 1 mg ml I mg
440,21 - - 0,02 281,09 - - 0,07 176,91 I-}- 0,06 93,87 , i 0 46,76
440,30 271,66 187,21 92,90 45,79
49,97 30,86 21,28 10,56 5,21
Abweichung
%
440,04 - - 0,07 271,75 -~ 0,05 187,39 -~ 0,10 92,99 ~ 0,09 45,79 -~ 0
Z u s a m m e n f a s s u n g
Es w e r d e n A s c o r b i n s i ~ u r e b e s t i m m u n g e n m i t C h l o r a m i n T - , J o d - ,
Kaliumjodat-, KMiumbijodat-, Eisen(Ill)-ammoniumsulf~t- und Eisen-
(III)-chloridmal3lSsungen unter Verwendung yon Variaminblau als
Indicator beschrieben. Variaminblau besitzt mehrere Vorteile gegenfiber
den bisher iiblichen Indicatoren. Die Verfahren eignen sich auch zur
Ascorbins~urebestimmung in Arzneimitteln und Pflanzenextrakten.
L i t e r a t u r
i BALLENTINE, I~.: In4. Engng. Chem., an~l. Edit. 13, 89 (1941). 2 B~ss~Y, O. A., u. C. G. Kr~G: J. biol. Chemistry 103, 687 (1933); vgl. diese Z. 102, 151 (1935). - - a E~D~.Y, L., u. A. ]~oDo~: diese Z. 137, 410 (1952/53). - - 4 Em)~Y, L., A. BODOa u. H. BvzAs: diese Z. 134, 22 (1951/52). - - 5 E~D~r, L., A. BODO~ u. M. P/~PAr: Acta chim. Acad. Sci. hung. 5, 235 (1955); vgl. diese Z. 148, 141 (1955/56). - - 6 E~D~Y, L., A. Z+mAr u. A. BoDo~: Vcgyip. Kut. Int. KSzl. 4, 16 ( 1 9 5 4 ) . - ~ I~I~ORMUTOVA, M. A. : Gigiena i Sanitarii 12, 30 ( 1 9 4 7 ) . - s LEO~HARDT, H., u. W. MOESER: diese Z. 122, 3 (1941). - - 9 LOnENZ, A. J. : Vortrag am 6.9. 1934, American Public Health Assoc. -- i0 Mll~iu I~. S., u. C. IZ. VlSWAi~A~]=~A~: Proc. nat. Inst. 8ci. India 10, 217 (1944). - - n NASCIMENTO, t{.: t~ev. soc. brasil. Quim. 16, 165 (1947). - - 12 PITT~d~ELLI, E., u. )/I. PrrT~aELLI: Biochim. Terap. sperim. 22,

J. MO~VAY, J. CsAszA~ und M. ALM_~Su Re~ktion yon Adrenalin 187
t00 (1938). - - 13 SA~AL~VA, G. I. : Dokl. Akad. Nauk USSt~ 67, 226 (1946). - - it SCKt~LWK, E., J. Kov~cs u. P. R6ZSA: diese Z. 121, 17 (1941). - - 1~ STEVENS, J. W. : Ind. Engng. Chem., anal. Edit. 10, 269 (1938). - - 16 STRom~cK~, R., u. Y. M~T: diese Z: 133, 342 (!951). - - 17 STROK~C~:~, t~., U. E. S I ~ e : Z. Lebens- mittel-Unters, u. -Yorsch. 90, 93 (1950). - - x s TAvBER, H.: Mikrochemie 17, 111 (1935); vgl. 4iese Z. 108, 69 (1937). - - ~ TILL~A~S, J., ~. HII~SCH u. E. ~:~EI~S- ]~AGE~: Z. Unters. Lebensmittel 56, 272 (1928); vgl. diese Z. 78, 315 (1929). - - ~o Z~KA, J.: Pharmazie 9, 812 (1954); vgl. diese Z. 147,439 (1955).
Prof. Dr. L. ERDE:~, Budapest XI., Gell6r-t~r 4 (Ungarn) L. KIPLin, Budapest XI., Gell6rt-t~r 4 (Ung~rn)
Pharmazeutiseh-chemisehes Institut und Institut ftir allgemeine und physikalisehe Chemic, Universit~t Szeged (Ungarn)
~ber die Reaktion des Adrenalins mit Dikalium- Quecksilber(II)-tetrarhodanid
Identifizierung des entstehenden Adrenochroms auf spektroskopischem und papierchromatographischem Wege
Von J. MORVAY, J. CS~SZ~R und M.ALM]SY
Mit 2 Textabbildungen
(Eingegangen am 15. November 1957)
Uber die F a r b r e a k t i o n des Adrena l ins m i t Dika l ium-quecks i lbe r ( I I ) - t e t r a r h o d a n i d ( D H T R ) b e r i c h t e t e E. SALG5 ~ im J a h r e 1953. W i r d die w~Brige L6sung des Adrena l ins m i t D t t T R 20 rain lang auf dem Wasser- b a d erwi~rmt, dann en t s t eh t en t sprechend der K o n z e n t r a t i o n eine rosa- bis rub in ro te Fi~rbung, die sich nach ]~. SALG0, J . KIS u. J . Mo~vAu la a]s Grundlage ffir eine pho tomet r i sche Be s t immungsme thode yon Adrena l in eignet. Die e ingehendere Un te r suchung dcr farbigen Verbin- dung des Adrena l ins mi t D t t T R und die E r m i t t l u n g ihrer K o n s t i t u t i o n war schon damal s geplant .
Unsere urspri ing]iche Vors te l lung war, dab en tweder das AdrenMin den farbigen K o m p l e x mi t D H T R bi ldet , oder da~ das Adrena l in durch D H T R zu Adrenoch rom oxydier t , bzw. dieser R e a k t i o n s a b l a u f durch das Reagens gefSrder t wird.
I m Gange unserer Vorversuche konn te folgendes beobach te t werden : a) Die wal~rige L6sung der Adrena l inbase verfi~rbt sich in b e k a n n t e r
Weise in Abh/ tngigkei t yon der T e m p e r a t u r und A r t der A ufbe w a hrung in ki i rzerer oder l~tngerer Zei t nach Rosa, sparer nach Braun.





























