Embed Size (px)
Citation preview

Beuth Kommentar
Qualitätsmanagement für Hersteller von Medizinprodukten
Kommentar und Praxisleitfaden zur dritten Ausgabe der DIN EN ISO 13485
Bearbeitet vonDipl.-Ing. Randolph Stender, DIN e.V.
1. Auflage 2017. Buch. 182 S. SoftcoverISBN 978 3 410 25696 0
Format (B x L): 14,8 x 21 cmGewicht: 282 g
Wirtschaft > Management > Qualitätsmanagement
schnell und portofrei erhältlich bei
Die Online-Fachbuchhandlung beck-shop.de ist spezialisiert auf Fachbücher, insbesondere Recht, Steuern und Wirtschaft.Im Sortiment finden Sie alle Medien (Bücher, Zeitschriften, CDs, eBooks, etc.) aller Verlage. Ergänzt wird das Programmdurch Services wie Neuerscheinungsdienst oder Zusammenstellungen von Büchern zu Sonderpreisen. Der Shop führt mehr
als 8 Millionen Produkte.

Randolph Stender
Qualitätsmanagement für Hersteller von MedizinproduktenKommentar und Praxisleitfaden zur dritten Ausgabe der DIN EN ISO 13485

V
Inhaltsverzeichnis
Inhaltsverzeichnis
Danksagung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . VII
1 Vorwort . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
2 Gesetzliche Anforderungen an Medizinprodukte . . . . . . . . . . . . . . . . . . . . . 32.1 Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) . . . . 72.2 Deutsches Institut für Medizinische Dokumentation und Information
(DIMDI) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92.3 Überwachungsbehörden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 102.4 Benannte Stellen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.5 Beratungsunternehmen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
3 Grundlagen Qualitätsmanagement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 153.1 DIN EN ISO 9001 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173.2 DIN EN ISO 13485:2016 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223.3 21 CFR 820 Quality System Regulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243.4 Einführung in die DIN EN ISO 13485 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 273.5 Anwendungsbereich der DIN EN ISO 13485 . . . . . . . . . . . . . . . . . . . . . . . . . . 283.6 Normative Verweisungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 293.7 Begriffe und Definitionen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
4 Qualitätsmanagementsystem . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 314.1 Allgemeine Anforderungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 314.2 Dokumentationsanforderungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
5 Verantwortung der Leitung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 585.1 Verpflichtung der Leitung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 585.2 Kundenorientierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 605.3 Qualitätspolitik . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 625.4 Planung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 635.5 Verantwortung, Befugnisse und Kommunikation . . . . . . . . . . . . . . . . . . . . . 665.6 Managementbewertung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
6 Management von Ressourcen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 746.1 Bereitstellung von Ressourcen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 746.2 Personelle Ressourcen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 756.3 Infrastruktur . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 776.4 Arbeitsumgebung und Beherrschung der Kontamination . . . . . . . . . . . . . 79

Qualitätsmanagement für Hersteller von Medizinprodukten
VI
7 Produktrealisierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 837.1 Planung der Produktrealisierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 837.2 Kundenbezogene Prozesse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 857.3 Entwicklung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 897.4 Beschaffung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1107.5 Produktion und Dienstleistungserbringung . . . . . . . . . . . . . . . . . . . . . . . . . . 1167.6 Lenkung von Überwachungs- und Messmitteln . . . . . . . . . . . . . . . . . . . . . . . 135
8 Messung, Analyse und Verbesserung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1388.1 Allgemeines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1388.2 Überwachung und Messung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1398.3 Lenkung nichtkonformer Produkte . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1508.4 Datenanalyse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1568.5 Verbesserung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 157
9 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
10 Begriffe und ihre Definitionen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164
11 Abbildungsverzeichnis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 171
12 Quellenangaben . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172

1
1 Vorwort
1 VorwortOrganisationen, die als Hersteller Medizinprodukte im Europäischen Wirt-schaftsraum erstmalig unter ihren Namen in Verkehr bringen, müssen nach den gesetzlichen Vorschriften für die Entwicklung und Herstellung ihrer Produkte neben einer Vielzahl von regulatorischen Anforderungen, abhängig vom Kon-formitätsbewertungsverfahren, ein vollständiges Qualitätsmanagementsystem einrichten und aufrechterhalten.
Die für die Implementierung eines vollständigen Qualitätssicherungssystems zurzeit einzige harmonisierte Norm ist die EN ISO 13485 – Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke. Die Norm wurde in der letzten Ausgabe als EN ISO 13485:2012+AC:2012 ver-öffentlicht und mit den Mitteilungen als harmonisierte Norm für die Richtlinien 93/42/EWG, 90/385/EWG und 98/79/EG bekannt gemacht.
Die internationale Norm ISO 13485 ist ein eigenständiger Qualitätsmanage-mentstandard, der sich von der internationalen Normenfamilie ISO 9000 für Qualitätsmanagementsysteme ableitet. Beide Normen basieren auf dem pro-zessorientierten Modell für den gesamten Produkt-Lebenszyklus. Obwohl die ISO 13485 auf der ISO 9001 und dem PDCA-Zyklus (plan – do – check – act) basiert, wurde sie in erster Linie für regulatorische Zwecke für Medizinprodukte entwickelt. Dadurch stellt die ISO 13485 weitaus mehr Anforderungen und fordert dadurch ein umfangreicheres dokumentiertes Qualitätsmanagement-system als die ISO 9001.
Obgleich die EN ISO 13485-Zertifizierung keine direkte Anforderung für die Konformitätsbewertungsverfahren von Medizinprodukten gemäß den europäi-schen Medizinprodukterichtlinien ist, wird sie als einzige harmonisierte Norm von der Europäischen Kommission anerkannt.
Am 1. März 2016 wurde die dritte Ausgabe der ISO 13485 veröffentlicht. Diese Veröffentlichung umfasst die Arbeit einer Vielzahl von internationalen Experten. Über vier Jahre wurde daran gearbeitet, die Norm sowohl an die regulatorischen internationalen als auch an die europäischen Anforderungen anzupassen, die seit der letzten Veröffentlichung 2003 stattgefunden haben.
Im August 2016 wurde schließlich die nationale Ausgabe der DIN EN ISO 13485 veröffentlicht und ist Gegenstand für die nun folgenden Kapitel dieses Buches.

Qualitätsmanagement für Hersteller von Medizinprodukten
2
Der Autor dieses Buches hat aktiv an der Entstehung dieser ISO-Norm mit-gewirkt und gibt Ihnen in diesem Buch eine detaillierte Übersicht über die be-währten, verbesserten, gelöschten und insbesondere neuen Anforderungen an ein Qualitätsmanagementsystem nach DIN EN ISO 13485:2016. Er gibt Ihnen praxiserprobte Hinweise zur Implementierung der Anforderungen, beschreibt aber auch bestehende Hindernisse, die bei der Interpretation auftreten können.

3
2 Gesetzliche Anforderungen an Medizinprodukte
2 Gesetzliche Anforderungen an MedizinprodukteMedizinprodukte im europäischen Wirtschaftsraum unterliegen einem inein-andergreifenden System von europäischen Regularien, verbunden mit der jeweiligen Umsetzung in nationales Recht in den Mitgliedsstaaten, zum Zwecke der Realisierung eines geregelten Warenverkehrs und der gegenseitigen An-erkennung eines gemeinsamen Ansatzes für diese besonderen Produkte.
In diesem System müssen in den Rechtsvorschriften für die jeweiligen Medizin-produkte das zu erreichende Niveau beim Schutz der Allgemeinheit sowie die grundlegenden Sicherheitsmerkmale festgelegt sein. Vorzuschreiben sind auch die Pflichten und Anforderungen für die Wirtschaftsakteure, wie Her-steller, Vertriebsorganisationen, Importeure, Bevollmächtigte etc. und – soweit dies erforderlich ist – das Kompetenzniveau der Benannten Stellen, die Konfor-mitätsbewertungen von Medizinprodukten oder Zertifizierungen von Qualitäts-managementsystemen durchführen, sowie die Kontrollmechanismen für diese Stellen. Außerdem sind Bestimmungen über die anzuwendenden Konformitäts-bewertungsverfahren (Module, die auch die Konformitätserklärung der Herstel-ler umfassen) in die Rechtsvorschriften aufzunehmen und es sind Regelungen für die internen und externen Marktüberwachungsmechanismen zu treffen, damit das gesamte Rechtsinstrument wirksam und reibungslos funktioniert.
Im Allgemeinen sind Medizinprodukte im Markt befindliche Produkte mit einer medizinischen Zweckbestimmung, wie zum Beispiel Therapie oder Diagnose, die vom Hersteller für die Anwendung am Menschen bestimmt sind. Es gibt aber auch Produkte, die keine medizinische Zweckbestimmung haben, aber vom Gesetzgeber als Medizinprodukt klassifiziert werden, wie zum Beispiel farbige Kontaktlinsen oder Desinfektionsmittel für Endoskope. Die genaue gesetzliche Definition eines Medizinprodukts und des Herstellers sind in § 3 des Gesetzes über Medizinprodukte (MPG) beschrieben.
Im Europäischen Wirtschaftsraum regeln maßgeblich drei Richtlinien das In-verkehrbringen bzw. die Herstellung von Medizinprodukten. Als ausführendes Organ der Europäischen Union erarbeitet die Europäische Kommission notwendige Harmonisierungen der gesetzlichen Anforderungen, welche durch den Europäischen Rat oder gegebenenfalls durch das Europäische Parlament verabschiedet werden. Jede dieser Richtlinien beschreibt die Übereinkunft, die erzielt wurde, und enthält eine Frist für die Umsetzung dieses europäischen Konsenses in das nationale Recht der Mitgliedsstaaten.
Regulatorische Anforderungen gelten auf verschiedenen Ebenen, der gesetz-lichen und der untergesetzlichen Ebene. Die auf gesetzlicher Ebene von der Europäischen Kommission verabschiedeten Richtlinien werden auf Landes-ebene in nationale Gesetze und Verordnungen umgesetzt.

Qualitätsmanagement für Hersteller von Medizinprodukten
4
Richtlinien auf untergesetzlicher Ebene sind nicht rechtsbindend, sondern haben nur einen hinweisenden Charakter. Sie werden als Begleitdokumente bezeichnet und sind als Hilfestellung zur Umsetzung von Gesetzen, Verord-nungen und Normen zu betrachten.
Die Anforderungen der europäischen Richtlinien für Medizinprodukte finden sich in Deutschland in erster Linie im Medizinproduktegesetz (MPG) wieder.
Das deutsche Medizinproduktegesetz wird durch verschiedene Verordnungen ergänzt, welche ebenfalls die europäischen Anforderungen wiedergeben. Hierzu gehören unter anderem die Sicherheitsplanverordnung, die Medizinpro-dukteverordnung und die Medizinproduktebetreiberverordnung.
Folgerichtig unterliegen Medizinprodukte in Deutschland dem Medizinpro-duktegesetz (MPG), das zusammen mit seinen Verordnungen und Bekannt-machungen die drei europäischen Richtlinien:
– 90/385/EWG (über aktive implantierbare medizinische Geräte)
– 98/79/EG (über In-vitro-Diagnostika)
– 93/42/EWG (über Medizinprodukte)
in nationales Recht umsetzt.
Nach § 6 Abs. 1 MPG dürfen Medizinprodukte nur in Verkehr gebracht oder in Betrieb genommen werden, wenn sie mit einer CE-Kennzeichnung versehen sind. Ausnahmen sind Medizinprodukte aus In-Haus-Herstellung, Sonderanfer-tigungen, Medizinprodukte für klinische Prüfungen und In-vitro-Diagnostika für die Leistungsbewertungsprüfung.
Anders als bei Arzneimitteln, die pharmakologisch, immunologisch oder metabolisch wirken, wird die bestimmungsgemäße Hauptwirkung bei Medizin-produkten primär auf, zum Beispiel, physikalischem Weg erreicht.
Organisationen, die als Hersteller Medizinprodukte erstmalig unter ihren Namen in Verkehr bringen, müssen nach den gesetzlichen Vorschriften für die Herstellung ihrer Produkte neben vielen anderen Anforderungen, abhängig vom Konformitätsbewertungsverfahren, ein vollständiges Qualitätssicherungs-system einrichten und aufrechterhalten.
Medizinprodukte dürfen im Europäischen Wirtschaftsraum (EWR) nur dann in Verkehr gebracht oder in Betrieb genommen werden, wenn sie mit der CE-Kennzeichnung versehen sind. Die CE-Kennzeichnung darf angebracht werden, wenn die Medizinprodukte die Grundlegenden Anforderungen einer Richtlinie erfüllen, das vorgeschriebene Konformitätsbewertungsverfahren durchgeführt wurde und der Hersteller des Medizinprodukts die Konformität erklärt hat.

5
2 Gesetzliche Anforderungen an Medizinprodukte
Im September 2012 veröffentlichte die Europäische Kommission den ersten Vor-schlag zweier Verordnungen, und zwar der MDR (Medical Devices Regulation) und IVDR (In-vitro-Diagnostic Medical Device Regulation). Nach der Europawahl im Mai 2014 hat der federführende ENVI-Ausschuss am 5. November 2014 den Berichterstattern das Mandat für die sogenannten Triolog-Gespräche mit dem Rat erteilt.
Nach einer Vielzahl von Ratsarbeitsgruppen- und Expertensitzungen wurde im Juni 2016 ein politischer Kompromiss zwischen dem Europaparlament und dem Rat gefunden. Der Umfang der beiden Verordnungen hat sich, gegenüber den ersten Vorschlägen der Kommission, mehr als verdoppelt.
Im Juni 2016 wurde die konsolidierte Fassung der Medizinprodukteverordnung (MDR) und Verordnung über In-vitro-Diagnostika (IVDR) veröffentlicht. Der Text beruht auf den Ergebnissen des „informellen Trilogs“ von Parlament, Rat und Kommission vom 25. Mai 2016.
Zu den neuen Regelungen der Medizinprodukteverordnung gehören unter anderem die Verschärfung der klinischen Bewertung, die Einführung des Systems der einmaligen Produktnummer (Unique Device Identification), detail-lierte Anforderungen an Software und das „Scrutiny-Verfahren“. Des Weiteren die Pflicht einer Haftpflichtversicherung beziehungsweise die ausreichende Bildung von Rücklagen nach dem Vorbild der deutschen Betriebshaftpflichtver-sicherung und die Verschärfung des Schutzes der Patientendaten.
Sobald die rechtlich geprüften Übersetzungen in allen Sprachen vorliegen, wird der Rat, frühestens Anfang 2017, eine Entscheidung bezüglich der beiden Verordnungen treffen. Im Anschluss wird das Europäische Parlament (zwei bis drei Monate später) zu einer abschließenden Entscheidung kommen. Das Datum des Inkrafttretens der beiden Verordnungen erfolgt 20 Tage nach Ver-öffentlichung im Europäischen Amtsblatt.
Die neuen Regeln der Medizinprodukteverordnung werden drei Jahre und der In-vitro-Diagnostika-Verordnung fünf Jahre nach der Veröffentlichung im Euro-päischen Amtsblatt in Kraft treten. Mit der Veröffentlichung der beiden Verord-nungen wird nicht vor Sommer 2017 gerechnet.
Des Weiteren gibt es spezielle Übergangsregelungen für Themen wie Benannte Stellen, nationale Behörden, Koordinierungsgruppe Medizinprodukte, Her-steller- und Produktregistrierungspflichten, UDI und der Kooperation zwischen den Mitgliedsstaaten und der Europäischen Kommission.
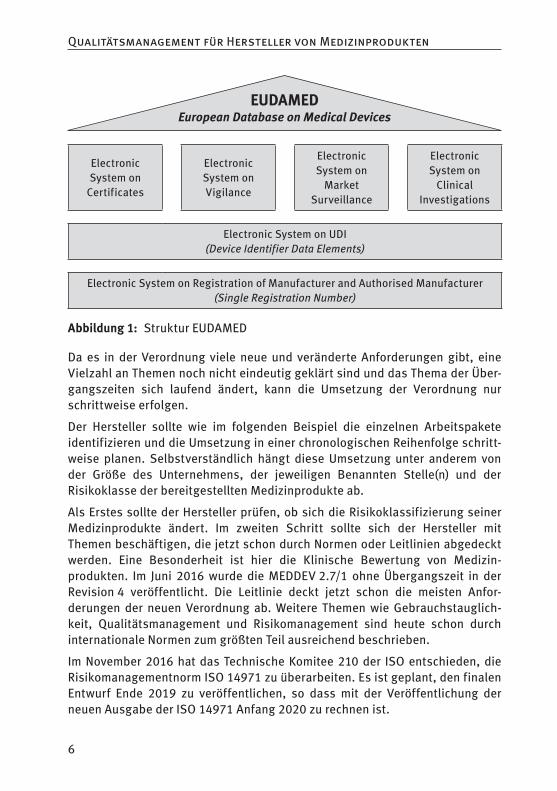
Alle Artikel der Verordnungen, die ein funktionierendes Datenbanksystem (EUDAMED) voraussetzen, werden erst anwendbar, wenn die Europäische Kommission eine Mitteilung über die volle Funktionstüchtigkeit von EUDAMED im Europäischen Anzeiger veröffentlicht hat.
Qualitätsmanagement für Hersteller von Medizinprodukten
6
EUDAMEDEuropean Database on Medical Devices
ElectronicSystem on
Certificates
ElectronicSystem onVigilance
Electronic System on
Market Surveillance
Electronic System on
Clinical Investigations
Electronic System on UDI(Device Identifier Data Elements)
Electronic System on Registration of Manufacturer and Authorised Manufacturer(Single Registration Number)
Abbildung 1: Struktur EUDAMED
Da es in der Verordnung viele neue und veränderte Anforderungen gibt, eine Vielzahl an Themen noch nicht eindeutig geklärt sind und das Thema der Über-gangszeiten sich laufend ändert, kann die Umsetzung der Verordnung nur schrittweise erfolgen.
Der Hersteller sollte wie im folgenden Beispiel die einzelnen Arbeitspakete identifizieren und die Umsetzung in einer chronologischen Reihenfolge schritt-weise planen. Selbstverständlich hängt diese Umsetzung unter anderem von der Größe des Unternehmens, der jeweiligen Benannten Stelle(n) und der Risikoklasse der bereitgestellten Medizinprodukte ab.
Als Erstes sollte der Hersteller prüfen, ob sich die Risikoklassifizierung seiner Medizinprodukte ändert. Im zweiten Schritt sollte sich der Hersteller mit Themen beschäftigen, die jetzt schon durch Normen oder Leitlinien abgedeckt werden. Eine Besonderheit ist hier die Klinische Bewertung von Medizin-produkten. Im Juni 2016 wurde die MEDDEV 2.7/1 ohne Übergangszeit in der Revision 4 veröffentlicht. Die Leitlinie deckt jetzt schon die meisten Anfor-derungen der neuen Verordnung ab. Weitere Themen wie Gebrauchstauglich-keit, Qualitätsmanagement und Risikomanagement sind heute schon durch internationale Normen zum größten Teil ausreichend beschrieben.
Im November 2016 hat das Technische Komitee 210 der ISO entschieden, die Risikomanagementnorm ISO 14971 zu überarbeiten. Es ist geplant, den finalen Entwurf Ende 2019 zu veröffentlichen, so dass mit der Veröffentlichung der neuen Ausgabe der ISO 14971 Anfang 2020 zu rechnen ist.

7
2 Gesetzliche Anforderungen an Medizinprodukte
Behörden & NB
EUDAMED
Regulatory Affairs
Marktbeobachtung
Technische Dokumentation
Unique Device Identification (UDI)
Sicherheit & Leistung (z. B. IEC 60601)
Risikomanagement (ISO 14971:2019)
Qualitätsmanagement (ISO 13485:2016)
Gebrauchstauglichkeit (IEC 62366-1:2015)
Klinische Bewertung (MEDDEV 2.7/1 Rev.4)
Risikoklassifizierung von Medizinprodukten (I, Ila, Ilb & III)
Medizinprodukterichtlinie
Medizinprodukte-verordnung
Übertragung
Abbildung 2: Beispiel zur Umsetzung der MDR
Zu vielen Themen, wie zum Beispiel die eindeutige Kennzeichnung von Medizin-produkten (UDI), Anforderungen an die Technische Dokumentation, Markt-beobachtung, Regulatory Affairs, EUDAMED, Behörden und Benannte Stellen (NB), fehlen noch die konkreten Anforderungen und Übergangszeiten sowie die Umsetzungsstrategien.
2.1 Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM)
Das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) ist eine selb-ständige Bundesoberbehörde im Geschäftsbereich des Bundesministeriums für Gesundheit (BMG).
Im BfArM arbeiten rund 1 100 Mitarbeiterinnen und Mitarbeiter – darunter Ärzte, Apotheker, Chemiker, Biologen, Juristen, Ingenieure, technische Assis-tenten und Verwaltungsmitarbeiter – an der Zulassung, der Verbesserung der Sicherheit von Arzneimitteln, der Risikoerfassung und -bewertung von Medizinprodukten und der Überwachung des Betäubungsmittel- und Grund-stoffverkehrs. Oberstes Ziel aller Maßnahmen ist die Erhöhung der Patienten-sicherheit. Auf diese Weise leistet das BfArM einen wichtigen Beitrag zur Abwehr von Gesundheitsgefahren für die Bürgerinnen und Bürger.

Qualitätsmanagement für Hersteller von Medizinprodukten
8
Das BfArM hat seinen Sitz in Bonn. Mit dem Regierungsumzug von Bonn nach Berlin wurde im Berlin/Bonn-Gesetz festgelegt, dass der Sitz der Behörde als Ausgleichsmaßnahme für die Bundesstadt Bonn von Berlin nach Bonn verlegt wird. Die Einnahmen und Ausgaben des BfArM sind im Kapitel 1510 des Bun-deshaushaltplans beschrieben, die ordnungsgemäße Haushaltsführung wird vom Bundesministeriums für Gesundheit und vom Bundesrechnungshof über-prüft. Die Einnahmen bestehen im Wesentlichen aus Gebühren für Amtshand-lungen. Zusätzliche Einnahmen entstehen aus Aufträgen der Europäischen Arzneimittel-Agentur EMA und anderer Einrichtungen auf dem Gebiet des Gesundheitswesens. Das BfArM betreibt keinerlei Werbung und hat keine Ein-nahmen durch Werbung.
Die Aufgaben des BfArM im Bereich der Medizinprodukte ergeben sich durch das Medizinproduktegesetz (MPG), die Medizinproduktesicherheitsverord-nung (MPSV) und die Verordnung über klinische Prüfung von Medizinprodukten (MPKPV).
Sie liegen insbesondere in der Bewertung sogenannter Vorkommnismeldungen. Dabei handelt es sich um Meldungen zu Ereignissen, die bei Produkten auftre-ten, die sich bereits auf dem Markt befinden und bei denen ein Produktmangel als ursächlich für einen Todesfall oder eine schwerwiegende Verschlechterung des Gesundheitszustandes, z. B. eines Patienten, angesehen wird.
Diese Meldungen erhält das BfArM von Anwendern (z. B. Ärzten, Kliniken) und Herstellern der Medizinprodukte. Es führt dann eine Risikobewertung des Vorkommnisses durch. Kommt das BfArM zu dem Ergebnis, dass aus Sicher-heitsgründen Änderungen am Produkt erforderlich sind und veranlasst der Her-steller dies nicht bereits gemäß seiner gesetzlichen eigenen Verantwortung, spricht es eine Empfehlung an den Hersteller bzw. die für die Überwachung zuständige Landesbehörde aus. Bei den letztgenannten Stellen liegen die gesetzlichen Möglichkeiten, um diese Empfehlungen nötigenfalls anzuordnen und ihre Umsetzung zu überwachen.
Das BfArM ist darüber hinaus zuständig für die Genehmigung klinischer Prüfungen von Medizinprodukten. Eine klinische Prüfung wird an freiwilligen Probanden vorgenommen, um Daten zu den Aspekten der Sicherheit und/oder Leistungsfähigkeit eines Medizinproduktes zu erheben, die sich nur in der kli-nischen Praxis überprüfen lassen. Eine solche Prüfung ist erforderlich, wenn klinische Daten aus der Literatur, aus klinischer Erfahrung oder aus bisher durchgeführten klinischen Prüfungen nicht ausreichen.

9
2 Gesetzliche Anforderungen an Medizinprodukte
Das BfArM entscheidet außerdem auf Antrag einer zuständigen Landes-behörde, einer Benannten Stelle oder des Herstellers über die Klassifizierung einzelner Medizinprodukte und deren Abgrenzung zu anderen Produkten.
[Quelle: BfArM]
2.2 Deutsches Institut für Medizinische Dokumentation und Information (DIMDI)
Das DIMDI erfüllt als Behörde im Ressort des Bundesministeriums für Gesund-heit (BMG) Aufgaben an zentralen Schaltstellen des deutschen Gesundheits-systems.
Das Medizinproduktegesetz (MPG) schreibt in § 33 die Einrichtung und den Betrieb eines datenbankgestützten Informationssystems für Medizinprodukte sowie die Bereitstellung von Daten für die europäische Datenbank für Medizin-produkte EUDAMED vor. Die Einzelheiten des Informationssystems sind in der Verordnung über das datenbankgestützte Informationssystem über Medizin-produkte des Deutschen Instituts für Medizinische Dokumentation und Infor-mation (DIMDI-Verordnung) geregelt.
Vom DIMDI werden Online-Erfassungssysteme, automatische Benachrichti-gungssysteme und Datenbanken für die Anzeigepflichtigen und die beteiligten nationalen und internationalen Institutionen zu folgenden Teilen des MPG be-reitgestellt:
– Anzeigen über das erstmalige Inverkehrbringen und zum Sicherheitsbeauf-tragten gemäß §§ 25 und 30 MPG
– Bescheinigungen der Benannten Stellen gemäß § 18 MPG
– Medizinprodukte-Beobachtungs- und Meldesystem mit Daten nach § 29 MPG
– Anzeigen über klinische Prüfungen/Leistungsbewertungsprüfungen mit Medizinprodukten gemäß §§ 20 und 24 MPG
– Mitteilungen über Einstufung oder Entscheidung zur Klassifizierung eines Medizinproduktes bzw. Abgrenzung zu anderen Produkten gemäß § 33 in Verbindung mit § 13 MPG
Das DIMDI ermöglicht auch zuständigen Behörden anderer Mitgliedstaaten sowie Institutionen der Europäischen Gemeinschaft und anderer Vertragsstaa-ten des Abkommens über den Europäischen Wirtschaftsraum den Zugang zu den Medizinprodukte-Datenbanken. Darüber hinaus stellt das DIMDI Literatur-datenbanken mit Informationen zu Medizinprodukten bereit.

Qualitätsmanagement für Hersteller von Medizinprodukten
10
Zur Unterstützung des eindeutigen Informationsaustausches über Medizinpro-dukte gibt das DIMDI die deutsche Fassung der Nomenklatur für Medizinpro-dukte (Universal Medical Device Nomenclature System – UMDNS) heraus.
[Quelle: DIMDI]
2.3 ÜberwachungsbehördenDas Medizinproduktegesetz unterscheidet zwischen zuständigen Landes-behörden und Bundesoberbehörden. Hierzu muss erwähnt werden, dass, wenn im Gesetz nicht die Landesbehörde genannt wird, immer die Bundesober-behörde zuständig ist.
Zuständige Bundesoberbehörden
Die zuständigen Bundesoberbehörden führen selbst keine eigenen Inspektio-nen bei den Herstellern durch, dürfen aber, meistens in Zusammenarbeit mit den zuständigen Landesbehörden, Dokumente, Aufzeichnungen oder Stellung-nahmen einfordern.
Ein Besonderheit ist die Aufgabe der zuständigen Bundesoberbehörde bei kli-nische Prüfungen: Mit der klinischen Prüfung eines Medizinproduktes darf in Deutschland erst begonnen werden, wenn die zuständige Ethik-Kommission diese nach Maßgabe des § 22 MPG zustimmend bewertet und die zuständige Bundesoberbehörde diese nach Maßgabe des § 22a MPG genehmigt hat.
Grundsätzlich ist als Bundesoberbehörde das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) für klinische Prüfungen von Medizinprodukten und für Leistungsbewertungsprüfungen zuständig. Nur bei Leistungsbewer-tungsprüfungen von Hochrisiko-In-vitro-Diagnostika ist das Paul-Ehrlich-Insti-tut (PEI) zuständig.
[Quelle: DIMDI]
Zuständige Landesbehörden
Die Begrifflichkeiten „oberste Landesbehörden“, „Landesoberbehörden“ und „untere Landesbehörden“ wirken missverständlich. Kennzeichnend für untere Landesbehörden ist nicht, dass sie „unten“ sind, sondern dass sie nur für einen Teil des Landes (lokal) zuständig sind.
Der Vollzug des Medizinprodukterechts ist grundsätzlich erst einmal Länder-sache, es sei denn, das Gesetz weicht explizit von dieser Regel ab. Das bedeutet, dass jedes Bundesland für die Umsetzung seine eigenen Behörden festlegen darf. Somit kann es sein, dass in einigen Bundesländern nur eine

11
2 Gesetzliche Anforderungen an Medizinprodukte
Behörde zuständig ist und in anderen Bundesländern mehrere Behörden zu-ständig sind. Allerdings darf für einen bestimmten Raum nur eine Behörde ver-antwortlich sein.
Je nach Sitz des Medizinprodukteherstellers oder seines Bevollmächtigten muss das erstmalige Inverkehrbringen eines Medizinproduktes bei der jeweils zuständigen Landesbehörde angezeigt werden.
Eine detaillierte Übersicht über die zuständigen Landesbehörden ist auf der Homepage des Deutschen Instituts für Medizinische Dokumentation und Infor-mation (DIMDI) zu finden.
Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln (ZLG)
Das Abkommen über die Zentralstelle der Länder für Gesundheitsschutz bei Arzneimitteln und Medizinprodukten gibt für die Tätigkeit der ZLG das Ziel vor, den in Deutschland erreichten Stand an Qualität und Sicherheit von Medizin-produkten im Rahmen und auf der Grundlage der Richtlinien zu halten und zu verbessern.
Die ZLG vollzieht im Bereich Medizinprodukte die Aufgaben der Länder hin-sichtlich der Anerkennung und Benennung. Darunter sind insbesondere die An-erkennung und Überwachung von Prüflaboratorien und Zertifizierungsstellen in den Bereichen der Medizinprodukte und In-vitro-Diagnostika zu verstehen.
Anerkennung, Benennungund Überwachung von
LaboratorienZertifizierungs-
stellen fürMedizinprodukte
Zertifizierungs-stellen für
Qualitätssiche-rungssysteme
Zertifizierungs-stellen fürPersonal
Benannten Stellen
Konformitätsbewertungsstellen
Abbildung 3: Aufgaben der ZLG

Qualitätsmanagement für Hersteller von Medizinprodukten
12
Bis Ende 2009 war die ZLG unter anderem auch zuständig für die Akkredi-tierung der Benannten Stellen. Aufgrund des § 1 Abs. 1 Akkreditierungs-stellengesetz (AkkStelleG) ist die Zuständigkeit für alle Akkreditierungen auf die Deutsche Akkreditierungsstelle GmbH (DAkkS) übergegangen, jedoch führt die ZLG gemäß § 2 Abs. 3 Akkreditierungsstellengesetz in Verbindung mit § 4 AkkStelleG-Beleihungsverordnung Begutachtungen und Überwachungen im Bereich Medizinprodukte im Auftrag der DAkkS durch.
Die ZLG ist außerdem nationale Kontaktstelle für die zuständigen Behörden anderer Staaten und für die EU-Kommission.
[Quelle: ZLG]
2.4 Benannte StellenDie Benannten Stellen sind vorgesehene Stellen, die durch die Kommission, die Europäische Union und die Vertragsstaaten des Abkommens über den Euro-päischen Wirtschaftsraum benannt worden sind.
Ihre Aufgaben sind die Durchführung von Prüfungen und die Erteilung von Be-scheinigungen, im Zusammenhang mit Konformitätsbewertungsverfahren nach Maßgabe der Rechtsverordnung nach § 37 Abs. 1.
Die Benannten Stellen sind, nach den Vorgaben der Normenserie EN 45000 und der zutreffenden Anhänge der jeweiligen EG-Harmonisierungsrichtlinien, natio-nal akkreditierte Auditierungs- und Zertifizierungsstellen.
So ist zum Beispiel nach Anhang II der Richtlinie 93/42 EWG bei allen Medizin-produkten, mit Ausnahme von Produkten mit dem geringsten Risikopotenzial (Klasse I), der Hersteller verpflichtet, vor dem Inverkehrbringen sein Qualitäts-managementsystem durch eine Benannte Stelle auditieren und zertifizieren zu lassen.
Der Erstzertifizierung folgen jährliche Wiederholungs- bzw. Überwachungs-Audits, die im Allgemeinen einen reduzierten Umfang haben. Nur soweit es sich um Medizinprodukte der Klasse I handelt, erklärt der Hersteller die Konformität in eigener Verantwortung. In allen Fällen unterliegt der Hersteller bzw. die Her-stellung und das Inverkehrbringen der Medizinprodukte auch der Überwachung der zuständigen Landesbehörde (§ 26 MPG).
Die Benannte Stelle ist durch den Hersteller oder Bevollmächtigten, im Rahmen der Befähigung der Benannten Stelle, frei zu wählen. Da alle Benannten Stellen des Europäischen Wirtschaftsraums den gleichen europäischen Mindestanfor-derungen entsprechen und somit rechtlich als gleichwertig gelten, sind diese innerhalb des Europäischen Wirtschaftsraums frei wählbar.

13
2 Gesetzliche Anforderungen an Medizinprodukte
Eine Liste der Benannten Stellen kann auf der Webseite des Deutschen Instituts für Medizinische Dokumentation und Information eingesehen werden. Herstel-ler können sich aber nur an eine Benannte Stelle ihrer Wahl wenden, die für das entsprechende Verfahren und die betreffende Produktkategorie benannt ist. Diese Eignung der nationalen Benannten Stellen kann auf der Webseite des ZLG eingesehen werden und ist zusammen mit dem Geltungsbereich der Benannten Stellen gelistet.
2.5 BeratungsunternehmenIm Allgemeinen ist Beratung oder auch „consulting“ eine individuelle Dienst-leistung zur Aufarbeitung von identifizierten oder potenziellen Problemstel-lungen durch Interaktion zwischen externen, unabhängigen Beratern oder Beratungsunternehmen und einem um Rat oder Information nachsuchenden Kunden oder Klienten.
Auch wenn das Beratungskonzept immer an die Aufgabenstellung und die Er-wartungshaltung des Kunden oder Klienten angepasst wird, ist die Beratung in der Medizintechnik eine branchenspezifische Dienstleistung und muss sich immer auch an den regulatorischen Anforderungen orientieren. Typische Beratungskonzepte decken neben den erforderlichen regulatorischen Rahmen, auch die Themen Strategie, Nachhaltigkeit und Management ab.
Die Unternehmensberatung ist ein Teilbereich der Beratung und deckt in erster Linie den betriebswirtschaftlichen Teil in einer Organisation ab. Sie ist somit kein Bereich, der Einfluss auf die regulatorischen Anforderungen an den Her-steller von Medizinprodukten hat. Somit spielt die Unternehmensberatung keine tragende Rolle in der Beratung im streng regulierten Bereich der Medizin-produkte.
In der Medizintechnik stehen daher unter anderem folgende Themengebiete im Fokus der Beratung:
– nationale und internationale regulatorische Anforderungen
– nationale und internationale Registrierung und Zulassung (Regulatory Affairs)
– Projektmanagement
– Entwicklung und Produktion
– Marktbeobachtung
– Qualitätsmanagement
– Klinische Bewertung

Qualitätsmanagement für Hersteller von Medizinprodukten
14
– Risikomanagement
– Gebrauchstauglichkeit
– Software
– elektrische Sicherheit
– Biokompatibilität
Es hat sich in der Praxis herausgestellt, dass Beratungsunternehmen in der Medizintechnik den kompletten Produktlebenszyklus von Medizinprodukten abdecken und die nötigen Kompetenzen und Erfahrungen nachweisen sollten.
Auftraggeber von Beratungsunternehmen sind überwiegend die Hersteller, also die Organisationen, die ein Medizinprodukt unter ihren Namen erstmalig in Verkehr bringen. Aber auch Zulieferer und externe Parteien sind typische Organisationen, die verstärkt Beratung in Anspruch nehmen.
Im Gegensatz zu den Benannten Stellen werden Beratungsunternehmen weder benannt noch überwacht. Somit sollte der Hersteller Beratungsunternehmen wie einen Lieferanten oder noch besser wie eine externe Partei behandeln.
Beratungsunternehmen sollen bei der Identifizierung und Lösung von poten-ziellen Problemen unterstützen und den Klienten oder Kunden bei der Ableitung von möglichen Verbesserungen, Korrekturen, Korrektur- und Vorbeugungsmaß-nahmen beraten.
Die Organisation von Beratungsunternehmen zeichnet sich generell durch eine flexible Struktur und flache Hierarchien aus.
In der Beratung wird zwischen Werks- und Dienstleistungsverträgen unter-schieden.
Die meisten Beratungsaufträge, die über einen Werksvertrag laufen, sind kom-plex, zeitlich begrenzt, risikobehaftet, zielorientiert und haben somit Projekt-charakter.
Hinsichtlich der Einbindung des Beraters in die Organisation des Klienten sind je nach Typ des Einsatzes alle Formen der Projektorganisation denkbar.

15
3 Grundlagen Qualitätsmanagement
3 Grundlagen QualitätsmanagementDas Wort Qualität wird von dem lateinischen Wort „qualitas“ abgeleitet und bedeutet „wie beschaffen“. Für den Begriff Qualität gibt es in der Medizintech-nik keine feste Definition. Das heißt, es ist kein feststehender Begriff, der eine absolute Größe bezeichnet. Dieser Begriff „der Qualität“ muss daher immer spezifisch von der Organisation definiert werden. Er beinhaltet nicht nur einen Aspekt des Produkts, sondern bezieht sich auf verschiedene Aspekte des Pro-dukts und des gesamten Produktrealisierungsprozesses.
Beispiele für die Definition des Begriffs „Qualität“:
– „Qualität ist, wenn der Kunde zurückkommt und nicht das Medizinprodukt!“
[Quelle: unbekannt]
– „Qualität“ ist die Gesamtheit von Merkmalen einer Einheit oder Dienstleis-tung bezüglich ihrer Eignung, festgelegte und vorausgesetzte Erfordernisse zu erfüllen.
[Quelle: DIN EN ISO 8402 – Achtung: Diese Norm wurde inzwischen zurück-gezogen.]
– „Qualität ist das Anständige“
[Quelle: T. Heuss]
– Quality means the totality of features and characteristics that bear on the
ability of a device to satisfy fitness-for-use, including safety and perfor-
mance.
[Quelle: 21 CFR 820.3(s) QSR]
– Grad, in dem ein Satz inhärenter Merkmale Anforderungen erfüllt.
[Quelle: DIN EN ISO 9000:2015-11]
Anmerkung 1: „Inhärent“ bedeutet, im Gegensatz zu „zugeordnet“, „einem
Produkt innewohnend, das Zusammengehören von Ding und Eigenschaft“,
insbesondere als ständiges Merkmal.
Anmerkung 2: Die Benennung „Qualität“ kann zusammen mit Eigenschafts-
worten schlecht, gut oder ausgezeichnet verwendet werden.

Qualitätsmanagement für Hersteller von Medizinprodukten
16
Die DIN EN ISO 9000:2015 beschreibt den Begriff „Qualität“ als ein grund-legendes Konzept wie folgt:
„Eine auf Qualität ausgerichtete Organisation fördert eine Kultur, die zu Verhaltensweisen, Einstellungen, Tätigkeiten und Prozessen führt, die Wert schaffen, indem sie die Erfordernisse und Erwartungen von Kunden und anderen relevanten interessierten Parteien erfüllen.
Die Qualität der Produkte und Dienstleistungen einer Organisation wird durch die Fähigkeit bestimmt, Kunden zufrieden zu stellen sowie durch die beabsichtigte und unabsichtliche Auswirkung auf relevante interessierte Parteien.
Die Qualität von Produkten und Dienstleistungen umfasst nicht nur deren vorgesehene Funktion und Leistung, sondern auch ihren wahrgenommenen Wert und Nutzen für den Kunden.“
Der Begriff Qualität wird vermutlich so alt sein wie die Herstellung und der Tausch von Waren: Denn wer Waren eintauschte, dürfte sich für die Qualität der erhaltenen Gegenleistung interessiert haben. Mit zunehmendem Handel entstanden die ersten Institutionen, die die Qualität von Produkten sichern sollten, etwa die Vorschriften und Kontrollen der Zünfte im Mittelalter.
Eine ganz neue Dimension gewann das Thema aber mit der industriellen Revo-lution, und hier mit der Fließbandfertigung – waren es doch oftmals ungelernte Arbeiter, die da – auch noch bezahlt im Akkord – am Fließband komplexe Pro-dukte wie etwa ein Auto herstellen sollten.
Das Qualitätsmanagement in der Medizintechnik wurde normativ mit der ISO 9001 in der Ausgabe von 1994 eingeführt. Diese Ausgabe hatte 20 Ele-mente, wie zum Beispiel Vertragsprüfung, Wartung oder statistische Metho-den, und folgte der Philosophie der „vorbeugenden Maßnahmen“.
Im gleichen Zeitraum hat die amerikanische Überwachungsbehörde Food and Drug Administration (FDA) für die Medizinprodukte einen eigenständigen regulierten Bereich geschaffen. Dieser hat unter anderem die Quality System Regulation veröffentlicht, in der erstmalig die Anforderungen an ein Qualitäts-managementsystem nur für Medizinprodukte gestellt wurden. Diesem Vorbild folgend wurden auch in Europa die Forderungen nach einer eigenständigen QM-Norm für Medizinprodukte lauter.

17
3 Grundlagen Qualitätsmanagement
ISO 9001:2000
ISO 9001:1994
DIN EN ISO 13485:2016
ISO 13485:2016
EN ISO 13485:2012Norm zurück-
gezogen
Übergangszeit
Norm in Kraft
8/94 9/96 12/00 2/01 7/03 12/03 3/04 7/06 9/12 3/16 8/16
ISO 13485:1996
ISO 46001:1996
ISO 13485:2003
ISO 13485:2001
Abbildung 4: Qualitätsmanagement in der Medizintechnik
Schließlich wurde im Jahre 2003, nach vielen Diskussionen, Abstimmungen und Fehlentscheidungen in den zuständigen Gremien, die erste eigenständige QM-Norm für Medizinprodukte mit einer Übergangszeit von drei Jahren harmo-nisiert. Seit dem Ende der Übergangszeit im Jahre 2006 hat sich der normative Teil der zweiten Ausgabe nicht verändert.
3.1 DIN EN ISO 9001Im Jahr 1979 entwickelte die British Standards Institution (BSI) mit dem BS 5750 den ersten Standard zum Thema Qualitätsmanagementsysteme, welcher als Vorläufer der ISO 9000er Serie gilt. Auf dessen Basis wurde 1987 die ISO 9000-Normenreihe veröffentlicht und international anerkannt.
In den Jahren 1994, 2000, 2008 und 2015 wurde die Norm regelmäßig von der zuständigen Normungsgruppe überarbeitet und an die aktuellen Anforde-rungen angepasst. In der letzten Ausgabe der ISO 9001 wurde zusätzlich die Struktur überarbeitet und unter anderem an den PDCL- oder Demingkreis angepasst.
Die DIN EN ISO 9001 legt im Allgemeinen, also nicht branchenspezifisch, die Mindestanforderungen an ein Qualitätsmanagementsystem fest, damit die Organisation Produkte und Dienstleistungen bereitstellen kann, welche die Kundenanforderungen nachweislich erfüllen.

Qualitätsmanagement für Hersteller von Medizinprodukten
18
ISO 9001:1987 dokumentierte
Verfahren
ISO 9001:1994 vorbeugende Maßnahmen
ISO 9001:2000 prozessorientierter
Ansatz
ISO 9001:2015 Risikien und
Chancen
Abbildung 5: Die Historie des ISO 9001-Zertifizierungsstandards
Nach Angaben der Internationalen Organisation für Normung (ISO) wurden bis einschließlich 2009 in 178 Ländern und Wirtschaftsgebieten mehr als eine Million ISO 9001-Zertifikate ausgestellt.
Die europäische Norm EN ISO 9000 wurde offiziell in englischer, deutscher und französischer Sprache veröffentlicht. Grundlage bei der Entstehung der Norm ist, seit der Ausgabe im Jahre 2000, der prozessorientierte Ansatz, basierend auf dem nach William Edwards Deming benannten Demingkreis.
Bei der Implementierung der DIN EN ISO 9001 und DIN EN ISO 13485 hat sich sowohl bei der Beschreibung von Prozessen als auch bei der Entwicklung von dokumentierten Verfahren die Berücksichtigung des PDCL- oder Deming-Krei-ses bewährt.
ständige Verbesserung
planen (plan)
ausführen (do)
überprüfen
(check)
verbessern (act)
Abbildung 6: PDCL- oder Deming-Kreis

19
3 Grundlagen Qualitätsmanagement
Im Jahr 2012 wurde nach einer Überprüfung der ISO 9001:2008 beschlossen, die Norm zu überarbeiten. Das Ergebnis dieser Revision ist im September 2015 veröffentlicht worden.
Somit gilt die neue ISO 9001 seit dem 15. September 2015. Die Übergangsfrist endet nach drei Jahren, also am 14. September 2018. Die Zertifizierungsstellen gewähren üblicherweise diese Übergangsfrist innerhalb der Organisation, um die QM-Systeme der Organisationen auf die neue Ausgabe umzustellen. Hierzu sollte sich die Organisation mit ihrem Zertifizierer bezüglich der Termine ab-stimmen, da ein verspätetes Zertifikat zu Problemen, wie zum Beispiel bei einer Ausschreibung oder Lieferantenbewertung, führen kann.
Bei der Überarbeitung der Struktur wurden die Titel der Kapitel und Abschnitte sowie die Reihenfolge der Klauseln und Absätze vollständig überarbeitet.
Diese neue Struktur hat jedoch keinerlei Auswirkung auf die Inhalte oder Anfor derungen der Norm. Bei genauer Prüfung des Textes stellt man jedoch fest, dass die Struktur dahingehend verändert wurde, dass sie den von der International Organization for Standardization (ISO) vorgegebenen neuen Text-gestaltungsrichtlinien und Themensequenzen entspricht.
ISO 9001:2008 ISO 9001:2015
1 Scope (Anwendungsbereich) 1 Scope
2 Normative references (Normative Verweise) 2 Normative references
3 Terms and definitions (Begriffe) 3 Terms and definitions
4 Quality management system (Qualitätsmanagementsystem)
4 Context of the organization(Kontext der Organisation)
5 Management responsibility (Verantwortung der Leitung)
5 Leadership(Führung)
5.4 Planning (Planung)
6 Planning(Planung)
6 Resource management (Management von Ressourcen)
7 Support(Unterstützung)
7 Product realization (Produktrealisierung)
8 Operation (Betrieb)
8 Measurement, analysis and improvement (Messung, Analyse und Verbesserung)
9 Performance evaluation(Bewertung der Leistung)
8.5 Improvement(Verbesserung)
10 Improvement(Verbesserung)
Abbildung 7: Kapitelstruktur der ISO 9001 in der Version 2008 und 2015

Qualitätsmanagement für Hersteller von Medizinprodukten
20
Diese Veränderung reflektiert eine strategische Entscheidung der ISO, die Schritt für Schritt auf alle ISO-Managementnormen, wie zum Beispiel die Norm-familien ISO 27000 und ISO 14000, angewandt werden soll.
Mit dieser neuen gemeinsamen Struktur will ISO Unternehmen und Organi-sationen die vollständige oder teilweise Integration ihrer unterschiedlichen Manage mentsysteme vereinfachen, um letztendlich über ein wirklich einheit-liches Managementsystem in der Organisation zu verfügen.
Leider wurde diese strategische Entscheidung zu einem, für die dritte Ausgabe der ISO 13485, ungünstigen Zeitpunkt getroffen. Die Arbeitsgruppe hat sich gegen die neue Struktur entschieden, da die Entwicklung der ISO 13485 nach der alten Struktur schon zu weit fortgeschritten war, dass eine Umstellung nicht mehr möglich war. Das bedeutet, dass die ISO 13485 erst mit der vierten Ausgabe an die neue Struktur angepasst wird.
Aus Sicht der ISO 9001-Gruppe macht die neue Struktur es den Unternehmen leichter, Teile anderer Normen, die sie als relevant erachten, einzubeziehen. Hierzu gehören zunächst Teile der Umweltnorm ISO 14001:2015, der IT-Sicher-heitsnorm 27001:2015, der Energiemanagementnorm ISO 50001 und zukünftig die Norm ISO 45001 für Gesundheit und Sicherheit am Arbeitsplatz.
Jede umfassende Normänderung bringt ein Konzept mit sich, das der zertifizierten Organisation zu einem höheren Reifegrad verhelfen soll. Das Risiko management mit seinem „risikobasierten“ Ansatz ist grundlegend für die Neuerung: Identifizierung, Qualifizierung und Management der Risiken. Qualität ist das Ergebnis des angemessenen Managements dieser Risiken, die über den Umfang des gelieferten Produkts oder der erbrachten Leistung hinausgehen. Von Qualität kann nur die Rede sein, wenn die Organisationen in der Lage sind, ihren Kunden auf lange Sicht konforme Produkte oder Dienstleis-tungen zur Verfügung zu stellen.
Hierzu ist zu beachten, dass die ISO 9000 den Begriff Risiko als „Auswirkung von Ungewissheit“ definiert und somit nicht mit der Definition der ISO 13485 beziehungsweise der ISO 14971 übereinstimmt. Nach ISO 9001 steht dem Risiko die Chance gegenüber und umfasst somit auch das Konzept der positi-ven Unsicherheit.
Durch die unterschiedlichen Definitionen unterscheidet sich auch der risiko-basierte Ansatz in den beiden Normen, da es in der Medizintechnik in erster Linie um den Patient, Anwender und Dritte im Zusammenhang mit der Sicher-heit und Leistung von Medizinprodukten geht und nicht die Zufriedenheit des Kunden im Vordergrund steht. Dadurch ist aus Sicht der ISO 13485, also aus regu latorischer Sicht, nicht der Kunde im Fokus, sondern der Patient, Anwender und Dritte und, wenn zutreffend, Umwelt und Güter.





























