Embed Size (px)
Citation preview

LETTERS
Reversible tuning of a block-copolymernanostructure via electric fieldsKRISTIN SCHMIDT1, HEIKO G. SCHOBERTH1, MARKUS RUPPEL2, HEIKO ZETTL1, HELMUT HANSEL1,THOMAS M. WEISS3†, VOLKER URBAN4, GEORG KRAUSCH1‡ AND ALEXANDER BOKER1*1Lehrstuhl fur Physikalische Chemie II, Universitat Bayreuth, 95440 Bayreuth, Germany2Lehrstuhl fur Makromolekulare Chemie II, Universitat Bayreuth, 95440 Bayreuth, Germany3European Synchrotron Radiation Facility (ESRF), 38043 Grenoble, France4Oak Ridge National Laboratory (ORNL), Oak Ridge, Tennessee 37831, USA†Present address: Stanford Linear Accelerator Center (SLAC), Menlo Park, California 94025, USA‡Present address: Universitat Mainz, Forum 2, 55128 Mainz, Germany*e-mail: [email protected]
Published online: 25 November 2007; doi:10.1038/nmat2068
Block copolymers consisting of incompatible componentsself-assemble into microphase-separated domains yieldinghighly regular structures with characteristic length scales ofthe order of several tens of nanometres. Therefore, in the pastdecades, block copolymers have gained considerable potentialfor nanotechnological applications, such as in nanostructurednetworks and membranes, nanoparticle templates and high-density data storage media1–4. However, the characteristic sizeof the resulting structures is usually determined by molecularparameters of the constituent polymer molecules and cannoteasily be adjusted on demand. Here, we show that electricd.c. fields can be used to tune the characteristic spacing of ablock-copolymer nanostructure with high accuracy by as muchas 6% in a fully reversible way on a timescale in the rangeof several milliseconds. We discuss the influence of variousphysical parameters on the tuning process and study the timeresponse of the nanostructure to the applied field. A tentativeexplanation of the observed effect is given on the basis ofanisotropic polarizabilities and permanent dipole moments ofthe monomeric constituents. This electric-field-induced effectfurther enhances the high technological potential of block-copolymer-based soft-lithography applications5,6.
In the past, electric fields have successfully been used toachieve long-ranged order in block-copolymer nanostructures7–12.The aligning effect is based on the differences in the dielectricconstants 1ε between the blocks. The microdomains tend toorient parallel to the electric-field vector, thereby lowering thefree energy of the system7. The parameters governing the kineticsand mechanisms of the reorientation process have been studiedexperimentally both with in situ13–16 and ex situ methods7–9,11,12,17,leading to increasing control over the domain orientation inblock copolymers, rendering them into highly valuable systems forvarious nanotechnological applications6.
For many potential applications, however, the dimensionsof the nanostructures need to be tuned precisely as well.Therefore, tools to systematically vary the characteristic spacingof the nanostructures in a predictable and simple manner areindispensable. For microphase-separated copolymers, tuning ofthe morphology and size of the nanoscopic patterns formed istypically achieved by changing the molecular weights or the block
ratio of the polymers used. However, this approach only allowscontrol of the characteristic spacing on coarse scales, whereasprecise adjustment of the spacing is impossible. The addition of ahomopolymer corresponding to one or both of the polymer blocksor the introduction of a non-selective solvent has successfully beenused to fine-tune block-copolymer nanostructures18–21. However,an exact adjustment to within a per cent or so of the characteristicspacing seems barely possible. Moreover, these approaches arenot reversible.
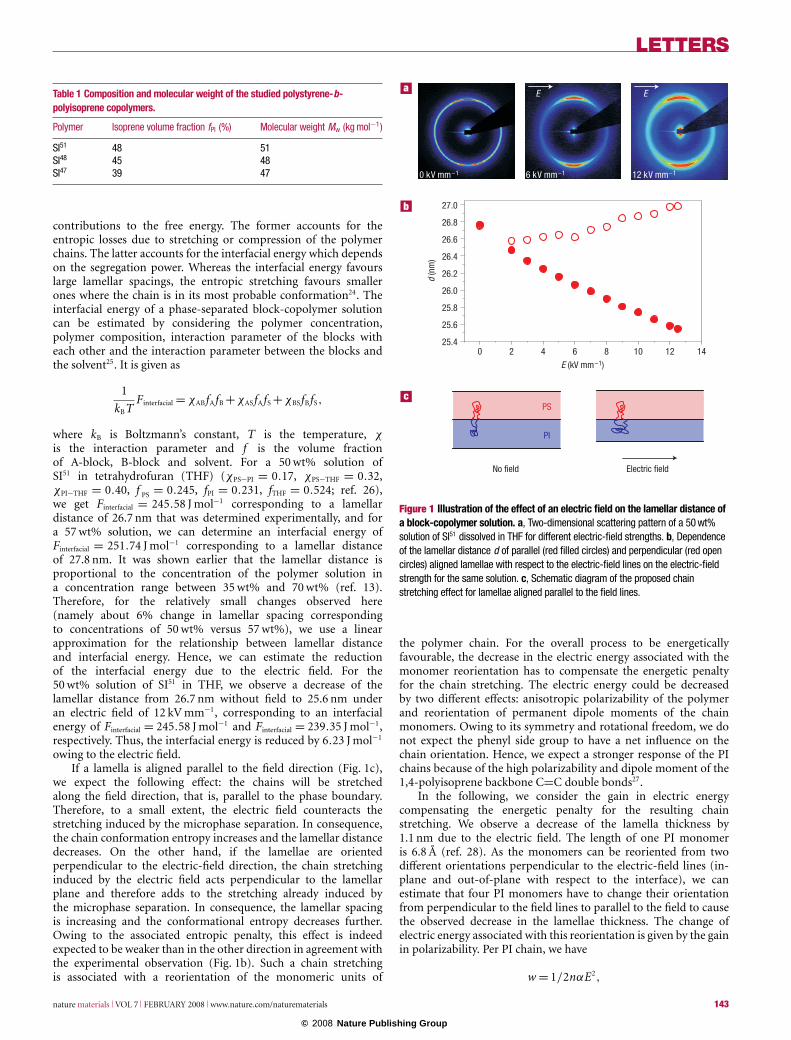
Here, we studied three different lamellar polystyrene-b-polyisoprene (SI) diblock copolymers (see Table 1) with molecularweights of around 50,000 g mol−1 in a home-built capacitor withsynchrotron small-angle X-ray scattering (SAXS) at the ID2beamline at the European Synchrotron Radiation Facility (ESRF).To exclude effects that may arise from the reorientation processitself, all samples were pre-aligned; therefore, most of the lamellaeare oriented along the electric-field lines. Figure 1a shows thescattering patterns for different electric-field strengths. We observehighly anisotropic SAXS patterns at higher electric fields. Theposition of the first-order Bragg peak is different for lamellaethat are aligned in the field direction and those that are alignedperpendicular to the field direction. Figure 1b shows the effectof the electric-field strength on the lamellar spacing calculatedfrom the position of the first-order Bragg peak for the lamellaealigned in the field direction (red filled circles) and the lamellaealigned perpendicular (red open circles). The lamellar distancefor the lamellae oriented along the field lines decreases rapidlywith increasing electric-field strength, whereas the lamellar distancefor the lamellae oriented perpendicular to the field only slightlyincreases. This behaviour may be explained by stretching of thepolymer chains. The only theoretical prediction of chain stretchingof block copolymers under the influence of an electric field wasdescribed by Gurovich22,23. His results based on self-consistentfield theory calculations indicate that the electric field polarizesthe monomers, interacts with the induced polar moments andeventually orients them. In consequence, chains are elongatedparallel or perpendicular to an applied field depending on theanisotropic polarizability of the monomers.
The equilibrium microphase structures of block copolymersresult from a competition between entropic and enthalpic
142 nature materials VOL 7 FEBRUARY 2008 www.nature.com/naturematerials
© 2008 Nature Publishing Group
LETTERS
Table 1 Composition and molecular weight of the studied polystyrene-b-polyisoprene copolymers.
Polymer Isoprene volume fraction fPI (%) Molecular weight Mw (kg mol−1)
SI51 48 51SI48 45 48SI47 39 47
contributions to the free energy. The former accounts for theentropic losses due to stretching or compression of the polymerchains. The latter accounts for the interfacial energy which dependson the segregation power. Whereas the interfacial energy favourslarge lamellar spacings, the entropic stretching favours smallerones where the chain is in its most probable conformation24. Theinterfacial energy of a phase-separated block-copolymer solutioncan be estimated by considering the polymer concentration,polymer composition, interaction parameter of the blocks witheach other and the interaction parameter between the blocks andthe solvent25. It is given as
1
kBTFinterfacial = χABfAfB +χASfAfS +χBSfBfS,
where kB is Boltzmann’s constant, T is the temperature, χis the interaction parameter and f is the volume fractionof A-block, B-block and solvent. For a 50 wt% solution ofSI51 in tetrahydrofuran (THF) (χPS−PI = 0.17, χPS−THF = 0.32,χPI−THF = 0.40, f PS = 0.245, fPI = 0.231, fTHF = 0.524; ref. 26),we get Finterfacial = 245.58 J mol−1 corresponding to a lamellardistance of 26.7 nm that was determined experimentally, and fora 57 wt% solution, we can determine an interfacial energy ofFinterfacial = 251.74 J mol−1 corresponding to a lamellar distanceof 27.8 nm. It was shown earlier that the lamellar distance isproportional to the concentration of the polymer solution ina concentration range between 35 wt% and 70 wt% (ref. 13).Therefore, for the relatively small changes observed here(namely about 6% change in lamellar spacing correspondingto concentrations of 50 wt% versus 57 wt%), we use a linearapproximation for the relationship between lamellar distanceand interfacial energy. Hence, we can estimate the reductionof the interfacial energy due to the electric field. For the50 wt% solution of SI51 in THF, we observe a decrease of thelamellar distance from 26.7 nm without field to 25.6 nm underan electric field of 12 kV mm−1, corresponding to an interfacialenergy of Finterfacial = 245.58 J mol−1 and Finterfacial = 239.35 J mol−1,respectively. Thus, the interfacial energy is reduced by 6.23 J mol−1
owing to the electric field.If a lamella is aligned parallel to the field direction (Fig. 1c),
we expect the following effect: the chains will be stretchedalong the field direction, that is, parallel to the phase boundary.Therefore, to a small extent, the electric field counteracts thestretching induced by the microphase separation. In consequence,the chain conformation entropy increases and the lamellar distancedecreases. On the other hand, if the lamellae are orientedperpendicular to the electric-field direction, the chain stretchinginduced by the electric field acts perpendicular to the lamellarplane and therefore adds to the stretching already induced bythe microphase separation. In consequence, the lamellar spacingis increasing and the conformational entropy decreases further.Owing to the associated entropic penalty, this effect is indeedexpected to be weaker than in the other direction in agreement withthe experimental observation (Fig. 1b). Such a chain stretchingis associated with a reorientation of the monomeric units of
a E E
0 kV mm–1 6 kV mm–1 12 kV mm–1
0 2 4 6 8 10 12 1425.4
25.6
25.8
26.0
26.2
26.4
26.6
26.8
27.0
E (kV mm–1)
d (n
m)
PS
PI
No field Electric field
b
c
Figure 1 Illustration of the effect of an electric field on the lamellar distance ofa block-copolymer solution. a, Two-dimensional scattering pattern of a 50 wt%solution of SI51 dissolved in THF for different electric-field strengths. b, Dependenceof the lamellar distance d of parallel (red filled circles) and perpendicular (red opencircles) aligned lamellae with respect to the electric-field lines on the electric-fieldstrength for the same solution. c, Schematic diagram of the proposed chainstretching effect for lamellae aligned parallel to the field lines.
the polymer chain. For the overall process to be energeticallyfavourable, the decrease in the electric energy associated with themonomer reorientation has to compensate the energetic penaltyfor the chain stretching. The electric energy could be decreasedby two different effects: anisotropic polarizability of the polymerand reorientation of permanent dipole moments of the chainmonomers. Owing to its symmetry and rotational freedom, we donot expect the phenyl side group to have a net influence on thechain orientation. Hence, we expect a stronger response of the PIchains because of the high polarizability and dipole moment of the1,4-polyisoprene backbone C=C double bonds27.
In the following, we consider the gain in electric energycompensating the energetic penalty for the resulting chainstretching. We observe a decrease of the lamella thickness by1.1 nm due to the electric field. The length of one PI monomeris 6.8 A (ref. 28). As the monomers can be reoriented from twodifferent orientations perpendicular to the electric-field lines (in-plane and out-of-plane with respect to the interface), we canestimate that four PI monomers have to change their orientationfrom perpendicular to the field lines to parallel to the field to causethe observed decrease in the lamellae thickness. The change ofelectric energy associated with this reorientation is given by the gainin polarizability. Per PI chain, we have
w = 1/2nαE2,
nature materials VOL 7 FEBRUARY 2008 www.nature.com/naturematerials 143
© 2008 Nature Publishing Group
LETTERS
c
0
0
2
4
6
2 4 6 8 10 12 14E (kV mm–1)
Δd (%
)
0
0
2
4
6
2 4 6 8 10 12 14E (kV mm–1)
E (kV mm–1)
Δd (%
)Δd
(%)
a
b
0 1 2 3 4 5 6 7 8 9 10 11 12 13
0
1
2
3
4
5
6
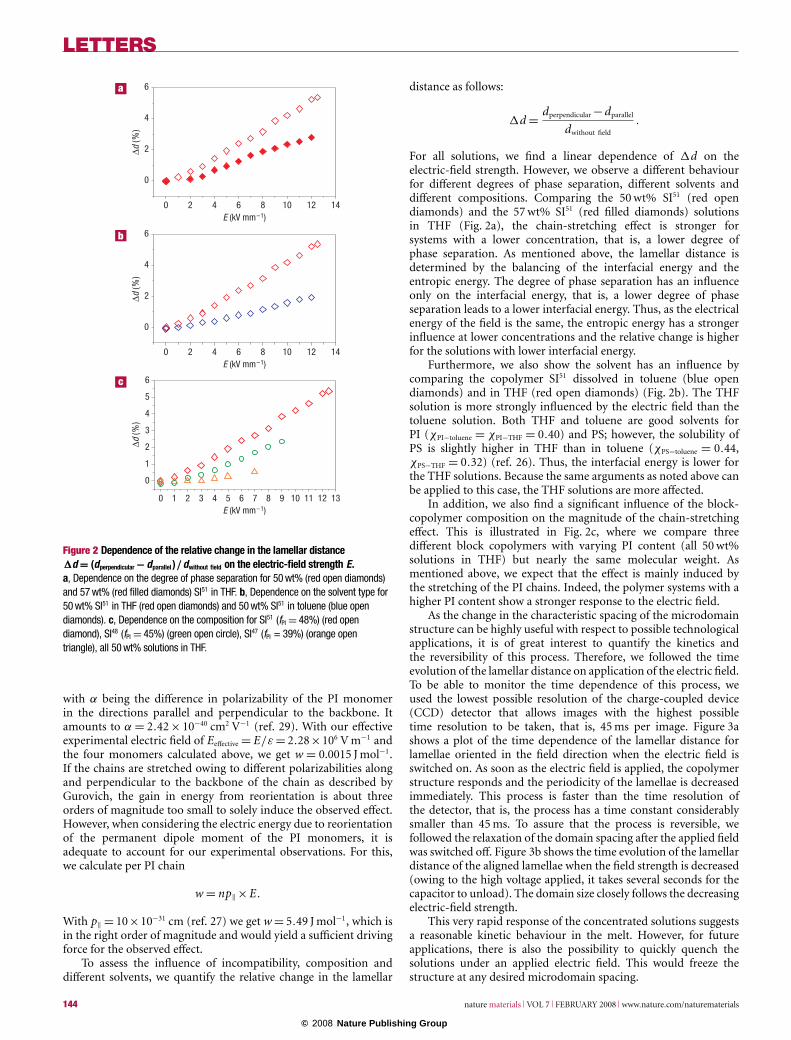
Figure 2 Dependence of the relative change in the lamellar distance1d = (dperpendicular − dparallel )/dwithout field on the electric-field strength E.a, Dependence on the degree of phase separation for 50 wt% (red open diamonds)and 57 wt% (red filled diamonds) SI51 in THF. b, Dependence on the solvent type for50 wt% SI51 in THF (red open diamonds) and 50 wt% SI51 in toluene (blue opendiamonds). c, Dependence on the composition for SI51 (fPI = 48%) (red opendiamond), SI48 (fPI = 45%) (green open circle), SI47 (fPI = 39%) (orange opentriangle), all 50 wt% solutions in THF.
with α being the difference in polarizability of the PI monomerin the directions parallel and perpendicular to the backbone. Itamounts to α = 2.42 × 10−40 cm2 V−1 (ref. 29). With our effectiveexperimental electric field of Eeffective = E/ε = 2.28×106 V m−1 andthe four monomers calculated above, we get w = 0.0015 J mol−1.If the chains are stretched owing to different polarizabilities alongand perpendicular to the backbone of the chain as described byGurovich, the gain in energy from reorientation is about threeorders of magnitude too small to solely induce the observed effect.However, when considering the electric energy due to reorientationof the permanent dipole moment of the PI monomers, it isadequate to account for our experimental observations. For this,we calculate per PI chain
w = np‖ ×E.
With p‖ = 10×10−31 cm (ref. 27) we get w = 5.49 J mol−1, which isin the right order of magnitude and would yield a sufficient drivingforce for the observed effect.
To assess the influence of incompatibility, composition anddifferent solvents, we quantify the relative change in the lamellar
distance as follows:
1d =dperpendicular −dparallel
dwithout field
.
For all solutions, we find a linear dependence of 1d on theelectric-field strength. However, we observe a different behaviourfor different degrees of phase separation, different solvents anddifferent compositions. Comparing the 50 wt% SI51 (red opendiamonds) and the 57 wt% SI51 (red filled diamonds) solutionsin THF (Fig. 2a), the chain-stretching effect is stronger forsystems with a lower concentration, that is, a lower degree ofphase separation. As mentioned above, the lamellar distance isdetermined by the balancing of the interfacial energy and theentropic energy. The degree of phase separation has an influenceonly on the interfacial energy, that is, a lower degree of phaseseparation leads to a lower interfacial energy. Thus, as the electricalenergy of the field is the same, the entropic energy has a strongerinfluence at lower concentrations and the relative change is higherfor the solutions with lower interfacial energy.
Furthermore, we also show the solvent has an influence bycomparing the copolymer SI51 dissolved in toluene (blue opendiamonds) and in THF (red open diamonds) (Fig. 2b). The THFsolution is more strongly influenced by the electric field than thetoluene solution. Both THF and toluene are good solvents forPI (χPI−toluene = χPI−THF = 0.40) and PS; however, the solubility ofPS is slightly higher in THF than in toluene (χPS−toluene = 0.44,χPS−THF = 0.32) (ref. 26). Thus, the interfacial energy is lower forthe THF solutions. Because the same arguments as noted above canbe applied to this case, the THF solutions are more affected.
In addition, we also find a significant influence of the block-copolymer composition on the magnitude of the chain-stretchingeffect. This is illustrated in Fig. 2c, where we compare threedifferent block copolymers with varying PI content (all 50 wt%solutions in THF) but nearly the same molecular weight. Asmentioned above, we expect that the effect is mainly induced bythe stretching of the PI chains. Indeed, the polymer systems with ahigher PI content show a stronger response to the electric field.
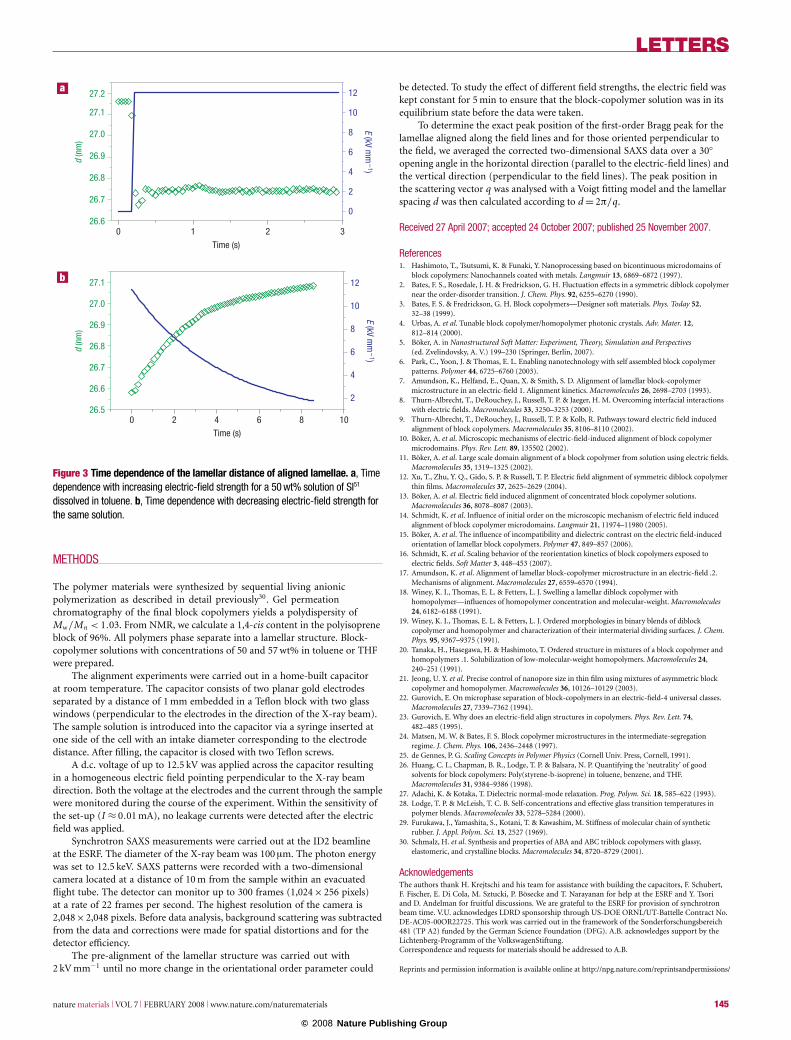
As the change in the characteristic spacing of the microdomainstructure can be highly useful with respect to possible technologicalapplications, it is of great interest to quantify the kinetics andthe reversibility of this process. Therefore, we followed the timeevolution of the lamellar distance on application of the electric field.To be able to monitor the time dependence of this process, weused the lowest possible resolution of the charge-coupled device(CCD) detector that allows images with the highest possibletime resolution to be taken, that is, 45 ms per image. Figure 3ashows a plot of the time dependence of the lamellar distance forlamellae oriented in the field direction when the electric field isswitched on. As soon as the electric field is applied, the copolymerstructure responds and the periodicity of the lamellae is decreasedimmediately. This process is faster than the time resolution ofthe detector, that is, the process has a time constant considerablysmaller than 45 ms. To assure that the process is reversible, wefollowed the relaxation of the domain spacing after the applied fieldwas switched off. Figure 3b shows the time evolution of the lamellardistance of the aligned lamellae when the field strength is decreased(owing to the high voltage applied, it takes several seconds for thecapacitor to unload). The domain size closely follows the decreasingelectric-field strength.
This very rapid response of the concentrated solutions suggestsa reasonable kinetic behaviour in the melt. However, for futureapplications, there is also the possibility to quickly quench thesolutions under an applied electric field. This would freeze thestructure at any desired microdomain spacing.
144 nature materials VOL 7 FEBRUARY 2008 www.nature.com/naturematerials
© 2008 Nature Publishing Group
LETTERS
27.2
26.6
26.7
26.8
26.9
27.0
27.1
E (kV mm
–1)
d (n
m)
26.6
26.5
26.7
26.8
26.9
27.0
27.1
d (n
m)
a 12
10
8
6
4
2
0
E (kV mm
–1)
12
10
8
6
4
2
0
0 2 4 6 8 10
1 2
Time (s)
Time (s)
3
b
Figure 3 Time dependence of the lamellar distance of aligned lamellae. a, Timedependence with increasing electric-field strength for a 50 wt% solution of SI51
dissolved in toluene. b, Time dependence with decreasing electric-field strength forthe same solution.
METHODS
The polymer materials were synthesized by sequential living anionicpolymerization as described in detail previously30. Gel permeationchromatography of the final block copolymers yields a polydispersity ofMw/Mn < 1.03. From NMR, we calculate a 1,4-cis content in the polyisopreneblock of 96%. All polymers phase separate into a lamellar structure. Block-copolymer solutions with concentrations of 50 and 57 wt% in toluene or THFwere prepared.
The alignment experiments were carried out in a home-built capacitorat room temperature. The capacitor consists of two planar gold electrodesseparated by a distance of 1 mm embedded in a Teflon block with two glasswindows (perpendicular to the electrodes in the direction of the X-ray beam).The sample solution is introduced into the capacitor via a syringe inserted atone side of the cell with an intake diameter corresponding to the electrodedistance. After filling, the capacitor is closed with two Teflon screws.
A d.c. voltage of up to 12.5 kV was applied across the capacitor resultingin a homogeneous electric field pointing perpendicular to the X-ray beamdirection. Both the voltage at the electrodes and the current through the samplewere monitored during the course of the experiment. Within the sensitivity ofthe set-up (I ≈ 0.01 mA), no leakage currents were detected after the electricfield was applied.
Synchrotron SAXS measurements were carried out at the ID2 beamlineat the ESRF. The diameter of the X-ray beam was 100 µm. The photon energywas set to 12.5 keV. SAXS patterns were recorded with a two-dimensionalcamera located at a distance of 10 m from the sample within an evacuatedflight tube. The detector can monitor up to 300 frames (1,024×256 pixels)at a rate of 22 frames per second. The highest resolution of the camera is2,048×2,048 pixels. Before data analysis, background scattering was subtractedfrom the data and corrections were made for spatial distortions and for thedetector efficiency.
The pre-alignment of the lamellar structure was carried out with2 kV mm−1 until no more change in the orientational order parameter could
be detected. To study the effect of different field strengths, the electric field waskept constant for 5 min to ensure that the block-copolymer solution was in itsequilibrium state before the data were taken.
To determine the exact peak position of the first-order Bragg peak for thelamellae aligned along the field lines and for those oriented perpendicular tothe field, we averaged the corrected two-dimensional SAXS data over a 30◦
opening angle in the horizontal direction (parallel to the electric-field lines) andthe vertical direction (perpendicular to the field lines). The peak position inthe scattering vector q was analysed with a Voigt fitting model and the lamellarspacing d was then calculated according to d = 2π/q.
Received 27 April 2007; accepted 24 October 2007; published 25 November 2007.
References1. Hashimoto, T., Tsutsumi, K. & Funaki, Y. Nanoprocessing based on bicontinuous microdomains of
block copolymers: Nanochannels coated with metals. Langmuir 13, 6869–6872 (1997).2. Bates, F. S., Rosedale, J. H. & Fredrickson, G. H. Fluctuation effects in a symmetric diblock copolymer
near the order-disorder transition. J. Chem. Phys. 92, 6255–6270 (1990).3. Bates, F. S. & Fredrickson, G. H. Block copolymers—Designer soft materials. Phys. Today 52,
32–38 (1999).4. Urbas, A. et al. Tunable block copolymer/homopolymer photonic crystals. Adv. Mater. 12,
812–814 (2000).5. Boker, A. in Nanostructured Soft Matter: Experiment, Theory, Simulation and Perspectives
(ed. Zvelindovsky, A. V.) 199–230 (Springer, Berlin, 2007).6. Park, C., Yoon, J. & Thomas, E. L. Enabling nanotechnology with self assembled block copolymer
patterns. Polymer 44, 6725–6760 (2003).7. Amundson, K., Helfand, E., Quan, X. & Smith, S. D. Alignment of lamellar block-copolymer
microstructure in an electric-field 1. Alignment kinetics. Macromolecules 26, 2698–2703 (1993).8. Thurn-Albrecht, T., DeRouchey, J., Russell, T. P. & Jaeger, H. M. Overcoming interfacial interactions
with electric fields. Macromolecules 33, 3250–3253 (2000).9. Thurn-Albrecht, T., DeRouchey, J., Russell, T. P. & Kolb, R. Pathways toward electric field induced
alignment of block copolymers. Macromolecules 35, 8106–8110 (2002).10. Boker, A. et al. Microscopic mechanisms of electric-field-induced alignment of block copolymer
microdomains. Phys. Rev. Lett. 89, 135502 (2002).11. Boker, A. et al. Large scale domain alignment of a block copolymer from solution using electric fields.
Macromolecules 35, 1319–1325 (2002).12. Xu, T., Zhu, Y. Q., Gido, S. P. & Russell, T. P. Electric field alignment of symmetric diblock copolymer
thin films. Macromolecules 37, 2625–2629 (2004).13. Boker, A. et al. Electric field induced alignment of concentrated block copolymer solutions.
Macromolecules 36, 8078–8087 (2003).14. Schmidt, K. et al. Influence of initial order on the microscopic mechanism of electric field induced
alignment of block copolymer microdomains. Langmuir 21, 11974–11980 (2005).15. Boker, A. et al. The influence of incompatibility and dielectric contrast on the electric field-induced
orientation of lamellar block copolymers. Polymer 47, 849–857 (2006).16. Schmidt, K. et al. Scaling behavior of the reorientation kinetics of block copolymers exposed to
electric fields. Soft Matter 3, 448–453 (2007).17. Amundson, K. et al. Alignment of lamellar block-copolymer microstructure in an electric-field .2.
Mechanisms of alignment. Macromolecules 27, 6559–6570 (1994).18. Winey, K. I., Thomas, E. L. & Fetters, L. J. Swelling a lamellar diblock copolymer with
homopolymer—influences of homopolymer concentration and molecular-weight. Macromolecules24, 6182–6188 (1991).
19. Winey, K. I., Thomas, E. L. & Fetters, L. J. Ordered morphologies in binary blends of diblockcopolymer and homopolymer and characterization of their intermaterial dividing surfaces. J. Chem.Phys. 95, 9367–9375 (1991).
20. Tanaka, H., Hasegawa, H. & Hashimoto, T. Ordered structure in mixtures of a block copolymer andhomopolymers .1. Solubilization of low-molecular-weight homopolymers. Macromolecules 24,240–251 (1991).
21. Jeong, U. Y. et al. Precise control of nanopore size in thin film using mixtures of asymmetric blockcopolymer and homopolymer. Macromolecules 36, 10126–10129 (2003).
22. Gurovich, E. On microphase separation of block-copolymers in an electric-field-4 universal classes.Macromolecules 27, 7339–7362 (1994).
23. Gurovich, E. Why does an electric-field align structures in copolymers. Phys. Rev. Lett. 74,482–485 (1995).
24. Matsen, M. W. & Bates, F. S. Block copolymer microstructures in the intermediate-segregationregime. J. Chem. Phys. 106, 2436–2448 (1997).
25. de Gennes, P. G. Scaling Concepts in Polymer Physics (Cornell Univ. Press, Cornell, 1991).26. Huang, C. I., Chapman, B. R., Lodge, T. P. & Balsara, N. P. Quantifying the ‘neutrality’ of good
solvents for block copolymers: Poly(styrene-b-isoprene) in toluene, benzene, and THF.Macromolecules 31, 9384–9386 (1998).
27. Adachi, K. & Kotaka, T. Dielectric normal-mode relaxation. Prog. Polym. Sci. 18, 585–622 (1993).28. Lodge, T. P. & McLeish, T. C. B. Self-concentrations and effective glass transition temperatures in
polymer blends. Macromolecules 33, 5278–5284 (2000).29. Furukawa, J., Yamashita, S., Kotani, T. & Kawashim, M. Stiffness of molecular chain of synthetic
rubber. J. Appl. Polym. Sci. 13, 2527 (1969).30. Schmalz, H. et al. Synthesis and properties of ABA and ABC triblock copolymers with glassy,
elastomeric, and crystalline blocks. Macromolecules 34, 8720–8729 (2001).
AcknowledgementsThe authors thank H. Krejtschi and his team for assistance with building the capacitors, F. Schubert,F. Fischer, E. Di Cola, M. Sztucki, P. Bosecke and T. Narayanan for help at the ESRF and Y. Tsoriand D. Andelman for fruitful discussions. We are grateful to the ESRF for provision of synchrotronbeam time. V.U. acknowledges LDRD sponsorship through US-DOE ORNL/UT-Battelle Contract No.DE-AC05-00OR22725. This work was carried out in the framework of the Sonderforschungsbereich481 (TP A2) funded by the German Science Foundation (DFG). A.B. acknowledges support by theLichtenberg-Programm of the VolkswagenStiftung.Correspondence and requests for materials should be addressed to A.B.
Reprints and permission information is available online at http://npg.nature.com/reprintsandpermissions/
nature materials VOL 7 FEBRUARY 2008 www.nature.com/naturematerials 145
© 2008 Nature Publishing Group





























