Embed Size (px)
Citation preview

28 Z. Anal. Chem., Band 257, Heft 1 (1971)
4. Covell, D. F. : Anal. Chem. 31, 1785 (1959) ; cf. Z. Anal. Chem. 176, 126 (1960).
5. Geochimia, mineralogia i gene~itsheskie tipi mestorosh- denij redkich elementov, Moskow: Nauka 1964.
6. Gordeev, I. V., Kardashev, D. A., Malishev, A. V. : Ja- derno-fisitskie konstanti. Moskow: Gosatomizdat 1963.
7. Gunne, X. Z., Pelekis, L. L., Col.: Neitronoaktivazionnij analis, p. 5. Riga: Sinatne 1966.
8. Gussev, N. G., Mashkovitsh, V. P., Verbizkij, B. V. : Ra- dioaktivhie isotopi kak gamma-islutshatsli. Moskow: Atomisdat 1964.
9. H6gdahl, O.T.: Prec. Conf. Radiochem. Methods of analysis, vol. 1, p. 23. Vienna: IAEA 1964.
10. Lange, P. W. de, Wet, W. J. de, Venter, J. H. : Talanta 15, 1488 (1968).
11. Nesmejanov, A.N., Lapizkij, A.V., Rudenko, N. D.: Polutshenie radioaktivnich isotopov. Moskow: Goschi- mizdat 1954.
12. Osnovnie tsherti geochmia urana. Moskow 1963. 13. Polikarpov, Y., Kostadinov, K. : Internat. survey course
on economic and technical aspects of nuclear power, IAEA/NPH/SG, Vienna, 1--12 September 1969.
14. Turkowsky, C., St~rk, H.: Z. Anal. Chem. 2o.1, 205 (1966).
15. -- -- Born, H. J.: Radiochim. Acta 8, 27 (1967); cf. Z. Anal. Chem. 241, 286 (1968).
Dr. I. Kuleff Chem. Faculty, Univ. "Ohridsky" Boul. "Anion Iwanow" 1, Sofia, Bulgaria
Z. Anal. Chem. 257, 28--33 (1971) �9 by Springer-Verlag 1971
Versuche zur Anreicherung yon Edelmetallspuren in Reinstquecksilber durch partielles LSsen der Matrix
E. JACKWERTH und A. KULOX
Institut fiir Spektroehemie and angewandte Spektroskopie Dortmund
Eingegangen am 12. Mai 1971
Investigations on the Enrichment of Traces of Noble Metals in High.Purity Mercury by Partial Dissolution o] the Matrix. Trace impurities of gold and palladium in metallic mercury can be enriched in a simple way by partially dissolving the sample in nitric acid. Practically the whole trace content of the sample will be collected in the residue. Up to a t least 100 g Hg the quant i ty of the mercury sample has no influence on the trace enrichment. After the partial dissolution of the metal the enriched gold was determined photometrically with Rhodamine B as a reagent, Pd was determined as [Pdg4] 2- complex. For the analysis of metallic mercury containing 0.5 p p m of Au and 2 ppm of Pd the relative s tandard deviation is 0.046, respectively 0.037. The limit of detection was found to be a t 0.2 p p m for both the elements. Using this method, the enrichment of traces of silver in mercury is not possible.
Znsammen/assung. In Quecksilbermetall als Verunreinigung enthaltene Gold- und Palladiumspuren kSnnen in einfacher Weise dadurch angereichert werden, dab das eingewogene Quecksilber bis auf einen kleinen Rest in Salpeters~ure gelSst wird. Praktisch der gesamtc Spurengehalt der Analysenprobe befindet sieh danaeh im LSseriickstand. Bis zu mindestens 100 g Quecksflber ha t die HShe der Einwaage keinen Einflu$ auf die Anreicherung. Nach dem partiellen LSsen der lV[etallproben wurde das angereicherte Gold mit Rhodamin B, Palladium als [PdJt]~--Komplex photometrisch bestimmt. Fiir Queeksflbermetall mit 0,5 ppm Au bzw. 2 p p m Pd betrKgt die relative Standardabweichung 0,046 bzw. 0,037. Die Nachweisgrenze liegt fiir beide Elemente bei 0,2 ppm. Silberspuren kSnnen nach dieser Methode in Quecksilber nicht angereichert werden.
1. Einleitung Sorgf~ltig destilliertes Qneeksflbermetall ist im all- gemeinen so rein, da$ noeh vorhandene Spurenele- mente nur nach starker Anreicherung analy~isch
erfaBt werden kSnnen. Dabei ist das Problem der Abtrennung metalliseher Verunreinigungen mit Hilfe chemischer Verfahren bisher nut fiir solche Spuren befriedigend gelSst, die wesentlich unedler sind als

E. Jaekwerth und A. Kulok: Versuche zur Anreieherung yon Edelmetallspuren in Reinstqueeksflber 29
die Matrix Queeksflber [3,4,10]. Zur gemeinsamen Anreieherung der Edelmetalle dagegen ist die destilla- t i re Trennung yon t{auptbestandteil und Spuren die seit langem allein benutzte 1Vfethode.
Der Trenneffekt dieses Verfahrens h/ingt in starkem MaBe yon den Destillationsbedingungen ab, was man auch yon der Aufreinigung des Quecksilbers dureh Vakuumdestillation her weil~: Vor allem bei hoher Temperatur und Destfllationsgeschwindigkeit oder aber bei ungenfigendem Vakuum sowie bei vollst/~n- digem Verdampfen der Queeksilbermetall-Einwaage wird leicht ein Tell der Spuren yore Quecksflber mit- genommen und finder sich naeh der Destillation in der Vorlage [8,19]. Ffir die Edelmetalle wurde dies vor allem yon Riesenfeld und Haase untersucht [16]. Ein Vergleieh der sehr untersehiedlichen Dampfdrucke der Metalle ffihrte diese und andere Autoren zu der Auffassung [13], dab im wesentlichen mechanisches MitreiBen der Spuren die Ursache ffir einen aueh nach mehrmaliger Destillation in der Vorlage gefundenen Edelmetallgehalt sein mfisse. Entsprechend erh/~lt man aueh nur schleeht reproduzierbare Analysen- werte, wenn die Edelmetallspuren, wie fiblieh, durch schnelles Abdampfen des Quecksflbers im Rfickstand angereichert werden.
Eine auf solchen Analysenverfahren beruhende l%hl- interpretation yon Versuchsergebnissen ffihrte vor nicht ganz 50 Jahren zu der heftig diskutierten Annahme, man kSnne aus Quecksilber durch elektrisehe Entladungen Gold maehen [11,12,14]. Selbst Patente warden dazu erteilt [9].
Trotz der bekannten FehlermSglichkeiten wird die destillative Anreicherung auch heute noch zur Reinheitsprfifung angewandt, weil dieses Verfahren sehr einfach auch grSBere Metalleinwaagen abzutren- hen gestattet und well geeignete chemische Trenn- verfahren, die mehrere Edelmetallspuren gleichzeitig erfassen, bisher unbekannt sind. Auf die Schwierig- keiten, Spuren und Quecksilbermatrix mit I{ilfe chelatbildender Gruppenreagentien oder anderer charakteristischer Reaktionen zu trennen, haben wir bereits in einer frfiheren Arbeit hingewiesen [5]. Danach seheint eine Spurenabtrennung yon groBen Mengen Queeksflber am ehesten mit Verfahren mSg- lleh zu sein, bei denen der Hauptbestandteil in selek- river Reaktion umgesetzt wird. Die Entwicklung soleher Trennmethoden setzt jedoeh Untersehiede im Reaktionsverhalten von Spuren uncl Matrix voraus, die um so st/~rker ausgepr/~gt sein mfissen, je grSBer die Konzentrationsuntersehiede der Elemente im Analysenmaterial shad.
Im folgenden haben wit versueht, die Differenz in den Normalpotentialen der Edelmetalle mad des
Quecksilbers sowie das davon abh/~ngende Redox- verhalten der Elemente zur Spurenanreieherung aus- zunutzen.
2. Allgemeine Grundlagen des Trennverfahrens
Wird Queeksilbermetall, das andere Metalle in Form einer homogenen fifissigen Legierung gel6st enth/~lt, mit einem Oxydationsmittel wie Salpeters~ure angegriffen, so kann man erwarten, dab sich die Bestandteile der Probe in der Reihenfolge ansteigen- der Normalpotentiale in der S/~ure 16sen und ein gewisser Trenneffekt erzielt wird. Die Gfite der Trennung, insbesondere bei den inder Spurenanalyse vorkommenden groBen Konzentrationsuntersehieden yon Spur und Matrix, wird maBgebend dureh die Potentialuntersehiede zwischen den metallischen Elementen in Kontakt mit der umgebenden w/~Brigen L6sung ihrer Ionen bestimmt. Ffir die in dieser Arbeit untersuchten Elemente und Reaktionen sind die zugehSrigen Normalpotentiale in Tab. 1 zusam- mengestellt. Eine interessante Diskussion fiber das ehemisehe und elektrochemische Verhalten yon Amal- gamen ist in einer Arbeit yon Jangg [6] zu finden.
Enth/ilt Quecksilber Spuren eines edieren Metalls, etwa yon Palladium, so kann formal mit Hilfe der I~ernst-Gleichung ausgerechnet werden, dab sich das unedlere Quecksilber zun~chst allein in HI~O 3 auflSst und zwar so lange, his (unter st/indiger Erh6hung der Hg2+-Konzentration) das Potential des Palladiums erreicht ist. Entsprechend wiirde sieh bis dahin Pd ~+ aus der L5sung an Queeksilbermetall abseheiden. Tats/ichlieh ist dieser ffir eine saubere Trennung der 2 Elemente interessante Punkt der Potentialgleich- heir einer exakten Rechnung aus mehreren Grfinden nieht zug/~ngllch:
Im Verlaufe des L5sevorganges nimmt die Queek- silberionen-Konzentration st/~ndig zu und erreicht sehneU Werte, ffir die die Iqernst-Gleichung nur noch unter Zuhilfenahme yon Aktivit ~tskoeffizienten gfiltig ist. Da, je naeh S/iurekonzentration, neben Hg 2+- Ionen aueh Hg~ ~+ entsteht, sind solehe auf die hier vorliegende Problematik anwendbare Koeffizienten nut sehwierig zu erhalten. Ebenso unsicher ist jede Aussage fiber die Pd~+-Konzentration bzw. -Aktivit/~t w/~hrend des LSsens einer nur Spuren Palladium ent- haltenden Queeksilberprobe. Ffir den Nernst-Ansatz wiehtige GrSBen shad sehlieBlieh die Bindungs- parameter der Elemente sowie deren L6sliehkeit innerhalb der Metallproben. Sofern die in Queek- sflber enthaltenen Spuren Amalgame bilden, gleieh weleher Form, hat dies einen EinfluB auf das Poten- tial und dessen Verlauf w/ihrend des LSseprozesses.

30 Z. Anal. Chem., Band 257, Heft 1 (1971}
Die Amalgame jedes Spurenelements haben ein eigenes Potential, das yon denen der reinen Elemente abweieht, und d~s sich beim LSsen der Probe in Salpeters~ure in dem Mal~e ~ndert, in dem das Queek- sflber in LSsung geht und aus dem Gleiehgewieht der Amalgambildung herausgenommen wird. Noch kom- plizierter sind die Verh~Lltnisse bei Mehrmetall- amalgamen. Eine siehere Vorhersage fiber das Ver- halten der Spurenelemente beim AuflSsen der Matrix Queeksilber ist also wegen des Fehlens der dem Vor- gang zugrunde liegenden Potentialwerte heute praktiseh unm5glieh.
Da die Differenzen zwisehen dem Potential des Queeksilbers einerseits und denen yon Palladium, Platin und Gold andererseits sehr gro~ sind, k~nn man jedoeh eine Anreieherung der Spuren dureh partielles LSsen auch grS~erer Einwaagen yon Queek- sflber erwarten. Dagegen sollte Sflber als unedleres Element auf diese einfaehe Weise nieht anzureiehern sein.
3. HersteUung yon Quecksilberproben mit definiertem Spurengehalt
Die Untersuchungen dieser Arbeit wurden an Proben mit definiertem Edelmetallgehalt durchgeffihrt. Dazu wurden Matrix und Spur gemeinsam als Metalle dutch Zusatz eines Reduktionsmittels aus der LSsung ihrer Salze ausgef~llt. Das dureh einen solehen F~llungs- prozeB gewonnene Probenmaterial ist ffir systema- tisehe und vergleiehende analytische Untersuehungen vor ahem dann verwertbar, wenn die Metallspuren in der Quecksflbermatrix homogen 15slieh sind. Von Sflber und Gold ist die LSsliehkeit im Vergleieh zu anderen Met~llen relativ grol~ und deshalb seit lan- gem zur teehnisehen Gewinnung dieser EdelmetaUe aus Erzen und Konzentraten ausgenutzt worden. Sie betr~gt bei Raumtempera~ur naeh Literaturangaben 0,131 Gew.-~ Au bzw. 0,035 Gew.-~ Ag [19], wobei die LSsliehkeit bei TemperaturerhShung erheblich ansteigt. Die Platinmetalle dagegen 15sen sieh nur sehr wenig. Palladium etwa bildet in grS~erem Umfang erst beim Erhitzen mit Hg flfissige undfesteAmalgame [2], und besonders beim Platin ist Amalgambildung nur unter besonderen Bedingungen z.B. durch Elektrolyse erreichbar [15].
Um einer sp&teren Entmisehung der Elemente im Probenmaterial vorzubeugen, haben wir die Reduktion der Metallsalze erst unmittelbar vet Ansetzen jeder Versuehsreihe und ffir jede Quecksflbereinwaage gesondert durehgefiihrt.
Ffir die Reduktion der Nitrate des 1- und 2 werti- gen Queeksilbers ist u. a. Ameisens~ure gut geeigne~
[10]. HMogenid in mehr als Spurenmengen st6rt jedoch in der Weise, dab das ausgef~llte, zun~chst rein verteflte Metall gar nicht oder mlr unvollst~ndig zu einem Tropfen zusammenflieBt. Zusammen mit Queeksflber werden auch Au, Pd und Pt metallisch ausgef~llt; Ag dagegen f~llt als Spurenelement mit Ameisens~ure nur teflweise und zu wenig reprodu- zierbaren Anteflen aus. Verwendet man Aseorbin- s~ure als Reduktionsmittel, so erreicht man auch beim Sflber die gewiinschte quantitative Mitf~llung, was dureh Analyse der zuriickbleibenden LSsungen kontrolliert wurde. Allerdings ffihren Halogenid- spuren in den LSsungen zur Ausf~llung des Silbers als Sflberhalogenid, das bei der anschlieBenden Analyse nur zu einem sehr geringen Tell erfaBt wird.
Zur Herstellung yon 10 g-Quecksilberproben wurde folgende Arbeitsvorschrift benutzt:
Einwaagen yon 14 g Hg~(N03) ~ �9 2 H~O werden in 250 ml- Beehergl~sern in je 100 ml Wasser unter Zusatz yon 2 ml 65~ Salpeters~ure gelSst. Entspreehend dem ge- wiinschten Spurengehalt wird die aus einer StammlSsung friseh verdfinnte, m5gliehst wenig Chlorid enthaltende Edel- metall-LSsung dosiert. Unter Erw~rmen der LSsung wird das Metall durch Zusatz von insgesamt etwa 15 ml Ameisen- s~ure (98--100~ ausgefEllt, wobei die Reduktionsmittel- zugabe der Heftigkeit der Reaktion angepaflt wird. Zur Her- stellung yon silberhaltigen Quecksilberproben wird Ascorbin- sEure in fester Form als Reduktionsmittel benutzt (s. o.). Das zunEehst feinverteilt ausfallende Metall sammelt sieh beim Kochen der L6sung am Boden des Gef~Bes. Dutch Dekantieren und Wasehen wird das zusammengeflossene Queeksilber veto iiberschiissigen Reduktionsmittel getrennt.
Zur Reduktion grSBerer oder kleinerer Probenmengen als bier angegeben werden die in der Arbeitsvorsehrift ver- wendeten Reagenszus~tze entsprechend ver~ndert.
Das nach der obigen Vorschrift erhaltene Queek- sflber mit Spuren Ag, Pd und Au ist vSllig blank; lediglieh zugesetztes Platin lagert sich schon bei der Reduktion der Metalle als grauer Film auf der Ober- fl~che des Quecksflbers ab. Durch ~berprfifung mit dem Radioisotop 197p~ wurde fes~gestellt, dab in diesem Belag praktisch alles eingesetzte Platin (50 ~g) enthalten ist; in Quecksilber gelSst ist weniger als 1 ~ des Zusatzes. Es wurde night n~her unter- sueht, ob das Platin als reines Metal] oder als Amalgam auf der Quecksilberoberfl~ehe abgelagert wird; gltere Literaturangaben lassen eher das letztere vermuten [i7].
Die an solehen inhomogenen Hg/Pt-Proben entspr. Kap. 4 durehgeffihrten Versuehe zur Platinanreiehe- rung ergaben bisher vSllig unreproduzierbare Ergeb- nisse. Auf weitere Untersuehungen mit Platin als Spurenelement wurde in dieser Arbeit deshalb zu- n~ehst verzich~et.
E. Jaekwerth und A. Kulok: Versuche zur Anreieherung yon Edelme~allspuren in Reinstqueeksilber 31
4. Versuche zur Anre icherung yon Au, Pd und A g dutch part iel les L~isen der Quecksilberproben
4.1. Versuche mit A u und Pd
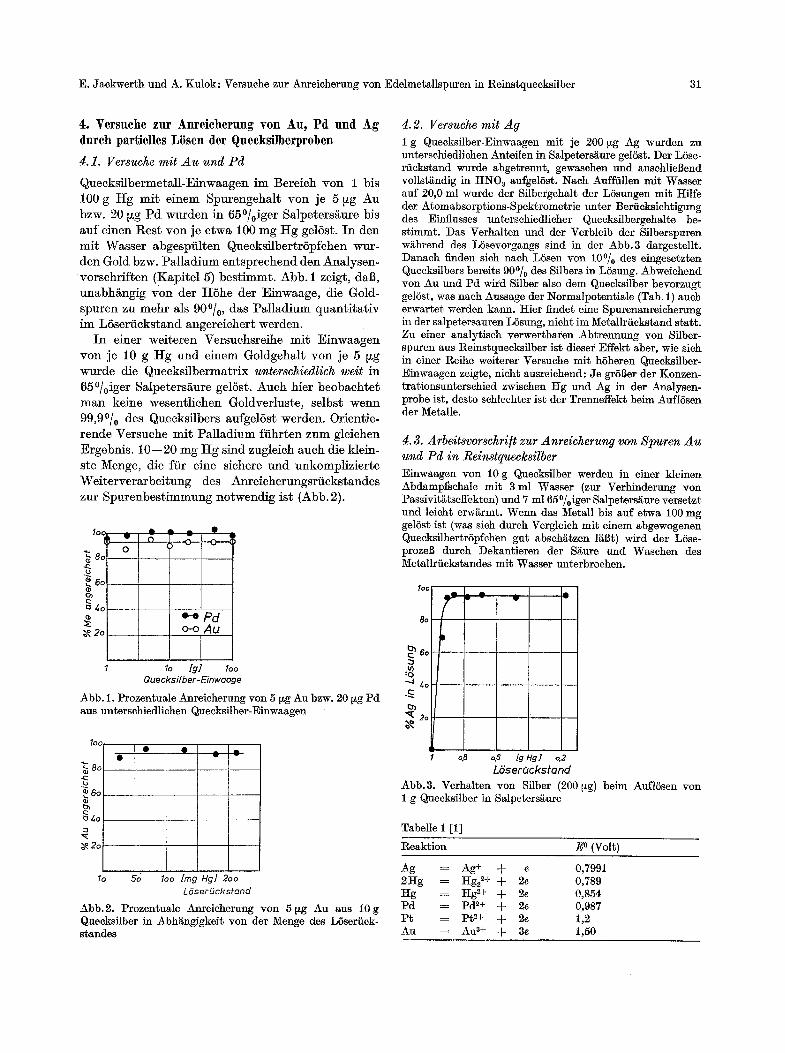
Quecks i lbe rmeta l l -E inwaagen im Bereich yon 1 bis 100 g H g m i t e inem Slourengehalt yon je 5 ~g Au bzw. 20 ~g P d wurden in 65~ Salper bis auf e inen Res t yon je e twa 100 mg H g gelSst. I n den m i t Wasse r abgesl~filten Quecks i lber t rSpfchen wur- den Gold bzw. P a l l a d i u m en t sp rechend den Analysen- vorschrff ten (Kaloitel 5) bes t immt . Abb . 1 zeigt, daB, unabh~ngig yon der I IShe der Einwaage , die Gold- Slouren zu mehr als 90~ das P a l l a d i u m q u a n t i t a t i v im L6ser f icks tand angere icher t werden.
I n einer wei te ren Versuchsreihe m i t E inwaagen yon je 10 g t t g und e inem Goldgeha l t yon je 5 ~zg wurde die Quecks i lbe rmat r ix unterschiedlich welt in 65~ Salloeters/iure gelSst. Auch hier beobachte~ m a n keine wesent l ichen Goldver lus te , selbst werm 99,90/0 des Quecksflbers aufgel6st werden. Orient ie- rende Versuche m i t P a l l a d i u m ff ihr ten zum gleichen Ergebnis . 10- -20 mg t t g s ind zugleich auch die klein- s te Menge, die ftir eine sichere und unkompl iz ie r t e W e i t e r v e r a r b e i t u n g des Anre icherungsr t i cks tandes zur Spu renbes t immung no twendig is t (Abb. 2).
1o~
8o L)
"~ 6O
C
N 20
0
Pd c-o Au
1o [g] 1oo Quecksilber-Einwaage
Abb. 1. Prozen~uale Anreieherung yon 5 ~g Au bzw. 20 ~zg Pd aus unterschiedliehen Queeksilber-Einwaagen
~ 8o
,<
20
1o 50
w v
1oo [mg Hg] 200 L 6 s e r C ~ c k s t a n d
Abb.2. Prozentuale Anreicherung yon 5 {zg Au aus 10 g Queeksilber in Abh~ngigkei~ yon der 5{enge des LSseriick- s~andes
4.2. Versuche mit Ag
I g Quecksilber-Einwaagen mit je 200 t~g Ag wurden zu unterschiedlichen Anteilen in Salpeters~ure gelSst Der LSse- rfickstand wurde abgetrennt, gewasehen und ansehlieBend vollst~ndig in H~q03 aufge15sk 5Tach Aufffillen mit Wasser auf 20,0 ml wurde der Silbergehalt der LSsungen mi~ Hilfe der A~omabsorptions-Spek~rometrie unter Beriicksichtigung des Einflusses un~ersehiedlicher Quecksilbergehalte be- s~imm~. Das Verhalten und der Verbleib der Silberspuren w~hrend des LSsevorgangs sind in der Abb.3 dargestellt. Danach finden sich nach LSsen yon 10~ des eingesetzten Quecksilbers bereits 900/0 des Silbers in LSsung. Abweiehend yon Au und Pd wird Silber also dem Quecksilber bevorzugt gelSst, was nach Aussage der ~Tormalpotentiale (Tab. 1) aueh erwartet werden kann. Hier finder eine Spurenanreicherung in der salpe~ersauren LSsung, nicht im Metallrfickstand start. Zu einer analytisch verwertbaren Abtrennung yon Silber- spuren aus Reinstqueeksilber is$ dieser Effekt aber, wie sieh in einer Reihe weiterer Versuehe mit hSheren Queeksilber- Einwa~gen zeigte, nieht ausreiehend: Je grSBer der Konzen- trationsunterschied zwischen t tg und Ag in der Analysen- probe ist, desto schlechter ist der Trenneffek~ beim AuflSsen der Metalle.
4.3. Arbeitsvorschri/t zur Anreicherung von Spuren A u uncl Pd in Reinstquecksilber
Einwaagen yon 10 g Quecksilber werden in einer k]einen Abdampfschale mi~ 3 ml Wasser (zur Verhinderung yon Passivit~tseffek~en) und 7 m165 ~ Salpeters~ure versetzt und leicht erw~rmt. Wenn das Metall bis auf etwa 100 mg gelSs~ ist (was sich durch Vergleich mi~ einem abgewogenen QueeksilbertrSpfchen gut absehi~tzen liil~t) wird der LSse- prozeB durch Dekantieren der Sgure und Waschen des Metallrfickstandes mit Wasser unterbrochen.
1oc
8o
• 6o
.M 4o
"~2o
- w
o~ o,6 [gHgl 0,2 L6sarQckstund
Abb.3. Verhalten yon Silber (200 ~tg) beim AuflSsen yon 1 g Quecksilber in Salpetersgure
Tabelle 1 [1]
Reaktion /~o (Volt)
Ag = Ag+ + e 0,7991 2Hg = Hg2 ~+ -t- 2e 0,789 Hg = t { g 2+ -]- 2e 0,854 Pd = Pd ~+ + 2e 0,987 p~ : p~2+ + 2e 1,2 Au = Au 3+ + 3e 1,50

32 Z. Anal. Chem., Band 257, Heft 1 (1971)
W/ihrend des Anreieherungsvorgangs ist unbedingt darauf zu achten, dab das eingewogene Quecksilber zusammen- h~ngend ,,als ein Tropfen" gelSst wird. Vorhandene kleine lY[etalltr6pfehen werden andernfalls vorzeitig aufgel6st, wo- bei die enthattenen Edelmetallspuren u. U. nieht mehr voll- st~indig an noch vorhandenem ungelSstem Quecksilbermetall abgesehieden werden.
Im Riiekstand angereiehertes Gold und Palladium werden photometrisch (Kap. 5.1) bestimmt.
5. Bestimmung yon Au und Pd im Spurenkonzentrat
Die Zahl der in der Li teratur zur photometrisehen Best immung yon Gold und Palladium in Gegenwart fibersehiissigen Queeksflbers zur Veffiigung stehenden Analysenverfahren ist sehr gering. Ursache dafiir ist im allgemeinen die bier unerwfinschte Komplex- bfldung des Queeksflbers mit den f/Jr die Photo- metrie der Edelmetallspuren gebr/iuchlichen Reagen- tien, wodurch die eharakteristische Reakt ion des Goldes bzw. Palladiums in den meisten F/~llen vSllig fiberdeekt wird. Auch bei den nach zahlreichen Vor- versuchen zur Analyse des Spurenkonzentrats aus- gew~hlten Best immungsmethoden (Goldbestimmung mit Rhodamin B, Best immung des Palladiums als [PdJ4]a--Komplex) muB ein gewisser stSrender Ein- flul3 des Quecksilbers in K a u f genommen werden. Beide Verfahren kSnnen deshalb nur als NotlSsung angesehen werden.
5.1. Bes t immung yon A u mi t Rhodamin B [18]
[AuC14]- reagiert, wie zahlreiche andere Chloro- metallat-Anionen, mi t Rhodamin ]3 zu einer extra- hierbaren roten Komplexverbindung, die bei 565 nm absorbiert. Die Molarextinktion des Diisopropyl- ~ther-Extraktes betr~gt bei dieser Wellenl/~nge 9,7 �9 1041. Mo1-1 �9 em -1. Da [HgC14] 2- den analogen Farbkomplex in geringem MaBe ebenfalls bfldet, bewirkt Quecksilber eine BlindwerterhShung bei der Goldbestimmung. Zur Korrek tur der Extinktions- mel]werte des Goldes in queeksflberhaltiger L6sung wurde die Kurve der Abb. 4 verwendet, die an gold- freien LSsungen mit ansteigender Hg2+-Konzentration ermittel t wurde. Die Korrek tur macht es notwendig, die bei der Anreieherung erhaltenen Hg-TrSpfchen zu wi~gen. Die der Queeksflberauswaage entspreehende Ext inkt ion wird bei der naehfolgenden Gold- best immung abgezogen.
Einen EinfluB auf die Bildung des Gold-Rhodamin- komplexes und auf das Verhalten des Queeksilbers bei der Goldbestimmung haben Aeidit/~t und Chlorid- ionenkonzentration der w/iBrigen Phase : Bei geringen HC1- und NH~C1-Konzentrationen f/~llt Quecksflber in Form einer nieht extrahierbaren blauvioletten
0.2,. E
o,15
o, lo
/ f
50 1oo 15o [mgitg] 250
Abb.4. Diagramm zur Korrektur der Gold-Extinktionen bei der Bestimmung in quecksilberhaltigen LSsungen
Rhodaminverbindung aus, die einen Teil des Goldes einsehliegt. Oberhalb best immter HC1- und NtI4C1- Konzentrat ionen bfldet sich der rote Farbkomplex des Goldes dagegen nur unvollsti~ndig, und man verliert an Empfindliehkeit bei der photometrisehen Bestimmung. Ms optimal haben sieh LSsungen er- wiesen, die 1 M an HC1 sowie etwa 2,5 M an NHaC1 sind. Unter diesen Bedingungen kann Gold bei entsprechender Korrektur der Extinktionswerte noch neben mehreren 100 mg Hg *+ best immt werden.
Arbeitsvorschri/t. Der gewogene aus etwa 100 mg Hg be- stehende Anreieherungsr/iekstand (s. Kap. 4.3) wird in 2,5 ml 65~ Salpeters~ure unter Zusatz yon 1 ml Wasser gelSst. Die L6sung wird zur Trockne eingedampft, der Riiek- stand wird mit 5 ml K6nigswasser aufgenommen und erneut auf etwa 0,5 ml eingeengt. Der beim Abkiihlen entstehende Kristallbrei wird mit 4,2 ml 6 M Salzs~ure und 10 ml ges~tt. Ammoniumehloridl6sung in einen Sch/itteltriehter iiberfiihrt und die L6sung mit Wasser auf 20 ml erg~nz$. Naeh Zusatz yon 5 ml 0,04~ w~Briger Rhodamin B-LSsung wird mit 10,0 ml Diisopropyl~ther 1 rain extrahiert. Die Extinktion der organischen Phase wird gegen das Extraktionsmittel als Vergleich gemessen (Zeiss-ELKO II, Filter Hg 546, 1 em- Kiivetten). Die MeBwerte werden mit Hilfe des Diagramms der Abb. 4 korrigiert.
5.2. Beatimmung yon Pd als [ P d J j ~- [18]
Pd ~+ bildet mit iiberschiissigem Jodid den roten Farb- komplex [PdJa] 2- mit Absorptionsmaxima bei 408 und 490 nm. In Gegenwart yon Quecksilber ist die Messung bei 408 nm wegen der gleichzeitigen Absorp- tion dureh [HgJa] 2- an dieser Stelle gest5rt. Bei 4 9 0 n m absorbiert der Tetrajodomercuratkomplex bis zu mindestens 50 mg/ml Hg ~+ jedoch nicht. Aller- dings ist die Empfindlichkeit der Palladiumbestim- mung gegenfiber lYIessungen bei 408 nm um den Fak tor 2,5 kleiner (Molarextinktion bei 490 nm: 4,3 �9 10 a 1 �9 Mo1-1 �9 cm-1).
Arbeitsvorschri/t. Der entspreehend derVorsehrift (Kap.5.1) nach der KSnigswasserbehandlung erhaltene Kristallbrei wird mit 5 ml 8 IV[ l~atriumjodidlSsung sowie 10 ml 25~

E. Jackwerth und A. Kulok: Versuehe zur Anreicherung yon Edelmetallspuren in Reinstquecksilber 33
AscorbinsgurelSsung versetzt und die LSsung mit Wasser auf 20,0 ml aufgefiillt. Die Extinktion wird gegen einen ent- sprechend behandelten Blindwert gemessen (Zeiss-ELKO II, Filter S 49, 5 cm-K/ivetten).
5.3. Statistische Daten [7]
Die relative Standardabweiehung des gesamten Anreieherungs- und Best immungsverfahrens wurde unter Verwenden yon Probenmater ia l mi t 0,5 ppm Au bzw. 2 ppm Pd ermittelt . Aus je 10 Analysen an 10 g Einwaagen Quecksilber ergeben sich Werte yon 0,046 ffir Au sowie 0,037 f/Jr Pd.
Die aus den Blindwerts t reuungen yon 25 Einwaagen mit je 10 g Reinstquecksilber errechnete Nachweis- grenze liegt ffir beide Elemente bei c ---- 0,2 ppm.
Wir danken tterrn Prof. Dr. H. Nickel sowie Herrn Dr. J. Rottmann, Kernforschungsanlage Jiilich, ffir ihre Hilfe bei radiochemischen Messungen.
L i t e r a t u r
1. Genshaw, M. A. : Electrochemical measurement. In: R. A. Rapp (Edit.): Physicochemieal measurements in metal research, Part 2. New York: Intersci. Publ. 1970.
2. Gillespie, L. J., Galstaun, L. S.: J. Am. Chem. Soc. 58, 2569 (1936).
3. gaekwerth, E. : diese Z. 202, 81 (1964). 4. -- diese Z. 206, 269 (1964). 5. -- Lohmar, J.: diese Z. 251, 353 (1970). 6. gangg, G.: Nfetall 19, 442 (1965). 7. Kaiser, H., Specker, H. : diese Z. 149, 46 (1956). 8. Kraus, R. : Angew. Chem. 50, 597 (1937); vgl. diese Z.
120, 33 (1940). 9. Nfachu, W.: 0sterr. Chem. Z. 41, 376 (1938).
10. Meyer, J. : diese Z. 219, 147 (1966). 11. YIiethe, A. : Naturwissenschaften 12, 597 (1924). 12. -- Naturwissenschaften 18, 635 (1925). 13. -- Stammreich, It.: Z. Anorg. Allg. Chem. 149, 263
(1925). 14. -- -- Z. Anorg. Allg. Chem. 158, 185 (1926). 15. Plaksin, I. N., Souvorovskaja, N. A. : C. R. Acad. USSR
[2] 27, 460 (1940). 16. Riesenfeld, E. I~., Haase, W.: Naturwissenschaften 18,
745 (1925). 17. Saha, H., Choudhury, K. N. : Z. Anorg. Allg. Chem. 86,
228 (1914). 18. Sandell, E. B.: Colorimetrie determination of traces of
metals. New York: Intersci. Publ. 1959, 19. Ullmanns EncyklopEdie d. techn. Chemic, 3. Aufl.
Miinchen-Berlin: Urban & Schwarzenberg 1963. 20. Venator, W. : Z. Angew. Chem. 89, 229 (1926).
Priv.-Doz. Dr. E. Jaekwerth Institut ffir Spektrochemie und angewandte Spektroskopie D-4600 Dortmund, Bunsen-Kirchhoff-StraBe 11 Deutschland
Z. Anal. Chem. 257, 33--36 (1971) �9 by Springer-Verlag 1971
Spectrophotometric Determination of Nickel(II), Palladium(II) and Copper(II) in Presence of Each Other and Other Ions with 1-(o-Carboxyphenyl)-3-hydroxy-3-phenyhriazene
A. K. I-~AJUMDAR and D. CHAKRABORTI
Department of Inorganic and Analytical Chemistry, Jadavpur University, Calcutta-32, India
Received May 24, 1971
Spektrophotometrische Bestimmung yon Nickel(II), Palladium(II) und Kup]er(II) nebeneinander und in Gegenwart anderer lonen mit Hil/e yon 1-(o-Carboxyphenyl)-3-hydroxy-3-phenyltriazen. Ni, Pd und Cu bilden mi t dem l~eagens bci p H 6,8--8,3, 2 ,4--3,5 bzw. 2,2--3,8 grfinlich-gelbe, gelblich-orange bzw. hellgr/ine Kom- plexe mi~ Absorpt ionsmaxima bei 410, 410 bzw. 400 nm. Das Beersche Gesetz wird in den Bereichen 0,25--2,0, 0 ,5--4,0 bzw. 0,5--4,0 ppm befolgt. Die Elemente bflden l : l - K o m p l e x e mit den Instabi l i t~tskonstanten 2,1 �9 10 -5, 1,5 �9 10 -5 bzw. 2 , 0 . 1 0 -5. Der Einflul3 anderer Kat ionen und Anionen wurde untersucht und ArbeiSs- vorsehrfften werden angegeben.
Summary. 1-(o-Carboxyphenyl)-3-hydroxy-3-phenyltr iazene was found to be an excellent spectrophotometr ie reagent for the determinat ion of nickel(II), pal ladium(II) and copper(II) . At p i t 6.8--8.3, 2 .4--3.5 and 2.2--3.8, nickel, pal ladium and copper form greenish yellow, yellowish orange and light green complexes with m a x i m u m
3 Z. Anal. Chem., Bd. 257





























