Embed Size (px)
Citation preview

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 1 von 14
Chronische immunvermittelte ZNS-Erkrankungen
Was gibt es Neues? • Für die Rasmussen-Enzephalitis existieren jetzt formale, auf einem europäischen Konsensus
basierende Diagnosekriterien (siehe Tab. 1). • In der Therapie der RE gewinnt „ vor“ der epilepsiechirurgisch begründeten Hemisphärektomie
bei „ ausgebrannten“ Fällen die langzeitige Immuntherapie zur Verbesserung des funktionellen Outcomes (Verhinderung der Hemisphärenatrophie und der damit assoziierten neurologischen Verschlechterung) an Bedeutung.
• Die limbische Enzephalitis (LE) wird zunehmend auch als nichtparaneoplastische Erkrankung beobachtet. In diesem Kontext wurden Autoantikörper gegen spannungsabhängige Kaliumkanäle (VKGC-Antikörper) beschrieben, die bei entsprechendem klinischem Bild die Diagnose LE belegen (hier üblicherweise mit guter Prognose) sowie eine nichtparaneoplastische Genese wahrscheinlich machen.
• Für die LE sind formelle Diagnosekriterien vorgeschlagen worden (siehe Tab. 3). • Für das früher „ Hashimoto-Enzephalopathie“ genannte Syndrom haben sich unter dem Begriff
„ Steroid-responsive Enzephalopathie mit assoziierter Autoimmun-Thyroiditis“ (SREAT) nunmehr formale diagnostische Kriterien etabliert.
• Die aggressive Therapie durch Kombination von Steroiden mit Immunsuppressiva (Methotrexat oder Azathioprin) scheint bei der Neurosarkoidose eine gute Langzeitprognose zu gewähren (allerdings gibt es dazu keine randomisierten kontrollierten Studien).
Die wichtigsten Empfehlungen auf einen Blick • Rasmussen-Enzephalitis: Eine frühzeitige Diagnose anhand der auf einem europäischen
Konsensus basierenden Diagnosekriterien sollte angestrebt werden, wenn anders (noch) nicht möglich, durch Hirnbiopsie. Ziel ist die möglichst frühzeitige Etablierung einer immuntherapeutischen Langzeittherapie.
• Limbische Enzephalitis: Bei klinisch-neuroradiologisch begründetem Verdacht ist 1. eine Tumorsuche und 2. eine Autoantikörperdiagnostik indiziert, um bei paraneoplastischen Formen (siehe Kapitel „ Paraneoplastische Syndrome“ ) frühzeitig eine Tumortherapie einleiten zu können und eine Prognoseabschätzung hinsichtlich der vermutlich zu erwartenden Wirksamkeit einer immuntherapeutischen Behandlung durchführen zu können.
• SREAT: Die Diagnose kann nach einem Kriterienkatalog verdachtsweise gestellt werden. Dies sollte zur Einleitung einer Steroidtherapie führen, deren Erfolg die Diagnose verbindlich bestätigt.
• Die Diagnose der Neurosarkoidose sollte wegen der notwendigen Langzeittherapie immer auf einer Histologie basieren; bei isoliertem ZNS-Befall gilt dies ganz besonders.
Definition In diesem Kapitel werden seltene und zum Teil erst kürzlich definierte Erkrankungen behandelt. Sie werden mit ausreichend hoher Wahrscheinlichkeit durch eine pathologische Interaktion von Elementen des Immunsystems mit dem ZNS verursacht, so dass ihre gemeinsame Behandlung unter diesem
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 2 von 14
Titel berechtigt erscheint. Häufigere und bereits seit längerem etablierte Entitäten (z. B. Multiple Sklerose oder paraneoplastische ZNS-Syndrome) werden in eigenen Kapiteln der Leitlinien behandelt. Pathogenetische Konzepte und diagnostische Kriterien für die hier behandelten Störungen und Syndrome etablieren sich in letzter Zeit. Es besteht aber noch keine belastbare Evidenz hinsichtlich der zu bevorzugenden Therapien. Allerdings können aus Fallstudien und aus pathogenetisch basierten Überlegungen vorläufige Therapieempfehlungen für diese beeinträchtigenden Erkrankungen abgeleitet werden.
Ziele und Anwendungsbereich Ziel dieser Leitlinie ist, Neurologen, Neuropädiatern und Psychiatern die behandelten Erkrankungen definitorisch-klassifikatorisch bekannt zu machen und mitzuteilen, welche diagnostischen und therapeutische Vorgehensweisen – vor dem Hintergrund des noch begrenzten Kenntnisstandes – als begründet und begründbar anzusehen sind.
Rasmussen-Enzephalitis
Definition Die Rasmussen-Enzephalitis (RE) ist eine durch zytotoxische, gegen Neurone und Astrozyten gerichtete T-Lymphozyten vermittelte Erkrankung (Bien et al. 2002a, Bauer et al. 2007), die fast immer nur eine Großhirnhemisphäre betrifft und diese in einem Monate bis Jahre dauernden Prozess in individuell unterschiedlichem Umfang zerstört. Klinisch ist die Erkrankung durch einen progredienten Verlust von Funktionen gekennzeichnet, die in der betroffenen Hemisphäre repräsentiert sind. Pharmakoresistente Anfälle, namentlich die Epilepsia partialis continua, sind typisch (Bien et al. 2002b). 85% der Betroffenen erkranken, bevor sie 10 Jahre alt sind (Oguni et al. 1992).
Untersuchungen Notwendig
• Neurologischer Status, vorzugsweise unter Verwendung einer quantitativen Motorikskala zur Dokumentation der Progredienz (z. B. Motricity Index nach Demeurisse; Demeurisse et al. 1980)
• Neuropsychologische Verlaufsdokumentation, namentlich der sprachlichen Funktionen bei Befall der (mutmaßlich) dominanten Hemisphäre
• Hirn-MRT und Verlaufsuntersuchungen zur Dokumentation der Progredienz der Hemiatrophia cerebri im Abstand von 2– 3 Monaten zu Erkrankungsbeginn, später in 6– 12-monatigen Abständen – zu diagnostischen Zwecken (Teil-B-Diagnosekriterien; Tab. 1) oder als Surrogatparameter für die Wirksamkeit immunsuppressiver Therapien (Bien et al. 2004)
• EEG-Diagnostik, ggf. Langzeit-Video-EEG-Monitoring (Teil-A-Diagnosekriterien; Tab. 1)
Im Einzelfall erforderlich
• Blut-/Liquoruntersuchungen zum Ausschluss von Differenzialdiagnosen (ausführliche Liste siehe Bien et al. 2005) sowie bei Immunsuppression zum Therapiemonitoring (in Abhängigkeit vom verwendeten Regime)
• Offene Hirnbiopsie bei frühen Verdachtsfällen, die die Teil-A-Diagnosekriterien (noch) nicht erfü
Archiv
- alte
Auflag
e
Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 3 von 14
llen (Tab. 1) • Wada-Test vor Indikation einer Hemisphärektomie
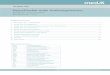
Tabelle 1 Diagnosekriterien für die Rasmussen-Enzephalitis
Teil A
1. Klinische Zeichen Fokale Anfälle (mit oder ohne Epilepsia partialis continua) und unilaterale kortikale Defizite
2. EEG Unihemisphärische Verlangsamung mit oder ohne epileptiforme Aktivität und unilateraler Anfallsbeginn
3. Hirn-MRT Unihemisphärische fokale kortikale Atrophie und mindestens einer der beiden folgenden Punkte: • Hyperintenses T2/FLAIR-Signal in der grauen oder weißen Substanz • Hyperintenses Signal oder Atrophie des ipsilateralen Kaudatuskopfes
Teil B
1. Klinische Zeichen Epilepsia partialis continua oder progredientes* unilaterales kortikales Defizit
2. Hirn-MRT Progrediente* unihemisphärische fokale kortikale Atrophie
3. Histopathologie T-Zell-dominierte Enzephalitis mit aktivierten Mikrogliazellen (typischer-, aber nicht notwendigerweise mit Formierung von Mikrogliaknötchen) Zahlreiche parenchymatöse Makrophagen, B-Zellen oder Plasmazellen oder virale Einschlusskörperchen schließen die Diagnose RE aus
„ Progredient“ bedeutet, dass mindestens zwei sequenzielle klinische oder MR-tomographische Untersuchungen nötig sind, um die entsprechenden Kriterien erfüllen zu können. Eine klinische Progredienz ist dann gegeben, wenn jede der Untersuchungen ein neurologisches Defizit zeigt und dieses über die Zeit zunimmt. Eine progrediente Hemiatrophie liegt vor, wenn jedes der MRTs eine Hemiatrophie zeigt und diese über die Zeit zunimmt.
Therapie Aufgrund der Seltenheit der Erkrankung existieren keine hochwertigen Therapiestudien. Insofern beruhen alle Empfehlungen auf unkontrollierten, jedenfalls nicht prospektiven Fallstudien und -serien (↔). Die antikonvulsive Pharmakotherapie folgt den üblichen Regeln. Für darüber hinausgehende therapeutische Vorgehen siehe das Flussdiagramm in Abbildung 1 (Bien et al. 2005, Bien u. Elger 2005).
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 4 von 14
Abbildung 1 Therapeutischer Pathway bei Rasmussen-Enzephalitis (RE).
Die folgenden Immuntherapien werden vorgeschlagen, um die Hemisphärenzerstörung aufzuhalten (Granata et al. 2003, Bien et al. 2005) (↔):
• Tacrolimus (Bien et al. 2004) • I.v. Immunglobuline (Hart et al. 1994) • Orale Langzeitsteroide (Chinchilla et al. 1994) • Plasmapherese/Immunadsorption (Antozzi et al. 1998) (alle B).
Der Effekt dieser Verfahren auf die Anfallsfrequenz ist inkonstant. Für Details siehe Tabelle 2.
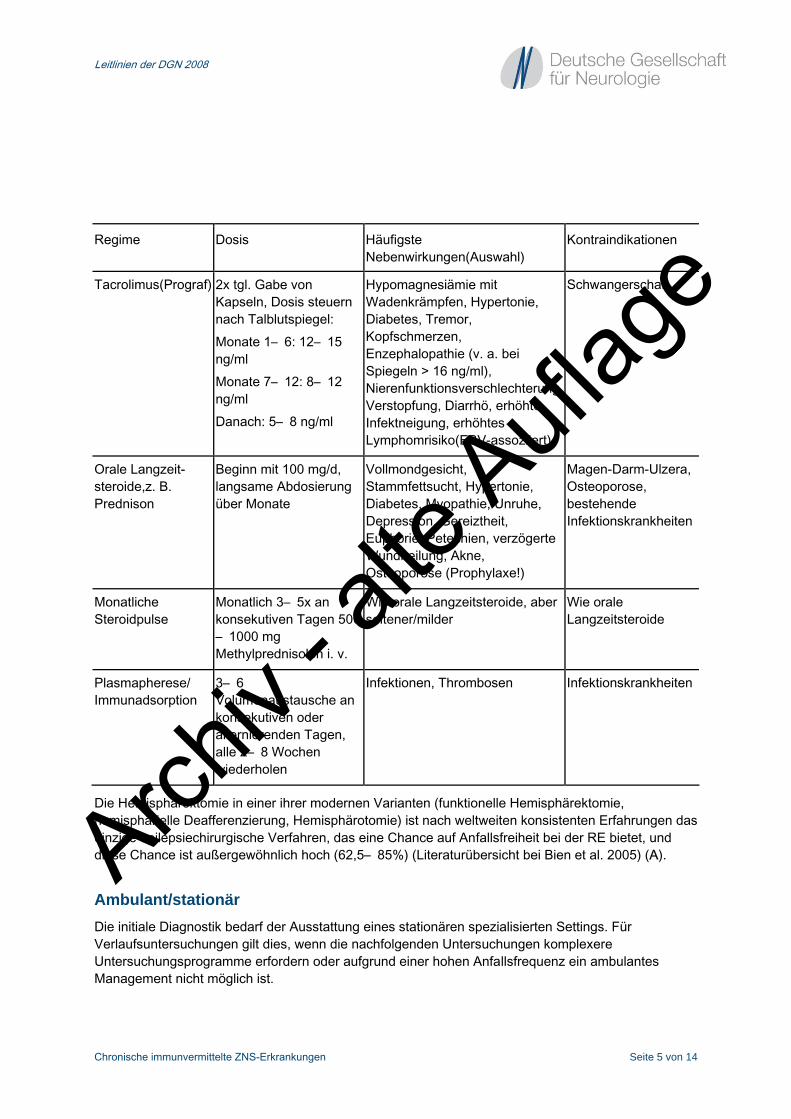
Tabelle 2 Therapieverfahren
Regime Dosis Häufigste Nebenwirkungen(Auswahl)
Kontraindikationen
I.v. Immun- globuline
Initial 3– 5 × 0,4 g/kg KG Danach: monatlich 0,4 g/kg – im Verlauf ggf. nach klinischem Bild Dosis schrittweise reduzieren oder Applikationsintervalle spreizen
Schüttelfrost, Kopfschmerz, Fieber, Übelkeit, Erbrechen, allergische Reaktionen
IgA-Mangel, wenn der Patient Antikörper gegen IgA aufweist
Archiv
- alte
Auflag
e
Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 5 von 14
Regime Dosis Häufigste Nebenwirkungen(Auswahl)
Kontraindikationen
Tacrolimus(Prograf) 2x tgl. Gabe von Kapseln, Dosis steuern nach Talblutspiegel: Monate 1– 6: 12– 15 ng/ml Monate 7– 12: 8– 12 ng/ml Danach: 5– 8 ng/ml
Hypomagnesiämie mit Wadenkrämpfen, Hypertonie, Diabetes, Tremor, Kopfschmerzen, Enzephalopathie (v. a. bei Spiegeln > 16 ng/ml), Nierenfunktionsverschlechterung, Verstopfung, Diarrhö, erhöhte Infektneigung, erhöhtes Lymphomrisiko(EBV-assoziiert),
Schwangerschaft
Orale Langzeit- steroide,z. B. Prednison
Beginn mit 100 mg/d, langsame Abdosierung über Monate
Vollmondgesicht, Stammfettsucht, Hypertonie, Diabetes, Myopathie, Unruhe, Depression, Gereiztheit, Euphorie, Petechien, verzögerte Wundheilung, Akne, Osteoporose (Prophylaxe!)
Magen-Darm-Ulzera, Osteoporose, bestehende Infektionskrankheiten
Monatliche Steroidpulse
Monatlich 3– 5x an konsekutiven Tagen 500– 1000 mg Methylprednisolon i. v.
Wie orale Langzeitsteroide, aber seltener/milder
Wie orale Langzeitsteroide
Plasmapherese/ Immunadsorption
3– 6 Volumenaustausche an konsekutiven oder alternierenden Tagen, alle 2– 8 Wochen wiederholen
Infektionen, Thrombosen Infektionskrankheiten
Die Hemisphärektomie in einer ihrer modernen Varianten (funktionelle Hemisphärektomie, hemisphärielle Deafferenzierung, Hemisphärotomie) ist nach weltweiten konsistenten Erfahrungen das einzige epilepsiechirurgische Verfahren, das eine Chance auf Anfallsfreiheit bei der RE bietet, und diese Chance ist außergewöhnlich hoch (62,5– 85%) (Literaturübersicht bei Bien et al. 2005) (A).
Ambulant/stationär Die initiale Diagnostik bedarf der Ausstattung eines stationären spezialisierten Settings. Für Verlaufsuntersuchungen gilt dies, wenn die nachfolgenden Untersuchungen komplexere Untersuchungsprogramme erfordern oder aufgrund einer hohen Anfallsfrequenz ein ambulantes Management nicht möglich ist.
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 6 von 14
Limbische Enzephalitis
Definition Bei der Limbischen Enzephalitis (LE) handelt es sich um eine meist im Erwachsenenalter auftretende, chronische, nicht infektiös bedingte, offenbar lymphozytär-mikroglial dominierte Hirnentzündung mit hauptsächlichem Befall temporomedialer Strukturen. Verschiedene Subsyndrome haben sich herauskristallisiert, welche wiederum unter die beiden Hauptkategorien „ paraneoplastisch“ und „nichtparaneoplastisch“ subsumiert werden können:
• Paraneoplastische LE: - Paraneoplastische LE mit Serum- (und Liquor-)Autoantikörpern gegen intrazelluläre Antigene
(Gultekin et al. 2000); als „ gut charakterisierte“ Antikörper gelten (Graus et al. 2004): Hu-, Ma-, CV2 / CRMP5-, Amphiphysin-Ak (siehe Kapitel „ Paraneoplastische Syndrome“ )
- Einige Fälle von „ Teratom-Enzephalitis“ (d. h. Enzephalitis assoziiert mit – überwiegend ovariell lokalisierten – Teratomen bei jungen Frauen) und Antikörpern gegen Heteromere von NMDA-Rezeptor-Untereinheiten identifiziert wurden (Dalmau et al. 2007). Tests sind noch nicht allgemein verfügbar.
- Antikörper-negative paraneoplastische LE • Nichtparaneoplastische LE:
- Subakute LE mit Serumautoantikörpern gegen spannungsabhängige Kaliumkanäle („ voltage gated potassium channel antibodies“ , VGKC-Ak), offenbar meistens nicht-paraneoplastisch (Vincent et al. 2004)
- „ Non-herpetische akute LE“ (Asaoka et al. 2004) - Bisher nicht durch Autoantikörper charakterisierte subakute nichtparaneoplastische
Enzephalitis (Bien et al. 2000)
Formelle Diagnosekriterien (Bien 2008) werden in Tabelle 3 vorgeschlagen. Für die Therapie ist die Identifikation eines zugrunde liegenden Tumors entscheidend. Für die Prognose (unter adäquater Therapie) hingegen scheint die Art der assoziierten Antikörper ausschlaggebend zu sein: Sie ist günstig bei Vorliegen von Antikörpern gegen Zellmembran-Antigene (VGKC oder NMDA-Rezeptor-Heteromere) und ungünstig bei Assoziation mit Antikörpern gegen intrazelluläre Antigene. Es wird auch auf das Kapitel „ Paraneoplastische Syndrome“ verwiesen.
Tabelle 3 Kriterien für die Diagnose „ limbische Enzephalitis“
Voraussetzungen („ Verdacht auf limbi-sche Enzephalitis“ )
Klinisch: Im Erwachsenenalter vor weniger als 5 Jahren aufgetretenes limbisches Syndrom, definiert durch ≥ 1 der folgenden 3 Symptome: • Neugedächtnisstörung • Temporallappenanfälle • Affektstörung Hirn-MRT: Nicht anders erklärbare temporomediale FLAIR-/T2-Signalanhebung
Definitive limbische Enzephalitis
Zusätzlich zu den o. g. „ Voraussetzungen“ : Nachweis von ≥ 1 der folgenden Punkte:
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 7 von 14
• Neuropathologie: chronische temporomediale Enzephalitis • Tumornachweis innerhalb von ± 5 Jahren bezogen auf den Beginn der
neurologischen Symptome • Antikörper-Nachweis: „ Well characterised“ onkoneurale Ak (Anti-Hu-Ak,
Ma/Ta-Ak, CV2 / CRMP5-Ak, Amphiphysin-Ak) oder Anti-VGKC
Mögliche limbische Enzephalitis
Bei Vorliegen der o. g. „ Voraussetzungen“ , aber fehlendem Nachweis einer der vorgenannten Punkte: Hirn-MRT-Verlauf: Temporomediale Schwellung und Signalanhebung mit nachfolgender Volumen- oder Signalabnahme (sofern dieser Verlauf nicht einem Status epilepticus zuzuschreiben ist; dies ist der Fall, wenn das EEG zum Zeitpunkt der Signalanhebung einen Status belegt und die Signal-Normalisierung innerhalb weniger Tage erfolgt)
Untersuchungen Notwendig
Bei Nachweis eines im Erwachsenenalter vor maximal 5 Jahren aufgetretenen „ limbischen Syndroms“ (Temporallappenanfälle, Neugedächtnisstörungen, affektive Symptome) sind erforderlich:
• Hirn-MRT • Liquoruntersuchung zum Ausschluss einer infektiösen Enzephalitis (namentlich HSV-, HHV-6-,
CMV-, und HIV-Enzephalitis) • Autoantikörperdiagnostik: „ gut charakterisierte“ onkoneuronale Antikörper (Hu, Ma, CV/CRMP5,
Amphiphysin) und VGKC-Antikörper. Cave: Die Bestimmung sollte in einem Labor durchgeführt werden, das international anerkannte Methoden verwendet (Details für die paraneoplastischen LE siehe Kapitel „ Paraneoplastische Syndrome“ , für die VGKC-Antikörper siehe die Originalpublikation [Vincent et al. 2004])
• Tumorsuche, adaptiert an das Risikoprofil des Patienten (siehe Kapitel „ Paraneoplastische Syndrome“ )
• Untersuchungen zur Dokumentation der Defizite des Patienten vor Therapiebeginn: - Eigen-/Fremdanamnese zur Ermittlung der Anfallsfrequenz, evtl. ergänzt durch 24-h- oder
48-h-Langzeit-EEG - Neuropsychologische Untersuchung (mindestens Bedside-Test wie MMSE) - Psychopathologische Exploration
Im Einzelfall erforderlich
• Klinische und apparative Diagnostik zur Erfassung von Ausfällen, die über das limbische System hinausgehen, z. B. zerebelläre oder peripherneurologische Störungen bei anti-Hu-Syndrom
Therapie Bereits bei klinisch ausreichendem Verdacht – insbesondere wenn dieser durch den Hirn-MRT-Befund unterstützt wird – ist die Einleitung einer Immuntherapie anzuraten (Darnell u. Posner 2005). In Frage kommen: Steroid-Pulstherapie, orale Langzeitsteroidtherapie, i. v. Immunglobuline, Plasmapherese (Tab. 2) (alle B).
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 8 von 14
Wenn alle Befunde, d. h. auch die der Autoantikörper und der Tumorsuche vorliegen, wird die Therapie aufgrund der subsyndromalen Zuordnung angepasst (↔): Bei Nachweis eines Tumors ist dessen onkologische Therapie vorrangig. Weitere Empfehlungen sind dem Kapitel „Paraneoplastische Syndrome“ zu entnehmen. Bei fehlendem Nachweis eines Tumors sollte der Erfolg der Immuntherapie nach 3 Monaten evaluiert werden (Anfallsfrequenz, neuropsychologische Performanz, psychopathologischer Befund). Bei fehlender Wirkung ist ein Wechsel der Therapie anzuraten. Sollte darunter nach weiteren 3 Monaten keine Besserung erkennbar sein, ist eine (intensivierte) Fortsetzung der Immuntherapie nur empfehlenswert, wenn es sich um eine (üblicherweise ja mit einer guten Prognose verbundene) VGKC-Antikörper-assoziierte LE handelt.
Ambulant/stationär Mindestens die initiale Diagnostik bedarf der Ausstattung eines stationären spezialisierten Settings. Für Verlaufsuntersuchungen gilt dies, wenn diese Untersuchungen komplexere Untersuchungsprogramme erfordern.
Steroid-responsive Enzephalopathie mit assoziierter Autoimmun-Thyroiditis (SREAT, „ Hashimoto-Enzephalopathie“ )
Definition Die SREAT ist definiert als eine nichtinfektiöse, vermutlich entzündliche Enzephalopathie, assoziiert mit einer durch Nachweis von Schilddrüsen-Autoantikörpern gesicherten autoimmunen Thyroiditis. Die Pathogenese ist unklar; möglicherweise wird sich dieses Syndrom – das gegenwärtig eine praktikable neurologische Entität darstellt – in der Zukunft in mehrere spezifischer definierte Erkrankungen „ auflösen“ (Chong et al. 2003). Die SREAT wurde früher „Hashimoto-Enzephalopathie/-itis“ genannt; dies implizierte einen kausalen Zusammenhang zwischen Hashimoto-Thyroiditis und Enzephalopathie. Eine solche Kausalität ist aber nicht nachzuweisen. Auch ein Zusammenhang der Schwere der SREAT mit dem Schilddrüsenhormonstatus besteht nicht.
Die SREAT manifestiert sich klinisch entweder mit Schlaganfall-ähnlichen, plötzlich einsetzenden fokalen, z. T. multiplen, neurologischen Ausfällen (ca. 10– 30% der Fälle) oder aber als schleichend progrediente Enzephalopathie (eventuell bis hin zum Koma) mit zunehmenden epileptischen Anfällen oder einem Status epilepticus (ca. 70– 90% der Fälle) (Kothbauer-Margreiter et al. 1996, Chong et al. 2003, Chaudhuri u. Behan 2003). Weiter müssen erhöhte Schilddrüsenautoantikörpertiter – typischerweise mikrosomale Antikörper (MAK, im Wesentlichen äquivalent zu thyroid peroxidase [TPO] antibodies) oder Thyreoglobulin-Antikörper (TAK, TG antibodies) – im Serum nachweisbar sein (Castillo et al. 2006). Andere Ursachen (infektiöse, toxische, medikamentöse, rheumatologisch-systemische, vaskulitische, Meningeosis, MS, Sarkoidose etc.) sind soweit wie möglich auszuschließen. Die Diagnose „ SREAT“ erfordert schließlich den Beleg einer Besserung auf Steroide (Schäuble et al. 2003, Castillo et al. 2006).
Untersuchungen Notwendig
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 9 von 14
• Bestimmung von Schilddrüsenhormonen und Schilddrüsenautoantikörpern • Labor- und Liquoruntersuchungen zum Ausschluss von Differenzialdiagnosen • Hirn-MRT
Im Einzelfall erforderlich
• EEG und andere elektroenzephalographische Untersuchungen • Ultraschalluntersuchungen im Rahmen der neurovaskulären Differenzialdiagnostik • EEG
Therapie Die Seltenheit des Krankheitsbildes verhindert eine auf hochwertiger Evidenz basierte Behandlungsstrategie (↔). Aus den vorhandenen Fallserien (Kothbauer-Margreiter et al. 1996, Chong et al. 2003, Schäuble et al. 2003, Chaudhuri u. Behan 2003, Castillo et al. 2006, Marshall u. Doyle 2006) können aber verwertbare Therapieempfehlungen abgeleitet werden. Medikamente der ersten Wahl bei der SREAT sind Steroide, unter denen eine ausgezeichnete, oft rasch (innerhalb von Tagen, ähnlich wie bei der Riesenzellarteriitis) einsetzende Wirkung beobachtet wird (A). Allerdings können auch bis zu 6 Wochen vergehen, ehe die Besserung eintritt (Kothbauer-Margreiter et al. 1996). Ein Therapie-Algorithmus ist in Abbildung 2 dargestellt. Epileptische Anfälle erfordern eine antiepileptische Therapie, die nach Abheilen der Erkrankung bzw. bei Normalisierung des EEG wieder ausgeschlichen wird. Eine zweite Episode wird wie eine erste behandelt; möglicherweise mit einem noch langsameren Ausschleichen der Steroide.
Arch
iv - a
lte Aufl
age

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 10 von 14
Abbildung 2 Therapie-Algorithmus für die Steroid-responsive Enzephalopathie mit assoziierter Autoimmun-Thyreoiditis.
Bei weiteren Episoden oder einem permanenten Aufflammen der Symptome nach Unterschreiten einer gewissen Steroiddosis sollten zusätzliche Steroid-sparende Immunsuppressiva zum Einsatz kommen. Einzelfallberichte/Mini-Serien berichten über gute Effekte von Azathioprin, Methotrexat, Ciclosporin A, Hydroxychloroquin und Mycophenolatmofetil. Pulstherapien mit intravenösen Immunoglobulinen und Cyclophyosphamid sowie Plasmapheresen wurden ebenfalls erfolgreich eingesetzt. Alle diese Therapien werden aber als zweite Wahl gegenüber den Steroiden angesehen (alle C).
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 11 von 14
Ambulant/stationär Wegen der Schwere der Symptomatik und des differenzialdiagnostischen Aufwandes ist mindestens initial eine stationäre Untersuchung und Behandlungseinleitung erforderlich.
Neurosarkoidose
Definition Die Sarkoidose ist definiert als multisystemische Granulomatose immer noch unbekannter Ätiologie, die vor allem junge Erwachsene befällt. Die charakteristische Histologie ist durch nicht verkäsende epitheloidzellige Granulome gekennzeichnet. Die Prävalenz klinisch manifester Fälle beträgt 5– 20 pro 100000, diejenige autoptisch verifizierter Fälle 35 pro 100000. Hauptmanifestationsorte sind die Lymphknoten der Lungenhili, das Lungenparenchym, die Haut und die Augen. Ein Befall des Nervensystems (Neurosarkoidose, NS) findet sich klinisch manifest in 5% (2– 16) und autoptisch in bis zu 27%. Dies ist eher im frühen Verlauf einer systemischen Sarkoidose und eher bei jüngeren Patienten (30– 40-jährig) der Fall. Der Befall des Nervensystems stellt bei ca. 50% die erste Manifestation der Sarkoidose überhaupt dar; bei Hirnnervenbefall sogar in 85%. In über 90% der Patienten mit NS finden sich auch andere Systemmanifestationen, Hiluslymphome in über 80% und Augenbefall (Uveitis, Keratokonjunktivitis) in ca. 50%.
Klinisches Bild Am häufigsten ist ein Hirnnervenausfall (50– 70%), typischerweise multiple Hirnnerven betreffend (über 50%) und oft rezidivierend. Zweithäufigste Manifestation ist eine aseptische Meningitis (18– 26%) vor allem der basalen Meningen, aber evtl. auch des Ependyms mit Kopfschmerzen, Erbrechen, Meningismus und Papillenödem. Dritthäufigste Manifestation ist ein Hydrozephalus (9– 17%), entweder obstruktiv bei Granulomen im Bereich des III. oder IV. Ventrikels oder kommunizierend bei entzündlichem Verschluss der Pacchionischen Granulationen. Gewisse intrakranielle Strukturen werden besonders häufig befallen: Hypothalamus und Hypophyse (15– 26%) mit Diabetes insipidus, bulimisches Verhalten und Hypersomnie. Zerebrale, subdurale und meningeale (en plaque) Massenläsionen können auch zu epileptischen Anfällen führen (ca. 20%). Eine Myelopathie infolge komprimierender extramedullärer oder auch intramedullärer Granulome ist in 6– 10% zu beobachten, eine periphere Neuropathie (vor allem Typ Mononeuritis multiplex) in 4– 14% und eine Myopathie in 7– 12% (Stern 2004).
Diagnostik Die Kernspintomographie hat die Diagnose des ZNS-Befalls wesentlich erleichtert. Der typische Befund ist eine knotige oder auch flächige Verdickung der Meningen, speziell an der Schädelbasis mit starker KM-Anreicherung (Smith et al. 2004). Der Kveim-Test wäre zwar sensitiv und spezifisch, leider ist aber zuverlässiges Antigenmaterial kaum erhältlich. Die Gallium-Szintigraphie und die Bestimmung des Angiotensin-Konversions-Enzyms haben eine sehr geringe Spezifität (Nowak u. Widenka 2001).
Je nach kernspintomographischem Befund ist die Differenzialdiagnose sehr breit:
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 12 von 14
• Bei parenchymatösen Massenläsionen: Lymphom, Gliom, Meningeom, Metastase • Bei vorwiegend periventrikulären herdförmigen Läsionen auch Multiple Sklerose • Bei vorwiegend menigealen Läsionen: bakterielle, mykotische oder tuberkulöse Infektionen,
leukämische oder karzinomatöse Infiltrationen
Der klinische Verlauf ist im Einzelfall nicht vorhersehbar, in zwei Drittel der Fälle ist er monophasisch, in einem Drittel rezidivierend. Die Prognose ist gut bei Hirnnervenläsionen, eher schlecht bei Hydrozephalus und zerebralen Massenläsionen.
Untersuchungen Notwendig
• Je nach klinischer Symptomatik: MRT von Kopf und/ oder Rückenmark • Zur Sicherung der Diagnose Sarkoidose: Suche nach Befall weiterer Systeme:
- Thorax- und Abdomen-CT - Augenärztliche Untersuchung - Untersuchung von Haut und Lymphknotenstationen und ggf. Biopsie (vor allem
Hiluslymphknoten) - Je nach Klinik: Muskelbiopsie oder Leberbiopsie - Lungenfunktionstest
• Liquoranalyse inklusive Bakteriologie und Zytologie (zum Ausschluss infektiöser oder neoplastischer Differenzialdiagnosen)
Im Einzelfall erforderlich
• Wenn isolierter ZNS-Befall (klinisch und bildgebend), dann – wenn immer möglich – meningeale oder zerebrale Biopsie je nach MRT-Befund
Therapie Angesichts der anerkannten Morbidität und Mortalität der NS empfehlen die meisten Autoren eine frühe und aggressive Therapie. Aber es gibt zurzeit weder klare Guidelines noch Indikationen und schon gar keine kontrollierte Studien. Die gängigen Therapieempfehlungen sind demzufolge erfahrungs- und nicht evidenzbasiert (↔) (Hoitsma et al. 2004). Therapeutikum erster Wahl sind orale Langzeit-Kortikosteroide in einer initialen Dosis von 1 mg/kg KG und Tag per os (Tab. 2) (B). In schweren Fällen können i. v. Steroide auch zu Beginn hochdosiert (Stoßtherapie) appliziert werden. Bei ungenügender Wirkung oder raschem Rezidiv bei Dosisreduktion bzw. hoher notwendiger Erhaltungsdosis werden verschiedene zytotoxische Immunsuppressiva empfohlen (Methotrexat, Azathioprin, Ciclosporin, Cyclophosphamid; alle C). Die größten Erfahrungen bestehen mit Methotrexat und Azathioprin. Einzelfälle wurden auch erfolgreich mit Mycophenolatmofetil, Hydrochloroquin und TNF-alpha-Blockern (Infliximab) behandelt. Eine Langzeitbeobachtung legt nahe, dass bei schweren neurologischen Symptomen der frühe Einsatz von Immunsuppressiva die Prognose verbessert (Scott et al. 2007). Ebenso wurde bei therapieresistenten Einzelfällen ein Entzündungsrückgang unter Strahlentherapie beobachtet. Bei Hydrozephalus ist die Shuntdrainage angezeigt. Zur Beurteilung der Therapieeffizienz dienen die Klinik, das MRT und der Liquorbefund.
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 13 von 14
Ambulant/stationär Wegen der oft unspezifischen Klinik, der Schwere der Symptomatik und des differenzialdiagnostischen Aufwandes, der – wenn immer möglich – eine Biopsie einschließen sollte, ist mindestens initial eine stationäre Untersuchung und Behandlungseinleitung erforderlich.
Expertengruppe
Dr. Stephan Rüegg, Universitätsspital Basel, Neurologische Klinik, Abteilung für Klinische Neurophysiologie
Univ.-Prof. Dr. Erich Schmutzhard, Medizinische Universitätsklinik für Neurologie, Innsbruck
Prof. Dr. Matthias Sturzenegger, Inselspital, Neurologische Klinik, Universitätsspital Bern
Federführend: PD Dr. Christian Bien, Universität Bonn, Klinik und Poliklinik für Epileptologie, Sigmund-Freud-Straße 25, 53105 Bonn
E-Mail: [email protected]
Literatur
Antozzi C, Granata T, Aurisano N, et al. Long-term selective IgG immuno-adsorption improves Rasmussen's encephalitis. Neurology 1988;51:302– 305. Asaoka K, Shoji H, Nishizaka S, et al. Non-herpetic acute limbic encephalitis: cerebrospinal fluid cytokines and magnetic resonance imaging findings. Intern Med 2004;43:42– 48. Bauer J, Elger CE, Hans VH, et al. Astrocytes are a specific immunological target in Rasmussen's encephalitis. Ann Neurol 2007;62: 67– 80. Bien CG, Bauer J, Deckwerth TL et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis. Ann Neurol 2002a;51:311– 318. Bien CG, Elger CE. Neue Erkenntnisse zur Rasmussen-Enzephalitis. Nervenarzt 2005;76:1470– 1487. Bien CG. ChronischeEnzephalitiden als Epilepsieursachen: Pathogenese, Diagnostik und Therapie. Akt Neurol 2008 (in press). Bien CG, Gleissner U, Sassen R, Widman G, Urbach H, Elger CE. An open study of tacrolimus therapy in Rasmussen encephalitis. Neurology 2004;62:2106– 2109. Bien CG, Granata T, Antozzi C, et al. Pathogenesis, diagnosis and treatment of Rasmussen encephalitis: a European consensus statement. Brain 2005;128:454– 471. Bien CG, Schulze-Bonhage A, Deckert M, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy. Neurology 2000;55:1823– 1828. Bien CG, Widman G, Urbach H, et al. The natural history of Rasmussen's encephalitis. Brain 2002b;125:1751– 1759. Castillo P, Woodruff B, Caselli R, et al. Steroid-responsive encephalopathy associated with autoimmune thyroiditis. Arch Neurol 2006;63:197– 202. Chaudhuri A, Behan PO. The clinical spectrum, diagnosis, pathogenesis and treatment of Hashimoto's encephalopathy (recurrent acute disseminated encephalomyelitis). Curr Med Chem 2003; 10:1945– 1953. Chinchilla D, Dulac O, Robain O, et al. Reappraisal of Rasmussen's syndrome with special emphasis on treatment with high doses of steroids. J Neurol Neurosurg Psychiatry 1994;57:1325– 1333. Chong JY, Rowland LP, Utiger RD. Hashimoto encephalopathy: syndrome or myth? Arch Neurol 2003;60:164– 171. Dalmau J, Tüzün E, Wu HY, et al. Paraneoplastic anti-N-methyl-D-aspartate receptor encephalitis associated with ovarian teratoma. Ann Neurol 2007;61:25– 36. Darnell RB, Posner JB. A new cause of limbic encephalopathy. Brain 2005;128:1745– 1746.
Archiv
- alte
Auflag
e

Leitlinien der DGN 2008
Chronische immunvermittelte ZNS-Erkrankungen Seite 14 von 14
Demeurisse G, Demol O, Robaye E. Motor evaluation in vascular hemiplegia. Eur Neurol 1980;19:382– 389. Granata T, Fusco L, Gobbi G, et al. Experience with immunomodulatory treatments in Rasmussen's encephalitis. Neurology 2003;61: 1807– 1810. Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004;75:1135– 1140. Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain 2000;123:1481– 1494. Hart YM, Cortez M, Andermann F, et al. Medical treatment of Rasmussen's syndrome (chronic encephalitis and epilepsy): effect of high-dose steroids or immunoglobulins in 19 patients. Neurology 1994;44:1030– 1036. Hoitsma E, Faber CG, Drent M, Sharma OP. Neurosarcoidosis: a clinical dilemma. Lancet Neurology 2004;3:397– 407. Kothbauer-Margreiter I, Sturzenegger M, Komor J, Baumgartner R, Hess CW. Encephalopathy associated with Hashimoto thyroiditis: diagnosis and treatment. J Neurol 1996;243:585– 593. Marshall GA, Doyle JJ. Long-term treatment of Hashimoto's encephalopathy. J Neuropsychiatry Clin Neurosci 2006;18:14– 20. Nowak DA, Widenka DC. Neurosarcoidosis: a review of its intracranial manifestation. J Neurol 2001;248:363– 372. Oguni H, Andermann F, Rasmussen TB. The syndrome of chronic encephalitis and epilepsy. A study based on the MNI series of 48 cases. Adv Neurol 1992;57:419– 433. Schäuble B, Castillo PR, Boeve BF, Westmoreland BF. EEG findings in steroid-responsive encephalopathy associated with autoimmune thyroiditis. Clin Neurophysiol 2003;114:32– 37. Scott TF, Yandora K, Valeri A, Chieffe C, Schramke C. Aggressive therapy for neurosarcoidosis: long-term follow-up of 48 treated patients. Arch Neurol 2007;64:691– 696. Smith JK, Matheus MG, Castillo M. Imaging manifestations of neurosarcoidosis. Am J Roentgenol 2004;182:289– 295. Stern BJ. Neurological complications of sarcoidosis. Curr Opin Neurol 2004;17:311– 316. Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004;127:701– 712.
Archiv
- alte
Auflag
e