Embed Size (px)
Citation preview

Zulassungsbehörden in der Kritik Seite 26
Die medikamentöse Behandlung der Endometriose Seite 28
Thrombozyten-Aggregationshemmer – eine Übersicht Seite 30
Osteoporosetherapie bei verschlechterter Nierenfunktion Seite 34
Sarkoidose – Erkennen und Behandeln Seite 36
Die Behandlung des primären Sjögren-Syndroms Seite 38
Gibt es neue Erkenntnisse zur Therapie der Arthrose mit Orthokin®? Seite 40
Yervoy® (Ipilimumab) Seite 42
Fampyra® (Fampridin) Seite 44
Wie gefährlich sind Rizinussamen? Seite 45
Schluckstörungen unter Neuroleptika Seite 46
Eine „Konjunktivitis“ durch Bisphosphonate Seite 47
Terminankündigung: Symposium „Personalisierte Medizin“ Seite 48
Arzneimittel – kritisch betrachtet
Das aktuelle Thema
Therapie aktuell
Neue Arzneimittel
Unerwünschte Arzneimittelwirkungen
Editorial
Aus der Praxis – für die Praxis
In eigener Sache
Arzneiverordnungin der Praxis
ImpressumHerausgeber:Arzneimittelkommission der deutschen Ärzteschaft Prof. Dr. med. W.-D. Ludwig (Vorsitzender)Wissenschaftlicher Beirat:Dr. med. J. Bausch,Dr. med. K. Ehrenthal,Frau Prof. Dr. med. U. Gundert-Remy,Prof. Dr. med. R. Lasek,Prof. Dr. med. B. Müller-Oerlinghausen,Prof. Dr. med. U. Schwabe,M. Voss, Arzt, Vorstand der Arzneimittelkommission derdeutschen ÄrzteschaftChefredakteur:Prof. Dr. med. D. Höffler Stellvertretender Chefredakteur:Dr. med. M. ZieschangAnschrift der Redaktion:Arzneimittelkommission der deutschen ÄrzteschaftPostfach 12 08 6410598 BerlinTelefon: 0 30 / 40 04 56-5 00Telefax: 0 30 / 40 04 56-5 55E-Mail: [email protected] 0939-2017Realisation und Vertrieb:Triple MPR Group Verlagsgesellschaft mbH, Postfach 19 01 30, D-53037 Bonn, E-Mail: [email protected],Telefax: 0228/224511Druck: Franz Paffenholz GmbH, BornheimAbonnement:Die Schutzgebühr des Jahresabonnements für4–6 x AVP einschl. Sonderhefte Thera pie emp -feh lungen beträgt EUR 39,– (für Stu den ten: EUR19,–; Nachweis erforderlich). Ihre Abo-Anfor-derung richten Sie bitte an die Arznei mittel -kommission [email protected]. Bezug im Jahres-abonnement, Kündigung zum Jahresende.Wir möchten darauf hinweisen, dass die in „Arznei ver -ordnung in der Praxis“ erscheinenden Publikationenprinzipiell den Charakter von Autorenartikeln – wie injeder anderen Zeitschrift – haben. Für die Richtigkeitund Vollständigkeit der Angaben zur Dosierung undauch zu den Preisen kann keine Gewähr übernommenwerden. Trotz sorgfältiger Recherche bitten wir Siedringend, die aktuellen Angaben des jeweiligen Her-stellers zu beachten. Die gemäß Arznei mittel-Richt -linien des Gemeinsamen Bundesausschusses zu ver-öffentlichenden Therapie empfeh lun gen in ihrer aktu-ellen Fassung werden als solche gekennzeichnet.© Alle Rechte vorbehalten. AkdÄ, Berlin 2012
Herausgegeben von der Arzneimittelkommission der deutschen Ärzteschaft Band 39 · Ausgabe 2 · März 2012
Als Anfang des 20. Jahrhunderts die pharmazeutischeIndustrie entstand und begann, für ihre Produkte zuwerben, wurde 1911 auf dem Kongress für InnereMedizin der Grundstein für die Arzneimittelkommissionder deutschen Ärzteschaft gelegt. Die Aufgabe derseinerzeit berufenen Kommission sollte es sein, dieÄrzteschaft durch Ärzte unabhängig und objektiv zuinformieren. Dieses Ziel verfolgen wir bis zum heu tigenTag, u. a. mit diesem Heft.
Arzneiverordnung in der Praxis ist Mitglied der International Society of Drug Bulletins(www.isdbweb.org)

26 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Zulassungsbehörden in der Kritik
Als im Jahre 2006 der einflussreicheU.S.-Politiker Henry A. Waxman der Fe-deral Drug Administration (FDA) zum100-jährigen Geburtstag gratulierte, dawar dies ein eher bittersüßer Gruß. DemLob „Unsere Zulassungsverfahren sindimmer der weltweite ‚Goldstandard’ ge-wesen.“ folgte wenige Zeilen danach:„Aber heute, am 100. Geburtstag derFDA sehe ich eine ziellose Behörde, (….)Die FDA [...], die einst die Arzneimittel-behörde der Welt mit dem größten Anse-hen war, hat in den letzten 5 Jahreneinen Verfall ihrer Kraft und einen Ab-sturz ihrer Reputation hinnehmen müs-sen. Heutzutage steht das Engagementder Behörde für die öffentliche Gesund-heit in Frage. Der Abstieg der Behördehat viele Facetten. Eine Hauptursacheist die Erhebung der Politik über die Wis-senschaft. [...[ Die Regierung hat wissen-schaftliche Erkenntnisse missachtet undverdreht, um vorherbestimmte Ergeb-nisse zu erzielen.“ (1)
Zulassungsbehörden sind zweifelloskeine inerten, apolitischen Institutio-nen, sondern befinden sich schon wegender enormen finanziellen Bedeutungeiner Arzneimittelzulassung immer ander Nahtstelle zwischen staatlichem Ver-waltungsakt, unabhängiger wissen-schaftlicher Begutachtung und dem pri-vaten wirtschaftlichen Interesse der An-tragsteller.
Wie sieht es nun mit unsererEMA aus?Ähnlich wie die FDA vor einigen Jahrenscheint jetzt auch die European Medici-nes Agency (EMA) in das Fadenkreuz deröffentlichen Debatten zu geraten:
– aus der Wissenschaft ist harte Kritikan der Verweigerung des Zugangs zuklinischen Studiendaten durch dieEMA laut geworden, ein Zugang, der ineinem bekannt gewordenen Fall erst
nach 7 Jahre langen Mühen nur unterEinschaltung des Europäischen Om-budsmannes erreicht werden konnte(2);
– die fachliche Qualität des von der EMAherausgegebenen European PublicAssessment Reports (EPAR) für Arz-neimittel ist in erhebliche Zweifel ge-zogen worden (3);
– das Europäische Parlament (EP) hatsich „ernsthaft besorgt“ gezeigt „überdie Antworten der Agentur auf wesent-liche, vom Rechnungshof und vom In-ternen Auditdienst (IAS) angespro-chene Fragen“, nämlich die Verwal-tung von Ausschreibungsverfahren,die fehlende Einhaltung von Verfah-rensgrundsätzen bei der Feststellungvon und zum Umgang mit Interes-senskonflikten bei Personal und beiSachverständigen sowie bei den Krite-rien für die Einstellung von Personal.“(4)
EMA und Europäisches Parla-ment (EP)InteressenkonflikteDer Haushaltskontrollausschuss hatfestgestellt, dass bereits der Interne Au-ditdienst der EMA „Unzulänglichkeiten“bei der „wissenschaftlichen Beratung fürHumanarzneimittel in der Agentur“festgestellt habe. Insbesondere hält esder Haushaltskontrollausschuss für „un-vertretbar“, dass die EMA bei Interessen-konflikten die entsprechenden Regelun-gen nicht effektiv anwendet. Dies habezur Folge, „dass es keine Gewähr dafürgibt, dass die Bewertung von Humanarz-neimitteln durch unabhängige Sachver-ständige vorgenommen wird.“ Bereitsaus mehreren früheren jährlichen Prüf-berichten des Internen Auditdienstesseien „zwölf ‚sehr wichtige’ Empfehlun-gen und eine ‚kritische’ Empfehlung“,„von denen die meisten die Unabhängig-keit von Sachverständigen betrafen“, imJahr 2009 noch immer nicht umgesetzt
gewesen, wobei die älteste Empfehlungaus dem Jahr 2005 stamme.
Der Haushaltskontrollausschuss des EPfordert weiter eine Transparenz der In-teressenverflechtung bei der Zuweisungdurch die EMA von Projektleitern zuProdukten und eine Veröffentlichungvon Zuweisungsentscheidungen auf derWebsite der EMA. Gleichermaßen hieltes der Haushaltskontrollausschuss desEP „für unvertretbar, dass die Zuverläs-sigkeitserklärung des Exekutivdirektorsvom 13. Mai 2010 keinerlei Vorbehaltenthält und dementsprechend nicht dieim Verhaltenskodex, der von der Agenturmit Blick auf die Zuverlässigkeitser-klärungen des Internen Auditdienstesund des Rechnungshofes angenommenwurde, eingegangene Verpflichtung er-füllt.“
Der Fall Mediator® und unvoll-ständige ProduktunterlagenDer Haushaltskontrollausschuss des EPhat eine Entlastung der EMA verwei-gert, bis eine vollständige Klärung derVorgänge bei der sich über 10 Jahre hin-ziehenden Marktrücknahme des inFrankreich vertriebenen AppetitzüglersBenfluorex (Mediator®) vorliege. DerHaushaltskontrollausschuss des EP warauch nicht bereit hinzunehmen, dassdie EMA unvollständige Informationenin Produktunterlagen zu Humanarz-neimitteln bereitstellt. Gefordert wurdevon der EMA die leichte Zugänglichkeitvon Schlüsselinformationen und Be-achtung der Leitlinien für das Ablage -system.
Gebührenzahlungen der Phar-maindustrie und EMA-HaushaltDer Haushaltskontrollausschuss des EPhat bei der Beurteilung des EMA-Haus-haltes betont, dass nur noch 18,7 % desEMA-Haushaltes in Höhe von rund 194
Editorial

Mill. Euro durch die EU bereitgestelltwerden.
Beim kritischen Punkt der Finanzierungder EMA ist man unwillkürlich erinnertan die Vorgänge rund um das DAMA-Er-richtungsgesetz in der 15. und 16. Legis-laturperiode des Deutschen Bundesta-ges. Die letzte rot-grüne RegierungSchröder wollte zur sogenannten Stei-gerung der internationalen Konkur-renzfähigkeit eine gebührenfinanzierte„Deutsche Arzneimittel- und Medizin-produkte Agentur“ (DAMA) einführen.Es sollten also die zulassungsbegehren-den pharmazeutischen Unternehmendurch ihre Gebühren die Zulassungs-behörde weitgehend finanzieren,während die gemeinwohlrelevantenAusgaben (z. B. BtM-Verkehr, Pharma-kovigilanz) auf einen konstanten Sockel-betrag von nur 10,61 Mill. Euro festge-schrieben wurden.
Die Große Koalition aus CDU/CSU undSPD führte dieses Vorhaben zunächstweiter. Erst nach negativer Beurteilungfast aller angehörten Sachverständigen(zu denen auch die AkdÄ gehörte) warauch in den Koalitionsfraktionen keineUnterstützung mehr vorhanden und dasVorhaben wurde nicht weiter verfolgt.
Mangelnde Transparenz derEPARsDer European Public Assessment Report(EPAR) findet regelmäßig Beachtung alseine Art Referenzinformation über neueArzneimittel und wird auch in „Arznei-verordnung in der Praxis“ in der Rubrik„Neuzugelassene Arzneimittel“ als Basisfür eine „Information der Arzneimittel-kommission der deutschen Ärzteschaft“verwendet.
Im British Medical Journal haben un-längst drei italienische Wissenschaftlerdie Qualität des EPAR am Beispiel vonPsychopharmaka deutlich in Zweifel ge-zogen. Anhand der untersuchten EPARsei eine Beurteilung der klinischen Da-tengrundlage und damit also der Zulas-sungsentscheidung kaum möglich (3).Die Autoren kamen nach Auswertungder 70 randomisierten Phase III-Studienvon 8 im Zeitraum von 2004 bis 2009 zu-
gelassenen Psychopharmaka zum Er-gebnis, dass nur 49 % die Zahl der Pati-enten im jeweiligen Behandlungsarmangaben, nur 27 % nannten die Gründebei Studienabbrechern, nur 43 % derStudien nannten die Zahl der Patienten,die den primären Endpunkt erreichtenund nur 13 % die Efficacy in absolutenZahlen. Die Autoren fordern für denEPAR eine klare tabellarische Darstel-lung jeder einzelnen Phase-III-Studie,die die Patientendaten und die Behand-lungsergebnisse zusammen mit eindeu-tigen Identifikationsnummern der be-treffenden klinischen Studien ähnlichder „clinicaltrials.gov“-Website enthält.Das mag aber ein recht frommer Wunschsein, denn an gleicher Stelle kritisierenForscher des Instituts für Qualität undWirtschaftlichkeit im Gesundheitswesen(IQWiG) die fortbestehenden Mängel desklinischen Studienregisters der EMA(EU Clinical Trials Register, EU-CTR),vor allem eine fehlende funktionale alp-hanumerische Suchoption und einenicht vorhandene Synonymsuche (5).
Zweifellos ist es vor allem der Zähigkeitzweier dänischer Forscher vom NordicCochrane Center zu verdanken, dass dieEMA einen offenen Datenzugang undeine verbesserte Datentransparenz beiklinischen Studiendaten zugesagt hat(6). Gøtzsche und Jørgensen (2) gebendetailliert Aufschluss über ihre sieben-jährige Odyssee, bei der erst die öffentli-che Anschuldigung der EMA durch denEU-Ombudsmann, die EMA leide an ma-ladministration, die Herausgabe der
Daten zu Rimonabant und Orlistat be-wirkte. Die zuvor vorgebrachten Ein-wände der EMA gegen die Dateneinsichtlauteten: „protection of commercial in-terests, no over-riding public interest,the administrative burden involved“ (2).Durch die EMA unbrauchbar gemachteDatenblätter und das stete Nichtbeant-worten der Anfragen des EU-Ombuds-mannes ergänzten das wenig attraktiveBehördenbild (2).
Übersetzung des englischen Originaltex-tes durch den Verfasser.
Literatur1. Congress of the United States, Houseof Representatives, Committee on Go-vernment Reform: Statement of Rep.Henry A. Waxman: FDA’s 100th Anni-versary: http://www.henrywaxman.house.gov/UploadedFiles/fda.pdf.Washington, 27. Juni 2006. Zuletztgeprüft: 1. Dezember 2011.
2. Gotzsche PC, Jorgensen AW: Openingup data at the European MedicinesAgency. BMJ 2011; 342: d2686.
3. Barbui C, Baschirotto C, Cipriani A:EMA must improve the quality of itsclinical trial reports. BMJ 2011; 342:d2291.
4. Stavrakakis G: Bericht betreffend dieEntlastung zur Ausführung des Haus -haltsplans der Europäischen Arznei-mittel-Agentur fur das Haushaltsjahr2009 (C7-0233/2010 . 2010/2173(DEC)). Plenarsitzungsdokument A7-0153/2011 vom 15. April 2011. Zu-letzt geprüft: 1. Dezember 2011.
5. Wieseler B, McGauran N, Kaiser T:Still waiting for functional EU Clini-cal Trials Register. BMJ 2011; 342:d3834.
6. Pott A: EMA’s response to articles.BMJ 2011; 342: d3838.
InteressenkonflikteEin Interessenkonflikt wird vom Autorverneint.
Prof. Dr. rer. nat. habil. Thomas [email protected]
27Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
FAZIT
1. Ein European Public Assessment Re-port (EPAR) eines Arzneimittels kanndeutliche methodische Mängel auf-weisen und sollte daher nicht ohneweitere Prüfung als Referenz zitiertwerden.2. Die Fachkreise müssen der Arbeitder Zulassungsbehörden beständigekritische Aufmerksamkeit schenken.3. Die Zulassungsbehörden müssenDatentransparenz bei ihren Entschei-dungen gewährleisten.

28 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Einleitung und Definition
Endometriose ist eine chronische Er-krankung der Frau, die durch Ansied-lung von Endometriumgewebe außer-halb der Gebärmutter gekennzeichnetist. Es handelt sich nach der Myomatosisum die zweithäufigste Frauenkrankheit.Etwa 10% der Frauen im reproduktivenAlter leiden an Endometriose. InDeutschland allein erkranken jedes Jahrca. 40.000 Frauen (1). Mindestens zweiMillionen deutsche Frauen bedürfenderzeit einer chirurgischen und/odermedizinischen Behandlung ihrer Endo-metriose.Wegen des Behandlungsaufwands unddes häufig resultierenden Arbeitsausfallshat die Endometriose eine erheblichevolkswirtschaftliche Bedeutung (1).Trotz ihres benignen Charakters kannEndometriose Organgrenzen durch-wachsen und z. B. Blase, Darm, Harnlei-ter oder sogar die Lunge befallen und zuzyklischer Hämaturie, Darmblutungenoder Hämoptysis führen. Die Erkran-kung kann bei betroffenen Frauen anvielen Tagen im Jahr zu schmerzbeding-ter Arbeitsunfähigkeit und häufig zumehrfachen Operationen führen.
Leitsymptome der Erkrankung sindchronischer Unterbauchschmerz undSterilität. Allerdings korreliert die Inten-sität der Beschwerden nicht immer mitdem Stadium der Erkrankung (2).Eine Behandlung sollte nur bei sympto-matischer Endometriose erwogen wer-den (1). Ziel der Therapie ist es, die Be-schwerden zu lindern, eine Progressionder Erkrankung zu verhindern und dieLebensqualität zu verbessern. Der Evidenzgrad vieler medikamentöserTherapiestrategien ist bzgl. Wirksamkeitund Sicherheit bisher nur gering,während es andererseits nach alleinigemoperativem Eingriff fast regelhaft zumRezidiv kommt. Daher gilt es, die thera-peutischen Optionen unter Berücksich-tigung der Stärke der Beschwerden, derAusbreitung der Erkrankung, des Kinder-
wunschs, des Alters der Patientin, der me-dikamentösen Nebenwirkungen, derchirurgischen Risiken und der Kosten derBehandlung adäquat zu kombinieren.
TherapieÄtiologie und Pathogenese der Endome-triose sind nicht geklärt. Eine kausaleTherapie ist daher bislang nicht möglich.
Chronischer Unterbauchschmerz ohneKinderwunschBei der Behandlung dieser chronischenErkrankung sollte die Indikation zumoperativen Eingriff restriktiv gestellt wer-den, um Patientinnen nicht unnötig mitWiederholungseingriffen zu belasten. Diemedikamentöse Therapie sollte nachMöglichkeit im Vordergrund stehen.
1. SchmerztherapieFrauen mit mäßig starker Dysmenorr-hoe werden häufig vor einer definitivenhistologischen Sicherung der Erkran-kung zunächst empirisch mit Analgetikain Kombination mit oralen Kontrazepti-va, behandelt (2). Am häufigsten zumEinsatz kommen Nichtsteroidale Anti-phlogistika (NSAID) wie Ibuprofen oderDiclofenac.
2. Endokrine TherapieOrthotope wie ektope Endometrium-schleimhaut wird von Östrogen zur Pro-liferation angeregt, so dass endokrineTherapien, welche den hormonellen Zy-klus der Frau beeinflussen, die weitereAusbreitung der Erkrankung hemmenund bereits existierende Herde verklei-nern können.
• Kombinierte orale KontrazeptivaÖstrogen-Gestagen-Kombinations-präparate wie z. B. Valette® oder Maxim®
führen zu einer Dezidualisierung desEndometriums und der Endometriose-herde. Sie sind jedoch in der Indikation„Endometriose“ nicht zugelassen. FürFrauen, bei denen keine Kontraindikati-on für die Einnahme einer hormonellen
Kontrazeption besteht, und die eine Ver-hütung wünschen, ist der Einsatz einesmonophasisch kombinierten, oralenKontrazeptivums die erste therapeuti-sche Wahl. Führt dies nicht zu Beschwer-defreiheit, kann die Umstellung auf eineLangzykluseinnahme über drei bis vierMonate sinnvoll sein (2). Der Vorteil ge-genüber den Gestagenmonopräparatenliegt in der besseren Zyklus-Stabilität (3).
Eine Besserung der Symptomatik unteroraler Kontrazeption +/- NSAID ist nurfür die Zeit der Anwendung zu erwarten,da nach Absetzen der Therapie dieSchmerzen zumeist erneut auftreten. Kommt es nach 3–6 Monaten unter die-ser Kombinationstherapie nicht zu einerentscheidenden Beschwerdebesserung,sollte als Alternative eine Mono-Thera-pie mit Gestagenen in Betracht gezogenwerden. Ist die Diagnose der Endome-triose noch nicht gesichert, ist zu diesemZeitpunkt ein operativer Eingriff indi-ziert. Bei ihm sollte Material zur Gewin-nung einer histologischen Diagnose ge-wonnen werden. Gleichzeitig bestehtunter günstigen Umständen die Mög-lichkeit einer chirurgischen Sanierung.
• GestageneGestagene hemmen direkt die Prolifera-tion der Endometrioseherde durch eineDezidualisierung und spätere Atrophiederselben. Zusätzlich unterregulieren sieÖstrogenrezeptoren und hemmen zumTeil die Ausschüttung von hypophysärenGonadotropinen. Dadurch wird eine the-rapeutische Amenorrhoe erreicht, diesich für die Behandlung der Dysmenor -rhoe, Dyspareunie und des chronischenUnterbauchschmerzes bei der Endome-triose als effektiv erwiesen hat (3). Eine Therapie mit Gestagenen wird auchhäufig nach operativer Behandlung derEndometriose begonnen, um das be-schwerdefreie Intervall zu verlängern.Verschiedene Gestagenpräparate undApplikationen stehen zur Verfügung:Orale Gabe, (Cerazette®), Depot-Injekti-on (Depo-Clinovir®), subkutanes Im-
Die medikamentöse Behandlung der Endometriose
Das aktuelle Thema

29Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
plantat (Implanon®), Intrauterinpessar(Mirena®). Letzteres ist eine gute Optionfür die Behandlung von Dyspareunie auf-grund einer Endometriose des Septumrectovaginale, jedoch ebenfalls wie diegenannten Medikamente für diese Indi-kation ein Off-label-Use.
Im Mai 2010 erfolgte für den WirkstoffDienogest (Visanne®) die erste Zulas-sung eines Gestagenmonopräparates fürdie Behandlung der Endometriose inDeutschland. Es hat sich als effektive, si-chere und gut verträgliche Therapieopti-on für die Langzeitbehandlung der En-dometriose erwiesen. Wird die Gestagentherapie gut vertra-gen, darf sie für unbegrenzte Zeit ver-wendet werden. Allerdings führen hochdosierte Gestagenpräparate bei einigenFrauen zu Zwischenblutungen, depres-siver Verstimmung und Gewichtzunah-me. Diese Nebenwirkungen können einAbsetzen der Therapie notwendig ma-chen. Ein Beschwerderezidiv ist häufigdie Folge.
• GnRH-AnalogaAgonisten des Gonadotropin-ReleasingHormons (GnRH) hemmen die Produk-tion von Gonadotropinen in der Hypo-physe mit nachfolgender Hypoöstro-genämie, endometrialer Atrophie undAmenorrhoe. Sie sind effektiver als oraleKontrazeptiva oder Gestagene bei derBehandlung von Schmerzen bei fortge-schrittener Endometriose, aber auch ne-benwirkungsreicher (klimakterische Be-schwerden aller Abstufungen). In diesemStadium der Erkrankung sind GnRH-Agonisten postoperativ Mittel der Wahl,um die Rezidivrate zu verringern unddas beschwerdefreie Intervall zu verlän-gern (1). Ist die Diagnose der Endome-triose noch nicht gesichert, ist vor derGabe von GnRH-Analoga ein operativerEingriff zur Histologiegewinnung mitder Möglichkeit einer gleichzeitigen,chirurgischen Sanierung der Endome-triose empfehlenswert.Verschiedene für die Endometriose zu-gelassene Präparate stehen zur Verfü-gung: Täglich als Nasenspray (Metre-lef®), alle 4 Wochen als subkutanes Im-plantat (Zoladex-Gyn®Depot) oder alssubkutane oder intramuskuläre Injekti-on (Enantone-Gyn®, Decapeptyl – Gyn®),
auch drei-monatlich möglich. Die emp-fohlene Dauer der Therapie ist normaler-weise auf 6 Monate begrenzt, da die Ne-benwirkungen erheblich sein können.Bei starken Nebenwirkungen oder län-gerdauernder Therapie kann eine pro-tektive Begleitmedikation („add-back“)mit einer niedrig dosierten, kombinier-ten Hormontherapie sinnvoll sein. Sowerden Serumöstradiolwerte zwischen30–45 pg/ml erreicht, die eine ausrei-chende Knochenprotektion gewährlei-sten, ohne die Proliferation der Endome-triose zu sehr zu stimulieren (2). Eine Therapie über nur drei Monate istbezüglich der Schmerzhemmung genauso effektiv, jedoch ist das rezidivfreie In-tervall dann kürzer (1). Anschließendkann eine Erhaltungs-Therapie mit Ges-tagenen oder kombinierter oraler Kon-trazeption erfolgen. Eine präoperativeGnRH-Analoga-Gabe mit dem Ziel derVerkleinerung der Endometrioseherdeund Optimierung der Resektabilität hatsich als nicht hilfreich erwiesen und wirdnicht empfohlen (1).
3. Andere TherapienDanazol wurde früher für die Behandlungder Endometriose verwendet. AndrogeneNebenwirkungen wie Stimmveränderun-gen waren nicht selten. Nach Auslaufender Zulassung ist es seit Jahren inDeutschland nicht mehr auf dem Markt.
SterilitätFrauen mit Endometriose und Sterilitäthaben durch endokrine Therapien imVergleich zu Placebo keinen Vorteil inBezug auf die Rate spontaner Schwan-gerschaften (2). Daher werden diese fürdie Behandlung der endometriosebe-dingten Sterilität nicht empfohlen undsollten eine Kinderwunschbehandlungnicht verzögern (1).
Chirurgische IndikationenGrundsätzlich ist die Gewinnung einerHistologie anzustreben. Die Laparosko-pie gilt als Goldstandard, um die Diagno-se zu stellen und eine Sanierung der En-dometriose durchzuführen (1).In manchen Fällen ist eine primär chir-urgische Sanierung der Endometrioseangezeigt, beispielsweise bei akuten,schweren Schmerzzuständen, Hinweisauf Endometriosezysten im Ultraschall
und/oder zur Abklärung bei unklaremAdnextumor. Nicht alle endometriosebedingten Be-schwerden sprechen auf eine medika-mentöse Therapie an. Therapierefraktä-re Schmerzen müssen unter Umständenoperativ behandelt werden.
Literatur1. Deutsche Gesellschaft für Gynäkolo-gie und Geburtshilfe (DGGG), Deut-sche Gesellschaft für Allgemein- und Viszeralchirurgie e. V. (DGAV): Diagnostik und Therapie der Endo-metriose (S1-Leitlinie): http://www.awmf.org/leitlinien/detail/ll/015-045.html. AWMF-Leitlinien-RegisterNr. 015/045, Stand: 1. Mai 2010. Zu-letzt geprüft: 1. Dezember 2011.
2. Giudice LC: Clinical practice. Endo-metriosis. N Engl J Med 2010; 362:2389–2398.
3. Leidenberger FA, Strowitzki T, Ort-mann O (Hrsg.): Klinische Endokri-nologie für Frauenärzte. 4. Aufl.; Ber-lin, Heidelberg: Springer-Verlag,2009.
InteressenkonflikteEin Interessenkonflikt wird von der Au-torin und den KoautorInnen verneint.
Dr. med. Barbara de Oriol, MünchenPriv.-Doz. Dr. med. Vanadin Seifert-Klauss, MünchenUniv. Prof. Dr. med. Marion Kiechle,Mü[email protected]
FAZIT
Endometrioseschmerz lässt sich mitendokriner Therapie gut behandeln.Eine Besserung der Symptomatik ist al-lerdings nur für die Zeit der Anwendungzu erwarten. Bei endometriosebeding-ter Sterilität bringt die endokrine The-rapie keinen Vorteil und sollte eine Kin-derwunschbehandlung nicht verzö-gern. Die Rolle der Chirurgie besteht inder Diagnosesicherung sowie der The-rapie der fortgeschrittenen Endome-triose mit medikamentös nicht be-herrschbaren Beschwerden.

30 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Thrombozyten-Aggregationshemmer – eine ÜbersichtAcetylsalicylsäure • Clopidogrel • Prasugrel • Ticagrelor • Cangrelor
1. AcetylsalicylsäureAcetylsalicylsäure (ASS) wurde 1897 vonF. Hoffmann synthetisiert und 1899 vonBAYER als Aspirin® vermarktet – inhohen Dosen als Analgetikum. Die Wir-kung auf die Thrombozyten wurde 1954von Bounameaux erkannt. Die selektiveHemmung der Thromboxan (TXA2)-Synthese durch niedrige Dosen wurde1983 von FitzGerald beschrieben. Be-reits in der von 1981 bis 1983 durchge-führten Cottbus-Reinfarkt-Studie konn-te gezeigt werden, dass 30 mg ASS beider Reinfarktprophylaxe wirksamer undnebenwirkungsärmer sind als 60 oder1000 mg/Tag (1). Zwischen 1986 und1989 wurde demonstriert, dass Patien-ten mit transitorischen ischämischenAttacken (TIA) oder kleinen Schlaganfäl-len durch 30 mg ASS/Tag ebenso gut vorneuen vaskulären Ereignissen geschütztwurden wie durch 283 mg/Tag, und dassbei der geringeren Dosis natürlich auchweniger Nebenwirkungen auftraten (2).Als Thrombozyten-Aggregationshem-mer ist niedrig dosiertes ASS heute eta-bliert.
Trotzdem muss man akzeptieren, dass z.B. die Wirkung von 75 mg ASS/Tag imVergleich zu Placebo im Rahmen der Se-kundärprophylaxe nach cerebrovas-kulären ischämischen Ereignissen imErgebnis der SALT (Swedish AspirinLow-dose Trial)-Studie nur mäßig war(3). Der zusammengesetzte primäreEndpunkt (nichttödlicher oder tödlicherSchlaganfall, anderer Todesfall) tratnach 32 Monaten unter Placebo bei 25 %und unter ASS bei 20 % der Patienten auf(ARR = 5 %, NNT = 20). Das hat dieSuche nach anderen Thrombozyten-Ag-gregationshemmern beflügelt.
2. Clopidogrel, PrasugrelClopidogrel (z. B. Plavix®, zugelassen am15. Juli 1998) und Prasugrel (Efient®, zu-
gelassen am 25. Februar 2009) sind irre-versible Hemmer der Transmembran-domäne des P2Y12-ADP-Rezeptors. Siehemmen nicht kompetitiv die ADP-abhängige Thrombozytenaktivierung.Beide Substanzen sind „Prodrugs“, die inder Leber über Cytochromoxidasen (z. B.CYP1A2, CYP3A4, CYP2C19) erst zu denaktiven Wirkstoffen biotransformiertwerden müssen. Das führt zu einem ver-zögerten Wirkungseintritt, zu einer ge-netisch bedingten interindividuellen Va-riabilität der Thrombozytenhemmungund zu einer Vielzahl pharmakokine-tisch bedingter Interaktionen (Induktio-nen, Inhibitionen).
2.1. ClopidogrelIn der zulassungsrelevanten CAPRIE(Clopidogrel versus Aspirin in Patientswith Ischaemic Events)-Studie war Clo-pidogrel (75 mg/Tag) so effektiv wie ASS(325 mg/Tag) (4). Eingeschlossen waren19.185 Patienten mit ischämischem Hir-ninfarkt, Herzinfarkt oder symptomati-scher arterieller Verschlusskrankheitder Beine. Die Behandlungsdauer betrugzwischen einem Jahr und drei Jahren.Unter ASS traten 1.021 Ereignisse (töd-licher oder nicht- tödlicher Schlaganfalloder Herzinfarkt oder anderer vaskulä-rer Tod) auf, unter Clopidogrel 939 Er-eignisse. Dem entspricht eine jährlicheEreignisrate von 5,83 % bzw. 5,32 % unddamit einer absoluten Risikoreduktion(ARR) von 0,51 % (NNT = 196). Indu-strieabhängige Autoren verweisen gernauf die relative Risikoreduktion (RRR)von 8,7 % (0,51 % von 5,83 %) und aufdie statistisch signifikante Differenz p =0,045. Beide Aspekte sind jedoch klinischirrelevant. Da ASS und Clopidogrel aberunterschiedliche Wirkmechanismenhaben, könnte die Kombination beiderWirkstoffe hilfreich sein.
Es soll noch einmal explizit darauf ver-wiesen werden, dass Clopidogrel z. B.
durch das Cytochrom-P450-EnzymCYP2C19 biotransformiert und damitaktiviert wird (vgl. auch AVP 2009; 36/5:109–111). Bei Patienten, die Träger einesfunktionsreduzierten CYP2C19-Allelssind (in Europa etwa 30 % der Bevölke-rung), muss mit einer vermindertenWirksamkeit von Clopidogrel gerechnetwerden.
Patienten (< 45 Jahre, 92 % Männer) er-hielten nach einem ersten HerzinfarktClopidogrel (75 mg/Tag). Den primärenkombinierten Endpunkt (Tod, Herzin-farkt oder koronare Revaskularisation)erlitten 20,5 % der Träger des funktions-reduzierten Allels, während von denNicht-Trägern nur 5,9 % betroffen waren(5). Eine Hochdosistherapie (initial 600mg, gefolgt von 150 mg/Tag) ist jedocheiner Standardtherapie (75 mg/Tag) beiPatienten, die einer perkutanen Inter-vention (PCI) unterzogen wurden, nichtüberlegen (6). Neuerdings wurde aberauch beschrieben, dass bei Patienten mitakutem Koronarsyndrom oder Vor-hofflimmern Clopidogrel (im Vergleichzu Placebo) unabhängig vom CYP2C19-Polymorphismus wirkt (7). Es bleibenalso noch manche Fragen offen.
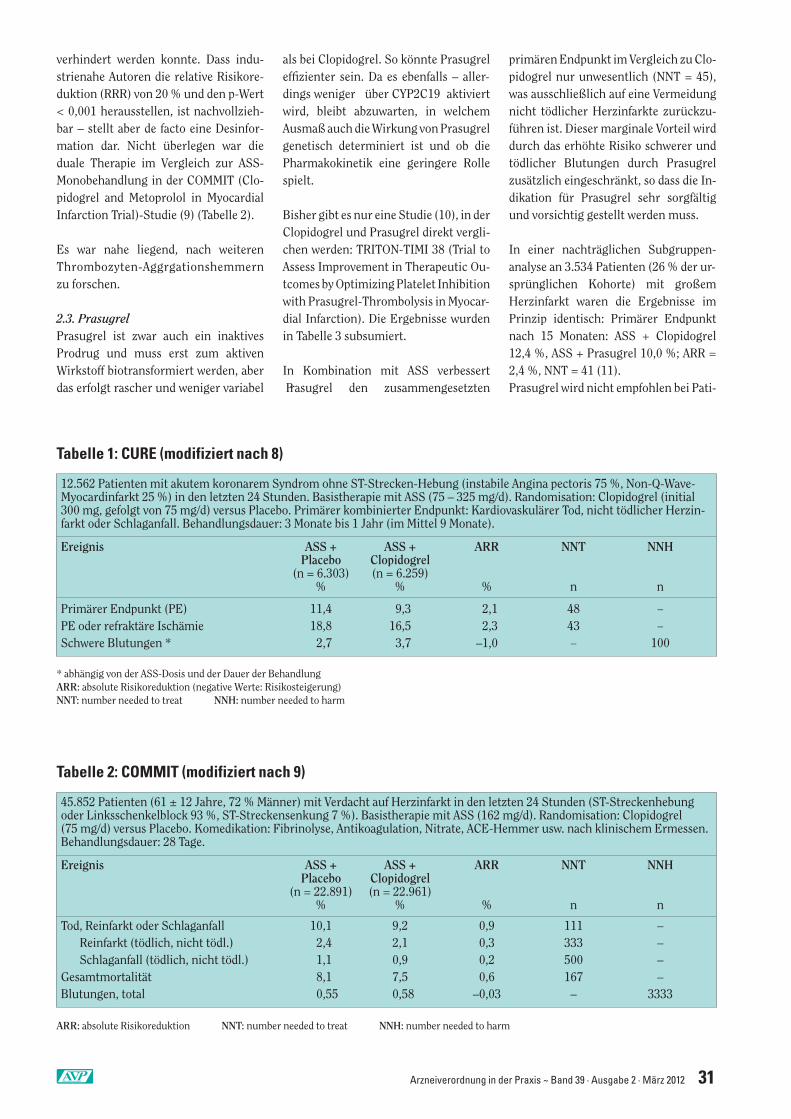
2.2. Clopidogrel plus ASSZur dualen Thrombozytenaggregations-hemmung wurden im letzten Jahrzehntetwa ein Dutzend großer Studien veröf-fentlicht. In der Regel war die Kombina-tion ASS plus Clopidogrel etwas wirksa-mer als die ASS-Monotherapie. Als Bei-spiel sei die erste einschlägige Studie (8)von 2001 hier aufgeführt: CURE (Clopi-dogrel in Unstable angina to prevent Re-current ischemic Events) (Tabelle 1).
Die ARR des primären Endpunktes be-trug durch die duale Therapie 2,1 %. Dasentspricht einem NNT-Wert von 48. Dasbedeutet, dass nur bei 1 von 48 behandel-ten Patienten ein zusätzliches Ereignis
Therapie aktuell
31Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
verhindert werden konnte. Dass indu-strienahe Autoren die relative Risikore-duktion (RRR) von 20 % und den p-Wert< 0,001 herausstellen, ist nachvollzieh-bar – stellt aber de facto eine Desinfor-mation dar. Nicht überlegen war dieduale Therapie im Vergleich zur ASS-Monobehandlung in der COMMIT (Clo-pidogrel and Metoprolol in MyocardialInfarction Trial)-Studie (9) (Tabelle 2).
Es war nahe liegend, nach weiterenThrombozyten-Aggrgationshemmernzu forschen.
2.3. PrasugrelPrasugrel ist zwar auch ein inaktivesProdrug und muss erst zum aktivenWirkstoff biotransformiert werden, aberdas erfolgt rascher und weniger variabel
als bei Clopidogrel. So könnte Prasugreleffizienter sein. Da es ebenfalls – aller-dings weniger � über CYP2C19 aktiviertwird, bleibt abzuwarten, in welchemAusmaß auch die Wirkung von Prasugrelgenetisch determiniert ist und ob diePharmakokinetik eine geringere Rollespielt.
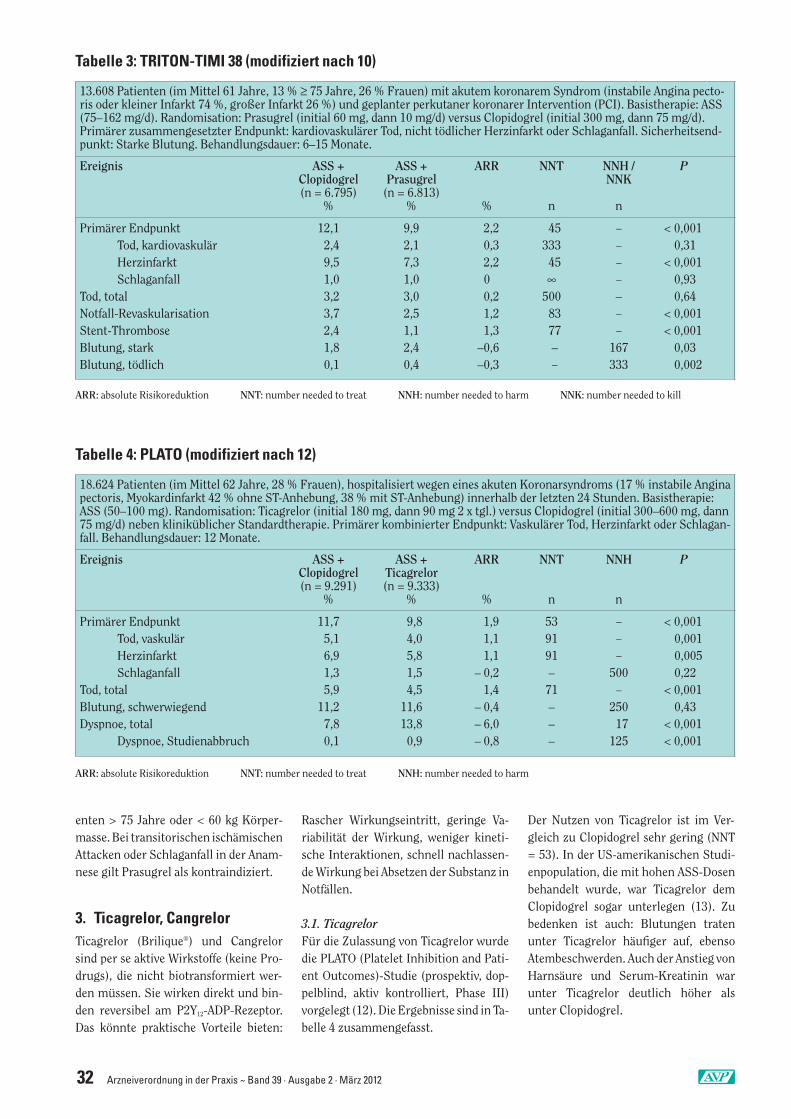
Bisher gibt es nur eine Studie (10), in derClopidogrel und Prasugrel direkt vergli-chen werden: TRITON-TIMI 38 (Trial toAssess Improvement in Therapeutic Ou-tcomes by Optimizing Platelet Inhibitionwith Prasugrel-Thrombolysis in Myocar-dial Infarction). Die Ergebnisse wurdenin Tabelle 3 subsumiert. In Kombination mit ASS verbessert Prasugrel den zusammengesetzten
primären Endpunkt im Vergleich zu Clo-pidogrel nur unwesentlich (NNT = 45),was ausschließlich auf eine Vermeidungnicht tödlicher Herzinfarkte zurückzu-führen ist. Dieser marginale Vorteil wirddurch das erhöhte Risiko schwerer undtödlicher Blutungen durch Prasugrelzusätzlich eingeschränkt, so dass die In-dikation für Prasugrel sehr sorgfältigund vorsichtig gestellt werden muss.
In einer nachträglichen Subgruppen-analyse an 3.534 Patienten (26 % der ur-sprünglichen Kohorte) mit großemHerzinfarkt waren die Ergebnisse imPrinzip identisch: Primärer Endpunktnach 15 Monaten: ASS + Clopidogrel12,4 %, ASS + Prasugrel 10,0 %; ARR =2,4 %, NNT = 41 (11).Prasugrel wird nicht empfohlen bei Pati-
Tabelle 1: CURE (modifiziert nach 8)
12.562 Patienten mit akutem koronarem Syndrom ohne ST-Strecken-Hebung (instabile Angina pectoris 75 %, Non-Q-Wave-Myocardinfarkt 25 %) in den letzten 24 Stunden. Basistherapie mit ASS (75 – 325 mg/d). Randomisation: Clopidogrel (initial300 mg, gefolgt von 75 mg/d) versus Placebo. Primärer kombinierter Endpunkt: Kardiovaskulärer Tod, nicht tödlicher Herzin-farkt oder Schlaganfall. Behandlungsdauer: 3 Monate bis 1 Jahr (im Mittel 9 Monate).
Ereignis ASS + ASS + ARR NNT NNHPlacebo Clopidogrel(n = 6.303) (n = 6.259)
% % % n n
Primärer Endpunkt (PE) 11,4 9,3 2,1 48 –PE oder refraktäre Ischämie 18,8 16,5 2,3 43 –Schwere Blutungen * 2,7 3,7 –1,0 – 100
* abhängig von der ASS-Dosis und der Dauer der BehandlungARR: absolute Risikoreduktion (negative Werte: Risikosteigerung)NNT: number needed to treat NNH: number needed to harm
Tabelle 2: COMMIT (modifiziert nach 9)
45.852 Patienten (61 ± 12 Jahre, 72 % Männer) mit Verdacht auf Herzinfarkt in den letzten 24 Stunden (ST-Streckenhebungoder Linksschenkelblock 93 %, ST-Streckensenkung 7 %). Basistherapie mit ASS (162 mg/d). Randomisation: Clopidogrel (75 mg/d) versus Placebo. Komedikation: Fibrinolyse, Antikoagulation, Nitrate, ACE-Hemmer usw. nach klinischem Ermessen.Behandlungsdauer: 28 Tage.
Ereignis ASS + ASS + ARR NNT NNHPlacebo Clopidogrel
(n = 22.891) (n = 22.961)% % % n n
Tod, Reinfarkt oder Schlaganfall 10,1 9,2 0,9 111 –Reinfarkt (tödlich, nicht tödl.) 2,4 2,1 0,3 333 –Schlaganfall (tödlich, nicht tödl.) 1,1 0,9 0,2 500 –
Gesamtmortalität 8,1 7,5 0,6 167 –Blutungen, total 0,55 0,58 –0,03 – 3333
ARR: absolute Risikoreduktion NNT: number needed to treat NNH: number needed to harm
32 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
enten > 75 Jahre oder < 60 kg Körper-masse. Bei transitorischen ischämischenAttacken oder Schlaganfall in der Anam-nese gilt Prasugrel als kontraindiziert.
3. Ticagrelor, CangrelorTicagrelor (Brilique®) und Cangrelorsind per se aktive Wirkstoffe (keine Pro-drugs), die nicht biotransformiert wer-den müssen. Sie wirken direkt und bin-den reversibel am P2Y12-ADP-Rezeptor.Das könnte praktische Vorteile bieten:
Rascher Wirkungseintritt, geringe Va-riabilität der Wirkung, weniger kineti-sche Interaktionen, schnell nachlassen-de Wirkung bei Absetzen der Substanz inNotfällen.
3.1. TicagrelorFür die Zulassung von Ticagrelor wurdedie PLATO (Platelet Inhibition and Pati-ent Outcomes)-Studie (prospektiv, dop-pelblind, aktiv kontrolliert, Phase III)vorgelegt (12). Die Ergebnisse sind in Ta-belle 4 zusammengefasst.
Der Nutzen von Ticagrelor ist im Ver-gleich zu Clopidogrel sehr gering (NNT= 53). In der US-amerikanischen Studi-enpopulation, die mit hohen ASS-Dosenbehandelt wurde, war Ticagrelor demClopidogrel sogar unterlegen (13). Zubedenken ist auch: Blutungen tratenunter Ticagrelor häufiger auf, ebensoAtembeschwerden. Auch der Anstieg vonHarnsäure und Serum-Kreatinin warunter Ticagrelor deutlich höher alsunter Clopidogrel.
Tabelle 3: TRITON-TIMI 38 (modifiziert nach 10)
13.608 Patienten (im Mittel 61 Jahre, 13 % ≥ 75 Jahre, 26 % Frauen) mit akutem koronarem Syndrom (instabile Angina pecto-ris oder kleiner Infarkt 74 %, großer Infarkt 26 %) und geplanter perkutaner koronarer Intervention (PCI). Basistherapie: ASS(75–162 mg/d). Randomisation: Prasugrel (initial 60 mg, dann 10 mg/d) versus Clopidogrel (initial 300 mg, dann 75 mg/d).Primärer zusammengesetzter Endpunkt: kardiovaskulärer Tod, nicht tödlicher Herzinfarkt oder Schlaganfall. Sicherheitsend-punkt: Starke Blutung. Behandlungsdauer: 6–15 Monate.
Ereignis ASS + ASS + ARR NNT NNH / PClopidogrel Prasugrel NNK(n = 6.795) (n = 6.813)
% % % n n
Primärer Endpunkt 12,1 9,9 2,2 45 – < 0,001Tod, kardiovaskulär 2,4 2,1 0,3 333 – 0,31Herzinfarkt 9,5 7,3 2,2 45 – < 0,001Schlaganfall 1,0 1,0 0 ∞ – 0,93
Tod, total 3,2 3,0 0,2 500 – 0,64Notfall-Revaskularisation 3,7 2,5 1,2 83 – < 0,001Stent-Thrombose 2,4 1,1 1,3 77 – < 0,001Blutung, stark 1,8 2,4 –0,6 – 167 0,03Blutung, tödlich 0,1 0,4 –0,3 – 333 0,002
ARR: absolute Risikoreduktion NNT: number needed to treat NNH: number needed to harm NNK: number needed to kill
Tabelle 4: PLATO (modifiziert nach 12)
18.624 Patienten (im Mittel 62 Jahre, 28 % Frauen), hospitalisiert wegen eines akuten Koronarsyndroms (17 % instabile Anginapectoris, Myokardinfarkt 42 % ohne ST-Anhebung, 38 % mit ST-Anhebung) innerhalb der letzten 24 Stunden. Basistherapie:ASS (50–100 mg). Randomisation: Ticagrelor (initial 180 mg, dann 90 mg 2 x tgl.) versus Clopidogrel (initial 300–600 mg, dann75 mg/d) neben kliniküblicher Standardtherapie. Primärer kombinierter Endpunkt: Vaskulärer Tod, Herzinfarkt oder Schlagan-fall. Behandlungsdauer: 12 Monate.
Ereignis ASS + ASS + ARR NNT NNH PClopidogrel Ticagrelor(n = 9.291) (n = 9.333)
% % % n n
Primärer Endpunkt 11,7 9,8 1,9 53 – < 0,001Tod, vaskulär 5,1 4,0 1,1 91 – 0,001Herzinfarkt 6,9 5,8 1,1 91 – 0,005Schlaganfall 1,3 1,5 – 0,2 – 500 0,22
Tod, total 5,9 4,5 1,4 71 – < 0,001Blutung, schwerwiegend 11,2 11,6 – 0,4 – 250 0,43Dyspnoe, total 7,8 13,8 – 6,0 – 17 < 0,001
Dyspnoe, Studienabbruch 0,1 0,9 – 0,8 – 125 < 0,001
ARR: absolute Risikoreduktion NNT: number needed to treat NNH: number needed to harm

33Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Zwei Subgruppenanalysen belegen, dasssich an dieser prinzipiellen Aussagenichts ändert, gleichgültig, ob Patientenuntersucht wurden, bei denen eine in-tensive Strategie vorgesehen war (14)oder die einem nicht-invasiven Manage-ment unterzogen wurden (15).
Ein wesentliches Problem bei der prakti-schen Anwendung dürfte die Tatsachesein, dass Ticagrelor auf Grund der rever-siblen Bindung an die Thrombozytenund wegen der kurzen Halbwertszeit (7 –8 Stunden) zweimal täglich eingenom-men werden muss. Im Vergleich zu Clo-pidogrel sind die Risiken für den Patien-ten bei Ticagrelor höher als die Chancen.
Trotzdem hat das Institut für Qualitätund Wirtschaftlichkeit im Gesundheits-wesen (IQWiG) dem Ticagrelor einen„beträchtlichen Zusatznutzen“ für Pati-enten mit Herzinfarkt ohne ST-Streckenhebung und für Patienten mitinstabiler Angina pectoris bescheinigt.
Ob diese Entscheidung angemessen ist,mag jeder Leser selbst entscheiden. Beidieser definierten Subgruppe wurde derprimäre Endpunkt unter Clopidogrelvon 11,8 % und unter Ticagrelor von 9,7% der Patienten erreicht. Das entsprichteiner ARR = 2,1 %, einem NNT = 48 undeinem NTN (number treated needlessly)= 47.
3.2. CangrelorCangrelor ist der erste direkte, reversibleP2Y12-Rezeptor-Antagonist, der intra-venös appliziert werden muss. Die Wir-kung tritt unmittelbar nach Applikationeines Bolus ein und ist etwa eine Stundenach Beendigung der Infusion (bis zu 72Stunden) wieder abgeklungen. Es mussabgewartet werden, ob der Wirkstoff wirk-lich gut geeignet ist zur Behandlung vonPatienten mit akutem Koronarsyndrom.
In zwei CHAMPION (Cangrelor versusStandard Therapy to Achieve OptimalManagement of Platelet Inhibition)-Stu-dien war Cangrelor dem Clopidogreloder dem Placebo nicht überlegen.
In CHAMPION PCI wurde Cangrelor(Bolus 30 µg/kg, Infusion 4 µg/kg/min)oder Clopidogrel (600 mg) vor der perku-
tanen koronaren Intervention appliziert(16). In CHAMPION PLATFORM erhiel-ten die Patienten während der Prozedurentweder Cangrelor- oder Placebo-Infu-sionen und anschließend entweder Pla-cebo bzw. Clopidogrel (17). Die weitereEntwicklung muss abgewartet werden.
Literatur1. Forster W: [Prevention of reinfarc-
tion with 100 mg or 30 mg ASSdaily?]. Z Kardiol 1995; 84: 335–343.
2. A comparison of two doses of aspirin(30 mg vs. 283 mg a day) in patientsafter a transient ischemic attack orminor ischemic stroke. The DutchTIA Trial Study Group. N Engl J Med1991; 325: 1261–1266.
3. Swedish Aspirin Low-Dose Trial(SALT) of 75 mg aspirin as secondaryprophylaxis after cerebrovascularischaemic events. The SALT Collabo-rative Group. Lancet 1991; 338:1345–1349.
4. A randomised, blinded, trial of clopi-dogrel versus aspirin in patients atrisk of ischaemic events (CAPRIE).CAPRIE Steering Committee. Lancet1996; 348: 1329–1339.
5. Collet JP, Hulot JS, Pena A et al.:Cytochrome P450 2C19 polymor-phism in young patients treated withclopidogrel after myocardial infarc-tion: a cohort study. Lancet 2009;373: 309–317.
6. Price MJ, Berger PB, Teirstein PS etal.: Standard- vs high-dose clopido-grel based on platelet functiontesting after percutaneous coronaryintervention: the GRAVITAS rando-mized trial. JAMA 2011; 305:1097–1105.
7. Pare G, Mehta SR, Yusuf S et al.: Ef-fects of CYP2C19 genotype on outco-mes of clopidogrel treatment. N EnglJ Med 2010; 363: 1704–1714.
8. Yusuf S, Zhao F, Mehta SR et al.: Ef-fects of clopidogrel in addition toaspirin in patients with acute coro-nary syndromes without ST-segmentelevation. N Engl J Med 2001; 345:494–502.
9. Chen ZM, Jiang LX, Chen YP et al.:Addition of clopidogrel to aspirin in45,852 patients with acute myocardi-al infarction: randomised placebo-controlled trial. Lancet 2005; 366:1607–1621.
10. Wiviott SD, Braunwald E, McCabeCH et al.: Prasugrel versus clopido-grel in patients with acute coronarysyndromes. N Engl J Med 2007; 357:2001–2015.
11. Montalescot G, Wiviott SD, Braun-wald E et al.: Prasugrel comparedwith clopidogrel in patients under-going percutaneous coronary inter-vention for ST-elevation myocardialinfarction (TRITON-TIMI 38):double-blind, randomised controlledtrial. Lancet 2009; 373: 723–731.
FAZIT
Acetylsalicylsäure in niedriger Dosie-rung (< 100 mg/Tag) ist ein nützlicher undpreiswerter Thrombozyten-Aggregati-onshemmer bei Patienten mit Herzinfarktoder Schlaganfall. Eine Clopidogrel-Mo-notherapie ist nicht effektiver. Die Kom-bination von ASS und Clopidogrel ist nurmarginal wirksamer und brachte nichtden erhofften Durchbruch. Eine definiti-ve Bewertung des Nutzens von Prasu-grel im Vergleich zu Clopidogrel ist nochnicht möglich, da bisher nur die TRITON-TIMI 38-Studie vorliegt, in der keine ent-scheidenden Vorteile von Prasugrel ge-zeigt werden konnten.
Der in der PLATO-Studie demonstriertegeringe Nutzen von Ticagrelor gegenü-ber Clopidogrel ist ebenfalls noch nichtendgültig zu bewerten. Vor allem sind die„überraschenden“ neuen Nebenwir-kungen zu beachten.
Cangrelor befindet sich noch in der Ent-wicklungsphase.
Die duale Thrombozyten-Aggregations-hemmung mit ASS plus Clopidogrelbleibt solange Goldstandard, bis in wei-teren Studien ein relevanter Nutzen vonPrasugrel und Ticagrelor belegt wird.

34 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Osteoporose und Niereninsuffizienzwerden mit zunehmendem Alter häufi-ger: Je älter der Patient desto größer dasAusmaß der Osteoporose und destogrößer die Gefahr von Knochenbrüchen.In ähnlichem Ausmaß steigt mit zuneh-mendem Alter auch die Wahrscheinlich-keit für Diabetes und Hypertonie, beidesRisikofaktoren für die Entstehung einerchronischen Nierenerkrankung.
In der Praxis steht man daher häufig vordem Problem, Patienten mit Osteoporo-se behandeln zu müssen, deren Nieren-funktion eingeschränkt ist (GFR < 60ml/Min = Chronische NiereninsuffizienzStadium 3–5). Über 60 % der Frauen mitOsteoporose haben eine Niereninsuffizi-enz im Stadium 3 (CKD1 3, glomeruläreFiltrationsrate (GFR) zwischen 60–30ml/Min), 23 % Stadium 4 (CKD 4, GFR30–15 ml/Min). In einem Übersichtsarti-kel (1) wird der gegenwärtige Wissens-stand zusammengefasst.
Osteoporose bedeutet eine Verringerungder Knochendichte (BMD), die Mikroar-chitektur der Trabekel ist gestört, Mengeund Zusammensetzung verschiedenerEiweiße im Knochen sind verändert.Insgesamt überwiegt der Knochenabbauden Knochenaufbau. Renal bedingteKnochenerkrankungen hängen zusam-men mit Veränderungen des Calcium-und Phosphathaushaltes, des Parathor-mons und des Vitamin D Metabolismus.Renal bedingte Knochenerkrankungenführen zu unterschiedlichen Verände-rungen des Knochenumsatzes, der Mi-neralisation und des Volumens. Sie wer-den daher knochenhistologisch nachdem Ausmaß dieser Veränderungen ein-geteilt (Tabelle 1). Besonders gefürchtetsind renal bedingte Knochenerkrankun-gen mit niedrigem Knochenumsatz, dadiese therapeutisch praktisch nicht be-einflussbar sind und eine sehr hoheFrakturrate bedingen.
Die Osteoporosetherapie zielt darauf ab,die Knochenmasse zu verstärken unddas Frakturrisiko zu vermindern,während die Behandlung der renalenOsteopathie darauf ausgerichtet ist, dasZusammenspiel von Parathormon, Vita-min D und den Calcium-Phosphathaus-halt zu normalisieren.
Therapiemöglichkeiten (sieheTabelle 2)
Veränderungen des Lebensstils
Körperliche AktivitätSelbst Laufen in geringem Tempo kanndie Knochendichte verstärken. Muskel -aufbautraining kann wahrscheinlichauch bei niereninsuffizienten Patientensinnvoll sein, wurde dafür aber bei dieserPatientengruppe nicht untersucht. DiePrävention von Stürzen, indem mansturzauslösende Medikamente vermei-det und die Umgebung verändert (Licht,Stolperfallen), ist bei allen Patienten-gruppen sinnvoll. Mehr als 30 % aller Pa-tienten über 60 stürzen einmal pro Jahr.
Osteoporosetherapie bei verschlechterter Nierenfunktion
12. Wallentin L, Becker RC, Budaj A etal.: Ticagrelor versus clopidogrel inpatients with acute coronary syndro-mes. N Engl J Med 2009; 361:1045–1057.
13. Arzneimittelkommission der deut-schen Ärzteschaft: Brilique™ (Ticag-relor). Arzneiverordnung in der Pra-xis (AVP) 2011; 38: 65–67.
14. Cannon CP, Harrington RA, James Set al.: Comparison of ticagrelor withclopidogrel in patients with a plan-ned invasive strategy for acute coro-nary syndromes (PLATO): a randomi-
sed double-blind study. Lancet 2010;375: 283–293.
15. James SK, Roe MT, Cannon CP et al.:Ticagrelor versus clopidogrel in pati-ents with acute coronary syndromesintended for non-invasive manage-ment: substudy from prospectiverandomised PLATelet inhibition andpatient Outcomes (PLATO) trial. BMJ2011; 342: d3527.
16. Harrington RA, Stone GW, McNultyS et al.: Platelet inhibition with can-grelor in patients undergoing PCI. NEngl J Med 2009; 361: 2318–2329.
17. Bhatt DL, Lincoff AM, Gibson CM etal.: Intravenous platelet blockadewith cangrelor during PCI. N Engl JMed 2009; 361: 2330–2341.
InteressenkonflikteEin Interessenkonflikt wird vom Autorverneint.
Prof. em. Dr. med. Frank P. Meyer, Groß[email protected]
Tabelle 1: Renal bedingte Knochenerkrankungen: Knochenhistolo-gische Einteilung
Knochenumsatz (T) Mineralisation (M) Volumen (V)
Hoch Normal Hoch
Normal Abnormal Normal
Niedrig Niedrig 1 Chronic kidney disease = CKD

35Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
ErnährungEine normale Kalorien- und Eiweißauf-nahme ist wichtig für den Erhalt derMuskelmasse und damit auch der Kno-chenmasse. Für niereninsuffiziente Pati-enten ist leider häufig eine Phosphatre-striktion notwendig. Dies bedeutet auto-matisch Eiweißrestriktion und damitVerlust an Muskelmasse und zunehmen-de Osteoporose.
Calcium und Vitamin D25-Hydroxyvitamin-D-Mangel ist beiniereninsuffizienten Patienten häufigerals in der Normalbevölkerung. Wird dasVitamin ersetzt, müssen aber nicht nurder Knochenhaushalt sondern bei Nie-renkranken auch Gewebscalcificationenbeachtet werden. Mögliche pleiotropeEffekte des Vitamin D sind noch nichtdurch randomisierte, prospektive Studi-en gesichert.
Eine adäquate Calciumzufuhr ist wich-tig für die Knochenmineralisation. Obdamit ein erhöhtes kardiovaskuläres Ri-siko einhergeht, wurde vermutet, ist
aber nicht gesichert. Niereninsuffizien-te sollten nicht mehr als 2 g Calcium proTag zu sich nehmen.
PharmakotherapieDie meisten Medikamente zur Therapieder Osteoporose sind für Patienten mitNiereninsuffizienz im Stadium 3 (GFR >30 ml/Min) untersucht. Allerdings nichtbei Vorliegen einer renal bedingten Kno-chenerkrankung, die leider auch schonbei diesem Grad der Nierenschwäche be-stehen kann.
Antiresorptive MedikamentePrinzipiell wird bei Patienten mit CKD3–5 vor Gabe dieser Medikamente beidem Verdacht auf das Vorliegen einer re-nalen Osteopathie eine Knochenbiopsiegefordert. Dieser Verdacht entsteht, wennlaborchemische Veränderungen des Parat -hormons, Calcium- und/oder Phosphat-haushaltes bestehen. Diese Parametersollten daher vor der Gabe antiresorptiverMedikamente bestimmt werden.
BisphosphonateBisphosphonate werden oral schlecht re-sorbiert. An den Knochen werden sie miteiner Halbwertszeit von mehr als 10 Jah-ren gebunden. Der größte Teil nichtge-bundenen Medikaments wird glome-rulär filtriert und tubulär sezerniert.Oral verabreichte Bisphosphonate sindoberhalb einer GFR von 30 ml/Min wohlnicht nephrotoxisch. Demgegenüberwird bei intravenöser Gabe diese uner-wünschte Arzneimittelwirkung durch-aus beschrieben. Ibandronat soll etwasweniger nephrotoxisch sein als die übri-gen intravenös verabreichbaren Bis-phosphonate. Außerdem besteht ein Ri-siko für Hypocalcämien.
Östrogene/HormontherapienDie Gabe von Östrogenen kann die Kno-chendichte vermehren und das Fraktur-risiko bei postmenopausalen Frauensenken. Demgegenüber muss allerdingsdas erhöhte Risiko für Brustkrebs, kar-diovaskuläre Erkrankungen und Schlag-anfall gesetzt werden. Die Medikamen-tenspiegel bei Niereninsuffizienten fürÖstrogene sind etwas erhöht und legeneine niedrige Dosierung nahe.
Selektive ÖstrogenrezeptorstimulatorenRaloxifen wird bei Niereninsuffizienzvermindert ausgeschieden. Dies führtaber nicht zu einer erhöhten Rate an un-erwünschten Arzneimittelwirkungen,behält man die normale Dosis bei. Dasdurch die Therapie generell erhöhte Ri-siko für thromboembolische Komplika-tionen ist jedoch zu beachten.
Calcitonin wurde bei Patienten mit Nie-renerkrankungen nicht speziell unter-sucht.
Anabole MedikamenteTeriparatidTeriparatid ist ein N-terminales Frag-ment des menschlichen Parathormonsund scheint dessen anabole Eigenschaf-ten zu besitzen. Untersucht wurde es biszu einer GFR von 30 ml/Min. Bei allenPatienten kam es zu vorübergehendenErhöhungen des Serumcalciums. DasVorliegen eines primären Hyperparathy-reoidismus muss vorher ausgeschlossenwerden.
Tabelle 2: Therapiestrategien
Intervention Kommentar zur Niereninsuffizienz
Lebensstilveränderungen
Ernährung
Phosphatrestriktion Je nach Grad der Niereninsuffizienz
Calcium und Vitamin-D Ersatz Alle Stadien
Körperliche Aktivität
Krafttraining????? Alle Stadien
Sturzprävention Alle Stadien
Pharmakotherapie
Antiresorptiv
Bisphosphonate CKD 3-5, Knochenbiopsie vor Einsatz?
Östrogene Risiko der UAW
Raloxifen CKD 1-3 mit normalen Laborparametern
Calcitonin Keine Studien
Alkali Salze Alle Stadien
Nitrate Alle Stadien?
Anabole Substanzen
Teriparatide CKD 1-3 mit normalen Laborparametern
Testosteron Keine Studien
Nandrolon Untersucht bei CKD 3-5 für Osteoporose, nicht inBezug auf renal bedingte Knochenerkrankungen
Nitrate (ISDN) Alle Stadien?

36 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Testosteron45 % der Patienten mit CKD 4 und mehrals 60 % der Patienten mit CKD 5 habenniedrig normale oder erniedrigte Testo-steronwerte. Da Testosteron nur fraglichauf die Knochendichte wirkt und zu ver-mehrter Flüssigkeitsretention führenkann, wird man es wohl besser nicht ver-wenden.
NandrolonNandrolon ist ein Abkömmling desTestosterons und kann bei CKD 4–5 dieKnochendichte verstärken.
Künftige Therapiemöglichkeiten
Korrektur der metabolischen AzidoseTierexperimentelle Untersuchungenund Studien an gesunden Patienten undbei Kindern mit distaler renaler tubulä-rer Azidose zeigen positive Auswirkun-gen auf den Knochenhaushalt.
NitrateIn einer kleineren Studie war Isosorbid-dinitrat 60 mg/Tag genauso wirksam fürdie Knochendichte wie Alendronat 70mg/Woche. Bei chronischer Nierenin-suffizienz ist die Wirksamkeit jedochnoch nicht untersucht.
Literatur1.Gordon PL, Frassetto LA: Manage-ment of osteoporosis in CKD Stages 3to 5. Am J Kidney Dis 2010; 55:941–956.
2.Kidney Disease: Improving Global Ou-tcomes (KDIGO): http://www.kdigo.org. Zuletzt geprüft: 20. September2011.
InteressenkonflikteEin Interessenkonflikt wird vom Autorverneint.
Dr. med. Michael [email protected]
FAZIT
Patienten oberhalb einer Nierenfunktionvon einer GFR > 30 ml/Min und normalemCalcium/Phosphathaushalt und Para-thormon können nach Standardvorga-ben behandelt werden. Für alle anderenPatienten sind aussagekräftige Studienselten und die Behandlung folgt weitge-hend Expertenempfehlungen (2). Liegtdie glomeruläre Filtrationsrate um 30ml/Min oder niedriger, so sollte dieOsteoporosetherapie mit der Behand-lung der renalen Osteopathie abge-stimmt werden, also in der Regel mit dembehandelnden Nephrologen. Bisphos-phonate sollten bei CKD 3 nur bei Vorlie-
gen normaler Laborparameter in Bezugauf Parathormon und Calcium- undPhosphathaushalt eingesetzt werden.Alle Patienten mit CKD 3 vor einer ge-planten Bisphosphonat-Therapie einerKnochenbiopsie zuzuführen, wird in derPraxis nicht möglich sein. Zu bedenkensind dennoch die sehr lange Halbwerts-zeit im Knochen sowie die Progressi-onstendenz der Nierenerkrankung. Ge-fürchtet ist eine adyname renale Kno-chenerkrankung, die praktisch nicht be-handelbar ist und evtl. durch Bisphos-phonate verstärkt werden kann.
Die Sarkoidose ist eine inflammatori-sche, granulomatöse Systemerkrankungunklarer Ätiologie. Aufgrund des vorwie-genden Befalls von Lunge, Haut undAugen wird eine umweltbedingte Anti-genaufnahme mit unkontrollierter zell-vermittelter Immunreaktion und Gra-nulombildung angenommen. Als rele-vante Antigene werden nichtinfektiöseBestandteile von Mykobakterien undPropionibakterien diskutiert. Die we-sentliche zugrundeliegende immunolo-gische Reaktion ist T-Zell-vermittelt.
Die Prävalenz beträgt etwa 10 –40/100.000, wobei die höchste Prävalenz
und ein eher chronischer Verlauf beiAfroamerikanern zu beobachten ist. DerAltersgipfel liegt zwischen dem 30. und40. Lebensjahr und eine familiäre Häu-fung ist möglich.
Manifestationen, Klinik und Erkennung der ErkrankungDie Symptomentrias aus bihilärerLymph adenopathie, Sprunggelenksar-thritis und Erythema nodosum kenn-zeichnet die akute Sarkoidose und wirdauch als Löfgren-Syndrom bezeichnet.Sie macht etwa 10 % bis höchstens 20 %der Sarkoidosen aus und betrifft vor-
nehmlich junge Frauen. Diese Form derManifestation hat eine gute Prognosemit hoher spontaner Remissionsrate.Eine histologische Sicherung ist in derRegel nicht notwendig, und die Therapiebeschränkt sich auf symptomatischeMaßnahmen mit Gaben von nichtstero-idalen Antiphlogistika.
Weitere Manifestationen der Sarkoidose,die sich nicht in dieser akuten Form prä-sentieren, können prinzipiell jedesOrgan betreffen. Ein thorakaler Befallfindet sich in 90 % der Patienten mit Sar-koidose, wobei eine bilaterale mediasti-nale und hiläre Lymphadenopathie mit
Sarkoidose – Erkennen und Behandeln

37Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
nahezu 98–100 % praktisch immer vor-handen ist. Ein zusätzlicher Befall desLungenparenchyms lässt sich mittelstransbronchialer Biopsie histologischmit epitheloidzelligen Granulomen inmehr als 50 % der Fälle nachweisen,ohne dass dies radiologisch (Röntgensta-dium II) auffällig sein muss. Lungen-funktionell sind dementsprechend eineEinschränkung der Diffusionskapaziätund eine restriktive Ventilationsstörungkennzeichnend, wenngleich sich jedochauch gelegentlich obstruktive Ventilati-onsstörungen finden lassen. Eine pul-monale Hypertonie kann in fortgeschrit-tenen Fällen bei schwerer Hypoxämieoder bei Einbezug der pulmonalarteriel-len Strombahn auftreten. Ein kutanerBefall, sei es isoliert oder multifokal,meist nodulär oder makulo-papillär, istin mindestens 30 % der Fälle vorhanden.Da diese Lokalisation für eine Biopsiemit histologischer Sicherung natur-gemäß am einfachsten zugänglich ist, isteine sorgfältige Hautinspektion beieinem entsprechenden Verdacht obligat.Mit einem Augenbefall ist in ca. 25 % derFälle zu rechnen, wobei eine chronischeanteriore Uveitis die häufigste okuläreManifestation ist und der allgemeinenManifestation und Diagnosestellungüber lange Zeit vorausgehen kann.Daher ist eine ophthalmologische Unter-suchung mit gezielter Fragestellung beientsprechendem Verdacht ebenfalls obli-gat. Eine kardiale Beteiligung der Sarko-idose findet sich autoptisch in wenig-stens 25 % der Sarkoidosefälle. Klinischstehen meist Herzrhythmusstörungenim Vordergrund. Man muss also den Pa-tienten genau befragen. Ein Ruhe- undLangzeit-EKG ist angezeigt. Umgekehrtsollte eine nichtischämische Kar-diomyopathie, einhergehend mit Rhyth-musstörungen, an eine Sarkoidose den-ken lassen. Unter den bildgebenden Ver-fahren ist in diesem Zusammenhang diekardiale MRT mit einer vergleichsweiseguten Spezifität einsetzbar. Die PETzeigt die höhere Sensitivität. Dieses Ver-fahren eignet sich, wie neuere Daten zei-gen, generell auch gut zum Nachweisokkulter anderweitiger inflammatori-scher Organbeteiligungen der Sarkoido-se. Ein Befall des Nervensystems findetsich zumindest in Obduktionsstudien inbis zu 25 %, klinisch in 10–17 % auch
ohne anderweitige Organmanifestation.Eine Beteiligung der Hirnnerven, insbe-sondere von Fazialis- und Optikusner-ven, ist am häufigsten. Das gesamteKrankheits-Spektrum bis hin zu Depres-sion und neuropsychiatrischen Sympto-men ist möglich. Dementsprechendschwierig gestalten sich manchmal dieDiagnostik und die ursächliche Zuord-nung.
Aufgrund der geschilderten möglichenOrganmanifestationen, ergibt sich, dassimmer zunächst eine genaue Anamneseund körperliche Untersuchung notwen-dig sind. Die Diagnose ist dann zu stel-len, wenn typische klinische und/oderradiologische Befunde vorliegen zusam-men mit dem histopathologischenNachweis von nichtverkäsenden Granu-lomen. Andere granulomatöse Erkran-kungen, insbesondere eine Tuberkulosemüssen ausgeschlossen werden. Da inden allermeisten Fällen eine pulmonaleBeteiligung vorliegt, kommt dem kon-ventionellen Röntgen-Thorax-Bild einebesondere Bedeutung zu, ggf. ergänztdurch eine Computertomographie. Fin-den sich anderweitig (Haut, periphereLymphknoten) keine geeigneten Biop-siestellen, muss bronchoskopiert undbiopsiert werden. Bei broncho-alveolä-rer Lavage findet sich dann bei einer er-höhten Gesamtzellzahl eine deutlicheLymphozytose. Ein erhöhter CD4/CD8-Quotient > 3,5 ist hoch spezifisch bei al-lerdings nur mäßiger Sensitivität. Er-gänzt wird die bronchoskopische Unter-suchung bei Nachweis von mediastina-len bzw. hilären Lymphknotenvergröße-rungen durch den endobronchialen Ul-traschall mit gezielter Punktion undNachweis von nichtverkäsenden epithe-loidzelligen Granulomen. Die Mediasti-noskopie ist heute meist entbehrlich. Zuden labortechnischen Untersuchungengehört die Bestimmung des Serum-Kal-ziums, der Nieren- und Leberwerte. Dasin den aktivierten Makrophagen gebilde-te Calcitriol steigert die enterale Kalzi-umresorption.
Verlauf, Prognose und TherapieGenerell ist die Sarkoidose eine progno-stisch gutartige Erkrankung, und dieMehrzahl der Patienten benötigt keine
spezielle Therapie. Bei mehr als der Hälf-te der Sarkoidosepatienten ist innerhalbvon drei Jahren nach Diagnosestellungeine Remission zu beobachten und in-nerhalb von zehn Jahren bei immerhinzwei Dritteln, und dies ohne relevanteFunktionseinbuße. Lediglich bei einemDrittel der Patienten ist ein chronischprogredienter Verlauf zu verzeichnen.Die Mortalität beträgt etwa 5 %, wobeidies dann auf eine chronisch respiratori-sche Insuffizienz, pulmonale Hyperto-nie, kardiale oder neurologische Beteili-gung zurückzuführen ist.
Kortikosteroide sind nach wie vor dieTherapie der Wahl, falls eine Therapiein-dikation besteht. Diese wird im Allge-meinen gesehen bei einem kardialen undrenalen Befall sowie bei einer Beteili-gung des zentralen Nervensystems. Beieinem okulären Befall ist in der Regeleine topische Therapie ausreichend.Gleiches gilt für den kutanen Befall. Beipulmonaler Sarkoidose bestehen keingenereller Konsensus oder durch größe-re Studien abgesicherte Empfehlungen,für welche Subgruppen eine Therapiein-dikation gegeben ist. Als Richtschnurkann jedoch gelten, dass bei progredien-ter Funktionseinbuße (Lungenfunktionbzw. Diffusionskapazität) ggf. in Kombi-nation mit entsprechender Symptoma-tik (Belastungsdyspnoe) die Indikationzur systemischen Kortikosteroidthera-pie besteht, seltener bei anderweitignicht behandelbarer Hustensymtoma-tik. Keinesfalls ist der radiologische Be-fund allein bei Diagnosestellung Anlasszur Therapieentscheidung. Ist die Ent-scheidung zur systemischen Therapie-einleitung gefallen, so beträgt die Dosisinitial zwischen 30–40 mg Prednison/dmit Dosisreduktion um 5 mg/Tag in 14-tägigen Abständen bis zur Erhaltungs-dosis von 10–20 mg/Tag über eine Ge-samttherapiedauer von 6–9 Monatenunter entsprechender klinischer undfunktioneller Kontrolle. Bei einer beglei-tenden Behandlung mit Vitamin D undKalzium ist auf eine initiale Hyperkalzä-mie zu achten.
Als mögliche Alternative zur Kortiko-steroidtherapie, als steroidsparende Al-ternative und als Verstärkung der Thera-pie bei geringem Ansprechen kann Aza-

38 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
thioprin in einer Tagesdosis von 2–3mg/kg Körpergewicht gegeben werden.Mit Methotrexat besteht eine weitere Alternative. Diese Substanz kann einenantiinflammatorischen Effekt durchTNF-Suppression haben. Wenngleichaus diesen theoretischen Überlegungender Einsatz spezieller TNF-Inhibitorenwie z. B. Infliximab durch ihren Angriffs-punkt im Entzündungsprozess unddamit der granulomatösen Reaktionsinnvoll und begründet erscheint, so istdie Datenlage hierzu derzeit noch unzu-reichend. Eine generelle Alternative zurSteroidtherapie ist hiermit noch nichtgegeben. Weitere Ergebnisse bleiben ab-zuwarten. Etanercept und Cyclosporin Awerden als ineffektiv angesehen.
Literatur1. Iannuzzi MC, Fontana JR: Sarcoidosis:clinical presentation, immunopatho-
genesis, and therapeutics. JAMA 2011;305: 391–399.
2.Morgenthau AS, Iannuzzi MC: Recentadvances in sarcoidosis. Chest 2011;139: 174–182.
InteressenkonflikteEin Interessenkonflikt wird vom Autorverneint.
Prof. Dr. med. H. Schäfer, Vö[email protected]
FAZIT
Die Sarkoidose, eine inflammatorisch-granulomatöse Multisystemerkrankungunklarer Ätiologie, befällt vorwiegenddie Lunge und mediastinale Lymphkno-ten, kann aber auch Herz, Nieren, Augenund das zentrale Nervensystem erfas-sen. Histopathologisches Korrelat ist derNachweis nichtverkäsender Granulo-me. Die Diagnosestellung erfordert je-doch auch immer den Ausschluss ande-rer granulomatöser Erkrankungen (M.Wegener, Tuberkulose u. a.). In der Auf-
klärung der Immunpathogenese wurdenFortschritte gemacht: die zentrale Rolleder CD4-T-Zellen und Zytokinaktivierungwurde erkannt. Dennoch existierenkeine prognostischen Biomarker. Wei-terhin sind Kortikosteroide die Therapieder Wahl bei gegebener Indikation. DerGroßteil der Patienten bedarf keinerTherapie bei einer spontanen Remissi-onsrate von bis zu zwei Dritteln innerhalbvon zehn Jahren.
Definition und PrävalenzHenrik Sjögren war ein schwedischerOphthalmologe und lebte von 1899–1986. Das von ihm beschriebene Syn-drom (SS) wird definiert durch ein au-toimmunallergisch verursachtes „Sicca-Syndrom“ an Schleimhäuten, das beson-ders als trockenes Auge durch Versiegender Tränenproduktion und als Mund-trockenheit durch Versiegen der Spei-chelproduktion vorkommt. Aber auch ananderen Stellen kann ein Sjögren-Syn-drom auftreten und sich mit weiterenimmunallergischen Krankheitsbildernkombinieren. Rheumatische Erkran-kungen, Lymphome, sowie neurologi-sche Manifestationen können beispiels-weise komplizierend hinzutreten. Alsprimäres Sjögren-Syndrom wird be-schrieben, wenn die Erkrankung beizuvor gesunden Patienten auftritt. Voneinem sekundären Sjögren-Syndromspricht man, wenn es im Rahmen eineranderen Autoimmunerkrankung auftritt
beispielsweise einer Rheumatoiden Ar-thritis. Das Sjögren-Syndrom gilt alseine der häufigsten Autoimmunerkran-kungen. In den USA sollen 0,5 bis 3 Mil-lionen Erkrankte leben. Betroffen sindmeist postklimakterische Frauen (Män-ner zu Frauen 1:9 bis 1:20) (1;2). FürDeutschland wird eine Prävalenz von 0,2 % in der erwachsenen Normalbevöl-kerung angegeben (2). Behandelt wird esvon Hausärzten, Internisten, Augen-und HNO-Ärzten und häufig von Rheu-matologen.
Systematische Übersicht übergesicherte Therapiemaßnahmen Da derzeit die Therapie des primären SSmeist auf einer Mischung von persönli-chen Erfahrungen, Expertenmeinungenund einzelnen Studienberichten basiertund entsprechende Leitlinien fehlen,haben Ramos-Casals et al. eine systema-tische Recherche in MEDLINE und EM-
BASE durchgeführt (1) mit dem Ziel, evidenzbasierte Behandlungsempfeh-lungen zu formulieren. Sie fanden dabei37 kontrollierte englischsprachige Stu-dien und 19 prospektive Kohortenstudi-en zur medikamentösen Behandlungvon erwachsenen Patienten mit einemprimären SS, die zwischen Januar 1986und April 2010 durchgeführt wordenwaren.
Ergebnisse:Insgesamt fanden die Autoren nur einensehr geringen Evidenzgrad für die Mehr-zahl der angewandten medikamentösenBehandlungen des primären Sjögren-Syndroms, zumal nur weniger als 10 %der Studien mit Vergleichen unter-schiedlicher Medikamente durchgeführtworden waren. Sie formulierten trotz-dem aufgrund der von ihnen gefundenenStudienergebnissen und Beobachtun-gen Behandlungsvorschläge für die fol-genden Hauptsymptome:
Die Behandlung des primären Sjögren-Syndroms

39Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Xerophthalmie (Trockenes Auge durchEpithelstörungen an Bindehaut undHornhaut):Behandelt wurde tagsüber mit Tränener-satzmittel ohne Konservierungsstoffeund nachts mit Gleitsalben. In kontrol-lierten Studien wurde bei Fällen mitschwerwiegendem trockenem Augeaußerdem 2x täglich 0,05 % Pilocarpinangewendet. Allerdings waren gerade diegrößten Studien nicht vorrangig an Pa-tienten mit primärem SS durchgeführtworden. Dabei wurde in einer Studietrotz 6-monatiger Anwendung von Pilo-carpin keine Besserung gesehen. Beischwerer therapierefraktärer Xeropht-halmie wurde gelegentlich die zusätzli-che Anwendung von lokalen NSAIDsoder Glukokortikoiden erforderlich, je-doch wegen der Nebenwirkungen nurfür eine möglichst kurze Dauer.
Xerostomie (Trockenheit der Mundhöh-le): Bei mildem oder nur mäßigem Befundkonnten Behandlungen mit Speicheler-satz-Präparaten und die Anwendung vonzuckerfreien Kaugummis helfen. Alko-holgebrauch und Tabakrauchen solltengemieden werden, eine optimierteMundhygiene wurde als essentiell ange-sehen. Bei Patienten, die erheblicheStörungen ihrer Speicheldrüsenfunkti-on entwickelt hatten, waren orales Pilo-carpin und Cevimeline (ein parasympa-thomimetischer Muskarin-Agonist, inden USA: Evoxac®) die Therapie der Wahl(in Deutschland Salagen® / Pilocarpinmono). Allerdings wurde die Wirksam-keit dieser beiden Stoffe nicht gegenein-ander verglichen. Die beste Dosis beimVerhältnis von Wirkung zu Nebenwir-kung war für Pilocarpin 5 mg alle sechsStunden und für Cevimeline 30 mg alleacht Stunden. Für Patienten mit Kontra-indikationen gegen diese beiden Stoffeoder mit Intoleranz gegenüber Muskarin-Agonisten wurde als Alternative die An-wendung von N-Acetylcystein erprobt.
Allgemeine Symptome (wie Muskel-, Ge-lenksschmerzen, Müdigkeit): Bei allgemeinen Symptomen des pri -mären SS konnte kein eindeutiger Nut-zen von Hydroxychloroquin (Quensyl®)gesehen werden. Es handelte sich umkontrollierte und prospektive Studien,
die alle nur klein waren. Ein Erfolgwurde nur in retrospektiven Studien ge-sehen.
Extraglanduläre Symptome (wie Vasku-litis, Neuropathie, Glomerulonephritis,Arthritis):Es fand sich nur eine begrenzte Evidenzfür die Anwendung von Glukokortikoi-den und Immunsuppressiva, da die kon-trollierten und prospektiven Studienklein waren und speziell auf Erfolgehauptsächlich beim Sicca-Syndrom hindurchgeführt worden waren. Rituximab(MabThera®) hatte in einer kontrollier-ten Studie bei Vaskulitis und in drei un-kontrollierten Studien (mit Fällen vonVaskulitis, Neuropathie, Glomerulo-nephritis und Arthritis) Symptomlinde-rung gezeigt. Da jedoch nach Meinungder Autoren erst größere Studien abge-wartet werden sollten, empfehlen sie, Ri-tuximab vorerst nur als Reservemedika-ment bei therapierefraktären Patienteneinzusetzen.
Lebensbedrohliche Situationen (beimschnell progressiven extraglandulärenSS wie z.B.bei Glomerulonephritis, Neuropathie,interstitieller Lungenerkrankung oderMyelitis):Solche Fälle wurden nur selten beschrie-ben. Es fanden sich nur vereinzelte re-trospektive Studien und isolierte Fallbe-richte. Die spärlichen Evidenzen hierzu,
die meist zusammen mit Expertenmei-nungen formuliert worden waren, lassenvermuten, dass hierbei Stossbehandlun-gen mit Methylprednisolon und Cyclo-phosphamid, auch möglicherweise mitPlasmaaustausch bei Patienten mitrasch progredienten extraglandulärenErscheinungen der SS (wie Glomerulo-nephritis, Neuropathie, interstitiellerLungenerkrankung, Myelitis oder einerschweren systemischen Vaskulitis) sinn-voll angewendet werden können. Rituxi-mab wird inzwischen zunehmend bei le-bensbedrohlichen Situationen und auchbei Fällen von B-Zell-Lymphom ange-wendet.
Literatur1.Ramos-Casals M, Tzioufas AG, StoneJH et al.: Treatment of primary Sjo-gren syndrome: a systematic review.JAMA 2010; 304: 452–460.
2.Westhoff G, Zink A: [Epidemiology ofprimary Sjorgren’s syndrome]. ZRheumatol 2010; 69: 41–49.
Interessenkonflikte:Ein Interessenkonflikt wird vom Autorverneint.
Dr. med. Klaus Ehrenthal, [email protected]
FAZIT
In den letzten drei Dekaden basierte dieBehandlung des primären Sjögren-Syn-droms auf der Substitution des Sicca-Syndroms (tagsüber Tränenersatz, zurNacht Gleitmittelersatz am Auge, imMund künstlicher Speichel) und der An-wendung von Glukokortikoiden und Im-munsuppressiva bei schwereren Fällensowie bei extraglandulären Krankheits-bildern.Die Anwendung neuer Immunsuppressi-va und Biologika hat die Beherrschbar-keit in lebensbedrohlichen Situationenverbessert. Aber nach wie vor fehlt es an
eindeutigen Daten für die Behandlungtherapieresistenter Fälle. Als zukünftigaussichtsreich wird besonders Rituxi-mab (inzwischen mit Berichten von über100 Fällen) angesehen. Außerdem könn-ten möglicherweise auch Hemmer vonB-Zell-aktivierenden Substanzen ausder Familie der Tumor-Nekrose-Fakto-ren-alpha zukünftige Therapien verbes-sern.Hierzu und zu den Anwendungsgefahrensind aber nach den Autoren dieser sy-stematischen Recherche zunächst wei-tere Untersuchungen erforderlich (1).

40 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Gibt es neue Erkenntnisse zur Therapie der Arthrose mit Orthokin®?
Seit mehr als zehn Jahren wird Ortho-kin® zur intraartikulären Therapie derArthrose eingesetzt. Dabei findet dieTherapie mit Orthokin® eine hohe öf-fentliche Beachtung. Die SuchmaschineGoogle findet mehr als 38.000 Einträge.Bei Orthokin® handelt es sich um ein ausEigenblut hergestelltes Serum, daseinen erhöhten Gehalt an Interleukin-1-Rezeptorantagonisten (IL-1Ra) und wei-teren, antiinflammatorisch wirksamenZytokinen haben soll. Diese Individu-alarznei soll somit bei der Volkskrank-heit Arthrose antiphlogistisch und anal-getisch wirken, wobei zusätzlich die Pro-gression der Knorpelzerstörung ge-hemmt werden soll. Wir berichteten be-reits im Jahr 2006 ausführlich über dieTherapie der Arthrose mit Orthokin® (1).Hierbei war ein wesentlicher Kri-tikpunkt, dass es keine nachvollziehba-ren Daten zu Wirksamkeit und Unbe-denklichkeit aus kontrollierten klini-schen Studien gibt (1).
Seit 2008 gibt es mittlerweile 2 klinischeStudien, die beide in der renommiertenorthopädischen Fachzeitschrift „Osteo-arthritis and Cartilage“ publiziert wur-den (2;3). Bei der in Düsseldorf durchge-führten Studie handelt es sich um einequalitativ gute, prospektive, randomi-sierte, doppeltblinde und Placebo- undHyaluronsäure-kontrollierte Studie, ander 376 Patienten mit einer Kniearthro-se teilnahmen (2). Danach entfaltete,über einen Zeitraum von bis zu 6 Mona-ten statistisch signifikant, die 6-maligeintraartikuläre Injektion von Orthokin®
bei Patienten mit relativ starken Knies-chmerzen (VAS > 50 mm) eine klinischrelevante, mindestens 20 %ige Linde-rung der Symptome wie Schmerz, Ge-lenksteifigkeit und Funktionsverlust.Diese wurden mit Hilfe des Western On-tario and McMaster Universities Osteoar-thritis Index (WOMAC)-Scores sowieeines VAS-Schmerzscores ermittelt.Auch verbesserten sich, jedoch etwasschwächer ausgeprägt, die Symptome
bei Patienten, die nur dreimal intraarti-kulär Hyaluronsäure oder ein Placeboinjiziert bekommen hatten. Da nur Pati-enten mit relativ starken Knieschmer-zen in die Studie eingeschlossen wur-den, lassen sich die Ergebnisse nichtohne weiteres auf Patienten mit gerin-gen Arthroseschmerzen übertragen. DieInjektionshäufigkeit kann jedoch einenerheblichen Einfluss auf das Studiener-gebnis gehabt haben, sofern man in Be-tracht zieht, dass bei jeder intraarti-kulären Injektion zunächst Synovial-flüssigkeit samt Knorpeldetritus undEntzündungsmediatoren aspiriert undverworfen wird, bevor Orthokin®, Hya-luronsäure bzw. das Placebo injiziertwurde. Hinzu kommt, dass bei einer un-vollständigen oder sogar fehlenden Aspi-ration von Synovialflüssigkeit diesedurch die intraartikuläre Injektion ver-dünnt wird, wodurch die Konzentrationan inflammatorisch wirksamen Bestand-teilen gesenkt wird. So liegt die Halb-wertszeit von Orthokin®, Hyaluronsäureund Placebo in den Kniegelenken ver-mutlich im Bereich von Stunden, wobeijedoch die Wirkung über Wochen bisMonate selbst in der Placebogruppe an-hielt. Dies deutet auf einen zusätzlichen,möglicherweise durch die Aspirationbzw. Verdünnung von arthrotischer Sy-novialflüssigkeit bedingten Effekt hin.Die unter Orthokin®-Therapie doppelt sohäufig durchgeführte Aspiration bzw.Verdünnung von noch vorhandenenoder neu synthetisierten Entzündungs-mediatoren kann somit möglicherweisefür den beobachteten besseren Therapie-erfolg von Orthokin® verantwortlich ge-macht werden.
Eine weitere Studie aus den Niederlan-den, die 2009 in demselben Journal pu-bliziert wurde und bei der Patienten mitähnlich starken Knieschmerzen (Visuel-le Analogskala (VAS) > 40 mm) Ortho-kin® erhielten, kommt jedoch nur zueinem enttäuschenden Ergebnis, ob-wohl der Titel viel verspricht (3). 167 Pa-
tienten erhielten über einen Zeitraumvon drei Wochen sechs intraartikuläreInjektionen von 2 ml Orthokin® oder 2ml physiologischer Salzlösung. Das nach3, 6, 9 und 12 Monaten gemesseneHauptzielkriterium, nämlich die Verbes-serung des die Arthrosesymptome erfas-senden WOMAC-Scores um klinisch re-levante 30 % wurde nicht erzielt, dage-gen zeigten zwei andere Scores eine sta-tistisch signifikante Verbesserung. Diesesind jedoch gering und wahrscheinlichklinisch nicht oder wenig relevant. Selt-sam erscheint, dass diese Erkenntnis dieAutoren in ihrem letzten Satz im Ab-stract („….although the improvement insymptomatology seems not clinically re-levant.“) auch als Schlußfolgerung zie-hen, aber einen Titel auswählten, derdem völlig widerspricht („Autologous in-terleukin-1 receptor antagonist impro-ves function and symptoms in osteoar-thritis…“).
Eine Reihe von Untersuchungen hat be-reits gezeigt, dass nur ein sehr hoherÜberschuss von IL-1Ra in der Lage ist,die Wirkung von IL-1 zu antagonisieren.Von Anakinra (Kineret®), einem weite-ren, jedoch rekombinant hergestelltenund zugelassenen IL-1-Ra, ist aus invitro-Untersuchungen bekannt, dass esdie Bindung von IL-1 an seinen Rezeptorähnlich stark hemmt wie der natürliche,glykosylierte IL-1Ra, der auch im Ortho-kin® im pg-Bereich vorhanden sein soll.Jedoch wurde mit 50 mg oder 150 mgAnakinra in einer randomisiert, doppelt-blind und placebokontrollierten klini-schen Studie nach vier Wochen im Ver-gleich zu Placebo keine statistisch signi-fikante Verbesserung der Arthrosesym-ptome, die mit Hilfe des WOMAC-Scoreserfaßt wurden, gefunden (4). An dieserMulticenterstudie nahmen 170 Patien-ten teil, wobei diese einmalig intraarti-kulär entweder Placebo oder Anakinra(Kineret®) in das arthrotische Kniege-lenk appliziert bekamen (4).
Arneimittel – kritisch betrachtet

41Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Angesichts des mittlerweile großen Ar-senals an preislich wesentlich günstige-ren Analgetika und Antiphlogistika istbesonders bei Arthrose die Frage von Be-deutung, inwiefern ein Arzneistoff einestrukturmodifizierende (chondropro-tektive) und somit basistherapeutischeWirkung besitzt. Unter einer strukturm-odifizierenden Wirkung versteht maneinen Arzneistoff, der in der Lage ist,beim Menschen die fortschreitende Zer-störung des Gelenkes zu verlangsamen,aufzuhalten oder sogar rückgängig zumachen (5). Die Individualarznei Ortho-kin® wäre aufgrund ihres postuliertenIL1-Ra-Antagonismus ein möglicherKandidat für eine derartige strukturmo-difizierende Wirkung. Jedoch fehlen ent-sprechende klinische Untersuchungen,so dass jegliche theoretisch hergeleitete,plausibel klingende Assoziationen zumjetzigen Zeitpunkt völlig spekulativ sind.Angesichts der Volkskrankheit Arthrosewäre die Durchführung derartiger Stu-dien wünschenswert.
Literatur1. Steinmeyer J: Orthokin® zur Therapieder Arthrose – eine kritische Betrach-tung eines Eigenblutpräparates. Arz-neiverordnung in der Praxis (AVP)2006; 33: 39–40.
2. Baltzer AW, Moser C, Jansen SA,Krauspe R: Autologous conditionedserum (Orthokine) is an effective tre-
atment for knee osteoarthritis. Osteo-arthritis Cartilage 2009; 17: 152–160.
3. Yang KG, Raijmakers NJ, van Arkel ERet al.: Autologous interleukin-1 recep-tor antagonist improves function andsymptoms in osteoarthritis whencompared to placebo in a prospectiverandomized controlled trial. Osteoar-thritis Cartilage 2008; 16: 498–505.
4. Chevalier X, Goupille P, Beaulieu AD etal.: Intraarticular injection of anakinrain osteoarthritis of the knee: a multi-center, randomized, double-blind, pla-cebo-controlled study. ArthritisRheum 2009; 61: 344–352.
5. Steinmeyer J, Konttinen YT: Oral tre-atment options for degenerative jointdisease – presence and future. AdvDrug Deliv Rev 2006; 58: 168–211.
InteressenkonflikteEin Interessenkonflikt wird von den Au-toren verneint.
PD Dr. med. J. Kordelle, Gießen, Prof. Dr. J. Steinmeyer, Gieß[email protected]
FAZIT
Seit 2008 gibt es zwei randomisierte,doppeltblinde und placebokontrollierteStudien zu Orthokin®, die widersprüch-lich sind. Obwohl in einer dieser beidenklinischen Studien eine statistisch signi-fikante Symptomlinderung nach intraar-tikulärer Injektion von Orthokin® gegen -über Placebo gemessen wurde, kanndiese Wirkung auch auf der doppelt sohäufigen intraartikulären Injektion vonOrthokin® im Vergleich zu Placebo undsomit auf einem falschen Studiendesignberuhen. In beiden Studien wurden nurPatienten mit relativ starken Knie -schmerzen eingeschlossen. Da diesePatienten nicht die große Gruppe aller
Arthrosepatienten repräsentieren, isteine Verallgemeinerung der Studien -ergebnisse nicht zulässig. Untersuchun-gen, die eine mögliche strukturmodifi -zierende (chondroprotektive) Wirkungbeschreiben, existieren derzeit nicht,sodass eine derartige Wirkung zwartheoretisch denkbar, aber zum jetzigenZeitpunkt völlig spekulativ wäre. Ange-sichts der hohen Kosten, die der Patientselbst zu tragen hat, und des enormenvolkswirtschaftlichen Schadens, den dieArthrose erzeugt, wären entsprechendeStudien mit korrektem Studiendesignvon besonderem Interesse.
Neue Arzneimittel
Hinweise zur Erstellung der Information „Neue Arzneimittel“
„Neue Arzneimittel“ ist eine Information der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ) zu neu zugelasse-nen Arzneimitteln/neu zugelassenen Indikationen.
Ziel ist es, den Vertragsärzten eine zeitnahe Information zu neu zugelassenen Arzneimitteln vor Markteinführung zur Ver-fügung zu stellen. Diese Information ist ebenfalls auf der Homepage der AkdÄ abrufbar (http://www.akdae.de/Arzneimitteltherapie/NA/index.html) und wird auch mittels elektronischem Newsletter aktiv versandt.
Dargestellt werden in der Information „Neue Arzneimittel“ von dem Committee for Medicinal Products for Human Use(CHMP) der European Medicines Agency (EMA) als positiv bewertete und von der Europäischen Kommission neu zugelas-sene Arzneimittel bzw. Indikationserweiterungen. Grundlage der Information und der Bewertung des Arzneimittels ist derEuropean Public Assessment Report (EPAR) der EMA.

42 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Yervoy® (Ipilimumab)
IndikationYervoy® ist zur Behandlung von fortge-schrittenen (nicht resezierbaren odermetastasierten) Melanomen bei Erwach-senen, die bereits zuvor eine Therapie er-halten haben, indiziert.
Pharmakologie und klinischeStudienIpilimumab ist ein monoklonaler Anti-körper. Er wirkt über eine Verstärkungder T-Zell-vermittelten Immunantwort.Es wird angenommen, dass Ipilimumabdas zyto¬toxische T-Lymphozyten Anti-gen 4 (CTLA-4) blockiert und so zur T-Zell-Aktivierung, Proliferation und Lym-phozyteninfiltration in Tumoren unddamit zum Tumorzelltod führt.
Für die Zulassung wurde eine dreiarmi-ge, randomisierte, doppelblinde Studie(MDX010-20) an 676 Patienten mit nichtresezierbarem malignen Melanom (aus-genommen okuläre Melanome) im Sta-dium III oder IV vorgelegt. Es wurdenausschließlich HLA-A2*0201-positivePatienten eingeschlossen, da im Ver-gleichsarm ein experimenteller Impf-stoff (gp100) eingesetzt wurde, der einauf diesen HLA-Marker restringiertesProtein enthält. Patienten mussten min-destens mit einem Zyklus eines odermehrerer der Wirkstoffe Interleukin-2,Dacarbazin (DTIC), Temozolomid, Fote-
mustin oder Carboplatin vorbehandeltworden sein und hatten entweder ein Re-zidiv nach erfolgtem Ansprechen erlit-ten, hatten nicht angesprochen oder dieBehandlung wegen Nebenwirkungennicht vertragen. Patienten erhielten eineInduktionstherapie (alle 3 Wochen, biszu 4 mal) und hatten die Möglichkeitweiterer Re-Induktionstherapien (mitjeweils 4 Dosen):
• Ip + gp: Ipilimumab 3 mg/kg plus experimenteller Peptid-Impfstoff(gp100); n = 403
• Ip: Ipilimumab 3 mg/kg plus Placebo-Impfstoff; n = 137
• gp: Placebo-Ipilimumab plus gp100; n = 136
Der primäre Endpunkt war das Gesamt -überleben (OS) unter Kombinations- (Ip + gp) verglichen mit Impfstoff-Thera-pie (gp); als sekundäre Endpunkte wur-den weitere Gruppenvergleiche und diekrankheitsbezogene Lebensqualität(HRQoL) ausgewertet. Bei Auswertungwaren die 151 noch lebenden Studien-teilnehmer über einen Zeitraum von imMedian mindestens 17 Monaten beob-achtet worden (Ip + gp = 21, Ip = 28, pg= 17 Monate). Das mediane Überlebenbetrug 10,0 Monate unter Kombinati-ons- (Ip + pg), 6,4 Monate unter Impf-stoff- (gp) und 10,1 Monate unter Ipili-mumab-Therapie (Ip). 5,7 % der Ip+gp-Gruppe hatten eine Re-Induktionsthera-pie erhalten versus 0,7 % der gp-Gruppe.Der Unterschied von Ip+gp versus gp warsignifikant (Hazard Ratio [HR] 0,68; 95 % Konfidenzintervall [CI] 0,55–0,85;p-Wert Log-Rank-Test 0,0004).
Die Ip-Behandlung ging mit erheblichen– v. a. immunvermittelten – UAW einherund 17 % der Sicherheitspopulationhatte unter einer Dosierung von 3 mg/kgschwerwiegende behandlungsbedingteUAW erlitten. Ob das unter Ip+gp versusgp um 3,6 Monate verlängerte OS unterBeibehaltung der HRQoL erreicht wer-den konnte, ist dem EPAR nicht zu ent-nehmen. Da Ip in der Zulassungsstudienicht gegen Placebo getestet wurde und
der mögliche Effekt des Komparators gpauf das Überleben nicht untersucht ist,bleibt die Größe des Behandlungseffek-tes von Ip verglichen mit einer Scheinbe-handlung unklar.
Daten aus einer weiteren doppelblindenRCT (CA184024) an Patienten mit nichtresezierbarem malignen Melanom imStadium III oder IV, die nicht vorbehan-delt und deren HLA-Status unbekanntblieb, wurden ebenfalls vorgelegt. DieWirkung von Ip 10 mg/kg + DTIC (n = 250) versus Placebo + DTIC (n = 252) gefolgt von einer Erhal-tungstherapie wurde verglichen. Ip +DTIC verlängerte das OS um 2,1 Monate,und betrug 11,2 Monate verglichen mit9,1 Monaten unter DTIC (HR 0,72; 95 %CI 0,59–0,87; p = 0,0009).
Unerwünschte Arzneimittelwir-kungenSehr häufig (≥ 1/10): Diarrhö, Erbre-chen, Übelkeit, Ausschlag, Pruritus, Ap-petitminderung, Müdigkeit, Fieber.
Häufig (≥ 1/100, < 1/10): Lymphopenie,Hypopituitarismus, Hypothyreose, Hy-pokaliämie, Verwirrtheit, periphere sen-sorische Neuropathie, Schwindel, Kopf-schmerzen, Hypotonie, Dyspnoe, Koli-tis, gastrointestinale Blutungen, Leber-funktionsstörungen, Dermatitis, Vitili-go, Alopezie, Arthralgie, Myalgie, Schüt-telfrost.
Gelegentlich (≥ 1/1000, < 1/100): septi-scher Schock, Meningitis, para neo plas -tisches Syndrom, hämolytische Anämie,Thrombozytopenie, Neutropenie, Ne-benniereninsuffizienz, Hyperthyreose,Hypogonadismus, Depression, Guillain-Barré-Syndrom, Synkope, Ataxie, Myo-klonie, Dysarthrie, Uveitis, Glaskörper-blutung, Iritis, Arrythmie, Vorhofflim-mern, akutes respiratorisches Distress-Syndrom, Pneumonitis, gastrointestina-le Perforation, Peritonitis, Pankreatitis,Enterokolitis, Ileus, Leberversagen, Hepatitis, toxische epidermale Nekroly-se, Vaskulitis, rheumatische Polymyal-
Bewertung
Yervoy® (Ipilimumab) in Kombinati-on mit einem experimentellen Impf-stoff verlängerte das Überleben vonPatienten mit fortgeschrittenem ma-lignen Melanom gegenüber alleinigerImpfstoff-Behandlung um ca. 4 Mo-nate. Eine zusätzlich eingereichteStudie mit anderen Einschlusskrite-rien und Behandlungsregimen zeigteeinen Überlebensvorteil von ca. 2 Mo-naten. Die Überlebensverlängerungmuss gegen die häufigen und schwe-ren Nebenwirkungen abgewogenwerden.
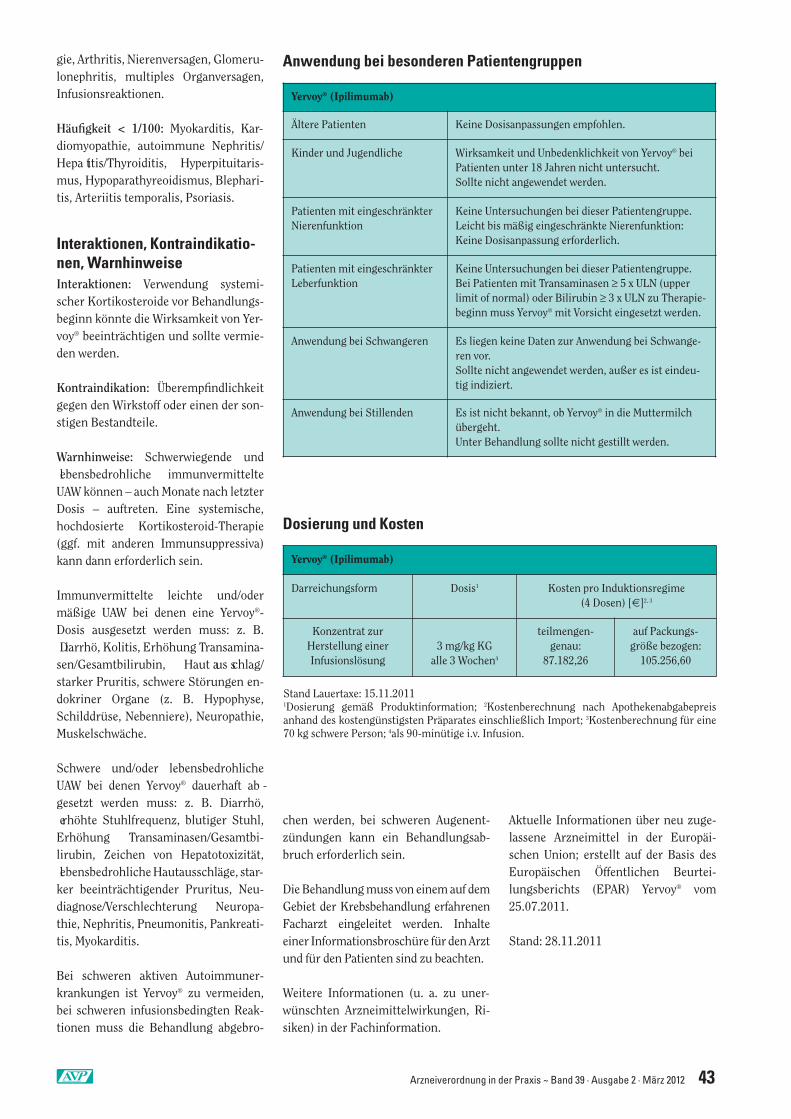
43Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
gie, Arthritis, Nierenversagen, Glomeru-lonephritis, multiples Organversagen,Infusionsreaktionen.
Häufigkeit < 1/100: Myokarditis, Kar-diomyopathie, autoimmune Nephritis/Hepa titis/Thyroiditis, Hyperpituitaris-mus, Hypoparathyreoidismus, Blephari-tis, Arteriitis temporalis, Psoriasis.
Interaktionen, Kontraindikatio-nen, WarnhinweiseInteraktionen: Verwendung systemi-scher Kortikosteroide vor Behandlungs-beginn könnte die Wirksamkeit von Yer-voy® beeinträchtigen und sollte vermie-den werden.
Kontraindikation: Überempfindlichkeitgegen den Wirkstoff oder einen der son-stigen Bestandteile.
Warnhinweise: Schwerwiegende und lebensbedrohliche immunvermittelteUAW können – auch Monate nach letzterDosis – auftreten. Eine systemische,hochdosierte Kortikosteroid-Therapie(ggf. mit anderen Immunsuppressiva)kann dann erforderlich sein.
Immunvermittelte leichte und/odermäßige UAW bei denen eine Yervoy®-Dosis ausgesetzt werden muss: z. B. Diarrhö, Kolitis, Erhöhung Transamina-sen/Gesamtbilirubin, Haut aus schlag/starker Pruritis, schwere Störungen en-dokriner Organe (z. B. Hypophyse,Schilddrüse, Nebenniere), Neuropathie,Muskelschwäche.
Schwere und/oder lebensbedrohlicheUAW bei denen Yervoy® dauerhaft ab -gesetzt werden muss: z. B. Diarrhö, erhöhte Stuhlfrequenz, blutiger Stuhl,Erhöhung Transaminasen/Gesamtbi-lirubin, Zeichen von Hepatotoxizität, lebensbedrohliche Hautausschläge, star-ker beeinträchtigender Pruritus, Neu-diagnose/Verschlechterung Neuropa-thie, Nephritis, Pneumonitis, Pankreati-tis, Myokarditis.
Bei schweren aktiven Autoimmuner-krankungen ist Yervoy® zu vermeiden,bei schweren infusionsbedingten Reak-tionen muss die Behandlung abgebro-
chen werden, bei schweren Augenent-zündungen kann ein Behandlungsab-bruch erforderlich sein.
Die Behandlung muss von einem auf demGebiet der Krebsbehandlung erfahrenenFacharzt eingeleitet werden. Inhalteeiner Informationsbroschüre für den Arztund für den Patienten sind zu beachten.
Weitere Informationen (u. a. zu uner-wünschten Arzneimittelwirkungen, Ri-siken) in der Fachinformation.
Aktuelle Informationen über neu zuge-lassene Arzneimittel in der Europäi-schen Union; erstellt auf der Basis desEuropäischen Öffentlichen Beurtei-lungsberichts (EPAR) Yervoy® vom25.07.2011.
Stand: 28.11.2011
Anwendung bei besonderen Patientengruppen
Yervoy® (Ipilimumab)
Ältere Patienten Keine Dosisanpassungen empfohlen.
Kinder und Jugendliche Wirksamkeit und Unbedenklichkeit von Yervoy® beiPatienten unter 18 Jahren nicht untersucht.Sollte nicht angewendet werden.
Patienten mit eingeschränkterNierenfunktion
Keine Untersuchungen bei dieser Patientengruppe.Leicht bis mäßig eingeschränkte Nierenfunktion:Keine Dosisanpassung erforderlich.
Patienten mit eingeschränkterLeberfunktion
Keine Untersuchungen bei dieser Patientengruppe.Bei Patienten mit Transaminasen ≥ 5 x ULN (upperlimit of normal) oder Bilirubin ≥ 3 x ULN zu Therapie-beginn muss Yervoy® mit Vorsicht eingesetzt werden.
Anwendung bei Schwangeren Es liegen keine Daten zur Anwendung bei Schwange-ren vor.Sollte nicht angewendet werden, außer es ist eindeu-tig indiziert.
Anwendung bei Stillenden Es ist nicht bekannt, ob Yervoy® in die Muttermilchübergeht.Unter Behandlung sollte nicht gestillt werden.
Dosierung und Kosten
Yervoy® (Ipilimumab)
Darreichungsform Dosis1 Kosten pro Induktionsregime(4 Dosen) [€]2, 3
Konzentrat zur Herstellung einer Infusionslösung
3 mg/kg KGalle 3 Wochen4
teilmengen-genau:87.182,26
auf Packungs-größe bezogen:105.256,60
Stand Lauertaxe: 15.11.20111Dosierung gemäß Produktinformation; 2Kostenberechnung nach Apothekenabgabepreis anhand des kostengünstigsten Präparates einschließlich Import; 3Kostenberechnung für eine70 kg schwere Person; 4als 90-minütige i.v. Infusion.

44 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Fampyra® (Fampridin)
IndikationFampyra® ist zur Verbesserung der Geh-fähigkeit von erwachsenen Patientenmit Multipler Sklerose (MS) mit Gehbe-hinderung zugelassen (EDSS 4–7).
Pharmakologie und klinischeStudienFampridin ist ein selektiver Kaliumka-nalblocker. Durch Blockade des Aus-stroms von Kaliumionen verlängert esdie Repolarisationszeit und soll die Akti-onspotentialbildung im demyelinisier-ten Axon verstärken.
Für die Zulassung wurden zwei rando-misierte, doppelblinde Studien vorge-legt. Patienten mit einer MS (McDonald-Kriterien), die beim Screening zweima-lig den 25-Fuß-Gehtest (T25FW1) ineiner durchschnittlichen Zeit von 8–45Sekunden absolviert hatten, wurden ein-geschlossen. Ausschlusskriterien warenu. a. Krampfanfälle, signifikante kardio-vaskuläre Erkrankungen, die Behand-lung mit Kortikosteroiden, Cyclophos-phamid oder Mitoxantron, eine kürzli-che Exazerbation sowie der Beginn einerimmunmodulatorischen MS-Therapie.Krankheitsdauer und MS-Verlaufsformwurden nicht berücksichtigt. Patientenerhielten Fampridin SR2 10 mg 2 x täg-lich oder Placebo.
Primärer Endpunkt war die Ansprechra-te im Gehtest (T25FW) in der ITT-Popu-lation. Responder waren definiert als Pa-tienten, die in drei von vier Kontrolleneine höhere Gehgeschwindigkeit übereine Distanz von 7,6 Metern zeigten, ver-glichen mit dem Höchstwert, den sieaußerhalb der Behandlung erzielt hat-ten. Unter den sekundären Endpunktenwaren Skalen, die die subjektive Geh-fähigkeit (MSWS-123), die Muskelkraft,den Muskeltonus und die subjektiv(SGI4) und objektiv (CGI5) empfundeneVeränderung erhoben.
In Studie MS-F203 erhielten PatientenFampridin (n = 224) oder Placebo (n = 72) über 14 Wochen (doppelblindeBehandlungsphase; Studiendauer 21 Wo -chen). In Studie MS-F204 erhielten Pati-enten Fampridin (n = 119) oder Placebo(n = 118) über 9 Wochen (doppelblindeBehandlungsphase; Studiendauer 14Wochen).
Ansprechraten waren unter Fampridinsignifikant höher als unter Placebo(F203: Fampridin 34,8 % vs. Placebo 8,3 %; F204: 42,9 % vs. 9,3 %; p < 0,001).Um zu prüfen, ob die zu Grunde liegendestatistisch signifikante Verbesserung derGehgeschwindigkeit auch in einem kli-nisch relevanten Ausmaß erzielt wurde,sah der Auswertungsplan vor, Verände-rungen der Gehfähigkeit (MSWS-12)von Respondern und Non-Respondern(Ergebnisse nicht im EPAR, Vorgehenwurde vom CHMP methodisch kritisiert)sowie der Gehgeschwindigkeit (T25FW)von Fampridin-Respondern und Patien-ten unter Placebo zu vergleichen. DieUnterschiede in der Gehgeschwindigkeitwaren gering. 7,6 Meter wurden in derPlacebo-Gruppe in 11,6/11,5 Sekunden(F203/F204) und in der Fampridin-Grup-pe in 10,8/10,2 Sekunden zurückgelegt.Für die Strecke benötigten Fampridin-Responder 9,8 Sekunden, Non-Respon-der 12,0 und Patienten mit Placebo 11,7.
Hinsichtlich des sekundären Endpunkts„Verbesserung der Gehfähigkeit“(MSWS-12) zeigte sich eine numerischeÜberlegenheit von Fampridin gegenüber
Placebo in Studie F203 (Placebo –0,08;Fampridin –2,84; p = n. s.) und eine sta-tistisch signifikante Überlegenheit inStudie F204 (Placebo 0,87; Fampridin –2,77; p = 0,006). Das Ausmaß der Verän-derungen wurde vom CHMP zunächst inseiner klinischen Relevanz angezweifelt.
Vom CHMP wurde initial angeführt, dassdie vorgelegten Ergebnisse im T25FW,inklusive der gewählten Responder-Ana-lysen die Frage nicht beantworteten, in-wieweit die gering verbesserte Gehge-schwindigkeit klinisch relevant sei, übergrößere Distanzen anhielte und miteiner verbesserten Gehqualität (MSWM-12) einherginge. Das CHMP kam daherzunächst zu einer negativen Nutzen-Ri-siko-Beurteilung von Fampridin.
In der Re-Evaluation der Daten kam dasCHMP dann zu der Auffassung, dass ein20-prozentiger Unterschied in der Geh-geschwindigkeit (T25FW) wahrschein-lich klinisch relevant sei. Dieses Respon-se-Kriterium erfüllten 31 % mit Fampri-din- und 13 % mit Placebo-Behandlung.Zudem präsentierte der Hersteller Datenzur Korrelation der Skalenwerte (0,4) fürdie Gehgeschwindigkeit (T25FW) und die Gehfähigkeit (MSWM-12), aufgrundderer das CHMP zu der Einschätzungkam, dass Veränderungen im T25FW Aus-sagen über die Gehfähigkeit erlauben.
Eine konditionale, positive Beurteilungwurde unter Formulierung von Kriteri-en für eine Verordnung erteilt (initialeVerschreibung auf 2 Wochen be-schränkt, Evaluierung des Therapieer-folgs mittels T25FW empfohlen, Weiter-verordnung nur bei – auch vom Patien-ten bestätigten – Therapieerfolg).
Die Behandlung mit Fampridin geht miteinem (dosisabhängig) erhöhten Risikofür Infektionen und Krampfanfälle ein-her. Unsicherheit besteht hinsichtlichder längerfristigen Sicherheit und Si-
Bewertung
Fampridin kann bei einigen MS-Pati-enten die Gehgeschwindigkeit ver-bessern. Gemessen wurde dieser Effekt über eine kurze Strecke von 7,6 Metern. Unsicherheit besteht da -rüber, ob der Effekt über längere Geh-strecken anhält, inwieweit er miteiner besseren Gehqualität einher-geht und welche Risiken Fampridinin Subpopulationen besitzt. Die Zu-lassung wurde nur unter der Ein-schränkung erteilt, dass die Verord-nung zunächst auf 2 Wochen be-schränkt bleibt und nur bei auch vonPatienten berichteter Besserung fort-geführt wird.
1 Timed 25 Feet Walking Test2 slow release3 12-Item MS Walking Scale4 Subject’s Global Impression5 Clinical Global Impression

45Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
cherheit bei kardiovaskulären Erkran-kungen, Krampfanfällen, leichter Nieren -insuffizienz und bei älteren Patienten.
Unerwünschte Arzneimittel -wirkungenSehr häufig (≥ 1/10): Harnwegsinfekte.
Häufig (≥ 1/100, < 1/10): Schwindel,Kopfschmerzen, Gleichgewichtsstö rung,Parästhesie, Tremor, Schlaflosigkeit,Angst, Dyspnoe, pharyngolaryngealeSchmerzen, Übelkeit, Erbrechen, Obsti-pation, Dyspepsie, Rückenschmerzen,Asthenie.
Gelegentlich (≥ 1/1000, < 1/100):Krampfanfälle.
Kontraindikationen, Warnhin-weiseKontraindikationen: Überempfindlichkeitgegen Fampridin oder einen der sonstigenBestandteile. Gleichzeitige Behandlungmit Arzneimitteln, die Fampridin (4-Ami-nopyridin) enthalten und mit Inhibitorendes organischen Kationentransporters 2(OTC2; Transporter für aktive Ausschei-dung von Fampridin), wie z. B. Cimetidin.Patienten mit Krampfanfällen (in der Vor-geschichte) oder mit Niereninsuffizienz.
Warnhinweise: Vorsicht bei: Faktoren,die die Krampfschwelle herabsetzenkönnen; bei gleichzeitiger Verordnungvon Arzneimitteln, die OTC2-Substratesind (z. B: Carvedilol, Propanolol, Met-formin); bei kardiovaskulären Risikofak-toren und Erregungsleitungsstörungen.
Fampridin sollte von einem Arzt ver-schrieben und überwacht werden, der
Erfahrung in der MS-Behandlung be-sitzt. Wenn nach zwei Wochen keineBesserung eingetreten ist, sollte Fampy-ra® abgesetzt werden.
Weitere Informationen (u. a. zu uner-wünschten Arzneimittelwirkungen, Ri-siken) in der Fachinformation.
Aktuelle Informationen über neu zuge-lassene Arzneimittel in der Europäi-schen Union; erstellt auf der Basis desEuropäischen Öffentlichen Beurtei-lungsberichts (EPAR) Fampyra® vom04.08.2011.
Stand: 30.11.2011
Anwendung bei besonderen Patientengruppen
Fampyra® (Fampridin)
Ältere Patienten Vor Beginn und während der Behandlung sollte dieNierenfunktion kontrolliert werden.
Kinder und Jugendliche Wirksamkeit und Unbedenklichkeit bei Patientenunter 18 Jahren nicht untersucht.
Patienten mit eingeschränkterNierenfunktion
Leichte, mittelschwere und schwere Niereninsuffizi-enz (Kreatinin-Clearance < 80 ml/min): Kontraindi-ziert.
Patienten mit eingeschränkterLeberfunktion
Keine Dosisanpassung erforderlich.
Anwendung bei Schwangeren Es liegen keine Daten bei Schwangeren vor.Fampyra® sollte nicht angewendet werden.Es gibt Hinweise auf Reproduktionstoxizität (tierexperimentell).
Anwendung bei Stillenden Es ist nicht bekannt, ob Fampridin in die Muttermilchübergeht.Die Anwendung wird nicht empfohlen.
Dosierung und Kosten
Fampyra® (Fampridin)
Darreichungsform Dosis pro Tag1 Kosten für 2 Wochen [€]2
Kosten pro Jahr[€]2, 3
Retardtabletten 2 x 10 mg 274,18 7027,17
Stand Lauertaxe: 15.11.20111Dosierung gemäß Produktinformation; 2Kostenberechnung nach Apothekenabgabepreis anhand des kostengünstigsten Präparates einschließlich Import (hier nur ein Präparat); 3Kostenberechnung für erstes Behandlungsjahr.
Wie gefährlich sind Rizinussamen?In einer der letzten Fallbesprechungender Arbeitsgemeinschaft Arzneimittel -therapie bei psychiatrischen Erkrankun-gen, (AGATE), wurde von einem Patien-ten berichtet, in dessen Schrank einige
Beutel mit Rizinussamen gefunden wur-den. Nach Angaben des Patienten beab-sichtigte er, die Samen zum Suizid zuverwenden.
Was sind „Rizinussamen“?Hierbei handelt es sich um die Samenvon Rizinus communis, der Rizinusstau-de, auch Wunderbaum oder Christuspal-me (Palma Christi) genannt, ein Strauch
Aus der Praxis – Für die Praxis

46 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
aus der botanischen Familie der Euphor-biaceae, der Wolfsmilchgewächse. Die inden Subtropen beheimatete Pflanze istbekannt aufgrund der laxierenden Wir-kung des aus ihren Samen gewonnenenÖles, des Rizinusöls. Sie wird heute welt-weit als Zierpflanze kultiviert und findetsich in vielen privaten Gärten. Die grau-braun marmorierten bohnenförmigenSamen sind in einer stacheligen Samen-kapsel zu finden. Im Unterschied zumRizinusöl sind die Samen der Pflanzetatsächlich extrem giftig.
GiftwirkungDie Toxizität der Rizinussamen beruhtauf dem in der Samenschale enthaltenenGlykoprotein Rizin, eines der stärkstenGifte im Pflanzenreich. Wird es in eineKörperzelle aufgenommen, kommt eszum Tod der Zelle (Apoptose) (1).
Bereits ein zerkauter Samen kannschwerwiegende Krankheitszeichen her-vorrufen. Zur typischen Symptomatikzählen nach Angaben der GiftzentraleBonn (2): Allgemeines Unwohlsein mitBlässe, Fieber und Zittern, Bauch-schmerzen, Übelkeit und Brechreiz. Inschwerwiegenden Fällen kommt es zuBewusstseinsverminderung, Krampfan-fällen, Herzrhythmusstörungen undTod. Bis zum Auftreten der ersten Sym-
ptome können mehreren Stunden ver-gehen. Der Tod tritt langsam und schlei-chend erst nach einigen Tagen als Folgeeines Nieren- oder Leberversagens ein.
Beim Pressen der Samen zur Gewinnungdes Rizinusöls verbleibt das giftige Rizinin den Samenschalen und gelangt nichtin den Presssaft, sondern bleibt im Press -rückstand. So ist zu erklären, dass Rizi-nusöl im Unterschied zu den Samen, ausdenen es gewonnen wird, keine eigeneGiftwirkung besitzt. Derzeit existiertweder ein Antidot noch eine andere ef-fektive Therapie zur spezifischen Be-handlung einer Vergiftung mit Rizinus-samen (3).
Literatur1.Horrix C, Raviv Z, Flescher E et al.:Plant ribosome-inactivating proteinstype II induce the unfolded protein re-sponse in human cancer cells. Cell MolLife Sci 2011; 68: 1269–1281.
2. Informationszentrale gegen Vergif-tungen Bonn: http://www.meb.uni-bonn.de/giftzentrale. Zuletzt geprüft:1. Dezember 2011.
3. Audi J, Belson M, Patel M et al.: Ricinpoisoning: a comprehensive review.JAMA 2005; 294: 2342–2351.
InteressenkonflikteProf. Haen gibt an: Vortragstätigkeit,Mitarbeit in wissenschaftlichen Beirätenund Teilnahme als Prüfarzt an Klini-schen Studien im Auftrag von Janssen-Cilag, Lilly, Pfizer, GlaxoSmithKli-ne,AstraZeneca, Bristol-Myers Squibb, Ot-suka, Bayer Vital, Servier, SüdmedicaGmbH.
Andrea Lamecker hat keine Kontakte,aus denen mögliche Interessenskonflik-te entstehen könnten.
cand. rer. nat. Andrea Lamecker,RegensburgProf. Dr. med. Dr. rer. nat. EkkehardHaen, Regensburg [email protected]
FAZIT
Die Rizinusstaude, eine mittlerweilebeliebte Zierpflanzen in unseren Gär-ten, ist auf-grund der hohen Toxizitäteine Gefahrenquelle für Kinder. Wegenihrer auffälligen Färbung und den sta-cheligen Samenkapseln kann sie einengroßen Anziehungseffekt ausüben.
Schluckstörungen unter NeuroleptikaSchlucken ist eine elementar lebens-wichtige Funktion. Bereits geringeStörungen können nicht nur zu subjek-tiven Beschwerden führen, sondern sieerhöhen auch das Risiko für lebensbe-drohliche Ereignisse (Aspirationspneu-monie, Bolustod). Sowohl die Anatomieals auch die Physiologie des Schluckak-tes sind sehr komplex.
In der unabhängigen Zeitschrift RevuePrescrire (1) ist kürzlich eine Über-sichtsarbeit zum Thema „Neuroleptikaund Störungen des Schluckens“ erschie-nen. Das Thema ist nicht nur theoretischinteressant, sondern auch praktisch
wichtig: Neuroleptika/Antipsychotikawerden häufig eingesetzt sowohl bei en-dogen-psychotischen Erkrankungen alsauch bei Verwirrtheitszuständen im Se-nium – hier oft als Off Label Use. Hierbeiwird die Möglichkeit, dass Schluck-störungen durch diese Medikamentehervorgerufen werden können, oft nichtbedacht. Die Erkenntnisse über den Zu-sammenhang von Neuroleptika undSchluckstörungen stammen aus epide-miologischen Daten und aus gut recher-chierten Fallberichten.
Trifirò et al (2) konnten in einer popula-tionsbasierten Fall-Kontrollstudie zei-
gen, dass Neuroleptika das Risiko fürambulant erworbene Pneumonien er-höhen. Das relative Risiko oder die oddsratio (OR) für atypische Neuroleptika be-trägt 2,61 (95 % CI: 1,48–4,61). und fürtypische Neuroleptika 1,76 (95 % CI:1,22-2,53). Lediglich für atypische Neu-roleptika ließ sich zusätzlich eine Risi-koerhöhung für tödliche Pneumoniennachweisen (OR 5,97; 95% CI:1,49–23,98).
Ruschena et al (3) zeigten, dass das Risi-ko für den Bolustod sowohl durch diepsychiatrische Grundkrankheit (Schizo-phrenie, hirnorganisches Psychosyn-
Unerwünschte Arzneimittelwirkungen

47Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
FAZIT
1. Neuroleptika können Schluck-störungen mit ernsten und tödlichenFolgen verursachen.2. Absetzen, Dosisreduktion, aberauch Wechsel des Neuroleptikumssind oft in der Lage, in wenigen Tagendie Störung zu beenden. 3. Insbesondere bei hirnorganischveränderten Patienten im höheren Le-bensalter ist die Indikation zum Einsatzvon Neuroleptika (insbesondere deratypischen Neuroleptika) besondersstreng zu stellen.4. Verschiedene pathophysiologischeMechanismen können an den pharma-kogenen Schluckstörungen beteiligtsein.
drom) erhöht wird, als auch durch ver-schiedene Psychopharmaka (in dieserStudie insbesondere Thioridazine alsNeuroleptikum (OR: 92,1) und Lithium(OR: 31,2). Antidepressiva, Anxiolytikaund Hypnotika hingegen erhöhten dasRisiko lediglich um das 2–3 fache.
In einer Vielzahl von Einzelfallberichtenzeigt sich, dass Neuroleptika (typischeund atypische) an Schluckstörungen be-teiligt sind und dass Absetzen, Dosisre-duktion oder Wechsel auf ein anderesNeuroleptikum innerhalb weniger Tagedie Schluckstörung zum Verschwindenbringen können. Diese Störung – aberauch ihre Reversibilität durch eine Ände-rung der Medikation – wurde nicht nurklinisch, sondern auch durch verschie-dene andere Verfahren (röntgenologi-sche Untersuchung des Schluckaktes, fi-beroptische Endoskopie, Ösophagusma-nometrie) objektiviert, z. B. (4–6).
Die pathophysiologischen Mechanismenfür den Zusammenhang zwischen Neu-roleptika und Schluckstörungen sind of-fensichtlich heterogen. Zu nennen sind:
– Extrapyramidale Nebenwirkungen,d.h. indirekte Effekte auf die querge-streifte Mund- und Schlundmuskula-tur (Frühdyskinesien, Parkinsonis-mus, tardive Hyperkinesen)
– Nebenwirkungen auf zentrale und pe-riphere Anteile des vegetativen Ner-vensystems mit Auswirkungen auf dieglatte Muskulatur des Ösophagus,aber auch auf die Speicheldrüsen (an-ticholinerge Wirkungen mit Austrock-nung des Mundes ebenso wie Sialorr-hoe durch Alpha-2-Blockade)
– Sedation als globaler, das Aspirations-risiko erhöhender Faktor
Diagnostisch relevant ist die Tatsache,dass Ösophagus-Motilitätsstörungenauch ohne wesentliche Sedation undohne gleichzeitige extrapyramidale Ne-benwirkungen an der Mund- undSchlundmuskulatur auftreten können.Von Bedeutung ist weiterhin, dass atypi-sche Neuroleptika, die im Allgemeinen alsbesser verträglich als typische Neurolepti-ka gelten, im Hinblick auf Schluckstörun-gen sogar gefährlicher sind, z. B. (6).
Wenn eine tardive Dyskinesie, d. h. eineSpätnebenwirkung der langfristigenNeuroleptika-Gabe, an der Schluck-störung beteiligt ist, besteht das thera-peutische Dilemma, dass eine Reduktiondes eingesetzten Neuroleptikums dietardive Störung zumindest für einigeZeit verschlechtern kann.
Literaturverzeichnis1.Neuroleptics: swallowing disorders.Prescrire Int 2011; 20: 15–17.
2. Trifiro G, Gambassi G, Sen EF et al.:Association of community-acquiredpneumonia with antipsychotic druguse in elderly patients: a nested case-control study. Ann Intern Med 2010;152: 418–440.
3. Ruschena D, Mullen PE, Palmer S etal.: Choking deaths: the role of antip-sychotic medication. Br J Psychiatry2003; 183: 446–450.
4.Dziewas R, Warnecke T, Schnabel M etal.: Neuroleptic-induced dysphagia:case report and literature review. Dys-phagia 2007; 22: 63–67.
5. Stewart JT: Dysphagia associated withrisperidone therapy. Dysphagia 2003;18: 274–275.
6.Maddalena AS, Fox M, Hofmann M,Hock C: Esophageal dysfunction onpsychotropic medication. A case re-port and literature review. Pharma-copsychiatry 2004; 37: 134–138.
InteressenkonflikteEin Interessenskonflikt wird vom Autorverneint.
Prof. Dr. med. Hans-Peter Vogel, [email protected]
Eine „Konjunktivitis“ durch Bisphosphonate
Kanadische Autoren (1) berichten übereine Patientin, bei der unter Alendronat,wöchentlich eine Tablette, eine Erkran-kung auftrat, die wie eine Konjunktivitisaussah.
Alendronat ist das in Deutschland mitAbstand am häufigsten eingesetzte Bis-
phosphonat. Laut Arzneiverordnungsre-port wurde es 2010 mit 138,8 Mio. DDDverordnet (2). Zu den häufigen uner-wünschten Arzneimittelwirkungen(UAW) von Alendronat gehören abdomi-nelle Beschwerden, Muskel- und Skelett-schmerzen sowie Kopfschmerzen undSchwindel. In der Datenbank des deut-
schen Spontanmeldesystems (gemeinsa-me Datenbank vom BfArM und der AkdÄ,Stand November 2011) sind insgesamt552 Verdachtsmeldungen zu Alendronaterfasst. Die am häufigsten gemeldetenReaktionen sind Osteonekrosen des Kie-fers und gastrointestinale Erkrankun-gen. Über unerwünschte Wirkungen am

48 Arzneiverordnung in der Praxis ~ Band 39 · Ausgabe 2 · März 2012
Auge wird in 25 Meldungen berichtet,dabei werden allgemein Sehverschlech-terung (6x), gefolgt von Uveitis (3x),Netzhautablösung und Konjunktivitis(je 2x) am häufigsten genannt.
Im vorliegenden Fall (1) handelt es sichum eine 65 Jahre Patientin, die fünf Mo-nate nach Behandlungsbeginn mit 70mg Alendronat pro Woche über Brennenund ein Fremdkörpergefühl in beidenAugen klagte, sodass zunächst eine Kon-junktivitis vermutet wurde. Eine topi-sche Behandlung mit Tobramycin undDexamethason brachte zunächst Besse-rung, nach Absetzen der Medikamentekehrten die Symptome jedoch zurück.Unter der Annahme einer allergischenKonjunktivitis wurde entsprechend be-handelt, jedoch ohne Erfolg. 19 Monatenach Beginn der Alendronat-Behand-lung verreiste die Patientin und vergaßihre Alendronat-Tabletten. 10 Tage nachder letzten Einnahme verschwand die„Konjunktivitis“. Wieder zu Hause ange-kommen setzte sie die Therapie mitAlendronat fort und die Symptome amAuge traten erneut auf. Auch unter Be-
handlung mit Risedronat und Etidronatkam es zu Beschwerden am Auge, sodassauf eine Behandlung der Osteoporosemit Bisphosphonaten verzichtet wurde.
Die Autoren geben einen Überblick überbekannt gewordene UAW von Bisphos-phonaten am Auge wie Uveitis, Episkle-ritis, Skleritis und Keratitis. Das höchsteRisiko eines langfristigen Sehverlustsgeht mit einer Skleritis und einer Kera-titis einher. In der Fachinformation vonAlendronat werden entzündliche Verän-derungen am Auge (Uveitis, Skleritis,Episkleritis) als „gelegentlich“ auftre-tend (≥1/1.000, <1/100) aufgeführt (3).Die kanadischen Autoren bedauern, dassim beschriebenen Fall erst vier Ärztekonsultiert werden mussten, bis ein Apo-theker auf die richtige Spur kam und denHausarzt informierte. Wie immer ist das„Daran-Denken“ der entscheidendePunkt.
Literatur1.McKague M, Jorgenson D, Buxton KA:Ocular side effects of bisphosphonates:
A case report and literature review.Can Fam Physician 2010; 56: 1015–1017.
2. Schwabe U, Paffrath D (Hrsg.): Arznei-verordnungs-Report 2010. Berlin,Heidelberg: Springer Medizin Verlag,2010.
3.MSD Sharp & Dohme GmbH: Fachin-formation „Fosamax® 10 mg Tablet-ten“. Stand: Oktober 2011.
InteressenkonflikteEin Interessenkonflikt wird von beidenAutoren verneint.
Prof. Dr. med. D. Höffler, DarmstadtDr. med. Th. Stammschulte, Berlin [email protected]
Terminankündigung118. Kongress der Deutschen Gesellschaft für Innere Medizin e.V.
Symposium der Arzneimittelkommission der deutschen Ärzteschaft„Personalisierte Medizin“
Termin Samstag, 14. April 2012, 14.00–15.30 UhrVeranstaltungsort Museum Wiesbaden, Friedrich-Ebert-Allee 2, 65185 Wiesbaden
(gegenüber Rhein-Main-Hallen)Vorsitzende Prof. Dr. med. Wolf-Dieter Ludwig, Berlin; Prof. Dr. med. Ulrich Schwabe, HeidelbergModeration Prof. Dr. med. Wolf-Dieter Ludwig, Berlin
Vorsitzender der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ)Programm Personalisierte Medizin in der Onkologie
Prof. Dr. med. Bernd Mühlbauer, Bremen, Vorstandsmitglied der AkdÄPersonalisierte Medizin in der KardiologieProf. Dr. med. Feraydoon Niroomand, MülheimNeue Arzneimittel 2011/2012 – eine kritische BewertungProf. Dr. med. Ulrich Schwabe, Heidelberg, Mitglied der AkdÄ
Auskunft Karoline Luzar, Arzneimittelkommission der deutschen Ärzteschaft, E-Mail: [email protected]; Telefon: 030 400456-500; Fax: 030 400456-555
FAZIT
Bisphosphonate können zu entzündli-chen Veränderungen am Auge führen.Bei jeder „Konjunktivitis“ eines älterenMenschen sollte nach der Einnahmevon Bisphosphonaten gefragt werden.
In eigener Sache





















