Embed Size (px)
Citation preview

Einführung
in die
Gentechnologie
Institut für Molekulargenetik, gentechnologische Sicherheitsforschung und Beratung Johannes Gutenberg-Universität Mainz Leiter: Prof. Dr. Erwin R. Schmidt Betreuer: Dr. Christiane Kraemer, Dr. Steffen Rapp
Dipl.Biol.s Nicholas Bachtadse, Sarah Brunck, Sabine Fischer, Tobias Lautwein, Romina Petersen
Dipl. Bioinformatiker (FH) Benjamin Rieger Rudolf Baader, Nicole Naumann

F1-Laborpraktikum SoSe 2013
2
Inhalt F1-Laborpraktikum Gentechnologie ....................................................................................................... 3
Zeitplan (nicht bindend) ...................................................................................................................... 8
Theorie Teil 1: Herstellen einer cDNA-Bibliothek für „Next-Generation Sequencing“ .................... 10
Praxis Teil 1: Herstellen einer cDNA-Bibliothek für die Sequenzierung ........................................... 12
Allgemeine Laborsicherheit und Verhaltensregeln ....................................................................... 12
Hinweise zum Arbeiten mit RNA ................................................................................................... 12
RNA-Isolierung: Guanidinthiocyanat (GTC) – Methode nach Chomczynski und Sacchi (1987) ... 12
Erstellen einer cDNA-Library mit dem IlluminaTruSeq RNA Kit .................................................... 15
Qualitätskontrolle und Quantifizieren der Bibliotheken ............................................................... 18
Vorbereitungen - cBot - Sequenzierung ........................................................................................ 21
Theorie Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-Daten ..................... 22
Sequenzalignment ......................................................................................................................... 22
Assemblierungsmethoden............................................................................................................. 22
BLAST ............................................................................................................................................. 23
De-Bruijn-Graph (DBG) .................................................................................................................. 25
Arbeitsablauf Sequenzverarbeitung .............................................................................................. 28
Möglichkeiten der Parallelisierung ................................................................................................ 28
Praxis Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-Daten ........................ 29
Sequenzierung und Basecalling ..................................................................................................... 30
Datenformat FASTQ....................................................................................................................... 31
Quality Scores ................................................................................................................................ 32
Bioinformatische Analyse der „Next-Generation Sequencing“ Daten mit Hilfe der „CLC Genomics
Workbench“ .................................................................................................................................. 34
Import der „Next-Generation Sequencing“-Daten........................................................................ 34
„Trimmen“ der Sequenzierungsreads ........................................................................................... 35
Assemblierung der Sequenzierungsreads ..................................................................................... 36
„De novo“ Assemblierung .............................................................................................................. 36
„Mapping“ der reads in einer Referenzsequenz ........................................................................... 37
„RNA-Seq“-Analyse ........................................................................................................................ 38
Interpretation der RNA-Seq-Analyse ............................................................................................. 42
Expressionsanalyse ........................................................................................................................ 44
Transformation und Normalisierung ............................................................................................. 47
Statistische Analyse: Identifizierung differenzieller Expression .................................................... 48
Anhang........................................................................................................................................... 49

F1-Laborpraktikum SoSe 2013
3
F1-Laborpraktikum Gentechnologie
Das FI-Praktikum „Methoden der Gentechnologie - NGS“ soll technische Fähigkeiten im Bereich der
Molekulargenetik sowie in der bioinformatischen Auswertung von Sequenzdaten vermitteln. Dabei
soll einerseits die Isolation von RNA aus Chironomus thummi Embryonen mit der anschließenden
Konstruktion einer cDNA-Bibliothek für die Sequenzierung auf dem HiSeq 2500 oder MiSeq erlernt
werden. Andererseits sollen bioinformatische Kenntnisse in der Analyse von NGS-Daten mit Hilfe von
gängiger Software vermittelt werden.
Die Familie der Chironomiden (Zuckmücken) gehört zur Ordnung der Diptera (Zweiflügler).
Chironomiden haben keine typischen Geschlechtschromosomen wie z.B. die X- und Y-Chromosomen
von Säugern. Trotzdem konnte durch genetische Analysen nachgewiesen werden, dass der
Geschlechtsbestimmungsmechanismus bei Chironomiden die männliche Heterogametie ist.
In C. thummi konnte darüber hinaus durch cytogenetische Analysen festgestellt werden, dass der
dominante männchenbestimmende Faktor M in einer Chromosomenregion nahe dem Telomer von
Chromosom III liegt.
In situ-Hybridisierungen an Polytänchromosomen von
Chironomiden mit hoch repetitiven Cla-Elementen als Sonde
zeigen ein hemizygotes Signal in genau dieser
Chromosomenregion bei männlichen Tieren der Art
C. thummi, welches in den Präparaten weiblicher Tiere fehlt.
Da dieses Cla-Element-Cluster in der geschlechts-
bestimmenden Chromosomenregion liegt und nur bei
männlichen Tieren und dort immer nur hemizygot zu finden
ist, muss dieses Cluster eng gekoppelt mit dem dominanten
Faktor M sein. Um einen molekularen Zugang zu dieser hochinteressanten Chromosomenregion zu
bekommen, wurden von unserer Arbeitsgruppe genomische Lambda-Bibliotheken von C. thummi mit
den hochrepetitiven Cla-Elementen als Sonde gescreent.
Alle positiven Klone wurden anschließend durch in situ-Hybridisierung an Polytänchromosomen von
C. thummi auf ihre chromosomale Herkunft hin überprüft. Die dabei gefundenen Lambda-Klone
λCla1.1Y und λCla1.8Z flankieren das männchenspezifische hemizygote Cla-Element-Cluster links und
rechts (siehe Abbildung 1).
Ausgehend von den genomischen Klonen λCla1.1Y und λCla1.8Z wurden sowohl mit λ- als auch
später mit BAC-Bibliotheken von C. thummi sowie C. piger „chromosomal walks“ durchgeführt. Auf
diese Art wurden insgesamt mehr als 200 kb aus der geschlechtsbestimmenden Region kloniert und
Abbildung 1: Geschlechtsbestimmende Region
Abbildung 2: Struktur der geschlechtsbestimmenden Region
F1-Laborpraktikum SoSe 2013
4
größtenteils sequenziert (Abbildung 2). Innerhalb dieser Chromosomenregion konnten repetitive
Hind-Elemente und Kopien von transposablen Elementen wie TFB1 oder CCE entdeckt werden.
Zudem zeichnet sich diese Region durch das Auftreten von zahlreichen invertierten
Sequenzwiederholungen (inverted repeats, IR) aus.
Um Genstrukturen innerhalb des ca. 200 kb großen Contigs zu identifizieren, wurden verschiedene
Programme (Genescan, PROSCAN, Orf-Finder) eingesetzt. Des Weiteren wurde die NCBI-Datenbank
mit Hilfe der BLASTn-, BLASTp- und BLASTx-Algorithmen auf mögliche Gene hin untersucht. Auf diese
Art und Weise konnten ca. 20 Gene identifiziert werden. Die Namensgebung erfolgte dabei nach den
Treffern in der NCBI-Datenbank.
Die Funktion der meisten dieser Gene bei C. thummi und C. piger ist bis jetzt unbekannt. Ob es sich
bei einem dieser Gene tatsächlich um den dominanten Männchenbestimmer M handelt kann also
nur vermutet werden. Teilweise ist die Funktion der homologen Gene z.B. in Drosophila
melanogaster bekannt. Da es aber nur abschnittsweise Übereinstimmungen zu der Sequenz in
C. thummi und C. piger gibt, können diese Informationen nur als Hinweise auf die Eigenschaften der
entsprechenden Gene in der geschlechtsbestimmenden Region dienen.
Gen Orient. Lage im Contig Funktion (in urspr. Organismus) Besonderheiten
GJ16627-like + 3116-3984 unbekannt (Drosophila virilis)
ACYPI006459 + 17794-20225 ähnl. Nuklearer Rezeptor Coaktivator GT198 (Acyrthosiphon pisum)
spitz-like 1 - 33015-20685
TGF-Alpha-Homolog (Drosophila melanogaster)
spitz-like 2 - 81050-70145
spitz-like 3 + 101929-107219
CENP-E-like - 111082-108639 Kinesin-ähnliches Motorprotein (Homo sapiens)
mi-er1-like - 114160-111216 Differenzierung zu mesodermalen Zellen (Xenopuslaevis)
LUC7-like + 114949-116987 Komponente des U1snRNPs
CG7845-like - 120124-117100 unbekannt (Drosophila melanogaster) WD40-Domäne
fs(1)K10-like + 120143-122339 Etablierung der dorso-ventralen Achse der Oocyte (Drosophila melanogaster)
bcn92-like + 122699-123309 evtl. Oxidoreduktase (Drosophila melanogaster)
d-Lactat-DH-like - 128444-122597 Oxidation von d-Lactat zu Pyruvat
polyhomeotic + 132545-139599 Kontrolle der homöotischen Genexpression (Drosophila melanogaster)
CG31635-like + 144342-145968 unbekannt (Drosophila melanogaster)
CG15207-like + 146638-147473 unbekannt (Drosophila melanogaster)
CG11203-like + 162169-171517 unbekannt (Drosophila melanogaster)
rpn5 + 173725-175435 Untereinheit und Regulator des 26S-Proteasoms
HPS-like - 180208-175467 Komponente des "biogenesis of lysosome-related organelles complex" (Dm)
rpS5-like + 180785-182242 28S ribosomales Protein S5 (Dm)
CtY - 196977-182308 DmX-Homolog (Drosophila melanogaster) WD-Repeat-Protein
CG2841(ptr)-like - 199998-197093 unbekannt (Drosophila melanogaster)
F1-Laborpraktikum SoSe 2013
5
Besonders interessant im Hinblick auf die Identität des Männchenbestimmers M sind
Sequenzunterschiede zwischen Proto-X- und Proto-Y-Chromosom. Ein solcher Unterschied konnte in
Form eines duplizierten Abschnitts im Bereich der Gene CG7845-like und fs(1)K10-like gefunden
werden. Des Weiteren wurden kurz hinter dem Gen D-Laktat-DH-like ein Hinf(lur)-Element und der
vermutliche Beginn des Cla-Element-Clusters identifiziert. Im Moment wird daran gearbeitet, mit
verschiedenen Methoden weitere Sequenzinformationen des Proto-Y-Chromosoms zu erhalten.
Zur näheren Analyse der Gene in der geschlechtsbestimmenden Region und einer möglichen
geschlechtsspezifischen Expression dieser Gene wird eines der modernsten Sequenzierverfahren
eingesetzt. Die Illumina HiSeq 2500-Plattform reiht sich ein in eine Reihe von neuen Technologien im
Bereich der Hochdurchsatzsequenzierung, dem sogenannten „Next-Generation Sequencing“ (NGS).
Die meist verbreiteten Technologien dieser Gruppe sind die SOLiDTM-, 454- und Illumina-Verfahren.
Sie machen es möglich, immer günstiger und in immer kürzerer Zeit große Mengen an
Sequenzinformation zu generieren. Hat die Sequenzierung des menschlichen Genoms im Rahmen
des humanen Genom-Projekts (1990-2003) noch über 3 Mrd. US$ und 13 Jahre Forschung benötigt,
so sind Resequenzierungen menschlicher Genome heute innerhalb von 1-2 Wochen und mit Kosten
im Bereich von 5.000-10.000 € möglich. Neben dem auf Ligation von Oligonukleotiden basierenden
SOLiDTM-System und der Pyrosequenzierung der 454 Roche-Plattform dürften die „Genome Analyzer
IIx“ und „HiSeq“-Systeme der Firma Illumina weltweit den größten Anteil an Sequenzier-Projekten
haben. Zurzeit sind hier mit bis zu 150 nt im Vergleich zur SOLiD-Platform wesentlich größere und im
Vergleich zur 454-Technologie (~800 nt) deutlich kürzere Leseweiten möglich. Allerdings bietet
Illumina mit wesentlich geringeren Fehlerraten und mehr Sequenzinformation eindeutige Vorteile
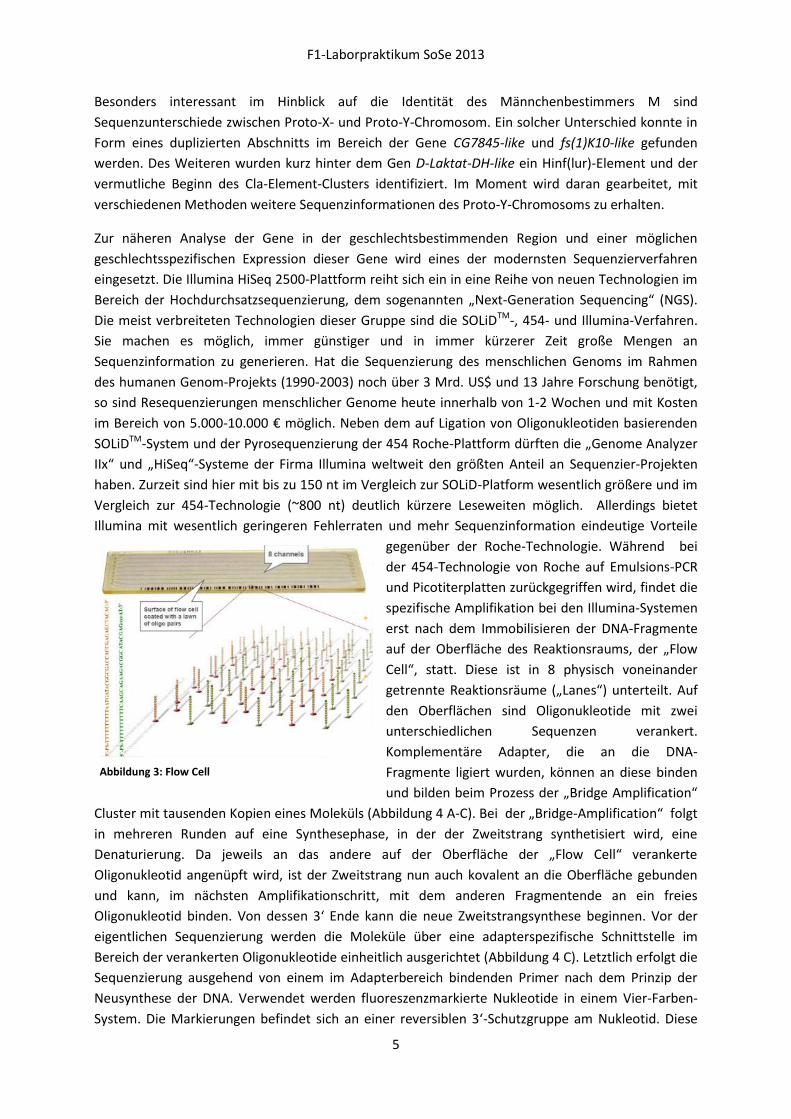
gegenüber der Roche-Technologie. Während bei
der 454-Technologie von Roche auf Emulsions-PCR
und Picotiterplatten zurückgegriffen wird, findet die
spezifische Amplifikation bei den Illumina-Systemen
erst nach dem Immobilisieren der DNA-Fragmente
auf der Oberfläche des Reaktionsraums, der „Flow
Cell“, statt. Diese ist in 8 physisch voneinander
getrennte Reaktionsräume („Lanes“) unterteilt. Auf
den Oberflächen sind Oligonukleotide mit zwei
unterschiedlichen Sequenzen verankert.
Komplementäre Adapter, die an die DNA-
Fragmente ligiert wurden, können an diese binden
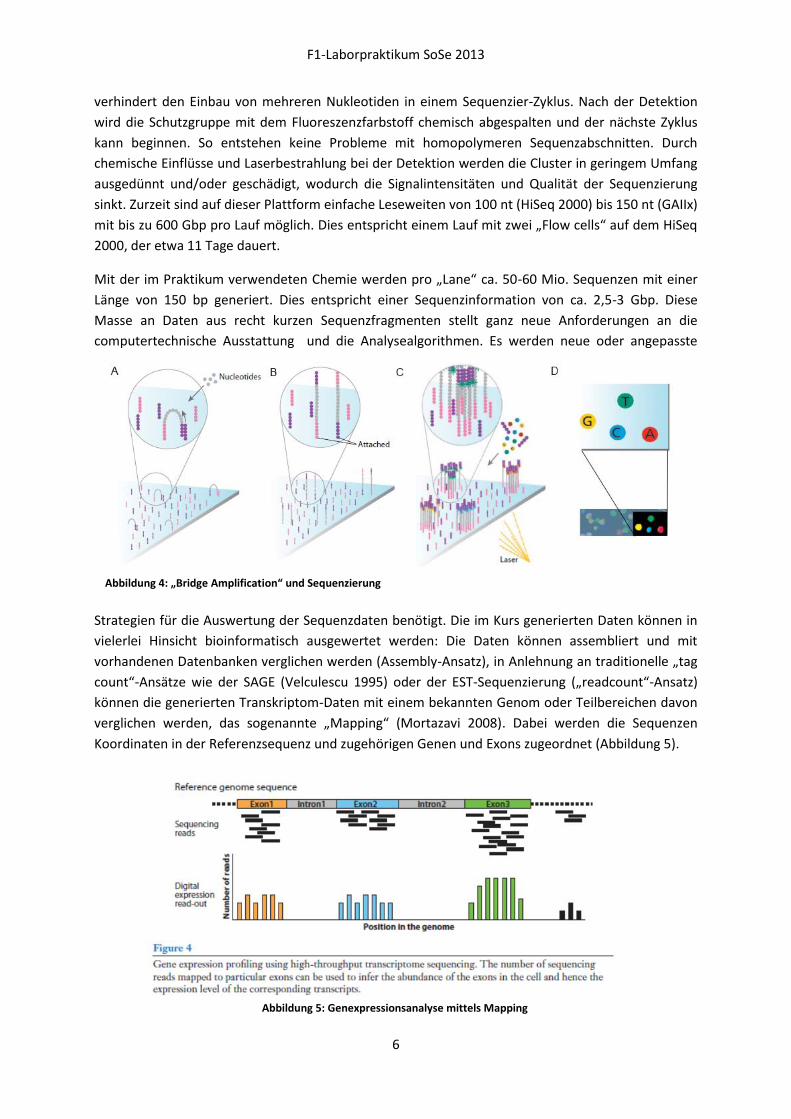
und bilden beim Prozess der „Bridge Amplification“
Cluster mit tausenden Kopien eines Moleküls (Abbildung 4 A-C). Bei der „Bridge-Amplification“ folgt
in mehreren Runden auf eine Synthesephase, in der der Zweitstrang synthetisiert wird, eine
Denaturierung. Da jeweils an das andere auf der Oberfläche der „Flow Cell“ verankerte
Oligonukleotid angenüpft wird, ist der Zweitstrang nun auch kovalent an die Oberfläche gebunden
und kann, im nächsten Amplifikationschritt, mit dem anderen Fragmentende an ein freies
Oligonukleotid binden. Von dessen 3‘ Ende kann die neue Zweitstrangsynthese beginnen. Vor der
eigentlichen Sequenzierung werden die Moleküle über eine adapterspezifische Schnittstelle im
Bereich der verankerten Oligonukleotide einheitlich ausgerichtet (Abbildung 4 C). Letztlich erfolgt die
Sequenzierung ausgehend von einem im Adapterbereich bindenden Primer nach dem Prinzip der
Neusynthese der DNA. Verwendet werden fluoreszenzmarkierte Nukleotide in einem Vier-Farben-
System. Die Markierungen befindet sich an einer reversiblen 3‘-Schutzgruppe am Nukleotid. Diese
Abbildung 3: Flow Cell
F1-Laborpraktikum SoSe 2013
6
verhindert den Einbau von mehreren Nukleotiden in einem Sequenzier-Zyklus. Nach der Detektion
wird die Schutzgruppe mit dem Fluoreszenzfarbstoff chemisch abgespalten und der nächste Zyklus
kann beginnen. So entstehen keine Probleme mit homopolymeren Sequenzabschnitten. Durch
chemische Einflüsse und Laserbestrahlung bei der Detektion werden die Cluster in geringem Umfang
ausgedünnt und/oder geschädigt, wodurch die Signalintensitäten und Qualität der Sequenzierung
sinkt. Zurzeit sind auf dieser Plattform einfache Leseweiten von 100 nt (HiSeq 2000) bis 150 nt (GAIIx)
mit bis zu 600 Gbp pro Lauf möglich. Dies entspricht einem Lauf mit zwei „Flow cells“ auf dem HiSeq
2000, der etwa 11 Tage dauert.
Mit der im Praktikum verwendeten Chemie werden pro „Lane“ ca. 50-60 Mio. Sequenzen mit einer
Länge von 150 bp generiert. Dies entspricht einer Sequenzinformation von ca. 2,5-3 Gbp. Diese
Masse an Daten aus recht kurzen Sequenzfragmenten stellt ganz neue Anforderungen an die
computertechnische Ausstattung und die Analysealgorithmen. Es werden neue oder angepasste
Strategien für die Auswertung der Sequenzdaten benötigt. Die im Kurs generierten Daten können in
vielerlei Hinsicht bioinformatisch ausgewertet werden: Die Daten können assembliert und mit
vorhandenen Datenbanken verglichen werden (Assembly-Ansatz), in Anlehnung an traditionelle „tag
count“-Ansätze wie der SAGE (Velculescu 1995) oder der EST-Sequenzierung („readcount“-Ansatz)
können die generierten Transkriptom-Daten mit einem bekannten Genom oder Teilbereichen davon
verglichen werden, das sogenannte „Mapping“ (Mortazavi 2008). Dabei werden die Sequenzen
Koordinaten in der Referenzsequenz und zugehörigen Genen und Exons zugeordnet (Abbildung 5).
Abbildung 4: „Bridge Amplification“ und Sequenzierung
Abbildung 5: Genexpressionsanalyse mittels Mapping

F1-Laborpraktikum SoSe 2013
7
Ziel ist es, das Vorkommen von Sequenzen in dieser Region zu quantifizieren und Unterschiede im
Vergleich verschiedener Zustände aufzuzeigen. In unserem Fall bedeutet dies, mögliche Unterschiede
in der Transkription der Gene aus der geschlechtsbestimmenden Region zwischen Männchen und
Weibchen zu entdecken. Dies kann sich beispielsweise in unterschiedlichen Transkriptionsraten
darstellen, aber auch mögliche geschlechtsspezifisch unterschiedlich gespleißte Transkripte können
auf diese Art und Weise detektiert werden.
Da die Determination des Geschlechts sehr früh in der Entwicklung eines Individuums stattfindet, ist
es sinnvoll, für die RNASeq –Analyse Embryonalstadien zu verwenden. Da in einem Chironomus
Gelege männliche und weibliche Embryonen nebeneinander vorkommen, ist es naturgemäß sehr
schwierig, das Geschlecht der Embryonen a priori zu bestimmen. Die Isolierung von RNA aus einem
einzelnen Embryo gestaltet sich ebenfalls als schwierig, da durch die notwendigen Reinigungsschritte
ein großer Teil der RNA aus dem Embryo verloren geht.
In dem diesjährigen Praktikum soll daher versucht werden, RNA aus einzelnen Chironomus
Embryonen zu isolieren und bei den notwendigen Aufreinigungsschritten durch Zugabe einer
heterologen „Carrier-RNA“ (z.B. von Vitis oder Malus) die Verluste an Chironomus-RNA zu
minimieren.
Nach erfolgreicher RNA-Isolierung wird eine cDNA-Bibliothek für die NGS-Sequenzierung konstruiert
und ein HiSeq- oder MiSeq-Lauf gestartet. Bei der anschließenden bioinformatischen Analyse der
Daten soll zunächst die heterologe Carrier-cDNA durch „Mapping“ aussortiert werden. Anschließend
soll analysiert werden, ob und in welchem Umfang es gelungen ist, die RNA eines einzelnen
Chironomus-Embryos zu sequenzieren. Diese Sequenzen sollen schließlich insbesondere in Hinblick
auf die Transkriptionsprofile der Gene aus der SDR analysiert werden.

F1-Laborpraktikum SoSe 2013
8
Zeitplan (nicht bindend)
Praxis Teil 1: Herstellen einer cDNA-Bibliothek für die Sequenzierung
Mo, 17.06.2013 - RNA-Isolierung - Bioanalyzer Di, 18.06.2013 - 2x polyA+ -Aufreinigung - cDNA-Synthese (Erst-und Zweitstrang) - AMPure-Aufreinigung - Endrepair - AMPure-Aufreinigung Mi, 19.06.2013 - A-Tailing des 3´Endes und Adapter-Ligation - AMPure-Aufreinigung (2x) - Anreicherung - AMPure-Aufreinigung - Q-Bit - Bioanalyzer Do, 20.06.2013 - qPCR (evtl.) - Vorbereitung c-Bot-Sequenzierung (ggfs) Fr, 21.06.2013 - Vorbereitung c-Bot-Sequenzierung

F1-Laborpraktikum SoSe 2013
9
Theorie Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-Daten
Mo, 24.06.2013 Di, 25.06.2013 Mi, 26.06.2013 Do, 27.06.2013 Fr, 28.06.2013

F1-Laborpraktikum SoSe 2013
10
Theorie Teil 1: Herstellen einer cDNA-Bibliothek für „Next-Generation Sequencing“
Zum Vergleich der Genexpression zwischen verschiedenen Zuständen wird zunächst die RNA dieser
Gewebe benötigt. Dazu muss zunächst ein Gewebeaufschluss erfolgen, der die Zellen aus dem
Verbund löst und somit besser erreichbar für die bei der Extraktion verwendeten Detergenzien
macht. Dies geschieht meist mechanisch durch Mörsern oder Pottern. Bei der RNA-Isolierung werden
Guanidinhydrochlorid, Guanidinthiocyanat oder sehr hohe Harnstoffkonzentrationen bevorzugt.
Diese zerstören gleichzeitig die Zellwand, lösen die Zellwandfragmente auf und denaturieren
endogene RNasen. Anschließend werden die Zelltrümmer und Proteine durch Phenol-Chloroform-
Extraktionen entfernt. Dabei verbleiben diese aufgrund ihrer hydrophoben Bestandteile in der
phenolischen bzw. Interphase. Nach mehrmaligen Extraktionen enthält die wässrige Phase die
hydrophilen Nukleinsäuren RNA und DNA. Zur Fällung von RNA werden im Wesentlichen
Natriumacetat oder Lithiumchlorid verwendet. Da RNA bei etwas höherem Ethanolgehalt (2,5faches
Volumen) gefällt wird, bietet die Verwendung von LiCl aufgrund des größeren Ionendurchmessers
etwas höhere Ausbeuten. Allerdings wird oftmals auch NaAc benutzt, um den Einfluss von LiCl auf
nachfolgende enzymatische Reaktionen auszuschließen. In diesem Fall sollte die Inkubationszeit bei -
20°C und die Zentrifugationszeit erhöht werden. Um Kontaminationen der RNA mit DNA zu
vermeiden kann anschließend ein DNase-Verdau durchgeführt werden. Da die isolierte RNA zu 98 %-
99 % aus ribosomaler RNA besteht, ist eine Anreicherung von mRNA für die meisten Experimente zur
Genexpressionsanalyse notwendig. Dabei macht man sich zu Nutze, dass mRNA am 3’ Ende
polyadenyliert ist. Durch Hybridisierung an ein Oligo-dT-Nukleotid, das an einen selektiv
abtrennbaren Stoff (Cellulose, Latex- oder magnetische Beads) gebunden ist, kann die nicht
gebundene rRNA weggewaschen und zum Großteil entfernt werden. Für die anschließende cDNA-
Synthese wird in der Regel ebenfalls ein Oligo-dT-Nukleotid als Primer verwendet. In diesem Fall
startet die Erststrangsynthese am 3’ Ende des Gens. Da die cDNA-Synthese häufig vorzeitig abbricht,
kommt es zu einer Anreicherung von 3‘-Sequenzen. Um dies zu vermeiden, werden in heutigen
Protokollen nach mehrmaliger polyA+-Aufreinigung „random“ Hexamere zum Priming der cDNA-
Synthese verwendet. In der Erststrangsynthese wird mittels einer Reversen Transkriptase ein zur RNA
komplementärer Strang synthetisiert. Der Zweitstrang wird meist mithilfe der T4-DNA-Polymerase
nach Zugabe von RNase H durchgeführt. RNase H in geringer Konzentration führt zu einem nur
teilweisen Abbau des RNA-Strangs des RNA-DNA-Hybrids. Die verbleibenden RNA Fragmente dienen
als Primer für die DNA-Polymerase. Vor der Ligation von Adaptern werden etwaige einzelsträngige
Enden mit Hilfe des Klenow-Fragments aufgefüllt. Um im Anschluss bessere Resultate bei der
Adapterligation zu erhalten, werden die doppelsträngigen DNA-Fragmente zusätzlich mit einem
A-Überhang versehen. Dies geschieht meist durch eine Taq-DNA-Polymerase, der zum Einbau nur
dATPs angeboten werden.
F1-Laborpraktikum SoSe 2013
11
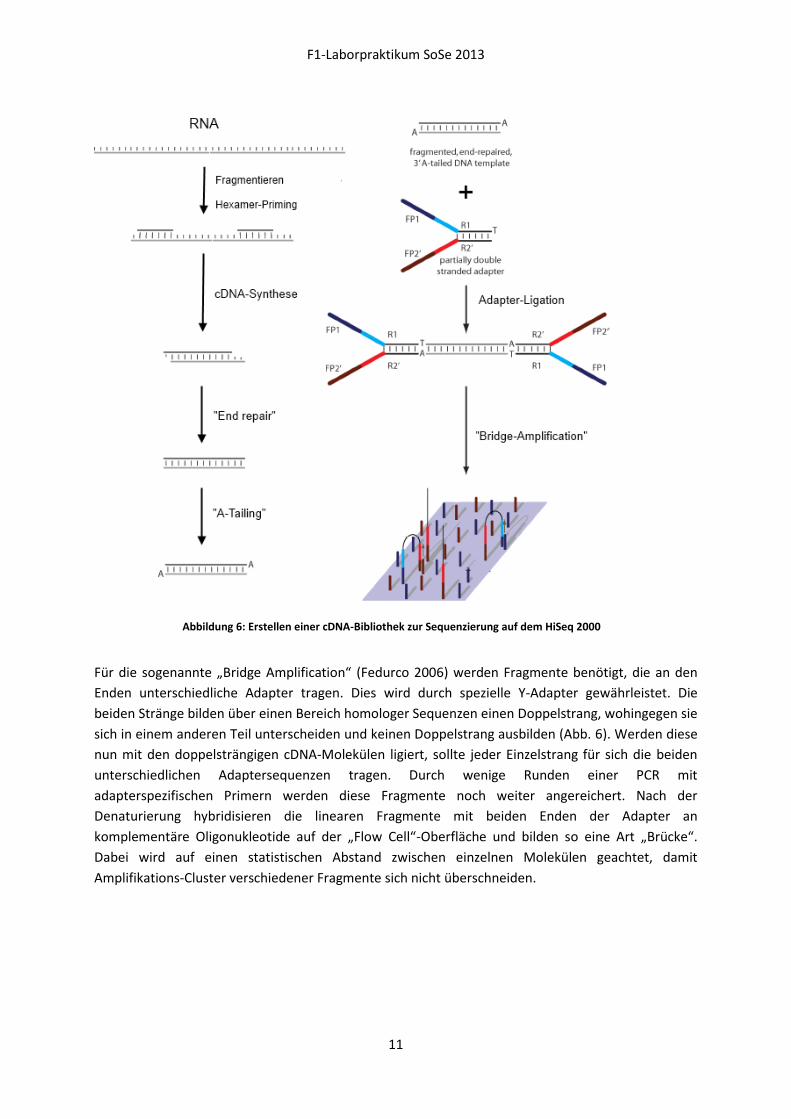
Für die sogenannte „Bridge Amplification“ (Fedurco 2006) werden Fragmente benötigt, die an den
Enden unterschiedliche Adapter tragen. Dies wird durch spezielle Y-Adapter gewährleistet. Die
beiden Stränge bilden über einen Bereich homologer Sequenzen einen Doppelstrang, wohingegen sie
sich in einem anderen Teil unterscheiden und keinen Doppelstrang ausbilden (Abb. 6). Werden diese
nun mit den doppelsträngigen cDNA-Molekülen ligiert, sollte jeder Einzelstrang für sich die beiden
unterschiedlichen Adaptersequenzen tragen. Durch wenige Runden einer PCR mit
adapterspezifischen Primern werden diese Fragmente noch weiter angereichert. Nach der
Denaturierung hybridisieren die linearen Fragmente mit beiden Enden der Adapter an
komplementäre Oligonukleotide auf der „Flow Cell“-Oberfläche und bilden so eine Art „Brücke“.
Dabei wird auf einen statistischen Abstand zwischen einzelnen Molekülen geachtet, damit
Amplifikations-Cluster verschiedener Fragmente sich nicht überschneiden.
Abbildung 6: Erstellen einer cDNA-Bibliothek zur Sequenzierung auf dem HiSeq 2000

F1-Laborpraktikum SoSe 2013
12
Praxis Teil 1: Herstellen einer cDNA-Bibliothek für die Sequenzierung
Allgemeine Laborsicherheit und Verhaltensregeln
1. Tragen Sie für alle Arbeiten im Labor einen Laborkittel 2. In den Laborräumen sollten nur Schreib- und Arbeitsmaterialien aufbewahrt werden.
Straßenkleidung und Taschen sind in die dafür vorgesehenen Spinde (SBI) einzuschließen. Bitte hierfür ein geeignetes Vorhängeschloss mitbringen. In den Laborräumen ist das Essen, Trinken und Rauchen verboten.
3. Arbeiten Sie umsichtig und bewusst. Gefährden Sie weder sich selbst noch Ihre Kommilitonen. 4. Studentinnen, die sich in anderen Umständen befinden, teilen dies dem Kursleiter mit. Er wird
diese Mitteilung vertraulich behandeln, eine erfolgreiche Kursteilnahme ermöglichen und Sie von gesundheitsschädlichen Stoffen fernhalten.
5. Wir arbeiten mit organischen Lösungsmitteln, Laugen und Säuren. Vermeiden Sie unbedingt Hautkontakt bzw. orale Aufnahme. Nutzen Sie die angebotenen Schutzmittel wie Handschuhe und Schutzbrillen.
6. Falls Sie sich verletzen sollten, informieren Sie sofort den Kursleiter. Im Bedarfsfall sind in jedem Laborraum Augenduschen vorhanden.
Hinweise zum Arbeiten mit RNA
Ribonukleasen (RNasen) sind sehr stabile und aktive Enzyme, die normalerweise auch ohne
Co-Faktoren ihre Funktion ausüben können. Da RNasen nur schwer zu inaktivieren sind und selbst
kleinste Mengen ausreichen um RNA zu zerstören, sind beim Arbeiten mit RNA besondere
Maßnahmen zu ergreifen. Aufgrund des ubiquitären Vorkommens von RNasen sollten bei Arbeiten
mit RNA-haltigen Lösungen Handschuhe getragen werden. RNA-Lösungen sollten zwischen den
Experimenten stets auf Eis gelagert werden. Sterile Polypropylen-Tubes sind für die Arbeit mit RNA
am besten geeignet, da diese Tubes normalerweise auch RNase-frei sind und daher keine besondere
Behandlung mehr benötigen. Alle Lösungen, auch Wasser, werden mit Diethylpyrocarbonat (DEPC)
behandelt, um evtl. enthaltene RNasen zu inaktivieren.
Achtung! DEPC gilt als carcinogen und ist leicht flüchtig! Alle Arbeiten mit DEPC werden daher
ausschließlich mit Handschuhen, sowie unter einem Abzug durchgeführt. Anschließend werden die
DEPC-behandelten Lösungen autoklaviert, um DEPC zu deaktivieren.
RNA-Isolierung: Guanidinthiocyanat (GTC) – Methode nach Chomczynski und Sacchi (1987)
1. Gewebeaufschluss
Zunächst müssen aus einem Gelege einzelne Embryonen isoliert werden:
- befruchtetes Gelege sammeln und in ein Eppendorfgefäß mit VE-Wasser geben
- einen Tropfen DanChlorix-Lösung (0,5% Natriumhypochlorit) zugeben, um die Gallerte
aufzulösen
- Embryonen mehrmals mit HPLC-Wasser waschen
- Embryonen in eine Petrischale überführen
2. - einen einzelnen Embryo vorsichtig mit einer Pipette mit weißer Spitze ansaugen, in 100 µl
Guanidinthiocyanat-Lösung überführen und einige Male auf- und abpipettieren
3. 1/10 Volumen 2M NaAcetat (pH 4,1) und Carrier-RNA zur Lösung geben.
F1-Laborpraktikum SoSe 2013
13
4. je eine Extraktion mit Phenol/Chloroform/Isoamylalkohol (PCI) und Chloroform/Isoamyalkohol
(CI) durchführen:
Hierzu 1 Volumen Tris-gesättigtes (pH 7,2) Phenol und 1/5 Volumen Chloroform/Isoamylalkohol
(24:1) zum Homogenisat geben. Danach mehrmals invertieren und anschließend 10 min auf Eis
inkubieren. Proben bei 4°C und 5000 g für 20 min zentrifugieren.
Überstand abheben und mit 1 Volumen Chloroform/Isoamylalkohol versetzen. Danach
mehrmals invertieren und anschließend 10 min auf Eis inkubieren. Proben bei 4°C und 5000 g
für 20 min zentrifugieren
5. Zur Fällung der RNA die wässrige Phase mit 1 Volumen Isopropanol (abs.) versetzen, mehrmals
invertieren und für 1 h bei -20°C lagern. Danach bei 4°C und 5000 g für 40 min zentrifugieren,
Überstand abnehmen und das Pellet mit 75 % Ethanol waschen, 5 min zentrifugieren (4°C, 5000
g), anschließend in einer Vakuum-Zentrifuge (Eppendorf „Concentrator 5301“) bei 30°C trocknen
und in 50 µl DEPC-Wasser/Nuklease freiem Wasser lösen.
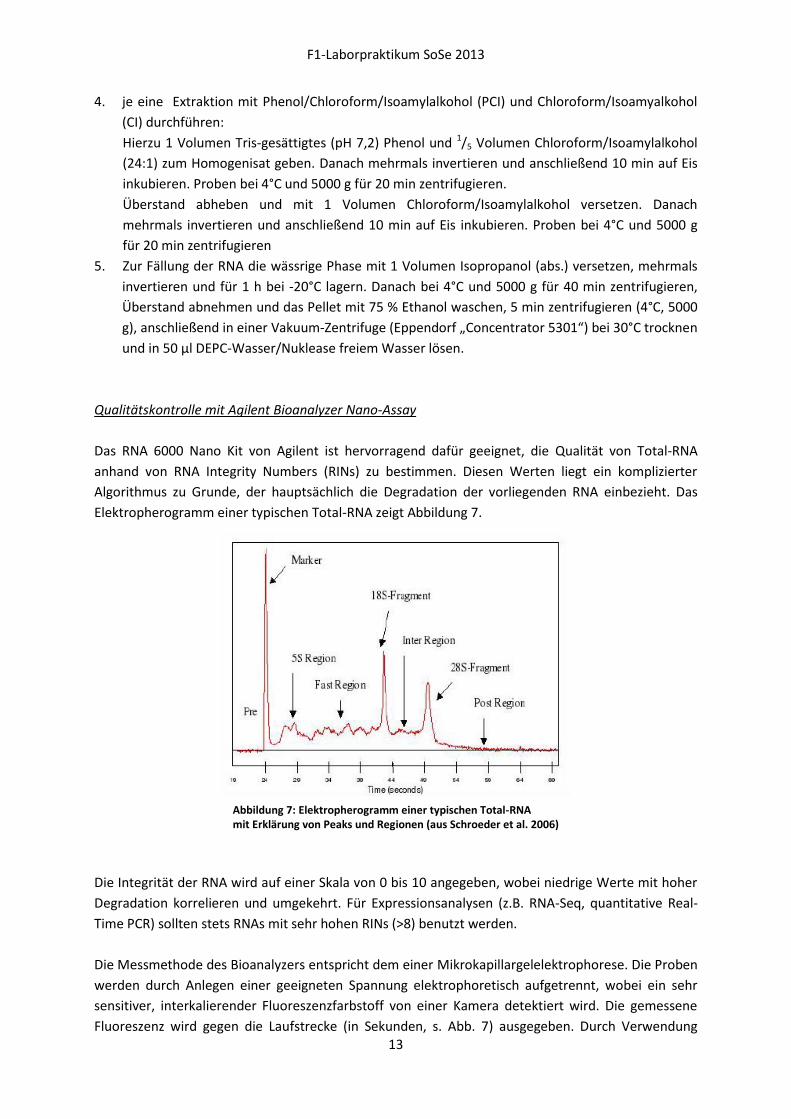
Qualitätskontrolle mit Agilent Bioanalyzer Nano-Assay
Das RNA 6000 Nano Kit von Agilent ist hervorragend dafür geeignet, die Qualität von Total-RNA
anhand von RNA Integrity Numbers (RINs) zu bestimmen. Diesen Werten liegt ein komplizierter
Algorithmus zu Grunde, der hauptsächlich die Degradation der vorliegenden RNA einbezieht. Das
Elektropherogramm einer typischen Total-RNA zeigt Abbildung 7.
Die Integrität der RNA wird auf einer Skala von 0 bis 10 angegeben, wobei niedrige Werte mit hoher
Degradation korrelieren und umgekehrt. Für Expressionsanalysen (z.B. RNA-Seq, quantitative Real-
Time PCR) sollten stets RNAs mit sehr hohen RINs (>8) benutzt werden.
Die Messmethode des Bioanalyzers entspricht dem einer Mikrokapillargelelektrophorese. Die Proben
werden durch Anlegen einer geeigneten Spannung elektrophoretisch aufgetrennt, wobei ein sehr
sensitiver, interkalierender Fluoreszenzfarbstoff von einer Kamera detektiert wird. Die gemessene
Fluoreszenz wird gegen die Laufstrecke (in Sekunden, s. Abb. 7) ausgegeben. Durch Verwendung
Abbildung 7: Elektropherogramm einer typischen Total-RNA mit Erklärung von Peaks und Regionen (aus Schroeder et al. 2006)

F1-Laborpraktikum SoSe 2013
14
eines Molekulargewichtsstandards (Leiter) kann dann aus der Laufstrecke das Molekulargewicht der
einzelnen Fragmente bestimmt werden.
Durchführung: Qualitätskontrolle mit Hilfe des Bioanalyzers
Richtiges Pipettieren: niemals an den Rand der Wells, sondern immer auf den Boden pipettieren.
1. Pipettieren Sie 2 µl Ihrer RNA (Konz. 25-500 ng/µl) in 0,2ml PCR Tubes und kennzeichnen Sie das
Gefäß sorgfältig.
2. Ein Gelaliquot (65 µl) wird Ihnen bereitgestellt. Pipettieren Sie 1 µl RNA 6000 Nano Dye (blau •)
hinzu. Der Gel-Dye-Mix muss vor der Benutzung sorgfältig gevortext und bei 13000 x g für
10 min zentrifugiert werden.
3. Den RNA 6000 Nano Chip in die Vorbereitungsstation legen und 9 µl Gel-Dye-Mix
in das mit gekennzeichnete Well (weißer Punkt in der unten aufgeführten
Grafik) pipettieren. Bitte entnehmen Sie das Gel aus dem Aliquot knapp
unterhalb des Meniskus, da sich am Boden Schwebstoffe absetzen, die den Lauf
beeinträchtigen können. Vermeiden Sie ebenfalls heftige Bewegungen und Stöße.
ACHTUNG: Lesen Sie an dieser Stelle bitte die Punkte 5-7 und bereiten Sie Pipetten und Lösungen
vor, bevor Sie weiterarbeiten
4. Die Spritze auf 1 ml einstellen, die Vorbereitungsstation schließen und die Spritze herunter
drücken, bis diese vom Clip gehalten wird, danach exakt 30 Sekunden (Timer) warten und den
Clip lösen.
5. Warten Sie 5 Sekunden und ziehen Sie dann die Spritze LANGSAM wieder in die
Ausgangsposition zurück.
6. Öffnen Sie die Vorbereitungsstation und pipettieren Sie 9 µl Gel-Dye-Mix in die
mit gekennzeichneten Wells.
7. Pipettieren Sie nun 5 µl RNA 6000 Nano Marker (grün •) in die verbleibenden 13
Wells.
8. Denaturieren Sie in der Zwischenzeit alle Proben und die RNA 6000 Nano Ladder bei 72°C für
2 min in einem Thermocycler.
9. Pipettieren Sie 1 µl der RNA 6000 Nano Ladder in das mit gekennzeichnete
Well.

F1-Laborpraktikum SoSe 2013
15
10. Pipettieren Sie nun jeweils 1 µl Ihrer Proben in die 12 vorhandenen Wells und notieren Sie die
Reihenfolge. In alle unbenutzten Wells pipettieren Sie 1 µl RNA 6000 Nano Marker (grün •).
11. Legen Sie den Chip in den IKA Vortexer (nur eine Orientierung ist möglich!) und starten Sie
diesen.
HINWEIS: Der IKA Vortexer stoppt automatisch nach einer Minute
12. Legen Sie den Chip in den Bioanalyzer (vermeiden Sie ruckartige Bewegungen) und beginnen Sie
den Lauf innerhalb von 5min nach Präparation des Chips durch Betätigung des Start-Knopfes
(wird grün, wenn der Chip erkannt wurde).
Erstellen einer cDNA-Library mit dem IlluminaTruSeq RNA Kit
Allgemeine Hinweise und Vorbereitungen:
1. Alle Puffer werden erst kurz vor Gebrauch aufgetaut und auf RT gebracht, danach sofort wieder
eingefroren, Enzyme stets auf Eis gelagert und nach Gebrauch sofort wieder bei -20°C gelagert.
2. Das 80 %ige EtOH wird am Ende des Arbeitstages bei -20°C gelagert
3. AMPure XP Beads werden 30 min vor Gebrauch auf RT gestellt
4. Thermocycler vorbereiten
polyA+-Aufreinigung
1. 1,5 µg (0,1-4 µg) der Gesamt-RNA in einem 0,2 ml Eppendorf Tube mit nukleasefreiem Wasser
auf 50 µl auffüllen
2. OligodT-beads vortexen bis alles gut resuspendiert ist und 50 µl Beads zur Total-RNA geben, gut
mischen durch vorsichtiges auf- und abpipettieren
3. Zur Denaturierung 5 min bei 65°C im Thermocycler inkubieren, auf 4°C herunterkühlen und
weitere 5 min bei RT inkubieren. Dabei bindet die RNA an die Beads.
4. Zum Waschen der Beads Eppi in den Magnethalter stellen, 5 min warten bis die Lösung klar ist.
Dann den Überstand vorsichtig abnehmen und verwerfen (Vorsicht: nicht die Beads ablösen!),
Eppi aus dem Magnethalter nehmen
200 µl Beadwashing-buffer zugeben, mit der Pipette vorsichtig resuspendieren und Tube in den
Magnethalter stellen, 5 min warten bis die Lösung klar ist, Überstand abnehmen und verwerfen
ohne die Beads zu lösen, anschließend Eppendorf Gefäß wieder aus dem Magnethalter nehmen.
5. RNA mit 50 µl Elution-Buffer eluieren. Dazu die Suspension mit der Pipette mischen und im
Thermocycler für 2 min bei 80°C inkubieren, auf 25°C herunterkühlen und halten. Dabei löst sich
die mRNA von den Beads. Die Beads verbleiben im Elutionspuffer.
6. Zweite Runde der Aufreinigung.
Dazu 50 µl Bead Binding Buffer zu der mRNA und den Beads geben, mit der Pipette mischen,
5 min bei RT inkubieren.
7. Beads waschen: Schritte 2 bis 4 wdh.
8. Elution und Fragmentierung der mRNA: 19,5 µl „Elute, Prime, Fragment Mix“ zugeben und mit
der Pipette gut mischen

F1-Laborpraktikum SoSe 2013
16
! Möglichkeit zum Lagern bei -20°C oder weiter mit der Erststrang-Synthese der cDNA !
Elution und cDNA-Synthese (Erst- und Zweitstrangsynthese):
1. Tube in den PCR-Cycler stellen, 94°C , 8 min, 4°C hold, danach kurz anzentrifugieren (die RNA
löst sich von den Beads)
2. Beads von der RNA separieren. Dazu Tube in den Magnethalter stellen, 5 min warten bis die
Lösung klar ist und 17 µl des Überstandes in ein neues 0,2 ml Eppi überführen
3. 1 µl SuperScript III zu 7 µl First Strand Master Mix und dieses zu den 17 µl Eluat geben, mit der
Pipette mischen, kurz anzentrifugieren. Danach Erststrangsynthese im Thermocycler:
Programm:
25°C für 10 min
42°C für 50 min
70°C für 15 min
4°C ∞
4. Für die Zweitstrang-Synthese der cDNA das Tube aus dem Cycler nehmen, 25 µl Second Strand
Master Mix zugeben, mit der Pipette mischen und im PCR-Cycler 1 h bei 16°C inkubieren.
AMPure-Aufreinigung:
5. Aufreinigung mit AMPure XP Beads. Diese gut vortexen und 90 µl zu den 50 µl ds cDNA geben,
mir der Pipette mischen, 10 min bei RT inkubieren
6. Eppendorf Tube in den Magnethalter stellen, 5 min warten bis die Lösung klar ist, 135 µl
Überstand abnehmen und verwerfen und 200 µl 80 % EtOH zugeben, ohne die Beads zu lösen
7. 30 sek inkubieren, Überstand abnehmen und verwerfen und nochmals mit 200 µl 80 % EtOH
waschen
8. 10 min bei RT im Magnetständer trocknen lassen
9. Tube aus dem Magnetständer nehmen, Beads in 62,5 µl Resuspensions-Puffer resuspendieren
und 2 min bei RT inkubieren
10. Tube in den Magnethalter stellen, 5 min warten bis die Lösung klar ist und 60 µl Überstand
(ds cDNA) in ein neues 0,2 ml Eppi überführen
! Möglichkeit zum Lagern bei -20°C oder weiter mit End Repair !
Endrepair
1. 40 µl End Repair Mix zugeben, mit der Pipette mischen und im PCR-Cycler 30 min bei 30°C
inkubieren
AMPure-Aufreinigung:
2. AMPure XP Beads gut vortexen und 160 µl zu der cDNA geben, mischen, 10 min bei RT
inkubieren
3. Eppi in den Magnethalter stellen, 5 min warten bis die Lösung klar ist, 255 µl (2x 127,5 µl)
Überstand (in 2 Schritten) abnehmen und verwerfen, ohne die Beads zu lösen

F1-Laborpraktikum SoSe 2013
17
4. 200 µl 80 % ETOH zugeben ohne die Beads zu lösen, 30 sek inkubieren, Überstand abnehmen
und verwerfen, Waschschritt 1x wiederholen
5. Eppi 10 min bei RT im Magnetständer trocknen lassen
6. Tube aus dem Magnetständer nehmen und das Pellet mit 20 µl Resupension Buffer lösen.
Danach 2 min bei RT inkubieren.
7. Eppi in den Magnethalter stellen, 5 min warten bis die Lösung klar ist
8. 17,5 µl des Überstandes in ein neues 0,2 ml Eppi überführen
! Möglichkeit zum Lagern bei -20°C oder weiter mit der A-Tailing des 3`Endes !
A-Tailing des 3´-Endes und Adapter-Ligation
1. Zu den 17,5 µl Probe 12,5 µl A-Tailing Mix zugeben und im PCR-Cycler 30 min bei 37°C
inkubieren
2. danach: + 2,5 µl DNA Ligase Mix
+ 2,5 µl verdünnte Ligase Control oder 2,5 µl Resuspensions-Puffer
+ 2,5 µl von jedem RNA Adapter Index, gut mischen
3. für 10 min bei 30°C im Cycler inkubieren, anschließend 5 µl StopLigase Mix zugeben und gut
mischen
AMPure-Aufreinigung:
4. AMPure XP Beads gut vortexen, 42 µl zur Probe geben, mit der Pipette mischen und 10 min bei
RT inkubieren
5. Eppi in den Magnethalter stellen, 5 min warten bis die Lösung klar ist, 79,5 µl Überstand
abnehmen und verwerfen ohne die Beads zu lösen
200 µl 80 % ETOH zugeben ohne die Beads zu lösen; 30 sek inkubieren, Überstand abnehmen
und verwerfen, Waschschritt 1x wiederholen
6. Pellet 10 min bei RT im Magnetständer trocknen lassen, aus dem Magnetständer nehmen
und getrocknetes Pellet mit 52,5 µl Resuspensions-Puffer mischen, dann 2 min bei RT inkubieren
7. Tube in den Magnethalter stellen, 5 min warten bis die Lösung klar ist und 50 µl des
Überstandes in ein neues 0,2 ml Eppi überführen
zweite AMPure-Aufreingung:
8. 50 µl Probe mit 50 µl AMPure XP Beads versetzen, mischen und 10 min bei RT inkubieren
9. Tube in den Magnethalter stellen, 5 min warten bis die Lösung klar ist, 95 µl Überstand
abnehmen und verwerfen ohne die Beads zu lösen
10. 200 µl 80 % ETOH zugeben ohne die Beads zu lösen, 30 sek inkubieren, Überstand abnehmen
und verwerfen, Waschschritt 1x wiederholen
11. Pellet 10 min bei RT im Magnetständer trocknen lassen, aus dem Magnetständer nehmen
getrocknetes Pellet mit 22,5 µl Resuspensions-Puffer mischen, 2 min bei RT inkubieren
12. Eppi in den Magnethalter stellen, 5 min warten bis die Lösung klar ist und 20 µl des Überstandes
in ein neues 0,2 ml Eppi überführen
! Möglichkeit zum Lagern bei -20°C oder weiter mit Anreicherung !

F1-Laborpraktikum SoSe 2013
18
Anreicherung 1. Ansatz (gut mischen): 20 µl Probe + 5 µl PCR Primer-Cocktail + 25 µl PCR Master-Mix 50 µl 2. Programm: 98°C 30 s 98°C 10 s
60°C 30 s 15x
72°C 30 s
72°C 5 min
4°C ∞
AMPure-Aufreingung:
3. 50 µl AMPure XP Beads zur Probe geben, mit der Pipette gut mischen und 10 min bei RT
inkubieren
4. Tube in den Magnethalter stellen, 5 min warten bis die Lösung klar ist, 95 µl Überstand
abnehmen und verwerfen ohne die Beads zu lösen
5. 200 µl 80 % EtOH zugeben ohne die Beads zu lösen, 30 s inkubieren, Überstand abnehmen und
verwerfen, Waschschritt 1x wiederholen
6. Pellet 10 min bei RT im Magnetständer trocknen lassen, aus dem Magnetständer nehmen und
getrocknetes Pellet mit 32,5 µl Resuspensions-Puffer mischen, 2 min bei RT inkubieren
7. Tube in den Magnethalter stellen, 5 min warten bis die Lösung klar ist und 30 µl des
Überstandes in ein neues 0,2 ml Eppi überführen
Qualitätskontrolle und Quantifizieren der Bibliotheken
QBit®HS Assay
Chemikalien ca. 30 min auf RT bringen und alle Proben des Kurses mit den gleichen Standards
messen.
1. Daher gemeinsamen Mastermix herstellen -> Probenzahl (4) + 2 Standards, dazu 1194 µl
(6x 199µl) „Qubit™buffer“ mit 6 µl „Qubit™reagent plus“ gut mischen und vortexen
2. Für die Standards 1 und 2 jeweils 10 µl der Standard-Lsg. mit 190 µl und je 1 µl der eigenen
Library mit 199 µl Mastermix mischen (! vortexen !)
3. 2 min bei RT inkubieren und erst Standards, dann Proben zügig durchmessen

F1-Laborpraktikum SoSe 2013
19
Qualitätskontrolle, Molekulargewichtsverteilung und Konzentrationsmessung der Banken mit Agilent
Bioanalyzer Nano-Assay
Die Messung von DNA funktioniert prinzipiell genau wie die der RNA, wobei hier zur Analyse der
ds cDNA ein doppelstrangspezifischer Farbstoff zum Einsatz kommt. Wie auch bei normalen
Gelelektrophoresen entscheidet die Konzentration des Gels über die Auftrennungseigenschaften. Mit
dem DNA High Sensitivity Kit können Fragmente zwischen 50 und 7000 Basenpaaren mit Quantitäten
bis zu 5 pg/µl detektiert werden.
Ein Gelaliquot versetzt mit dem Farbstoff (Gel-Dye-Mix) wird Ihnen bereitgestellt. Lassen Sie alle
Lösungen 30 min auf RT equilibrieren.
1. Nehmen Sie einen neuen DNA High Sensitivity Chip aus der Packung und legen Sie Ihn in die
Vorbereitungsstation.
2. Pipettieren Sie 9 µl Gel-Dye-Mix in das mit gekennzeichnete Well (weißer
Punkt in der unten aufgeführten Grafik). Bitte entnehmen Sie das Gel aus dem
Aliquot knapp unterhalb des Meniskus, da sich am Boden Schwebstoffe absetzen,
die den Lauf beeinträchtigen können.
ACHTUNG: Lesen Sie an dieser Stelle bitte die Punkte 5-7 und bereiten Sie Pipetten und Lösungen
vor, bevor Sie weiterarbeiten
3. Stellen Sie die Spritze auf 1 ml ein, schließen Sie die Vorbereitungsstation und drücken Sie die
Spritze herunter, bis diese vom Clip gehalten wird.
4. Warten Sie exakt 60 Sekunden (Timer) und lösen Sie den Clip.
5. Warten Sie 5 Sekunden und ziehen Sie dann die Spritze LANGSAM wieder in die
Ausgangsposition zurück.
6. Öffnen Sie die Vorbereitungsstation und pipettieren Sie 9 µl Gel-Dye-Mix in die
mit gekennzeichneten Wells.
7. Pipettieren Sie nun 5µl DNA High Sensitivity Marker (grün •) in die verbleibenden
12 Wells.
8. Pipettieren Sie 1 µl der DNA High Sensitivity Ladder (gelb •) in das mit
gekennzeichnete Well.
Pipettieren Sie nun jeweils 1 µl Ihrer Proben in die 11 vorhandenen Wells und
notieren Sie die Reihenfolge. In alle unbenutzten Wells pipettieren Sie 1 µl DNA
High Sensitivity Marker (grün •).
9. Legen Sie den Chip in den beigestellten IKA Vortexer (nur eine Orientierung ist
möglich!) und starten Sie diesen.
HINWEIS: Der IKA Vortexer stoppt automatisch nach einer Minute
F1-Laborpraktikum SoSe 2013
20
10. Legen Sie den Chip in den Bioanalyzer (vermeiden Sie ruckartige Bewegungen) und beginnen Sie
den Lauf innerhalb von 5 min nach Präparation des Chips durch Betätigung des Start-Knopfes
(wird grün wenn der Chip erkannt wurde).
qPCR-SYBR-Assay
Verdünnungen:
1. Bibliothek und PhiX-Standard auf ca. 20 pM mit 0,1 % Tween verdünnen.
In der Regel eine 1:500 Verdünnung: 2 µl Bibliothek + 998 µl 0,1 % Tween und gut vortexen
2. Den Schritt mit einer weiteren unabhängigen Verdünnung
3. Beide Proben nochmal um 1:10 (auf 1:5000) und 1:100 auf (1:50000) mit 0,1 % Tween
verdünnen
Vorbereiten der qPCR-Platte:
4. SYBR-Mastermix für 40 Wells herstellen und vorlegen. Dazu:
KAPA SYBR FAST Master Mix Universal (2x) 5 µl 200 µl
qPCR Primer 1.1 (10 μM) 0.1 µl 4 µl
qPCR Primer 2.1 (10 μM) 0.1 µl 4 µl
HPLC-Wasser 3,8 µl 152 µl
Gesamt 9 µl 360 µl
5. Mastermix gut mischen und jeweils 9 µl in ein Well der qPCR Platte geben
6. Proben nach folgender Plattenbelegung zugeben
1 2 3 4 5 6 7 8 9 10 11 12
A PhiX_1-Verdünnungen Probe1_1 Probe1_2 Probe2_1
B Probe2_2 Probe3_1 Probe3_2 Probe4_1
C Probe4_2 PhiX_2-Verdünnungen Negativkontrollen
D
E
F
G
H 1:500 1:5000 1:50000
7. Platte mit der dafür vorgesehenen Folie luftblasenfrei verschließen, in den qPCR-Cycler stellen
und folgendes Programm starten:
Hot start 95°C 3 min
95°C 3 sek
60°C 30 sek
40x

F1-Laborpraktikum SoSe 2013
21
Vorbereitungen - cBot - Sequenzierung
1. Bestimmen Sie die Konzentration und das durchschnittliche Molekulargewicht der DNA-
Fragmente
2. Bringen Sie die Bibliothek auf eine Endkonzentration von 10 nM. Sollte dies nicht möglich sein,
verdünnen Sie die Bibliothek auf 2 nM. Verwenden sie Qiagen EB Pufer oder 10 mM Tris-HCl
pH 8,5
3. Um optimale Clusterdichten zu erhalten, müssen die Bibliotheken nochmals auf 7 pM verdünnt
werden. Dies geschieht allerdings erst kurz vor der Sequenzierung und wird von den Betreuern
übernommen.

F1-Laborpraktikum SoSe 2013
22
Theorie Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-Daten
Sequenzalignment
In der Bioinformatik wird unter einem Alignment der Vergleich von zwei (paarweises Alignment) oder
mehreren (multiples Alignment, MSA) Zeichenketten oder Sequenzen verstanden. Voraussetzung für
einen Vergleich ist, dass beide Sequenzen derselben Notation (Alphabet) folgen. Beide Sequenzen
werden für das Alignment untereinander gelegt und die einzelnen Stellen der Zeichenkette
miteinander verglichen. Sind beide Stellen identisch, so spricht man von einem match, sind sie
ungleich von einem mismatch. Da auch ganze Abschnitte der einen Sequenz innerhalb der anderen
fehlen können, müssen diese Stellen im Alignment berücksichtigt werden. Diese Lücken (gaps)
stellen eine große Herausforderung in der Bioinformatik dar, da sie nicht direkt erkennbar sind.
Für die Visualisierung von Sequenzalignments gibt es verschiedene
Methoden. Die einfachste ist der sogenannte Dot-Plot (Abbildung 8).
Hier werden in einer Matrix, aufgespannt durch die ortogonal
zueinander liegenden Sequenzen, mögliche matches entsprechend
eingetragen, wodurch sich spezielle Muster ergeben. Diese Muster
deuten auf verschiedene Eigenschaften hin, wie z.B. längere
Abschnitte mit Übereinstimmung (lange Diagonalen),
Wiederholungen der Sequenz (repeats, horizontale oder vertikale
wiederkehrende Diagonalen), oder Poly-Nukleotid-Abschnitte bzw.
sehr kurze Wiederholungen (dunkle Blöcke).
Da es für zwei Sequenzen alleine schon mehrere Möglichkeiten des Alignments gibt, müssen die
jeweiligen Anordnungen einzeln bewertet werden. Hierbei vergibt man für matches, mismatches
sowie gaps Punkte, welche zusammengezählt am Ende zu einem score führen. Dieser stellt ein Maß
für die Güte des Alignments dar. Für die Berechnung des scores werden scoring-Matrizen benutzt.
Diese geben für jedes mögliche Alignment einen Wert aus. Für Nukleotidsequenzen sind diese
Matrizen sehr einfach gehalten. Auf Ebene der Aminosäuren gibt es komplexer aufgebaute Matrizen,
da ein Basenaustausch nicht zwangsläufig auch eine Veränderung der Aminosäuresequenz zufolge
hat. Sogenannte Substitutionsmatrizen spiegeln hier die Wahrscheinlichkeit einer Mutation durch
entsprechend angepasste scoring-Werte wider. (Dayhoff et al. 1978; Henikoff & Henikoff 1992).
Assemblierungsmethoden
MSAs bilden die Grundlage der ersten Assemblierungsmethoden. Mit dem Aufkommen der ersten
Sequenziertechniken war es nötig, die Ursprungssequenz(en), welche durch Scherung des
genetischen Materials zerstört wurden, wieder zu rekonstruieren. Das Scheren - ob chemisch oder
mechanisch - bildet bis heute einen Bestandteil der Herstellung von Sequenzbibliotheken, da keine
Technik über eine Leseweite von mehr als einer Kilobase (kb) verfügt und neuere Ansätze,
sogenannte next-generation sequencing - Methoden noch kürzere Sequenzschnipsel (reads) liefern.
Durch Amplifizierungschritte vor der Scherung entstehen reads, welche sich in der Ursprungsequenz
Abbildung 8: Dot Plot (http://upload.wikimedia.org/wikipedia/commons/3/33/Zinc-finger-dot-plot.png)

F1-Laborpraktikum SoSe 2013
23
überlagern und somit einen Überlapp (overlap) besitzen. Diesen macht man sich im
Assemblierungsprozess zunutze, indem man durch Sequenzalignments der Randregionen größere
Sequenzstücke produziert. Im optimalen Fall kann man so die Ursprungssequenz rekonstruieren.
Erschwert wird das Ganze durch eine, jeder Technik zugrunde liegende Fehlerrate, welche z.B. zu
falschen Basen innerhalb der reads führt.
Erste Assemblierungsalgorithmen aus den 70er und 80er Jahren beruhen auf der Tatsache, dass
relativ wenige, lange reads aus der damals am weitesten verbreiteten Technik, der Sanger-
sequenzierung vorliegen. Sogenannte greedy-Algorithmen, die schnelle, suboptimale Lösungen
lieferten, waren die ersten Ansätze für die Implementierung solcher Assemblierungsstrategien.
Obwohl die Hard- und Softwareindustrie in den letzten Jahrzehnten große Fortschritte gemacht hat,
konnten die Algorithmen mit den immer riesiger werdenden Datenmengen nicht mithalten, da ihre
Rechenzeit mit dem Wachstum der Daten exponentiell anstieg. Neuere Strategien bedienen sich
Methoden der dynamischen Programmierung, heuristischen Ansätzen sowie mathematischen
Methoden aus den Graphentheorien.
BLAST
Einer der bekanntesten Algorithmen, welcher sich dieser Methoden bedient, ist der BLAST-
Algorithmus („Basic Local Alignment Search Tool“, Altschul et. al. 1990). Er basiert auf der Annahme,
dass Sequenzen mit großer Ähnlichkeit kurze, identische Abschnitte besitzen.
Abbildung 9: BLAST Algorithmus

F1-Laborpraktikum SoSe 2013
24
Durch das Erstellen einer Liste mit kurzen Teilsequenzen (words) aus der Anfrage- (query) und
Zielsequenz (subject) und durch beidseitiges Verlängern dieser Übereinstimmungen werden so
gezielt kurze, lokale Alignments zwischen nahe verwandten Sequenzen erzeugt. Diese geben
Aufschluss über ihre Funktion oder die Beziehung zueinander. Mithilfe dieser Technik wird die Anzahl
der Sequenzvergleiche deutlich reduziert und somit eine Lösung in endlicher Zeit möglich gemacht.
Obwohl der BLAST-Algorithmus keine Assemblierungsmethode ist, findet man diese Technik in vielen
neueren kommerziellen Assemblierungsprogrammen (DNAStar, SeqMan, NGEN etc.) wieder. So
werden im Vorfeld schon gezielt Sequenzen für einen näheren Vergleich (Alignment) aufgrund
identischer Subsequenzen in Betracht gezogen.
Trotz dieser heuristischen Ansätze sind auch neuere Implementierungen bisweilen mit der Menge
der Daten überfodert, da ein simples Alignment zweier Sequenzen sehr speicherintensiv sein kann.
So wurden in den letzten Jahren verstärkt Ansätze aus den Graphentheorien zu Rate gezogen. Vor
allem Sequenzierungen aus NGS-Projekten erfordern eine veränderte Denkweise für
Assemblierungsstrategien. Da hier mehr qualitativ hochwertigere, kürzere reads mit einer höheren
Abdeckung (coverage) der Nukleotidpositionen erzeugt werden, rückt der Fokus weg von
rechenintensiven Alignments einzelner Sequenzpaare hin zu speicherentlastenden Vergleichen von
identischen Subsequenzen auf Nukleotidebene. Unterschiede, wie Punktmutationen (SNPs) spiegeln
sich hier in Aufgabelung der Graphen wider, sich wiederholende Abschnitte in Blasen innerhalb des
Graphen. Einer der erfolgreichsten dieser Methode ist der Ansatz des De-Bruijn-Graphen.

F1-Laborpraktikum SoSe 2013
25
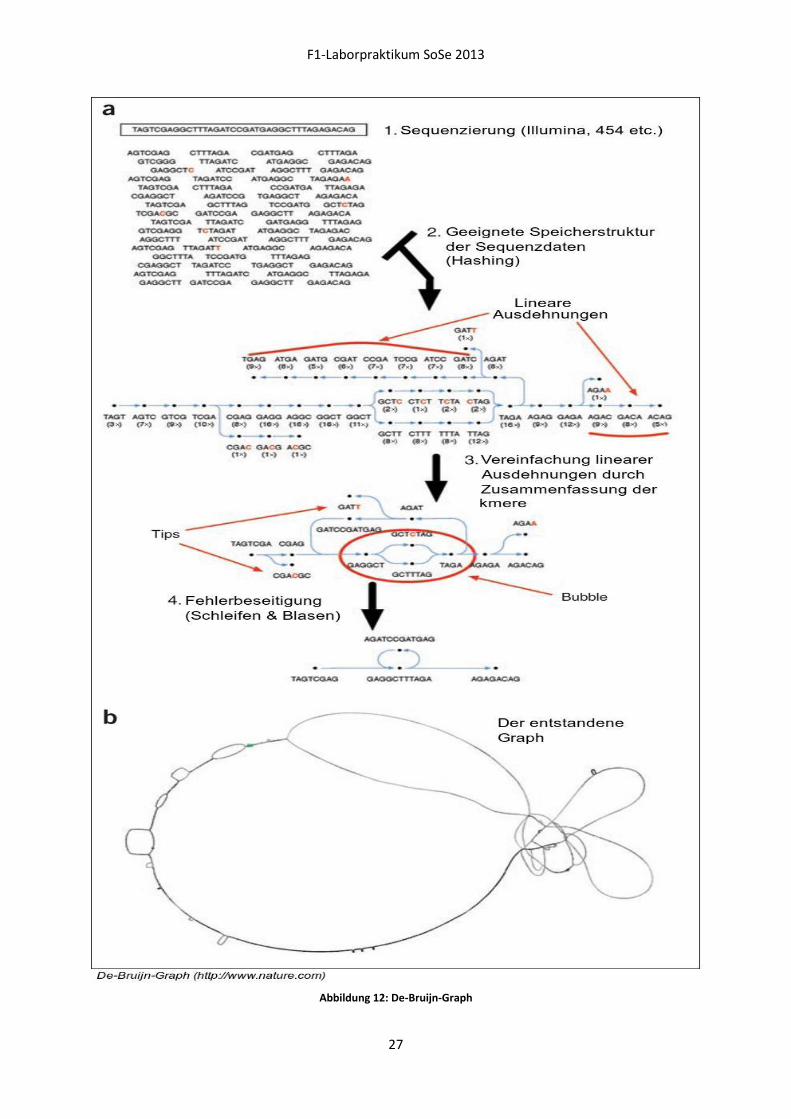
De-Bruijn-Graph (DBG)
Die Grundlage des DBG sind Datensätze, welche:
- Eine hohe Abdeckung der Ursprungssequenz besitzen,
- eindeutig in ihrer Notation sind (kein IUPAC-Code),
- reads gleicher Länge besitzen (wenn möglich).
Da der DBG keine Alignments im klassischen Sinne durchführt (und somit keine gaps einbaut), wird
der Überlapp durch eine Präprozessierung der Rohdaten gewährleistet. Dieser 'Rückwärtsschritt'
besteht in der Zerlegung der Eingangsdaten in eine Menge von Subsequenzen (kmers). Diese
Zerlegung geschieht in nukleotidlangen Schritten, wodurch sich die kmers eines einzelnen reads
überlappen und zwar mit der Länge kmer–1. Obwohl dieser Schritt eine Amplifizierung der
Datenmenge bedeutet, kann durch geschickte Implementierung die Menge des Speichers reduziert
werden, da Subsequenzen, die öfters auftreten, nur einmal gespeichert werden müssen.
Der Ablauf des Algorithmus sieht nun wie folgt aus:
1. Beginne bei einem beliebigen kmer.
2. Schneide am 5' Ende ein Nukleotid ab.
3. Erzeuge 4 mögliche Nachbar-kmere, indem am 3' Ende einzeln alle 4 Nukleotide anhängt werden.
4. Wiederhole die Schritte 1-3 mit dem Ursprungs-kmer mit dem 5' und dem 3' Ende vertauscht.
Abbildung 10: Mögliche Nachbar-kmere
Dadurch erhält man für ein einzelnes kmer 8 mögliche Nachbar-kmere (Abbildung 10). Für jedes
Nachbar-kmer, das im Datenpool existiert, wird im Graphen eine Kante zwischen den beiden kmeren
gezogen. Der so entstandene Graph zeichnet sich durch eine Vielzahl komplexer Strukturen (Blasen,
Schleifen) aus, welche auf verschiedene Charakteristiken der Ursprungssequenz wie SNPs oder
repeats hindeuten. Zur Auflösung dieser Strukturen werden Meta-Informationen über die Abdeckung
oder Qualität der Sequenzdaten hinzugezogen. Durch den Ablauf des Graphen können nun im letzten
Schritt die mögliche(n) Ursprungssequenz(en) ermittelt werden.

F1-Laborpraktikum SoSe 2013
26
Obwohl Ansätze aus der Graphentheorie sehr viele Probleme der klassischen Ansätze umgehen,
entstehen auch neue Schwierigkeiten, da z.B. Repeat-Regionen, die von den kmeren nicht
überspannt werden, nicht überwunden werden können. Um dieses Problem zu lösen, werden schon
während der Sequenzierung neue Techniken angewandt, welche Datensätze mit read-Paaren
erzeugen (paired-end-reads, mate-pairs). Der Abstand dieser Paare innerhalb der Ursprungssequenz
ist bekannt und kann somit im Assemblierungsprozess berücksichtigt werden.
Abbildung 11
F1-Laborpraktikum SoSe 2013
27
Abbildung 12: De-Bruijn-Graph

F1-Laborpraktikum SoSe 2013
28
Arbeitsablauf Sequenzverarbeitung
Aufgrund der immer stärker wachsenden Menge der Daten werden im Institut für Molekulargenetik
gezielt Methoden der parallelen Datenverarbeitung etabliert, sei es durch Nutzung neuer, bereits
parallel implementierter Algorithmen oder durch parallele Verarbeitung auf Ebene der voneinander
unabhängigen Eingangsdaten. Als Beispiel dient hierfür die Parallelisierung des Blastprozesses.
Die Blastoption des NCBI (National Center for Biotechnology Information) bietet nur beschränkte bis
keine Möglichkeiten der Hochdurchsatzdatenverarbeitung, da Sequenzen einzeln eingegeben
werden müssen oder nur kleinere Datensätze hochgeladen werden können. Durch die Verlagerung
der Berechnungen auf lokale Desktop-PCs, wird die Verarbeitung der Daten zwar unabhängig von
Aspekten wie Serverauslastung und Netzwerkverbindungen, jedoch bildet der PC selbst aufgrund der
(z. Zt.) geringen Ausstattung den Flaschenhals des Verarbeitungsprozesses. Des Weiteren müssen für
lokale Blastsuchen entsprechende Datenbanken zuerst heruntergeladen oder eigene
Sequenzdatenbanken erzeugt werden.
Möglichkeiten der Parallelisierung
Es gibt mehrere Ansätze der Parallelisierung der Blastsuche. Durch die Nutzung von Multicore-
Systemen oder HPC (High-Performance-Cluster, vernetzte Computer mit zentraler Steuerung und
Verwaltung durch entsprechende Software) ist es möglich, voneinander unabhängige Aufgaben
parallel zu verarbeiten, deren Ergebnisse zu speichern und am Ende wieder zusammenzuführen.
Durch gezielte parallel implementierte Algorithmen ist es möglich auf Softwareebene die vorhandene
Hardware maximal auszunutzen und somit eine (im optimalen Falle) lineare Skalierung zu erzielen.
Ein Ansatz für paralleles Arbeiten mit dem Blastprozess ist die Aufteilung der Datenbank. Dies
erfordert aber eine intensive Kommunikation zwischen den Prozessen, da dieselbe Sequenz
gleichzeitig in mehreren Datenbankuntereinheiten gesucht wird. Durch Aufteilung der Datenbank
wird darüber hinaus die Berechnung des E-value komplizierter, da dieser von der Datenbankgröße
abhängig ist. Ein anderer Ansatz ist die Aufteilung auf Sequenzebene. Das bedeutet, dass die
Eingangsdaten im Vorfeld in mehrere Datensätze aufgeteilt werden, um diese unabhängig
voneinander auf getrennten Systemen zu verarbeiten. Der Vorteil dieses Prozesses ist, dass die
Statistik nicht verändert wird. Nachteil dieser Art der Prozessierung ist allerdings, dass die Datenbank
für jeden einzelnen Prozess geladen werden muss und somit eine erhöhte Auslastung des
Arbeitsspeichers erfolgt.

F1-Laborpraktikum SoSe 2013
29
Praxis Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-Daten
In diesem praktischen Teil des Praktikums analysieren Sie Ihre in Teil 1 gewonnenen Transkriptom-
Daten im Hinblick auf folgende Fragestellungen:
Bestätigen Ihre Daten die bisher vorhergesagten Gene der geschlechtsbestimmenden
Genomregion? Eventuell können Sie die bestehende Annotation bestätigen oder erweitern.
Können neue Gene identifiziert werden?
Können unterschiedliche Transkriptvarianten durch alternatives Splicing charakterisiert
werden?
Welche Unterschiede gibt es zwischen dem weiblichen und männlichen Transkriptom im
Hinblick auf unterschiedliche Transkriptvarianten und Stärke der Expression?
Dabei sollen Sie verschiedene Analyseverfahren wie Mapping, „Read Count“ oder Assembly mit
verschiedenen Programmen kennen lernen. Zentrale, aber nicht ausschließliche, Analysesoftware soll
dabei die schon letzte Woche benutzte „CLC Genomics Workbench“ sein.

F1-Laborpraktikum SoSe 2013
30
Sequenzierung und Basecalling
Da auf der HiSeq Flow Cell lediglich 8 seperate Reaktionsräume, sogenannte Lanes vorhanden sind,
wurde zusätzlich zur eigentlichen Sequenzierung ein Index Read durchgeführt. Schon bei der
Konstruktion der Libraries wurden leicht variierte Adapter eingesetzt. Diese tragen noch vor der
Bindestelle für den Sequenzierprimer eine 6 Basenpaar lange Erkennungssequenz. Im Index Read
werden ausgehend vom revers-komplementären Sequenzierungsprimer diese 6 Nukleotide
ausgelesen. Dies wird notwendig, wenn verschiedene Libraries in einer Lane zusammen sequenziert
werden. Dies kann unterschiedliche Gründe haben. Beispielsweise benötigen viele Projekte nicht die
Masse an Daten einer kompletten Lane, oder - wie in diesem Fall - der Organismus besitzt einen
extremen A/T oder G/C –Gehalt. Nach dem Mischen der Libraries in den gewünschten Verhältnissen,
werden diese auf eine Endkonzentration von ca. 7 pM gebracht. Erfahrungsgemäß sollte diese
Konzentration eine Clusterdichte auf der Flow Cell-Oberfläche von ca. 450.000 bis 500.000 Cluster
pro mm2 ergeben. Mit dieser nahezu optimalen Clusterdichte sind ca. 80-100 Mio. Reads pro Lane
möglich.
Bei der Sequenzierung wird die Flow Cell zunächst in 16 Analysebereiche, sogenannte „Tiles“,
unterteilt. Dies sind zwei 8er Reihen mit Bildabschnitten, die zusammen die gesamte
Abbildung 13: Analysepipeline – vom Bild zur Sequenz

F1-Laborpraktikum SoSe 2013
31
Oberfläche der Flow Cell abdecken. Ausgehend von dem Bild im TIFF-Format jedes einzelnen Tiles
werden die Clusterintensitäten im „Firecrest“-Modul der RTA („Real Time Analysis“ Software)
verarbeitet (Abbildung 13). Die daraus resultierenden CIF-Dateien („cluster intensity files“) werden
dem „Bustard“-Modul zum eigentlichen Basecalling übergeben. Dieses erstellt zunächst binäre
Sequenz-Dateien, die BCL-Dateien („basecall files“). Diese werden separat in das Standardformat für
NGS-Daten, das FASTQ-Format, gebracht.
Datenformat FASTQ
Wichtigstes Dateiformat für NGS-Daten ist das FASTQ-Format. Dieses besteht immer aus 4 Zeilen pro
Sequenz (Abbildung 14). Die durch ein „@“ eingeleitete erste und die mit einem „+“ beginnende
dritte Zeile stellen dabei den Namen der Sequenz dar. In der zweiten Zeile folgt die Basensequenz. Im
Fall von „+“ folgen in der vierten Zeile die Qualitätswerte („ Quality Scores“) für jede einzelne Base
im ASCII-Code.
Abbildung 14: Aufbau des FASTQ-Formats
Im Namen der Sequenz finden sich weitere Informationen kodiert. Diese reichen vom Namen des
Geräts, auf dem sequenziert wurde bis zur Koordinate des Clusters auf der Flow Cell (Abbildung 15).
Abbildung 15: Aufbau des Sequenznamens der ersten und dritten Zeile

F1-Laborpraktikum SoSe 2013
32
Quality Scores
Vielen bekannt aus der herkömmlichen Sangersequenzierung, lassen sich auch die ASCII kodierten
Quality Scores der NGS-Daten in einen dezimalen Zahlenwert (Phred score) umrechnen und geben
dann eine Wahrscheinlichkeit für die fehlerhafte Sequenzierung der jeweiligen Base an (Abbildung
16).
Abbildung 16: Phred Score
Sie erhalten Ihre Daten als zusammengefasste FASTQ-Datei.
Jede Gruppe erhält zunächst den selbst generierten Datensatz, wobei der Austausch der
Rohdaten und von Teilergebnissen im weiteren Verlauf notwendig wird. Der Kursablauf
selbst wie auch die verschiedenen Analyseverfahren und Verwendung verschiedener
Programme werden sehr von den erarbeiteten Ergebnissen abhängen. Der schematische
Ablauf soll nur einige Möglichkeiten zusammenfassen und als Anhaltspunkt dienen.
F1-Laborpraktikum SoSe 2013
33
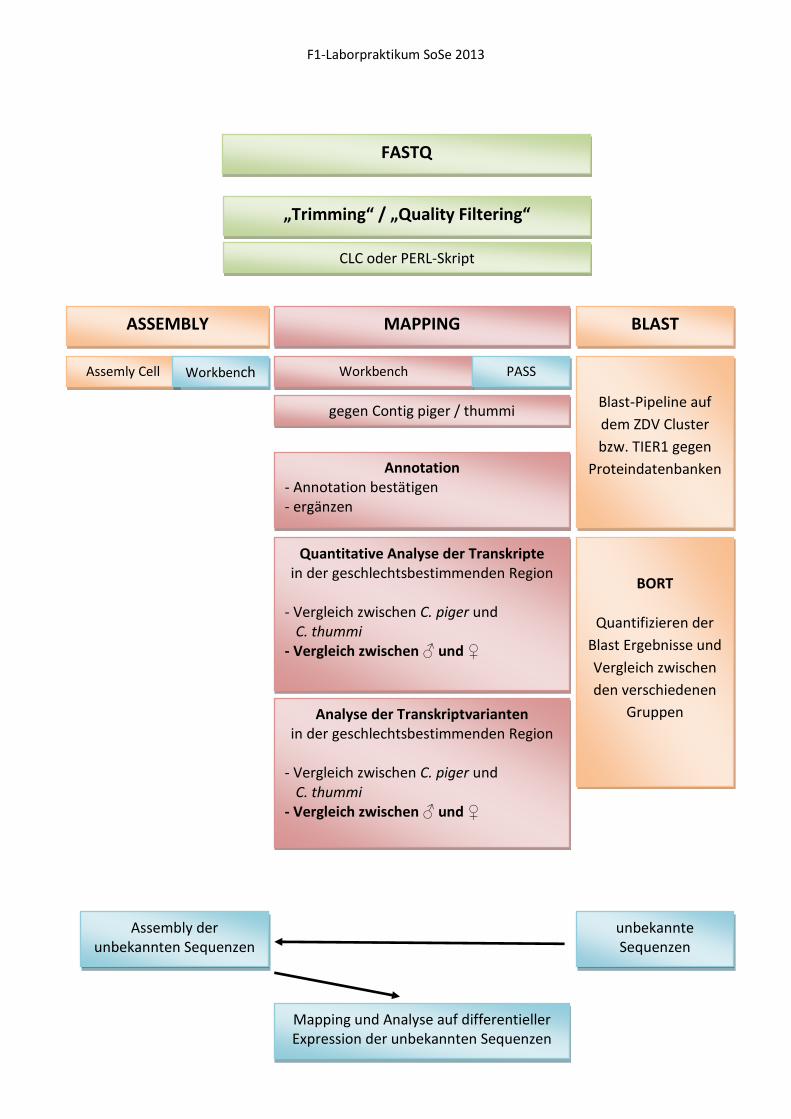
FASTQ
„Trimming“ / „Quality Filtering“
CLC oder PERL-Skript
ASSEMBLY MAPPING BLAST
Assemly Cell Workbench Workbench PASS
gegen Contig piger / thummi
Annotation - Annotation bestätigen - ergänzen
Quantitative Analyse der Transkripte in der geschlechtsbestimmenden Region
- Vergleich zwischen C. piger und C. thummi - Vergleich zwischen ♂ und ♀
Analyse der Transkriptvarianten in der geschlechtsbestimmenden Region
- Vergleich zwischen C. piger und C. thummi - Vergleich zwischen ♂ und ♀
Blast-Pipeline auf
dem ZDV Cluster
bzw. TIER1 gegen
Proteindatenbanken
BORT
Quantifizieren der
Blast Ergebnisse und
Vergleich zwischen
den verschiedenen
Gruppen
unbekannte Sequenzen
Assembly der unbekannten Sequenzen
Mapping und Analyse auf differentieller Expression der unbekannten Sequenzen

F1-Laborpraktikum SoSe 2013
34
Bioinformatische Analyse der „Next-Generation Sequencing“ Daten mit Hilfe der „CLC Genomics
Workbench“
Laden Sie sich bitte das ausführliche Manual der „CLC Genomics Workbench“ unter dem folgenden
Link herunter, da das Skript auf verschiedene Abschnitte innerhalb dieser Beschreibung verweisen
wird:
http://www.clcbio.com/files/usermanuals/CLC_Genomics_Workbench_User_Manual.pdf
Die Analyse beinhaltet folgende Schritte:
Import der „Next-Generation Sequencing“-Daten
„Trimming“ der Sequenzierungsreads
„Mapping“ der Sequenzierungsreads an eine genomische Referenzsequenz bzw. an die darin
bereits annotierten Transkripte
Vergleichende Analyse der „gemappten“ Sequenzierungsreads zwischen weiblichen und
männlichen Transkriptomen
Import der „Next-Generation Sequencing“-Daten
Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 2.1 Tutorial: Getting started S.42 2.1.1 Creating a folder S.42 2.1.2 Import data S.43 19.1 Import high-throughput sequencing data S.427
Öffnen Sie die „CLC Genomics Workbench 5“ durch die Verknüpfung auf Ihrem Desktop und machen
Sie sich mit der Softwareoberfläche vertraut, die im Wesentlichem aus 3 Bereichen besteht:
Oben links: die „Navigation Area“ zeigt alle importierten Daten an, die der Software zur
Verarbeitung zur Verfügung stehen
Unten links: in der „Toolbox“ befindet sich die Analysesoftware, durch Auswahl des Reiters
„Processes“ ist es später möglich, den Fortschritt einzelner Analysen nachzuverfolgen
Rechts: in der „View Area“ werden Daten durch eine grafische Oberfläche dargestellt
Erstellen Sie sich zunächst einen eigenen Ordner innerhalb der „Navigation Area“, indem Sie auf dem
Lokalen Datenträger (D:) einen Ordner „F1CLC“ erstellen und diesen in der Funktionsleiste der
„Navigation Area“ durch folgende Auswahl hinzufügen:
Add file location
Laden Sie Ihre Illumina-qseq-Dateien über den folgenden Pfad oder den Button „NGS Import“ der
oberen Funktionsleiste in Ihren neu erstellten Ordner ein:

F1-Laborpraktikum SoSe 2013
35
File | Import High-Throughput Sequencing Data
Beachten Sie hierbei, dass es sich eventuell um Sequenzierungsreads ohne „paired-end“-Information
handelt. Verwerfen Sie beim Import die reads ohne erfolgreiche Sequenzierungsreaktion. Erhalten
Sie dabei jedoch die Informationen über die automatisch ausgelesenen Qualitäts-scores.
Die importierten Sequenzen werden jetzt in der „Navigation Area“ in einer Liste zusammengefasst,
die Sie durch einen Doppelklick in der „View area“ öffnen können. Die einzelnen
Sequenzierungsreads enthalten außer der eigentlichen Sequenz auch die Information über die
Sequenzierungsqualität der einzelnen Basen („Quality score“), die für die weitere Bearbeitung der
reads genutzt werden kann.
Die Sequenzierungsreads enthalten noch Sequenzabschnitte, die bei der Assemblierung zu einem
nicht optimalen oder sogar falschen Ergebnis führen könnten.
Erkennen Sie in der Liste Ihrer Sequenzierungsreads solche Abschnitte, und wenn, welche
sind dies und wodurch werden diese verursacht?
Welche Auswirkungen hätten diese Abschnitte auf die Assemblierung?
„Trimmen“ der Sequenzierungsreads
Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 19.3 Trim sequences S.455 19.3.1 Quality trimming S.455 19.3.2 Adapter trimming S.457 19.3.3 Length trimming S.463 19.3.4 Trim output S.463
Die „CLC Genomics Workbench“ beinhaltet unterschiedliche Möglichkeiten, Sequenzierungsreads auf
den optimalen Bereich für eine Weiterverarbeitung in einer Assemblierung zu verkürzen:
Bewertung der Qualitätsangaben einzelner Basen
Erkennung uneindeutiger Basenfolgen
Entfernung der Adaptersequenz
o UNIV_adapter:
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
o TAG_adapter:
CAAGCAGAAGACGGCATACGAGAT*6bpINDEX*GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC
Eliminierung einer spezifischen Anzahl von Basen am 5’- bzw. 3’-Ende
Ausschluss einzelner Reads aufgrund von Längenbeschränkungen

F1-Laborpraktikum SoSe 2013
36
Lesen Sie Kapitel 19.3 im „CLC Genomics Workbench-Manual“ und bewerten Sie dabei, welche
Bereiche Ihrer Sequenzierungsreads im „Trimm“-Prozess berücksichtigt werden müssen und welche
Parametereinstellungen Sie hierfür verwenden.
Starten Sie dann den „Trim“-Prozess durch:
Toolbox | High-throughput Sequencing | Trim Sequences
Lassen Sie hierbei eine Übersicht des „Trim“-Prozesses erstellen und wählen Sie die Option zur
Generierung einer Liste der verworfenen Sequenzierungsreads. Nach der Fertigstellung öffnen Sie
beide Dokumente und bewerten, ob es sich hier um ein optimales „Trim“-Ergebnis handelt.
Wenn durch Ihre Bearbeitung kein optimales Ergebnis erzielt wurde, wiederholen Sie diesen Schritt.
Falls Sie nur eine zusätzliche Option hinzufügen möchten und dabei keine Parameter der bisherigen
Analyse verändern, können Sie die Bearbeitungszeit durch die Verwendung der bereits „getrimmten“
Sequenzen verkürzen.
Assemblierung der Sequenzierungsreads
Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 19.4 De novo assembly S.465 19.5 Map reads to reference S.479 19.6 Mapping reports S.488 19.7 Mapping table S.494
Folgende für unsere Fragestellung nutzbare Möglichkeiten der Assemblierung sind im
Programmpaket „CLC Genomics Workbench“ implementiert:
„De novo“-Assemblierung: Erstellung von „Contigs“ durch Assemblierung aller
Sequenzierungsreads nach „de Bruijn Graphen“, die reads werden danach in den „Contig“-
Referenzsequenzen abgebildet („gemapped“)
Abbildung/Kartierung der Sequenzierungsreads in einer genomischen Referenzsequenz
(„Map reads to reference“). Hierfür stehen 2 verschiedene Algorithmen für kurze und lange
(>55 bp) Sequenzierungsreads zur Verfügung.
Abbildung/Kartierung der Sequenzierungsreads in einer genomischen Referenzsequenz,
sowie in bereits annotierten Transkripten („RNA-Seq“-Analyse): Kalkulation der
Expressionsstärke, Ermittlung neuer Exons, alternativer Transkriptvarianten
„De novo“ Assemblierung
Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 2.5 Tutorial: De novo assembly and BLAST S.49 19.4 De novo assembly S.465

F1-Laborpraktikum SoSe 2013
37
Das „de novo“-Assembly der Transkriptomdaten wird durch die „CLC Genomics Workbench“ mit Hilfe
des „de Bruijn Graphen“ berechnet. Das Ziel ist dabei, die exprimierten Transkripte durch die
Assemblierung zusammenzusetzen. Dies wird dadurch erschwert, dass es durch alternatives Spleißen
unterschiedliche Transkripte eines Gens geben kann.
Welche Bereiche der cDNA enthält ein durch das „de novo“-Assembly zusammengesetzter
Contig optimalerweise?
Dieses „de novo“-Assembly kann als Referenz (Alternative zu genomischer Referenzsequenz) für die
Analyse der Expressionsstärke (RNA-Seq) verwendet werden. So ist es möglich, die Expression nicht
nur in einem beschränkten genomischen Bereich, sondern insgesamt zu analysieren.
Durch die BLAST-Analyse gegen Datenbanken ist es möglich, die zusammengesetzten Transkripte zu
charakterisieren.
Für eine Einführung in die „de novo“-Assemblierung führen Sie das Tutorial „De novo assembly und
BLAST“ durch und informieren sich über die verschiedenen Parameter im „CLC Genomics
Workbench-Manual“.
„Mapping“ der reads in einer Referenzsequenz
Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 19.5 Map reads to reference S.479 19.6 Mapping reports S.488 19.7 Mapping table S.494
Da es sich bei Ihren Sequenzreads um Transkriptomdaten handelt, die Sie gegen eine genomische
Referenzsequenz „mappen“, muss der Algorithmus das Einfügen langer Lücken (Introns)
ermöglichen. Das „Large Gap Plug-in“ der „CLC Genomics Workbench“ erreicht dies durch folgende
Schritte:
Für jeden Read wird der beste „match“ in der Referenzsequenz ermittelt („seed“ Segment)
Wenn der „match“ den definierten Parametern entspricht, wird die passende Region auf der
Referenzsequenz „gemappt“ und die nicht alignierten Enden werden bei ausreichender
Länge (>17 bp) erneut assembliert („non seed“ Segment)
Dabei sind zwei zusätzliche Parameter zu beachten:
Die maximale Anzahl der Treffer für ein Segment: wenn ein „non seed“ Segment diese
Anzahl überschreitet, wird der Read als „nicht gemappt“ klassifiziert
Maximale Distanz zwischen „seed“ Segment und „non seed“ Segment
Alternative Methoden zu dieser „Seed-extend“-Methode (b) nutzen den „Exon-first“-Ansatz (a), der
erst alle perfekten Übereinstimmungen zur Referenzsequenz mappt und dann die Reads untersucht,
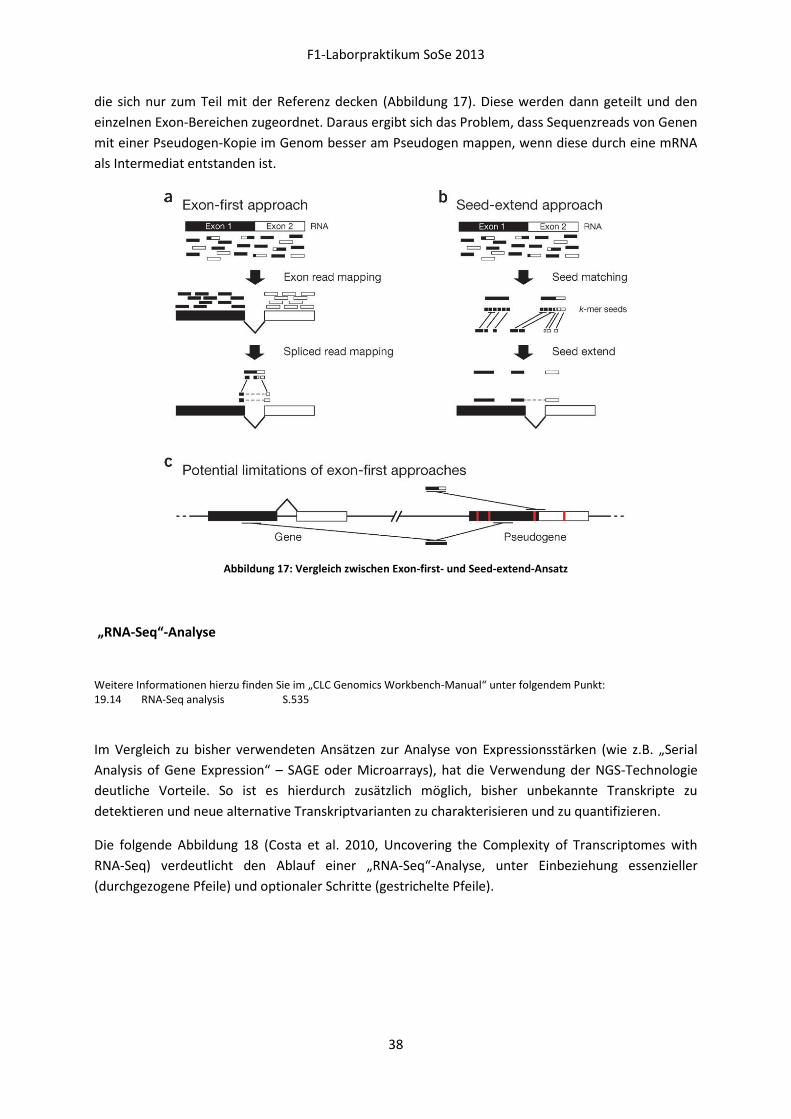
F1-Laborpraktikum SoSe 2013
38
die sich nur zum Teil mit der Referenz decken (Abbildung 17). Diese werden dann geteilt und den
einzelnen Exon-Bereichen zugeordnet. Daraus ergibt sich das Problem, dass Sequenzreads von Genen
mit einer Pseudogen-Kopie im Genom besser am Pseudogen mappen, wenn diese durch eine mRNA
als Intermediat entstanden ist.
Abbildung 17: Vergleich zwischen Exon-first- und Seed-extend-Ansatz
„RNA-Seq“-Analyse
Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgendem Punkt: 19.14 RNA-Seq analysis S.535
Im Vergleich zu bisher verwendeten Ansätzen zur Analyse von Expressionsstärken (wie z.B. „Serial
Analysis of Gene Expression“ – SAGE oder Microarrays), hat die Verwendung der NGS-Technologie
deutliche Vorteile. So ist es hierdurch zusätzlich möglich, bisher unbekannte Transkripte zu
detektieren und neue alternative Transkriptvarianten zu charakterisieren und zu quantifizieren.
Die folgende Abbildung 18 (Costa et al. 2010, Uncovering the Complexity of Transcriptomes with
RNA-Seq) verdeutlicht den Ablauf einer „RNA-Seq“-Analyse, unter Einbeziehung essenzieller
(durchgezogene Pfeile) und optionaler Schritte (gestrichelte Pfeile).
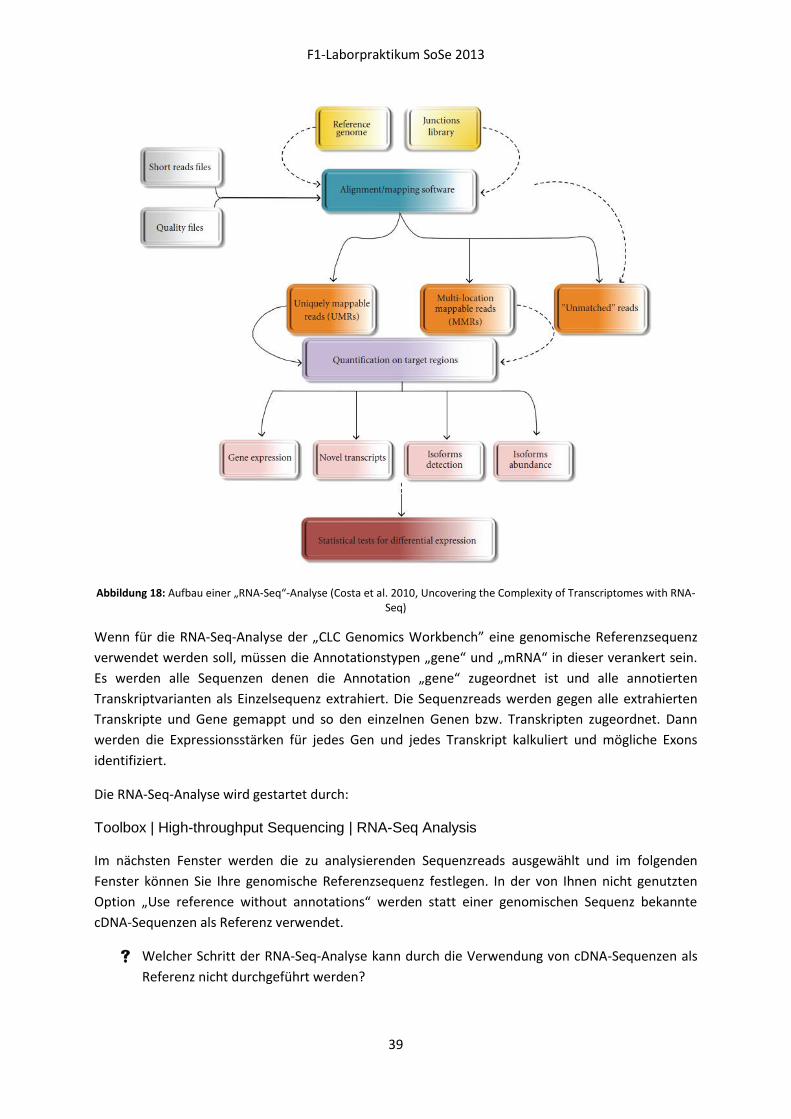
F1-Laborpraktikum SoSe 2013
39
Abbildung 18: Aufbau einer „RNA-Seq“-Analyse (Costa et al. 2010, Uncovering the Complexity of Transcriptomes with RNA-Seq)
Wenn für die RNA-Seq-Analyse der „CLC Genomics Workbench” eine genomische Referenzsequenz
verwendet werden soll, müssen die Annotationstypen „gene“ und „mRNA“ in dieser verankert sein.
Es werden alle Sequenzen denen die Annotation „gene“ zugeordnet ist und alle annotierten
Transkriptvarianten als Einzelsequenz extrahiert. Die Sequenzreads werden gegen alle extrahierten
Transkripte und Gene gemappt und so den einzelnen Genen bzw. Transkripten zugeordnet. Dann
werden die Expressionsstärken für jedes Gen und jedes Transkript kalkuliert und mögliche Exons
identifiziert.
Die RNA-Seq-Analyse wird gestartet durch:
Toolbox | High-throughput Sequencing | RNA-Seq Analysis
Im nächsten Fenster werden die zu analysierenden Sequenzreads ausgewählt und im folgenden
Fenster können Sie Ihre genomische Referenzsequenz festlegen. In der von Ihnen nicht genutzten
Option „Use reference without annotations“ werden statt einer genomischen Sequenz bekannte
cDNA-Sequenzen als Referenz verwendet.
Welcher Schritt der RNA-Seq-Analyse kann durch die Verwendung von cDNA-Sequenzen als
Referenz nicht durchgeführt werden?

F1-Laborpraktikum SoSe 2013
40
Sie können flankierende Bereiche von Genen, die „5’-upstream“ und „3’-downstream“ der
Genannotationen liegen, durch die Option „Extend annotated gene regions“ in die Analyse
miteinbeziehen.
In welchen Fällen ist diese Erweiterung der Referenzsequenzen sinnvoll?
Im nächsten Fenster können folgende Parameter für Sequenzreads <56 bp definiert werden:
„Maximum number of mismatches“: maximale Anzahl der zugelassenen Fehlpaarungen
(maximal: 3)
„Maximum number of hits for a read“: wenn ein Sequenzread mit mehreren Bereichen der
Referenzsequenz übereinstimmt, wird er nicht gemappt, wenn er diese Anzahl der „hits“
überschreitet. Wenn der Read mit mehreren Bereichen übereinstimmt und die maximale
Anzahl unterschreitet, wird er in Proportion zur Anzahl der eindeutigen „hits“ und der Länge
des Exons zufällig zugeordnet
“Strand specific alignment”: wird diese Option gewählt, werden die Sequenzreads nur in der
„forward“-Orientierung gemappt und nicht revers-komplementär.
Unter welchen Gegebenheiten wählen Sie ein „Strang-spezifisches Alignment“ aus? Welche
falsche Zuordnung von Sequenzread und Referenzsequenz schließt diese Option aus?
Für lange Sequenzreads (>56 bp) sind folgende zusätzliche Parameter verfügbar:
„Minimum length fraction“: definiert die Länge des Bereichs, der mit der Referenzsequenz
übereinstimmen muss (der Wert 0.9 bedeutet, dass der 90 % der Basen eines Sequenzreads
an der Referenzsequenz mappen muss)
„Minimum similarity fraction“: spezifiziert, wie exakt der Bereich eines langen Sequenzread-
Bereichs (Länge des Bereichs wird durch die „length fraction“ definiert) mit der
Referenzsequenz übereinstimmen muss.
Eine „length fraction“ von 0.9 und eine „similarity fraction“ von 0.8 definiert, dass ein 90 % eines
Sequenzreads zu 80 % mit der Referenzsequenz übereinstimmen müssen.
Im nächsten Eingabefenster erfolgen die Einstellungen zur Identifikation von annotierten und neuen
Exons.
Warum benötigt der Algorithmus die Angabe, ob die zu analysierenden Sequenz pro- oder
eukaryotischen Ursprungs ist? Welche Schritte sind bei einer Analyse prokaryotischer
Sequenzen abweichend?
Wenn das Feld „Exon discovery“ ausgewählt ist, erfolgt das Mapping über den gesamten Genbereich,
so dass auch bisher nicht definierte Exons als solche erkannt werden können. Dabei können folgende
Parameter festgelegt werden:

F1-Laborpraktikum SoSe 2013
41
„required relative expression level“: die Expressionsstärke dieses Exons muss mindestens zu
diesem Prozentsatz dem der bekannten Exons dieses Gens entsprechen
„minimum number of reads“: Die Mindestanzahl von Sequenzreads, die an ein neues Exon
mappen. Bei niedrig exprimierten Genen könnten sonst schon wenige Reads ein neues Exon
definieren.
„minimum length“: die minimale Länge eines neuen Exons, das durch überlappende Reads
definiert wird.
Im nächsten Fenster kann der Output der Analyse definiert werden. Der Standard-Output besteht
aus einer Tabelle der Gene und deren Statistik, sowie der Expression der einzelnen Transkripte.
Wenn Sie die Option zur Erstellung einer Sequenzliste der nicht-gemappten Reads wählen, kann
dieser Output zur Identifizierung neuer Transkripte bzw. Gene genutzt werden, die nicht auf der
Referenzsequenz gemappt werden konnten. Wählen Sie „Create report“, um eine detaillierte
Statistik der Analyse zu erhalten. In diesem Fenster kann auch die Normalisierung der
Expressionsstärke definiert werden.
Warum kann die Expressionsstärke nicht durch die absolute Anzahl der Sequenzreads
angegeben werden? Nehmen Sie hierzu die folgende Abbildung 19 zu Hilfe:
Abbildung 19: Problematik der absoluten Anzahl der gemappten Reads
Wählen Sie für die Normalisierung die Option „Genes RPKM“:
RPKM/FPKM: Sequenzreads/Fragmente pro Kilobase des Transkripts pro Millionen gemappter Reads
__________________alle gemappten Reads innerhalbe der Exons eines Gens____________________ gemappte Reads innerhalb des gesamten Genbereichs (Millionen) x Länge der Exons eines Gens (Kb)
Die folgende Abbildung 20 veranschaulicht den Einfluss der RPKM-Normalisierung auf die
Expressionsstärke:
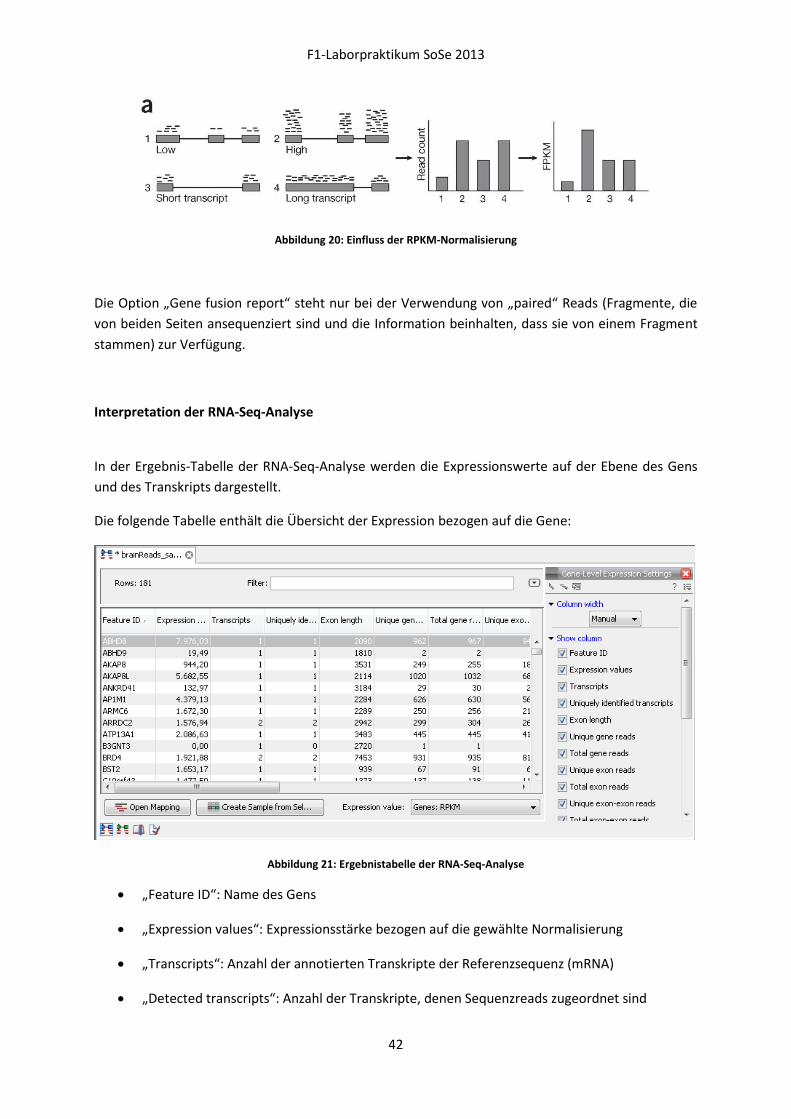
F1-Laborpraktikum SoSe 2013
42
Abbildung 20: Einfluss der RPKM-Normalisierung
Die Option „Gene fusion report“ steht nur bei der Verwendung von „paired“ Reads (Fragmente, die
von beiden Seiten ansequenziert sind und die Information beinhalten, dass sie von einem Fragment
stammen) zur Verfügung.
Interpretation der RNA-Seq-Analyse
In der Ergebnis-Tabelle der RNA-Seq-Analyse werden die Expressionswerte auf der Ebene des Gens
und des Transkripts dargestellt.
Die folgende Tabelle enthält die Übersicht der Expression bezogen auf die Gene:
Abbildung 21: Ergebnistabelle der RNA-Seq-Analyse
„Feature ID“: Name des Gens
„Expression values“: Expressionsstärke bezogen auf die gewählte Normalisierung
„Transcripts“: Anzahl der annotierten Transkripte der Referenzsequenz (mRNA)
„Detected transcripts“: Anzahl der Transkripte, denen Sequenzreads zugeordnet sind

F1-Laborpraktikum SoSe 2013
43
„Exon length“: Gesamtlänge aller Exons
„Unique gene reads“: Anzahl der Reads, die eindeutig diesem Gen zugeordnet werden
können
„Total gene reads“: Anzahl der Reads, die diesem Gen zugeordnet sind (eindeutig
zugeordnete und zufällig zugeordnete Reads, die mehreren Genen zuzuordnen sind)
„Unique exon reads“: Anzahl der Reads, die eindeutig den Exons des Gens zugeordnet sind
„Total exon reads“: eindeutig zugeordnete und mehreren Exons zugeordnete Reads
„Unique exon-exon reads“: Anzahl der Reads, die einer Exon-Exon-Grenze zugeordnet sind.
Dieser Read wird nur einmal in der gesamten Anzahl berücksichtigt, obwohl er zu zwei Exons
zugeordnet ist.
„Total exon-exon reads“: „Unique exon-exon reads“ und nicht eindeutig zugeordnete Reads.
„Unique intron-exon reads“: Anzahl der Reads, die eindeutig einer Exon-Intron-Grenze
zuzuordnen sind
Auf was könnte eine hohe Anzahl dieser Reads hindeuten?
„Total intron-exon reads“: „Unique intron-exon reads“ und nicht eindeutig zugeordnete
Reads.
„Exons“: Anzahl der Exons der mRNA-Annotation der Referenzsequenz
„Putative exons“: Anzahl der zusätzlichen Exons, die durch diese Analyse detektiert wurden
„RPKM“: wenn die RPKM-Normalisierung in der Analyse verwendet wurde, entspricht dieser
Wert dem „Expression value“. Bei einer Abänderung der Normalisierung können die Werte
mit dem RPKM-Wert dieser Spalte verglichen werden
„Median coverage“: durchschnittliche Abdeckung für alle Exons. Hierbei werden eindeutige
und zufällig zugeordente Reads berücksichtigt, Reads über Exon-Exon-Grenzen jedoch nicht.
„Chromosome region start“: Startposition des annotierten Gens
„Chromosome region end“: Endposition des annotierten Gens
Am Ende der Tabelle kann die Normalisierung der Expressionsstärke verändert werden. Bei einem
Vergleich von verschiedenen RNA-Seq-Analysen muss diese an diesem Punkt angeglichen werden.
Durch einen Doppelklick auf ein Gen öffnet sich die Übersicht der gemappten Reads an der
Referenzsequenz:

F1-Laborpraktikum SoSe 2013
44
Transkriptvarianten
Referenzsequenz
Abdeckung der Referenzsequenz mit Sequenzreads Gemappte Sequenzreads- Reads an Exon-Exon-Grenzen werden durch gestrichelte Linien dargestellt
Um zur Ansicht der Expression auf Transkript-Ebene zu gelangen, wählen Sie den zweiten Button
links unten in der Tabelle („Transcript-level expression“). Hier sind die Expressionsstärken in Bezug
auf die einzelnen Transkripte dargestellt. Unterschiedliche Transkripte eines Gens sind unter der
„Feature ID“ nummeriert. Die Berechnung der Reads, die einem Transkript insgesamt zugeordnet
werden, erfolgt erst nach der Bestimmung der Anzahl der eindeutigen Reads. Die Gesamtanzahl wird
dann durch proportionale Zuordnung der nicht eindeutigen Reads in Bezug auf die Anzahl der
eindeutig zugeordneten Reads berechnet. Die Angabe der „Ratio of unique to total transcript reads
kann dazu genutzt werden, um wenig zuverlässige Resultate auszusortieren, bei denen die Anzahl
der nicht eindeutig zugeordneten Reads sehr hoch ist.
Expressionsanalyse
Für eine Expressionsanalyse benötigt die „CLC Genomics Workbench“ die Definition eines
Experiments, bei dem die Expressionswerte der RNA-Seq-Analyse als Daten dienen:
Toolbox | Expression Analysis | Set Up Experiment
Wählen Sie Ihre selbst analysierten Datensätze von Chironomus thummi- bzw. piger und die
entsprechende Analyse des anderen Geschlechts aus und verschieben Sie es in die rechte Spalte.
Im nächsten Fenster können Sie festlegen, wie viele Gruppen verglichen werden sollen. Als „paired“
bezeichnete Analysen vergleichen miteinander in Verbindung stehende Daten, wie z.B. ein
Individuum in verschiedenen Zuständen oder Zeitabständen. An dieser Stelle kann die
Normalisierung der Expressionsstärke erneut festgelegt werden oder die zuvor berechneten Werte
werden in die Analyse einbezogen. Nach der Benennung der Gruppen können Sie Ihre Datensätze
(linke Spalte) mit den Gruppen (Auswahl in rechter Spalte) verknüpfen.
Abbildung 21: Mapping der Transkriptomdaten gegen eine genomische Referenz
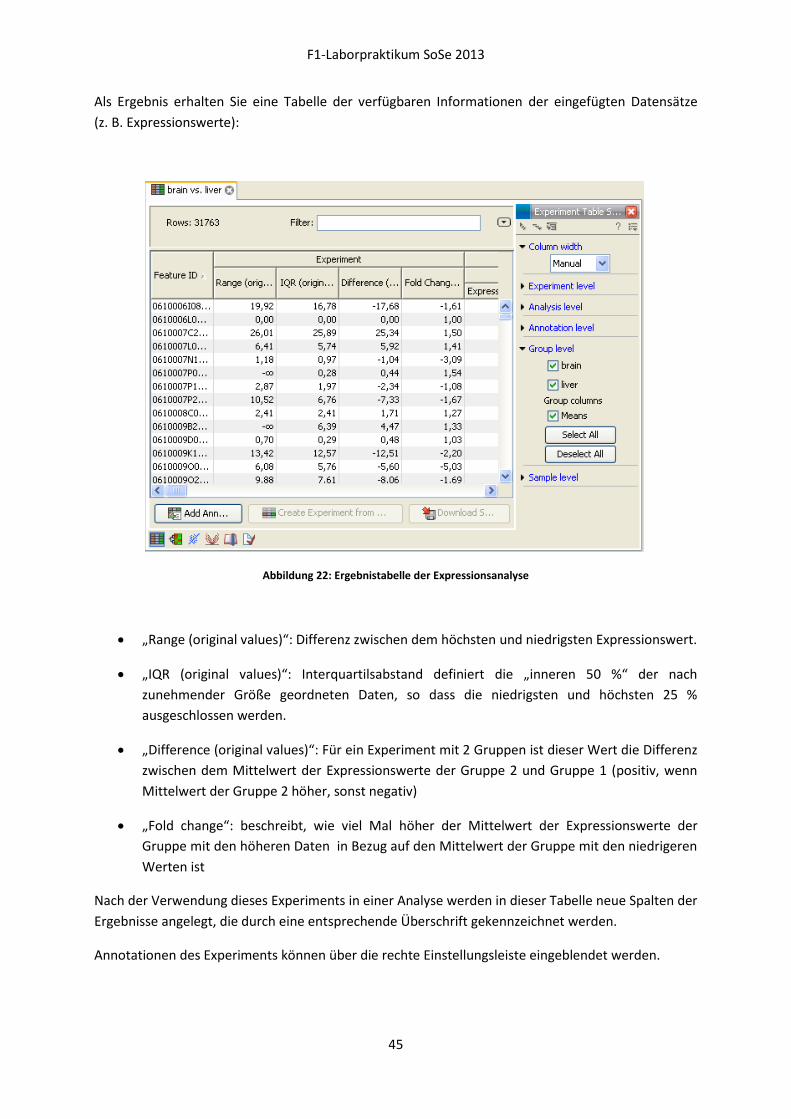
F1-Laborpraktikum SoSe 2013
45
Als Ergebnis erhalten Sie eine Tabelle der verfügbaren Informationen der eingefügten Datensätze
(z. B. Expressionswerte):
Abbildung 22: Ergebnistabelle der Expressionsanalyse
„Range (original values)“: Differenz zwischen dem höchsten und niedrigsten Expressionswert.
„IQR (original values)“: Interquartilsabstand definiert die „inneren 50 %“ der nach
zunehmender Größe geordneten Daten, so dass die niedrigsten und höchsten 25 %
ausgeschlossen werden.
„Difference (original values)“: Für ein Experiment mit 2 Gruppen ist dieser Wert die Differenz
zwischen dem Mittelwert der Expressionswerte der Gruppe 2 und Gruppe 1 (positiv, wenn
Mittelwert der Gruppe 2 höher, sonst negativ)
„Fold change“: beschreibt, wie viel Mal höher der Mittelwert der Expressionswerte der
Gruppe mit den höheren Daten in Bezug auf den Mittelwert der Gruppe mit den niedrigeren
Werten ist
Nach der Verwendung dieses Experiments in einer Analyse werden in dieser Tabelle neue Spalten der
Ergebnisse angelegt, die durch eine entsprechende Überschrift gekennzeichnet werden.
Annotationen des Experiments können über die rechte Einstellungsleiste eingeblendet werden.

F1-Laborpraktikum SoSe 2013
46
Wenn Sie Gene identifiziert haben, die differentiell exprimiert werden, können diese in einem
„Unter-Experiment“ gespeichert werden. Markieren Sie hierfür die entsprechenden Reihen und
wählen Sie folgende Option:
Create Experiment from Selection
Im unteren Bereich der Tabelle können Sie zwischen verschiedenen Ansichten des Experiments
wählen:
Abbildung 23: Auswahl der Ansichten des Experiments
„Experiment table“: Standardansicht der Tabelle des Experiments
„Heat map“ (Abbildung 24): grafische Darstellung der Daten durch Farbcodierung der
Expressionsstärke. Dabei werden ähnlich stark exprimierte Gene oder Transkripte
zusammengefasst (geclustert). Jede Reihe entspricht einem Gen oder Transkript, jede Spalte
einem Datensatz.
Um einen „Volcano plot“ darzustellen müssen Sie zuerst das Clustern der Daten durchführen:
Toolbox |Expression Analysis|Feature Clustering|Hierarchical Clustering of Features
Abbildung 24: „Heat Map“
„Scatter Plot“ (Abbildung 25): Vergleich von Expressionswerten von zwei 2 verschiedenen
Datensätzen. Jeder Punkt repräsentiert den Expressionswert eines Gens oder Transkripts
beider Datensätze, ein Expressionswert wird auf der X-Achse, der andere auf der Y-Achse
dargestellt. Die gleiche Expressionsstärke in beiden Datensätzen erzeugt einen Punkt auf der
diagonalen Achse. Abweichende Punkte zur Diagonalen entsprechen Genen oder
Transkripten, die in den Daten unterschiedliche Expressionsstärken aufweisen.
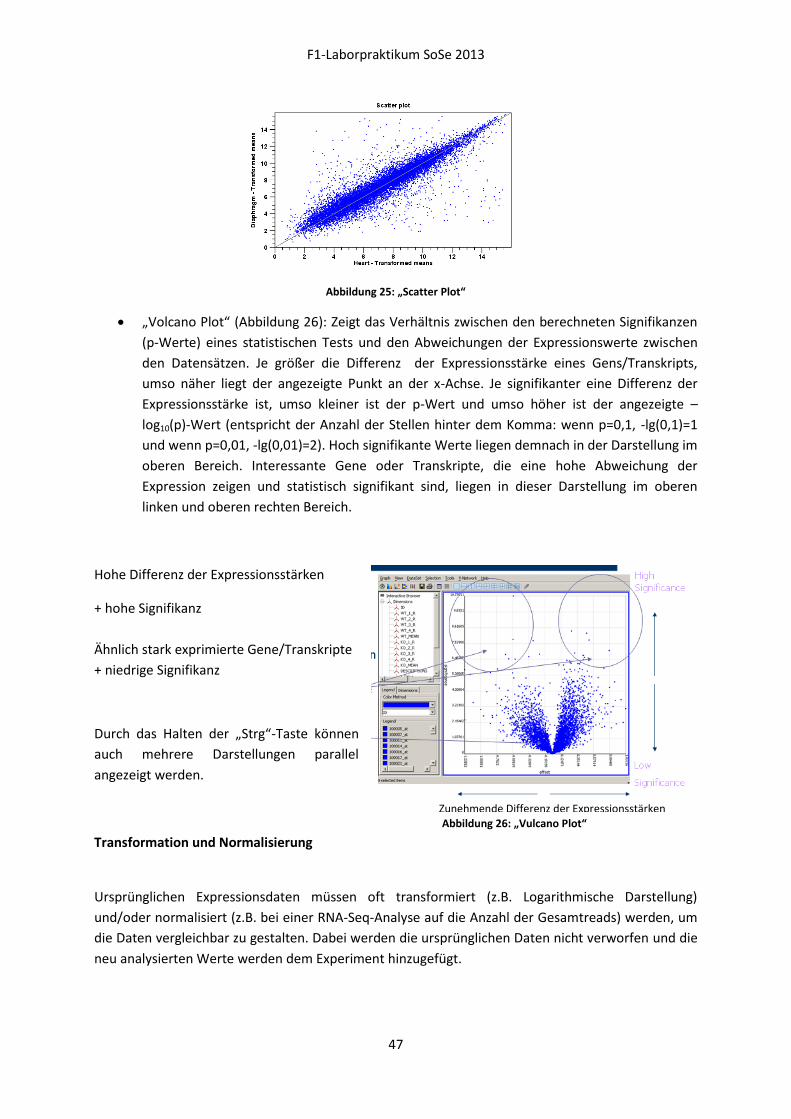
F1-Laborpraktikum SoSe 2013
47
Abbildung 25: „Scatter Plot“
„Volcano Plot“ (Abbildung 26): Zeigt das Verhältnis zwischen den berechneten Signifikanzen
(p-Werte) eines statistischen Tests und den Abweichungen der Expressionswerte zwischen
den Datensätzen. Je größer die Differenz der Expressionsstärke eines Gens/Transkripts,
umso näher liegt der angezeigte Punkt an der x-Achse. Je signifikanter eine Differenz der
Expressionsstärke ist, umso kleiner ist der p-Wert und umso höher ist der angezeigte –
log10(p)-Wert (entspricht der Anzahl der Stellen hinter dem Komma: wenn p=0,1, -lg(0,1)=1
und wenn p=0,01, -lg(0,01)=2). Hoch signifikante Werte liegen demnach in der Darstellung im
oberen Bereich. Interessante Gene oder Transkripte, die eine hohe Abweichung der
Expression zeigen und statistisch signifikant sind, liegen in dieser Darstellung im oberen
linken und oberen rechten Bereich.
Hohe Differenz der Expressionsstärken
+ hohe Signifikanz
Ähnlich stark exprimierte Gene/Transkripte
+ niedrige Signifikanz
Durch das Halten der „Strg“-Taste können
auch mehrere Darstellungen parallel
angezeigt werden.
Transformation und Normalisierung
Ursprünglichen Expressionsdaten müssen oft transformiert (z.B. Logarithmische Darstellung)
und/oder normalisiert (z.B. bei einer RNA-Seq-Analyse auf die Anzahl der Gesamtreads) werden, um
die Daten vergleichbar zu gestalten. Dabei werden die ursprünglichen Daten nicht verworfen und die
neu analysierten Werte werden dem Experiment hinzugefügt.
Abbildung 26: „Vulcano Plot“
Zunehmende Differenz der Expressionsstärken

F1-Laborpraktikum SoSe 2013
48
Die verschiedenen „Tools“ der Expressionsanalyse lassen eine Auswahl der zu verwendenden Daten
zu. So kann zwischen „Original“, „Transformed“ und „Normalized“ gewählt werden, wenn diese
Optionen durchgeführt wurden:
Abbildung 27: Analyse der originalen, transformierten oder normalisierten Daten
Müssen Sie mit Ihren Daten eine solche Normalisierung durchführen?
Statistische Analyse: Identifizierung differenzieller Expression
Es gibt zwei Kategorien der Analyse:
Tests, die eine Gauß‘sche Verteilung der Daten voraussetzen (Vergleich der Mittelwerte)
o t-Test (Experimente mit 2 oder mehr Gruppen)
o ANOVA (Experimente mit mehr als 2 Gruppen)
Toolbox | Expression Analysis | Statistical Analysis | On Gaussian Data
Tests, die das Verhältnis von verschiedenen Anzahlen in Datensätzen vergleichen (z.B.
Vergleich von Expressionswerten der RNA-Seq-Analyse)
o z-Test (Vergleich von 2 Datensätzen)
o Beta-Binominal-Test (Vergleich der Verhältnisse verschiedener Datensätze innerhalb
einer Gruppe mit den Verhältnissen einer weiteren Gruppe)
Toolbox | Expression Analysis | Statistical Analysis | On Proportions
Für beide Statistiken muss vorher ein Experiment generiert werden (siehe oben).
Sie führen mit Ihren Datensätzen einen z-Test durch, der das Verhältnis der Expressionsstärken
vergleicht. Der Test kann p-Werte und korrigierte p-Werte berechnen, die in der Tabelle des
Experiments hinzugefügt werden. Die Ergebnisse können dann z.B. in einem „Volcano plot“ angezeigt
werden.
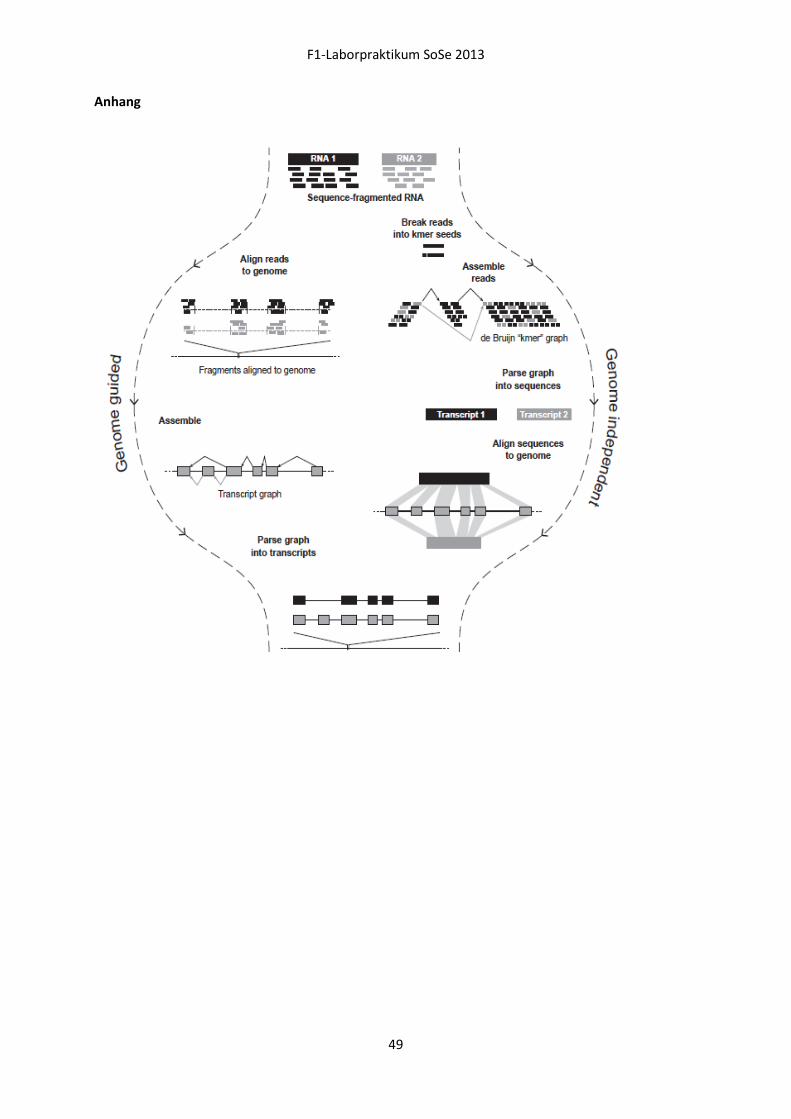
F1-Laborpraktikum SoSe 2013
49
Anhang
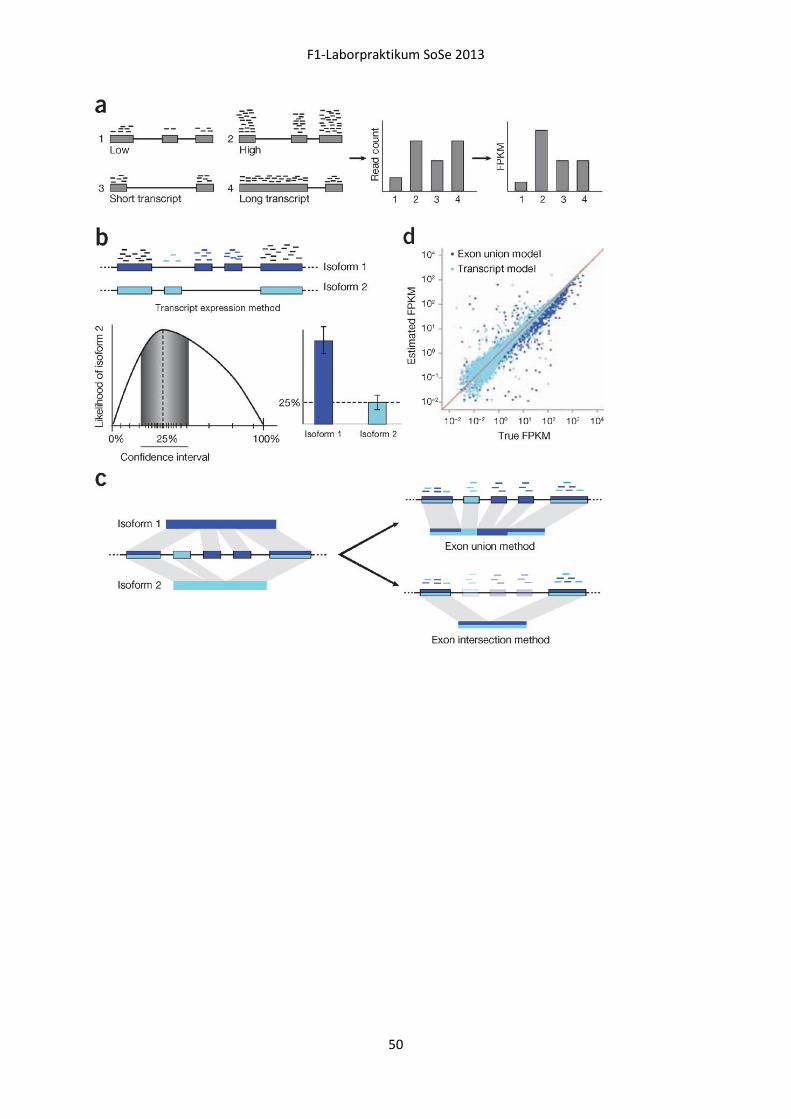
F1-Laborpraktikum SoSe 2013
50
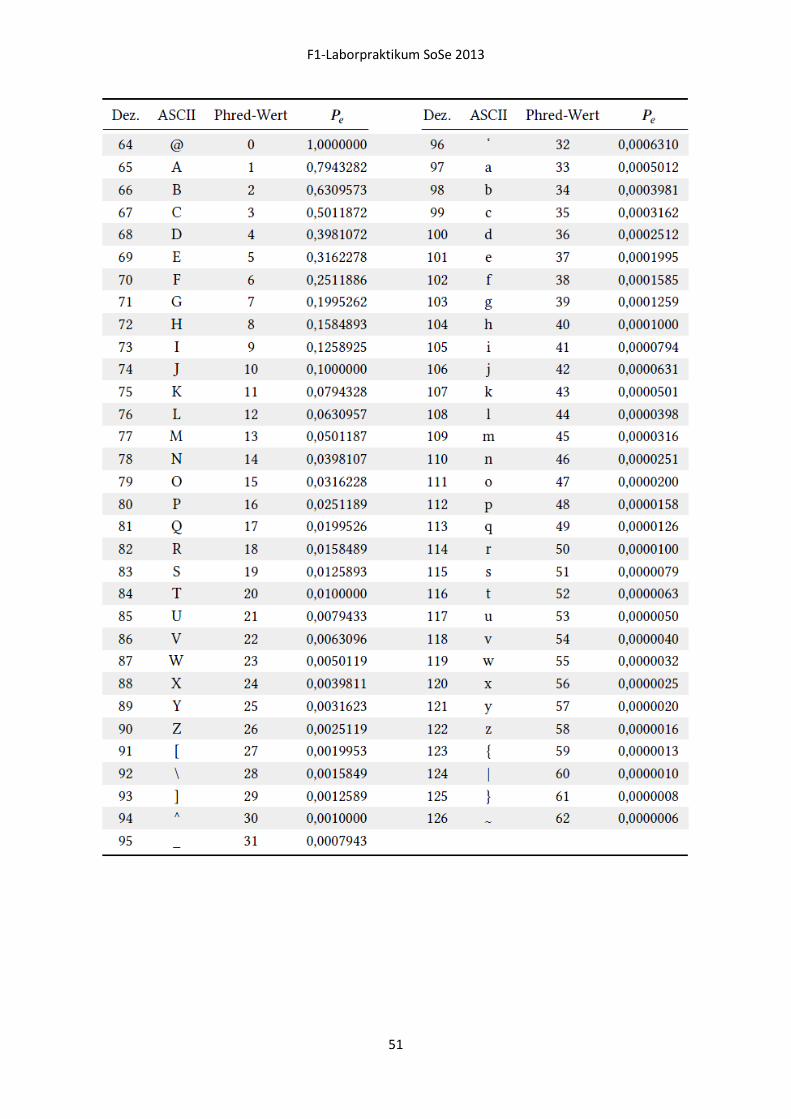
F1-Laborpraktikum SoSe 2013
51





























