Embed Size (px)
Citation preview

Einführung
in die
Gentechnologie
Institut für Molekulargenetik, gentechnologische Sicherheitsforschung und Beratung Johannes Gutenberg-Universität Mainz Leiter: Prof. Dr. Erwin R. Schmidt Betreuer: Dr. Christiane Kraemer, Dr. Steffen Rapp
Dipl.Biol.s Nicholas Bachtadse, Sarah Brunck, Birte Ding, Sabine Fischer, Tobias Lautwein, Regina Stiehl
Dipl. Bioinformatiker (FH) Benjamin Rieger Rudolf Baader

F1-Laborpraktikum SoSe 2015
2
Inhalt F1-Laborpraktikum Gentechnologie ....................................................................................................... 4
Zeitplan (nicht bindend) .................................................................................................................... 11
Teil 1: Herstellen einer DNA-Bibliothek für die Illumina-Sequenzierung ...................................... 11
Teil 2: Seminarvorträge ................................................................................................................. 11
Teil 3: Bioinformatische Analyse der „NGS“-Daten ....................................................................... 11
Theorie Teil 1: Herstellen einer DNA-Bibliothek für „NGS“ .................................................................. 12
Praxis Teil 1: Herstellen einer DNA-Bibliothek für die Sequenzierung ................................................. 14
Allgemeine Laborsicherheit und Verhaltensregeln ........................................................................... 14
DNA-Isolierung aus Chironomus-Larven ........................................................................................... 14
Agarose-Gelelektrophorese .............................................................................................................. 15
Herstellung eines 0,8%-igen Agarosegels ...................................................................................... 15
Vorbereitung der GENterphorese-Apparatur ............................................................................... 16
Durchführung der Elektrophorese ................................................................................................ 17
Erstellen einer DNA-Library mit dem Illumina TruSeq Nano DNA Kit ............................................... 17
Allgemeine Hinweise und Vorbereitungen ................................................................................... 17
Fragmentierung der DNA .............................................................................................................. 17
Aufreinigung der fragmentierten DNA .......................................................................................... 18
End Repair ..................................................................................................................................... 19
Größenselektion ............................................................................................................................ 19
Adenylierung der 3‘ Enden ............................................................................................................ 20
Adapterligation .............................................................................................................................. 20
Zweifache Aufreinigung ................................................................................................................. 20
Enrichment .................................................................................................................................... 21
Aufreinigung .................................................................................................................................. 21
Quantifizierung der Library mit Qubit HS Assay ............................................................................ 22
Qualitätskontrolle, Molekulargewichtsverteilung und Quantifizierung der Library mit Agilent
Bioanalyzer Nano-Assay ................................................................................................................ 23
Theorie Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-Daten ......................... 25
Sequenzalignment ............................................................................................................................. 25
Assemblierungsmethoden ................................................................................................................ 25
BLAST ................................................................................................................................................. 26
De-Bruijn-Graph (DBG) ...................................................................................................................... 27
Arbeitsablauf Sequenzverarbeitung .................................................................................................. 30
Möglichkeiten der Parallelisierung .................................................................................................... 30

F1-Laborpraktikum SoSe 2015
3
Praxis Teil 2: Bioinformatische Analyse von „NGS“-Daten .................................................................... 31
Sequenzierung und Basecalling ......................................................................................................... 32
Datenformat FASTQ .......................................................................................................................... 33
Quality Scores .................................................................................................................................... 34
Bioinformatische Analyse der „Next-Generation Sequencing“ Daten mit Hilfe der „CLC Genomics
Workbench“ ...................................................................................................................................... 36
Import der „Next-Generation Sequencing“-Daten........................................................................ 36
„Trimmen“ der Reads .................................................................................................................... 37
Assemblierung der Reads .............................................................................................................. 39
De novo-Assemblierung ................................................................................................................. 39
„Mapping“ der Reads gegen eine Referenzsequenz ..................................................................... 40
„Large Gap Read Mapping“ ........................................................................................................... 41
Anhang........................................................................................................................................... 44

F1-Laborpraktikum SoSe 2015
4
F1-Laborpraktikum Gentechnologie Das FI-Praktikum „Methoden der Gentechnologie - NGS“ soll technische Fähigkeiten im Bereich der
Molekulargenetik sowie in der bioinformatischen Auswertung von Sequenzdaten vermitteln. Dabei
soll einerseits die Isolation von DNA aus Larven der Arten Chironomus (C.) annularius, C. luridus und
C. piger mit der anschließenden Konstruktion einer DNA-Bibliothek für die Sequenzierung auf dem
Illumina HiSeq 2500TM erlernt werden. Des Weiteren sollen bioinformatische Kenntnisse in der
Analyse von NGS-Daten mit Hilfe von gängiger Software vermittelt werden.
Die Familie der Chironomiden (Zuckmücken) gehört zur Ordnung der Diptera (Zweiflügler; Abbildung
1). Chironomiden haben keine typischen Geschlechtschromosomen wie z. B. die X- und Y-
Chromosomen von Säugern. Trotzdem konnte durch genetische Analysen nachgewiesen werden,
dass der Geschlechtsbestimmungsmechanismus bei Chironomiden die männliche Heterogametie ist.
In C. thummi und C. piger konnte darüber hinaus durch zytogenetische Analysen festgestellt werden,
dass der dominante männchenbestimmende Faktor M in einer Chromosomenregion nahe dem
Telomer von Chromosom III liegt.
In situ-Hybridisierungen an Polytänchromosomen von
Chironomiden mit hoch repetitiven Cla-Elementen als Sonde
zeigen ein hemizygotes Signal in genau dieser
Chromosomenregion bei männlichen Tieren der Art
C. thummi, welches in den Präparaten weiblicher Tiere fehlt.
Da dieses Cla-Element-Cluster in der geschlechts-
bestimmenden Chromosomenregion liegt und nur bei
männlichen Tieren und dort immer nur hemizygot zu finden ist, muss dieses Cluster eng gekoppelt
mit dem dominanten Faktor M sein. Um einen molekularen Zugang zu dieser hochinteressanten
Chromosomenregion zu bekommen, wurden von unserer Arbeitsgruppe genomische Lambda-
Bibliotheken von C. thummi mit den hochrepetitiven Cla-Elementen als Sonde gescreent.
Alle positiven Klone wurden anschließend durch in situ-Hybridisierung an Polytänchromosomen von
C. thummi und piger auf ihre chromosomale Herkunft hin überprüft. Die dabei gefundenen Lambda-
Klone λCla1.1Y und λCla1.8Z flankieren das männchenspezifische hemizygote Cla-Element-Cluster
links und rechts (siehe Abbildung 2).
Abbildung 2 Geschlechtsbestimmende Region
Abbildung 1 Stammbaum der Diptera
F1-Laborpraktikum SoSe 2015
5
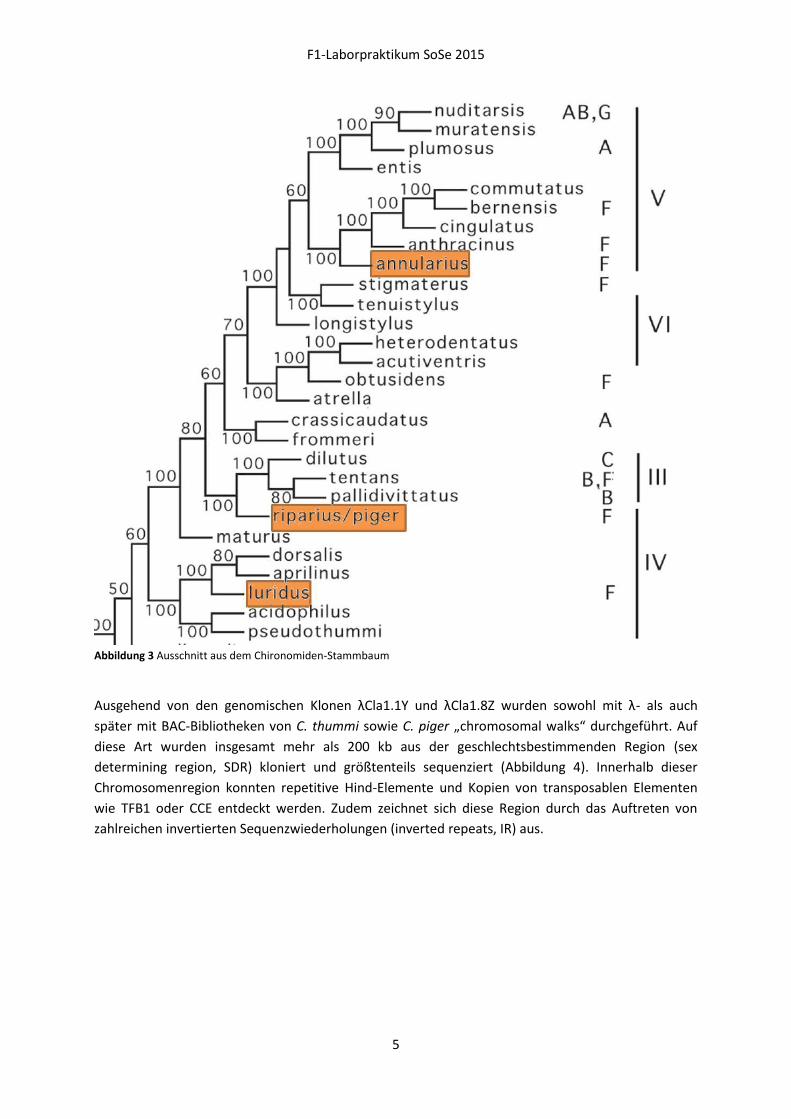
Abbildung 3 Ausschnitt aus dem Chironomiden-Stammbaum
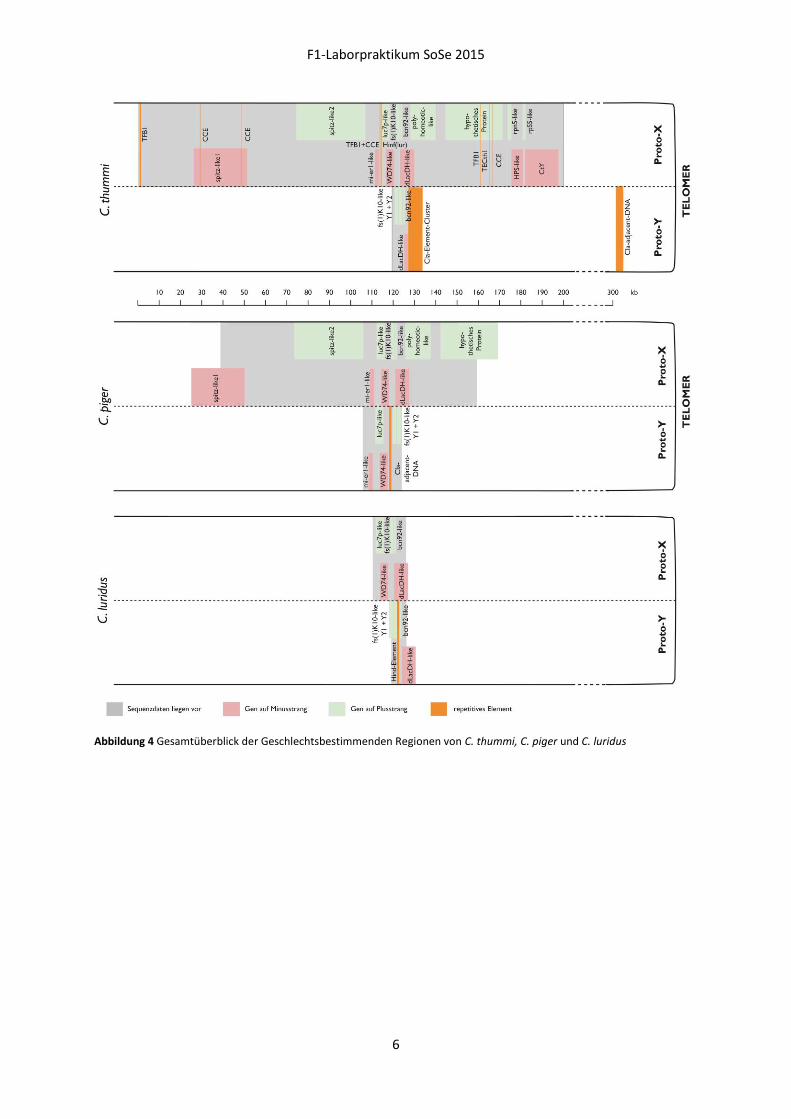
Ausgehend von den genomischen Klonen λCla1.1Y und λCla1.8Z wurden sowohl mit λ- als auch
später mit BAC-Bibliotheken von C. thummi sowie C. piger „chromosomal walks“ durchgeführt. Auf
diese Art wurden insgesamt mehr als 200 kb aus der geschlechtsbestimmenden Region (sex
determining region, SDR) kloniert und größtenteils sequenziert (Abbildung 4). Innerhalb dieser
Chromosomenregion konnten repetitive Hind-Elemente und Kopien von transposablen Elementen
wie TFB1 oder CCE entdeckt werden. Zudem zeichnet sich diese Region durch das Auftreten von
zahlreichen invertierten Sequenzwiederholungen (inverted repeats, IR) aus.
F1-Laborpraktikum SoSe 2015
6
Abbildung 4 Gesamtüberblick der Geschlechtsbestimmenden Regionen von C. thummi, C. piger und C. luridus
F1-Laborpraktikum SoSe 2015
7
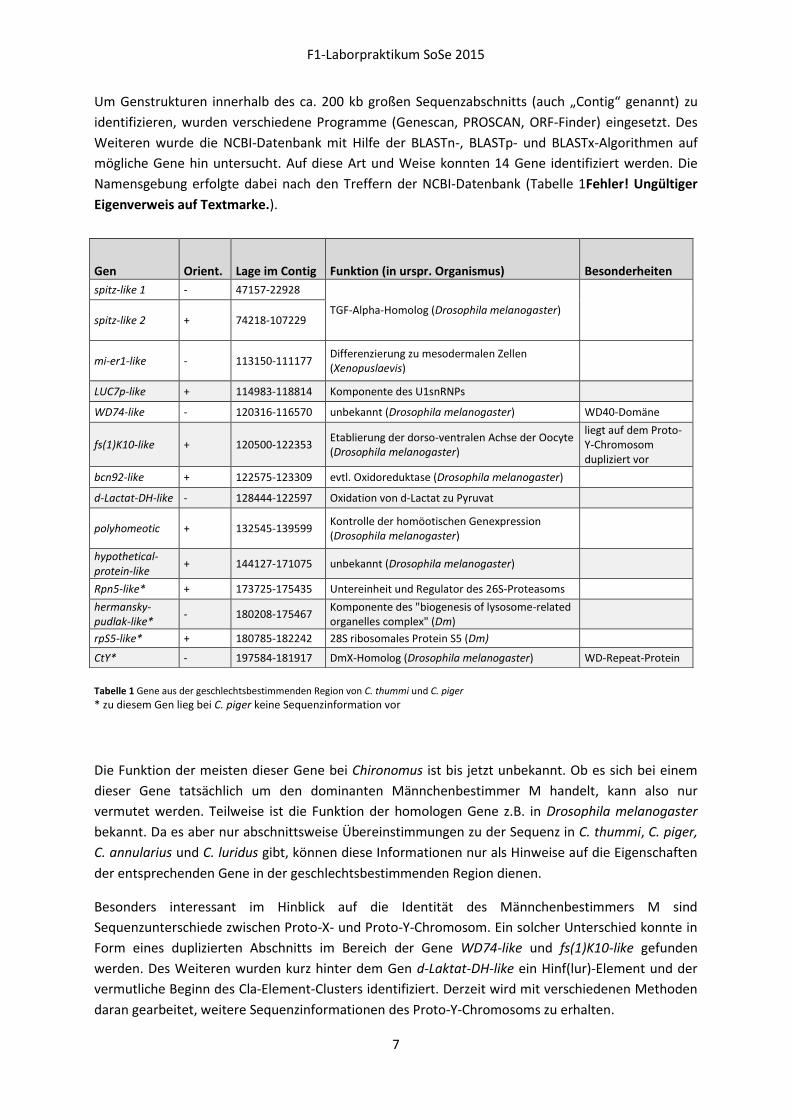
Um Genstrukturen innerhalb des ca. 200 kb großen Sequenzabschnitts (auch „Contig“ genannt) zu
identifizieren, wurden verschiedene Programme (Genescan, PROSCAN, ORF-Finder) eingesetzt. Des
Weiteren wurde die NCBI-Datenbank mit Hilfe der BLASTn-, BLASTp- und BLASTx-Algorithmen auf
mögliche Gene hin untersucht. Auf diese Art und Weise konnten 14 Gene identifiziert werden. Die
Namensgebung erfolgte dabei nach den Treffern der NCBI-Datenbank (Tabelle 1Fehler! Ungültiger
Eigenverweis auf Textmarke.).
Tabelle 1 Gene aus der geschlechtsbestimmenden Region von C. thummi und C. piger
* zu diesem Gen lieg bei C. piger keine Sequenzinformation vor
Die Funktion der meisten dieser Gene bei Chironomus ist bis jetzt unbekannt. Ob es sich bei einem
dieser Gene tatsächlich um den dominanten Männchenbestimmer M handelt, kann also nur
vermutet werden. Teilweise ist die Funktion der homologen Gene z.B. in Drosophila melanogaster
bekannt. Da es aber nur abschnittsweise Übereinstimmungen zu der Sequenz in C. thummi, C. piger,
C. annularius und C. luridus gibt, können diese Informationen nur als Hinweise auf die Eigenschaften
der entsprechenden Gene in der geschlechtsbestimmenden Region dienen.
Besonders interessant im Hinblick auf die Identität des Männchenbestimmers M sind
Sequenzunterschiede zwischen Proto-X- und Proto-Y-Chromosom. Ein solcher Unterschied konnte in
Form eines duplizierten Abschnitts im Bereich der Gene WD74-like und fs(1)K10-like gefunden
werden. Des Weiteren wurden kurz hinter dem Gen d-Laktat-DH-like ein Hinf(lur)-Element und der
vermutliche Beginn des Cla-Element-Clusters identifiziert. Derzeit wird mit verschiedenen Methoden
daran gearbeitet, weitere Sequenzinformationen des Proto-Y-Chromosoms zu erhalten.
Gen Orient. Lage im Contig Funktion (in urspr. Organismus) Besonderheiten
spitz-like 1 - 47157-22928
TGF-Alpha-Homolog (Drosophila melanogaster) spitz-like 2 + 74218-107229
mi-er1-like - 113150-111177 Differenzierung zu mesodermalen Zellen (Xenopuslaevis)
LUC7p-like + 114983-118814 Komponente des U1snRNPs
WD74-like - 120316-116570 unbekannt (Drosophila melanogaster) WD40-Domäne
fs(1)K10-like + 120500-122353 Etablierung der dorso-ventralen Achse der Oocyte (Drosophila melanogaster)
liegt auf dem Proto-Y-Chromosom dupliziert vor
bcn92-like + 122575-123309 evtl. Oxidoreduktase (Drosophila melanogaster)
d-Lactat-DH-like - 128444-122597 Oxidation von d-Lactat zu Pyruvat
polyhomeotic + 132545-139599 Kontrolle der homöotischen Genexpression (Drosophila melanogaster)
hypothetical-protein-like
+ 144127-171075 unbekannt (Drosophila melanogaster)
Rpn5-like* + 173725-175435 Untereinheit und Regulator des 26S-Proteasoms
hermansky-pudlak-like*
- 180208-175467 Komponente des "biogenesis of lysosome-related organelles complex" (Dm)
rpS5-like* + 180785-182242 28S ribosomales Protein S5 (Dm)
CtY* - 197584-181917 DmX-Homolog (Drosophila melanogaster) WD-Repeat-Protein
F1-Laborpraktikum SoSe 2015
8
Zur näheren Analyse der Gene in der geschlechtsbestimmenden Region und einer möglichen
geschlechtsspezifischen Expression dieser Gene wird eines der modernsten Sequenzierverfahren
eingesetzt. Die Illumina „HiSeq 2500“-Plattform ist Teil einer Reihe von neuen Technologien im
Bereich der Hochdurchsatzsequenzierung, dem sogenannten „Next-Generation Sequencing“ (NGS).
Die meist verbreiteten Technologien dieser Gruppe sind die SOLiDTM-, 454- und Illumina-Verfahren.
Sie machen es möglich, immer günstiger und in immer kürzerer Zeit große Mengen an
Sequenzinformation zu generieren. Hat die Sequenzierung des menschlichen Genoms im Rahmen
des humanen Genom-Projekts (1990-2003) noch über 3 Mrd. US$ und 13 Jahre Forschung benötigt,
so sind Resequenzierungen menschlicher Genome heute innerhalb von einiger Tage und mit Kosten
im Bereich von unter 10.000 € möglich. Neben dem auf Ligation von Oligonukleotiden basierenden
SOLiDTM-System und der Pyrosequenzierung der 454 Roche-Plattform dürften die „NextSeq“- und
„HiSeq“-Systeme der Firma Illumina weltweit den größten Anteil an Sequenzierprojekten haben.
Zurzeit sind hier mit bis zu 250 nt im Vergleich zur SOLiD-Platform größere und im Vergleich zur 454-
Technologie (~800 nt) deutlich kürzere Leseweiten möglich. Allerdings bietet Illumina mit wesentlich
geringeren Fehlerraten und mehr Sequenzinformation eindeutige Vorteile gegenüber der Roche-
Technologie. Während bei der 454-Technologie von
Roche auf Emulsions-PCR und Picotiterplatten
zurückgegriffen wird, findet die spezifische
Amplifikation bei den Illumina-Systemen erst nach
dem Immobilisieren der DNA-Fragmente auf der
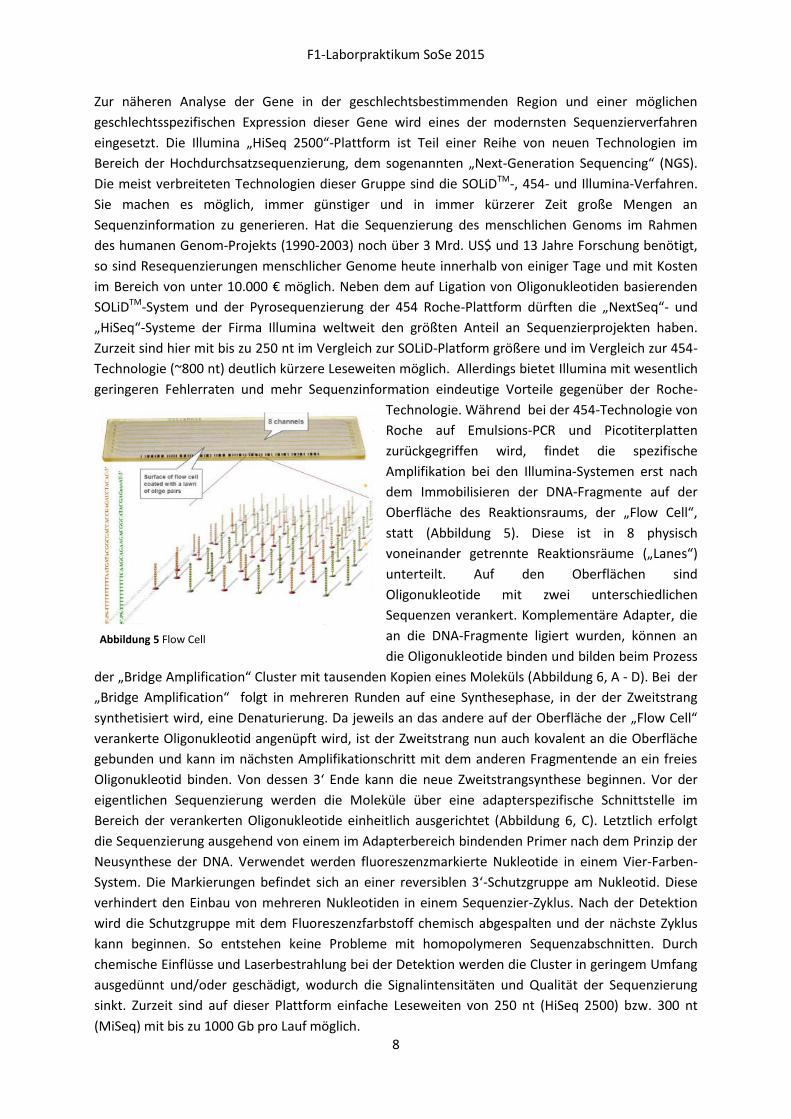
Oberfläche des Reaktionsraums, der „Flow Cell“,
statt (Abbildung 5). Diese ist in 8 physisch
voneinander getrennte Reaktionsräume („Lanes“)
unterteilt. Auf den Oberflächen sind
Oligonukleotide mit zwei unterschiedlichen
Sequenzen verankert. Komplementäre Adapter, die
an die DNA-Fragmente ligiert wurden, können an
die Oligonukleotide binden und bilden beim Prozess
der „Bridge Amplification“ Cluster mit tausenden Kopien eines Moleküls (Abbildung 6, A - D). Bei der
„Bridge Amplification“ folgt in mehreren Runden auf eine Synthesephase, in der der Zweitstrang
synthetisiert wird, eine Denaturierung. Da jeweils an das andere auf der Oberfläche der „Flow Cell“
verankerte Oligonukleotid angenüpft wird, ist der Zweitstrang nun auch kovalent an die Oberfläche
gebunden und kann im nächsten Amplifikationschritt mit dem anderen Fragmentende an ein freies
Oligonukleotid binden. Von dessen 3‘ Ende kann die neue Zweitstrangsynthese beginnen. Vor der
eigentlichen Sequenzierung werden die Moleküle über eine adapterspezifische Schnittstelle im
Bereich der verankerten Oligonukleotide einheitlich ausgerichtet (Abbildung 6, C). Letztlich erfolgt
die Sequenzierung ausgehend von einem im Adapterbereich bindenden Primer nach dem Prinzip der
Neusynthese der DNA. Verwendet werden fluoreszenzmarkierte Nukleotide in einem Vier-Farben-
System. Die Markierungen befindet sich an einer reversiblen 3‘-Schutzgruppe am Nukleotid. Diese
verhindert den Einbau von mehreren Nukleotiden in einem Sequenzier-Zyklus. Nach der Detektion
wird die Schutzgruppe mit dem Fluoreszenzfarbstoff chemisch abgespalten und der nächste Zyklus
kann beginnen. So entstehen keine Probleme mit homopolymeren Sequenzabschnitten. Durch
chemische Einflüsse und Laserbestrahlung bei der Detektion werden die Cluster in geringem Umfang
ausgedünnt und/oder geschädigt, wodurch die Signalintensitäten und Qualität der Sequenzierung
sinkt. Zurzeit sind auf dieser Plattform einfache Leseweiten von 250 nt (HiSeq 2500) bzw. 300 nt
(MiSeq) mit bis zu 1000 Gb pro Lauf möglich.
Abbildung 5 Flow Cell

F1-Laborpraktikum SoSe 2015
9
Mit der im Praktikum verwendeten Chemie werden pro „Lane“ ca. 250 - 400 Mio. Sequenzen mit
einer Länge von 200 bp generiert, welche als „Reads“ bezeichnet werden. Dies entspricht einer
Sequenzinformation von ca. 50-80 Gbp. Diese Masse an Daten aus recht kurzen Sequenzfragmenten
stellt ganz neue Anforderungen an die computertechnische Ausstattung und die Analysealgorithmen.
Es werden neue oder angepasste Strategien für die Auswertung der Sequenzdaten benötigt. Die im
Kurs generierten Daten können in vielerlei Hinsicht bioinformatisch ausgewertet werden: Die Daten
können assembliert und somit in einen genomischen Kontext gebracht werden (de novo Assembly),
das sogenannte „Mapping“ erlaubt es, einzelne Reads gegen eine bereits vorhandene
Referenzsequenz zu kartieren. Dabei werden die Reads Koordinaten in der Referenzsequenz
zugeordnet.
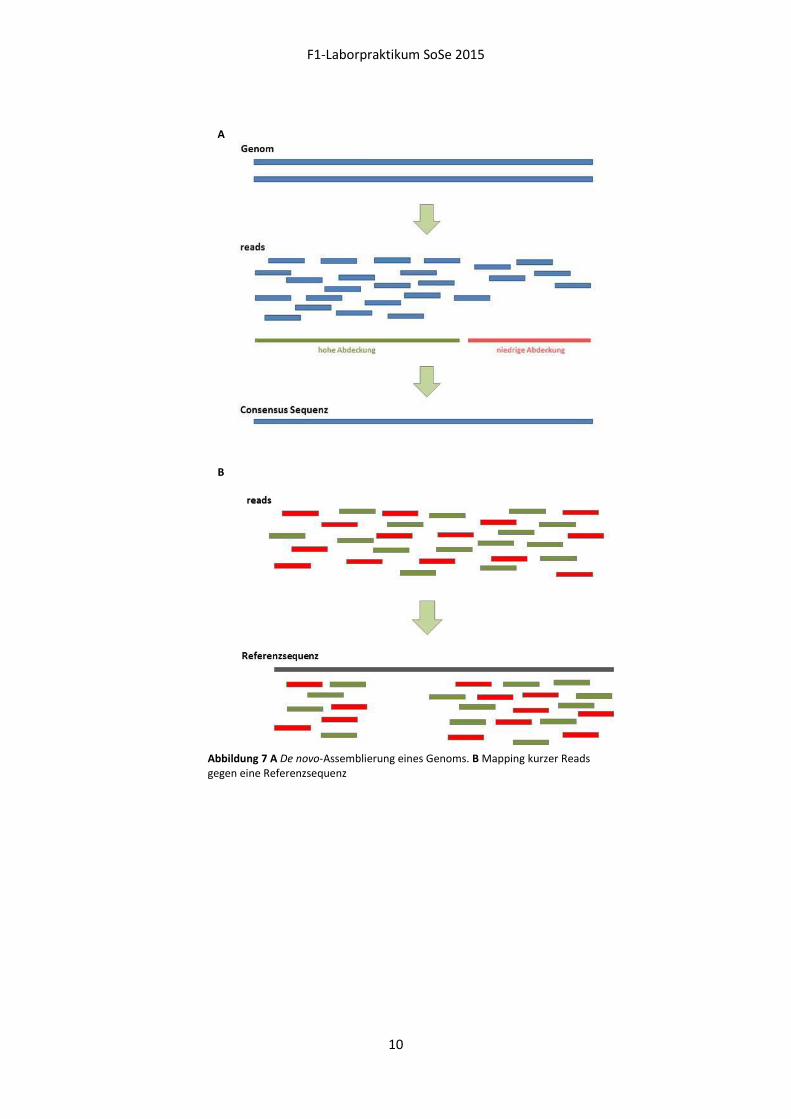
Ziel ist es, die einzelnen Datensätze nach erfolgreicher Assemblierung hinsichtlich art- und
geschlechtsspezifischer Unterschiede zu untersuchen (Abbildung 7). In unserem Fall bedeutet dies,
Assemblierungen aus selbst generierten Sequenzdaten der Chironomusarten C. piger, C. luridus und
C. annularius zu erstellen. Des Weiteren ist es möglich, die Daten gegen die bereits vorhandenen
Referenzen von C. thummi, C. piger und C. luridus zu mappen. Hierfür wurden im Vorfeld des
Praktikums jeweils Larven der drei zu untersuchenden Arten nach ihrem Geschlecht sortiert
(„gesext“). Es soll nun DNA aus Pools von je ca. 20 Larven gewonnen und aufgereinigt werden.
Nach erfolgreicher DNA-Isolierung wird eine DNA-Bibliothek für die Illumina-Sequenzierung
konstruiert und ein „HiSeq2500“-Lauf gestartet. Bei der anschließenden bioinformatischen Analyse
der Daten sollen die Sequenzen dann insbesondere in Hinblick auf Unterschiede in der SDR analysiert
werden.
Abbildung 6 „Bridge Amplification“ und Sequenzierung
F1-Laborpraktikum SoSe 2015
10
A
B
Abbildung 7 A De novo-Assemblierung eines Genoms. B Mapping kurzer Reads gegen eine Referenzsequenz

F1-Laborpraktikum SoSe 2015
11
Zeitplan (nicht bindend)
Teil 1: Herstellen einer DNA-Bibliothek für die Illumina-Sequenzierung
Mo, 08.06.2015 - DNA-Isolierung aus Chironomus-Larven
- Agarose-Gelelektrophorese - Konzentrationsbestimmung mit NanoDrop und Qubit Di, 09.06.2015 - Fragmentierung der DNA mit Covaris - Aufreinigung der fragmentierten DNA - End Repair - Größenselektion - optional: Größenbestimmung mit Bioanalyzer Mi, 10.06.2015 - A-Tailing des 3´Endes - Adapter-Ligation - Aufreinigung - Enrichment - Aufreinigung - Qualitätskontrolle mit Bioanalyzer - Quantifizierung mit Qubit Do, 11.06.2015 - Vorbereitung der Sequenzierung - Besichtigung des HiSeq2500 Fr, 12.06.201 - Vorbereitung der Sequenzierung
Teil 2: Seminarvorträge
Nach Terminvereinbarung Mo, 15.06. bis Fr, 19.06.2015
Teil 3: Bioinformatische Analyse der „NGS“-Daten
Mo, 22.06. bis Fr, 26.06.2015

F1-Laborpraktikum SoSe 2015
12
Theorie Teil 1: Herstellen einer DNA-Bibliothek für „NGS“ Zur Whole Genome Sequenzierung verschiedener Chironomus Spezies wird zunächst genomische
DNA dieser Zuckmücken benötigt. Als Ausgangsmaterial für die DNA-Isolierung dienen gesexte (d.h.
anhand der Genitalimaginalscheiben nach Geschlechtern getrennte) Larven des L4-Stadiums der
verschiedenen Spezies. In einem ersten Schritt wird das Gewebe mechanisch mit einem Potter sowie
chemisch mittels milder Detergenzien (Triton-X-100) aufgeschlossen. Dies dient dazu, die Zellen aus
dem Verbund zu lösen und sie zugänglich für die bei der folgenden Extraktion verwendeten
Detergenzien zu machen. Im Anschluss werden die Kerne mittels Zentrifugation isoliert und mit
scharfen Detergenzien (SDS) lysiert, sodass die DNA austritt. Vorhandene Proteine werden
denaturiert und mit organischen Lösungsmitteln (PCI) extrahiert. Dabei verbleiben Proteine aufgrund
ihrer hydrophoben Bestandteile in der phenolischen unteren Phase oder an der Grenzfläche, der so
genannten Interphase. Die obere wässrige Phase enthält die hydrophilen Nukleinsäuren. Im
Anschluss kann die DNA mit Hilfe von Ethanol gefällt werden. Die Qualität und Quantität der DNA
kann anhand einer Agarose-Gelelektrophorese abgeschätzt werden. Genauere Aussagen zur
Konzentration ermöglichen der NanoDrop und das Qubit Fluorometer.
Da mit dem verwendeten Protokoll zur DNA-Isolierung in der Regel recht hochmolekulare DNA
isoliert werden kann, die Leseweiten der Illumina-Sequenzierung jedoch sehr begrenzt sind (je nach
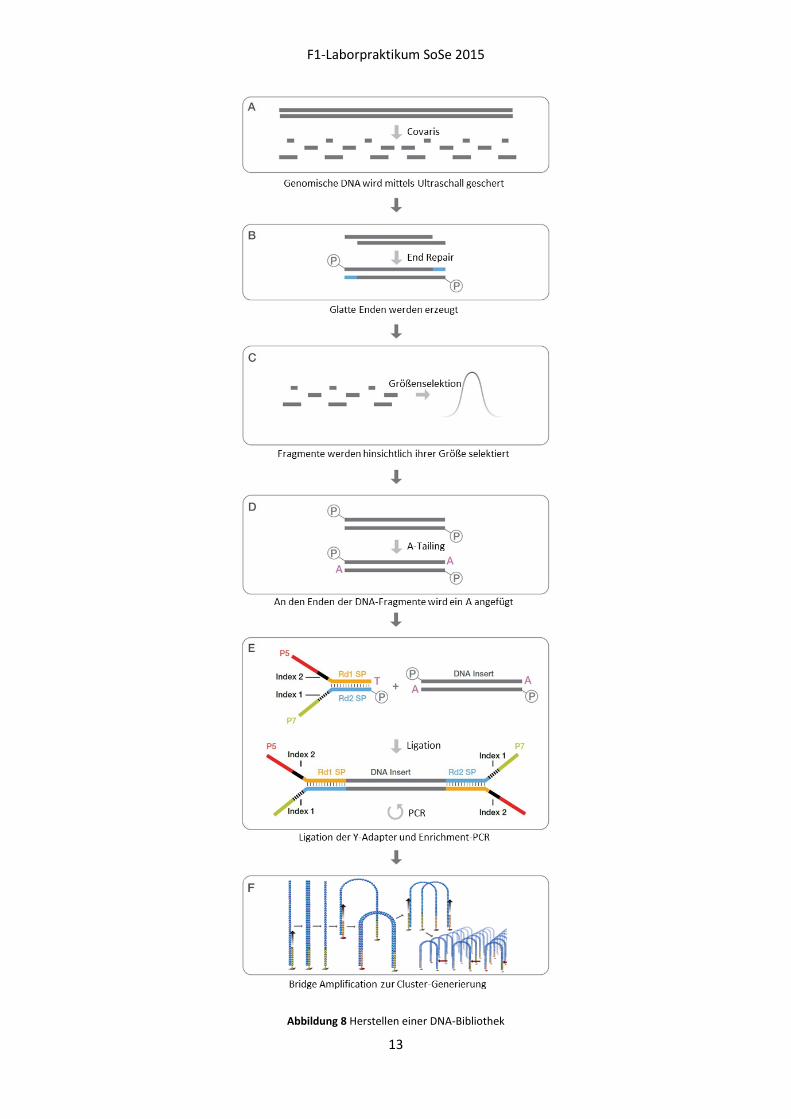
verwendeter Chemie 100-200 bp), wird die DNA zu Beginn der Library-Präparation mittels Ultraschall
geschert (Abbildung 8-A). Dabei entstehen dsDNA-Fragmente mit 3‘ oder 5‘ Überhangen. Diese
werden während des End Repair in glatte Enden umgewandelt (Abbildung 8-B). Zu große und zu
kleine Fragmente werden bei einer Größenselektion mit magnetischen Beads entfernt (Abbildung 8-
C). Im Anschluss werden die verbleibenden DNA-Fragmente beidseitig mit Adaptern versehen. Um
sicherzustellen, dass ein DNA-Fragment auf beiden Seiten einen Adapter erhält und zu verhindern,
dass zwei DNA-Fragmente oder zwei Adapter miteinander reagieren, werden beim vorangehenden
A-Tailing die Enden der Fragmente mit einem A-Überhang versehen (Abbildung 8-D). Die Adapter
tragen an einem Ende einen komplementären T-Überhang, wodurch die Ligation zwischen DNA-
Fragment und Adapter gefördert wird (Abbildung 8-E). Für die so genannte Bridge Amplification
werden Fragmente benötigt, die an den Enden unterschiedliche Adapter tragen. Dies wird durch
spezielle Y-Adapter gewährleistet. Die beiden Stränge eines Adapters bilden über einen Bereich
komplementärer Sequenzen einen Doppelstrang aus, wohingegen sie sich in einem anderen
Abschnitt unterscheiden und keinen Doppelstrang ausbilden. Werden diese Y-Adapter nun mit den
doppelsträngigen DNA-Fragmenten ligiert, sollte jeder Einzelstrang für sich betrachtet die beiden
unterschiedlichen Adaptersequenzen tragen. Durch wenige Runden einer Enrichment-PCR mit
adapterspezifischen Primern werden DNA-Fragmente, die beidseitig einen Adapter tragen, spezifisch
angereichert. Nach diesem Schritt erhält man eine fertige DNA-Bibliothek, die nach Verdünnung für
die Generierung der Cluster mittels Bridge Amplification auf dem cBot verwendet werden kann
(Abbildung 8-F). Hierbei hybridisieren die denaturierten linearen Fragmente mit den Enden der
Adapter an komplementäre Oligonukleotid-Anker auf der Flow Cell Oberfläche. Die Amplifikation
findet ausgehend von den Flow Cell-Ankern statt, wodurch die Fragmente kovalent an die Flow Cell
gebunden werden. Eine anschließende Denaturierung mit Wasch-Schritt entfernt den ursprünglichen
DNA-Strang. Die neu-synthetisierten, am Anker immobilisierten DNA-Moleküle hybridisieren über ihr
freies Ende mit benachbarten Ankern, die wiederum als Primer für die Amplifikation dienen. Da die
DNA-Moleküle dabei Brücken bilden, wird diese Technik auch als Bridge Amplification bezeichnet.
Durch die wiederholte Amplifikation mit anschließender Denaturierung und Bildung neuer Brücken
entstehen Cluster, die aus tausenden Kopien des zuvor gebundenen Fragments bestehen.
F1-Laborpraktikum SoSe 2015
13
Abbildung 8 Herstellen einer DNA-Bibliothek

F1-Laborpraktikum SoSe 2015
14
Praxis Teil 1: Herstellen einer DNA-Bibliothek für die Sequenzierung
Allgemeine Laborsicherheit und Verhaltensregeln
1. Tragen Sie für alle Arbeiten im Labor einen Laborkittel. 2. In den Laborräumen sollten nur Schreib- und Arbeitsmaterialien aufbewahrt werden.
Straßenkleidung und Taschen sind in die dafür vorgesehenen Spinde (SBI = benachbartes Gebäude) einzuschließen. Bitte hierfür ein geeignetes Vorhängeschloss mitbringen. In den Laborräumen ist das Essen, Trinken und Rauchen verboten.
3. Arbeiten Sie umsichtig und bewusst. Gefährden Sie weder sich selbst noch Ihre Kommilitonen. 4. Studentinnen, die sich in anderen Umständen befinden, teilen dies dem Kursleiter mit. Er wird
diese Mitteilung vertraulich behandeln, eine erfolgreiche Kursteilnahme ermöglichen und Sie von gesundheitsschädlichen Stoffen fernhalten.
5. Wir arbeiten mit organischen Lösungsmitteln, Laugen und Säuren. Vermeiden Sie unbedingt Hautkontakt bzw. orale Aufnahme. Nutzen Sie die angebotenen Schutzmittel wie Handschuhe und Schutzbrillen.
6. Falls Sie sich verletzen sollten, informieren Sie sofort den Kursleiter. Im Bedarfsfall sind in jedem Laborraum Augenduschen vorhanden.
7. Beschriften Sie alle Proben ausreichend mit einem wasserfesten Stift (z.B. Inhalt, Gruppennr., Datum)!
DNA-Isolierung aus Chironomus-Larven
Die DNA-Isolierung erfolgt in folgenden Schritten:
Gewebeaufschluss
Kernisolation
Kernlyse
Extraktion der Proteine
Ethanol-Fällung der DNA
Lösen der DNA
Bitte notieren Sie sich ihre Gruppennummer, die verwendete Chironomus Spezies und das
Geschlecht der Larven!
1. Larven in Potter (auf Eis) überführen.
2. 2 ml "Homogenisierungspuffer" hinzufügen (eiskalt), 1/10 Vol. 10% Triton-X-100 zugeben.
3. Unter Eiskühlung homogenisieren.
4. Homogenat durch Gaze filtrieren und mit 1 ml Homogenisierungspuffer Glas und Gaze
nachspülen.
5. Filtrat in kleinen Falcons 15 min., 5.000 Upm, 4 °C zentrifugieren.
6. Überstand sauber abheben, verwerfen und Sediment in 2 ml Homogenisierungspuffer
resuspendieren, zentrifugieren (wie 5), diesen Schritt gegebenenfalls ca. 3x wiederholen bis
Kern-Pellet „sauber“ erscheint.
7. Das Kern-Pellet in etwa 2 ml eiskaltem Homogenisierungspuffer resuspendieren.
8. Proteinase K (eine Spatelspitze) hinzufügen.
9. 1/10 Vol. 10% SDS und 1/10 Vol. 10x Dialysepuffer zur Kernlyse hinzufügen – durch Invertieren
vorsichtig mischen. (Lösung sollte viskos werden! Warum?)

F1-Laborpraktikum SoSe 2015
15
10. 1-2 Std. (evtl. auch über Nacht) bei 60 °C inkubieren (Kernlyse).
11. Inkubationsgemisch auf Raumtemperatur abkühlen (optional).
12. 0,1 Vol. gesättigtes Tris, pH 8,5 hinzufügen (zur pH-Einstellung!)
13. 0,25 Vol. 5 M NaClO4 (Natriumperchlorat) hinzufügen, gut invertieren (es bildet sich ein
weißlicher Niederschlag).
14. Gleiches Vol. Phenol/Chloroform/Isoamylalkohol-Gemisch (25:24:1) hinzufügen.
15. Mind. 5 min. durch Invertieren gut mischen.
16. 10 min., 5000 Upm bei RT zentrifugieren.
17. Wässrige Phase (immer „oben“!) ohne das Interphase-Material mit einer „weiten“ Pipette
(abgesägt) abheben und in ein neues Falcon-Röhrchen überführen (enthält die DNA), damit die
Schritte 14-17 ca. 3 bis 5x wiederholen (Betreuer fragen!).
18. Zuletzt die DNA-Phase 1x mit gleichem Vol. Chloroform-Isoamylalkohol (24:1) versetzen,
extrahieren und zentrifugieren (zur Entfernung sämtlicher Phenolreste aus der DNA-Phase).
19. Nach dem Zentrifugieren obere, wässrige Phase mit einer "weiten" Pipette (abgesägt) ohne
Interphase-Material abheben und in vorgekühltes (mögl. graduiertes) Falcon-Röhrchen
überführen - in Eis abkühlen lassen.
20. Noch einmal 1/10 Vol. 10x Dialysepuffer zugeben und mischen, dann mit 2 Vol. kaltem Ethanol
abs. überschichten und invertieren.
21. 30 min. bei -20°C inkubieren.
22. 45 min., Höchstgeschwindigkeit bei 4 °C zentrifugieren.
23. Überstand abheben, Pellet mit 1 ml 70% EtOH überschichten (nicht resuspendieren!)
24. 10 min., Höchstgeschwindigkeit bei RT zentrifugieren.
25. Überstand abheben, Pellet trocknen.
26. Pellet in 100-150 µl HPLC-H2O lösen (Betreuer fragen!)
27. 1/10 Vol. der Probe in ein neues Eppendorfgefäß überführen (Agarose-Gelelektrophorese)
28. Restliche genomische DNA bis zum Fragmentieren bei -20 °C wegfrieren. (Beschriftung!)
Agarose-Gelelektrophorese
Durch Agarose-Gelelektrophorese werden DNA-Fragmente entsprechend ihres Molekulargewichts
aufgetrennt. Die Gelelektrophorese wird mit Hilfe des Gelkammersystems GENterphorese der Firma
GENterprise durchgeführt. Die DNA kann im Gel durch den interkalierenden Fluoreszenzfarbstoff
Ethidiumbromid unter UV-Licht (312 nm) direkt sichtbar gemacht werden.
Herstellung eines 0,8%-igen Agarosegels
1. 0,8 g Agarose in Schott-Flasche einwiegen.
2. 100 ml 1x TBE zugeben.
3. In Mikrowellengerät bis zum Kochen erhitzen, ggf. mit VE-H2O auf 100 ml auffüllen.
4. Gelkassette in Ständer stellen, mit Tesafilm seitlich verschließen, Kamm einstecken.
5. „Sockel“ zur Abdichtung der Gelkassette gießen.
6. Gellösung auf ca. 60°C abkühlen lassen, dann luftblasenfrei mit Pipette einfüllen.
7. Während der Abkühlphase Agaroselösung nach Bedarf nachfüllen.
8. Nachdem die Agarose erstarrt ist, Kamm vorsichtig aus dem Gel ziehen.
9. Probentaschen mit Pasteurpipette ausspülen.

F1-Laborpraktikum SoSe 2015
16
Vorbereitung der GENterphorese-Apparatur
1. Füllen Sie die Kammer mit 1 x TBE-Laufpuffer bis etwa 3 mm über die obere Elektrode (Kathode).
2. Setzen Sie das Gel über die Führungsnut in die Kammer ein und drücken Sie es nach unten (Abb.). Wenn die Anschlüsse der Kammer nach rechts zeigen, muss sich die kurze Platte der Gelkassette vorne befinden.
3. Schieben Sie den GENterphorese-Deckel auf die Pufferkammer.
4. Verbinden Sie den roten Stecker mit dem roten Anschluss des Spannungsgerätes und den schwarzen Stecker mit dem schwarzen Anschluss. Prüfen Sie vor dem Auftragen der Proben, ob Strom (Einheit: mA) durch das Gel fließt (Netzgerät kurz einschalten; Betreuer fragen!).
5. Spannungsgerät ausschalten und Deckel entfernen.

F1-Laborpraktikum SoSe 2015
17
Durchführung der Elektrophorese
Achtung: EtBr ist mutagen! Alles, was mit diesem Stoff in Berührung kommt, nur mit Einweghandschuhen aus Nitril anfassen! Nicht direkt in das UV-Licht hineinschauen und UV-undurchlässige Schutzbrillen tragen oder hinter Glas arbeiten! 1. 1/10 Vol. der isolierten genomischen DNA in ein neues Eppendorfgefäß überführen und mit 1/6
Vol. Gelladepuffer versetzen. 2. Die mit Ladepuffer versetzen Proben auf das Agarosegel auftragen. Zusätzlich werden Spuren mit
Hind III-restringierter Lambda-DNA und einem weiteren Molekulargewichtsstandard für kleine DNA-Fragmente (z.B. 100 bp-Leiter) beladen.
3. Deckel der GENterphorese-Kammer schließen und Spannungsgerät anschließen. 4. Elektrophorese bei 100 V (ca. 20 min.). 5. Elektrophorese durch Ausschalten des Spannungsgeräts beenden, wenn die Bromphenolblau-
front ca. 2-3 cm vom unteren Gelrand entfernt ist. Kabelverbindungen lösen. 6. Gel mithilfe des Schiebers aus der Gelkassette herausschieben und in die EtBr-Färbelösung legen. 7. Gel für 2-5 min. färben und kurz wässern. 8. Gel auf UV-Tisch dokumentieren.
Erstellen einer DNA-Library mit dem Illumina TruSeq Nano DNA Kit
Allgemeine Hinweise und Vorbereitungen
1. Alle Puffer werden erst kurz vor Gebrauch aufgetaut und auf RT gebracht, danach sofort wieder
eingefroren.
2. Enzyme werden stets auf Eis gelagert und nach Gebrauch sofort wieder bei -20°C eingefroren.
3. Enzyme werden knapp unter dem Meniskus pipettiert. Es ist darauf zu achten, dass kein
überschüssiges Enzym an der Außenseite der Pipettenspitze hängt.
4. Bevor Enzymreaktionen inkubiert werden, werden sie kurz gemischt und abzentrifugiert.
5. Das 80%-ige EtOH wird am Ende des Arbeitstages bei -20°C eingefroren.
6. Die Beads werden 30 min. vor Nutzung auf RT gestellt und direkt vor dem Gebrauch gut
gevortext.
Fragmentierung der DNA
Achtung: Für die Library-Generierung sollten laut Illumina 100-200 ng DNA verwendet werden.
Jedoch kann maximal 50 µl fragmentierte DNA eingesetzt werden. Zum Scheren werden ca. 1 µg
genomische DNA in ca. 100 µl eingesetzt, damit im Anschluss ein Testgel durchgeführt und falls
nötig das Scheren wiederholt werden kann. Bitte Rücksprache mit dem Betreuer halten!
1. 100 µl genomische DNA (ca. 1 µg) in ein Covaris-Tube überführen.
2. Probe im Covaris-Tube abzentrifugieren.

F1-Laborpraktikum SoSe 2015
18
3. Scheren:
Setting 350 bp Insert 550 bp Insert
Duty cycle 10 %
Intensity 5,0 2,0
Cycles per burst 200
Duration 45 s
Mode Frequency sweeping
Displayed Power 23 W 9 W
Temperature 5,5 - 6,0 °C
4. Probe im Covaris-Tube abzentrifugieren.
5. Gescherte DNA in neues Gefäß überführen.
6. Teil der DNA (z.B. 1/10 Vol.) auf Gel testen (Agarose-Gelelektrophorese).
7. Falls Fragmentierung nicht ausreichend: Scheren wiederholen.
Aufreinigung der fragmentierten DNA
1. Sample Purification Beads gründlich vortexen.
2. 80 µl Sample Purification Beads zu 50 µl fragmentierter DNA geben und durch Auf- und
Abpipettieren gut mischen.
3. 5 min. bei RT inkubieren.
4. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
Folgende Schritte 5-10 werden im Magnetständer durchgeführt:
5. 125 µl Überstand entfernen ohne die Beads zu lösen.
6. 200 µl 80 % EtOH zugeben ohne die Beads zu lösen und 30 s inkubieren.
7. Überstand entfernen ohne die Beads zu lösen und nochmals mit 200 µl EtOH waschen.
8. Verbleibenden EtOH mit 10 µl Pipette entfernen.
9. 5 min. bei RT im Magnetständer trocknen lassen. (Beads sollten nicht „bröseln“)
10. 62,5 µl Resuspension Buffer über die Beads laufen lassen, dabei die Beads nicht mit der
Pipettenspitze berühren, da sie sonst an der Spitze hängen bleiben.
11. Probe aus dem Magnetständer nehmen und die Beads durch wiederholtes Auf- und
Abpipettieren resuspendieren.
12. 2 min. bei Raumtemperatur inkubieren
13. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
14. 60 µl klaren Überstand in ein neues Gefäß überführen, dabei die Beads nicht lösen.
Umgehend mit End Repair fortfahren!

F1-Laborpraktikum SoSe 2015
19
End Repair
1. 40 µl End Repair Mix 2 zur Probe geben und durch Auf- und Abpipettieren gut mischen.
2. Probe kurz anzentrifugieren.
3. Probe bei 30 °C für 30 min. im Thermocycler inkubieren.
Umgehend mit Größenselektion fortfahren!
Größenselektion
1. Sample Purification Beads gründlich vortexen.
2. Sample Purification Beads in Eppendorfgefäß mit HPLC-Wasser verdünnen:
350 bp Insert Size Formel
Sample Purification Beads # der Proben x 109,25 µl
HPLC-H2O # der Proben x 74,75 µl
550 bp Insert Size Formel
Sample Purification Beads # der Proben x 92 µl
HPLC-H2O # der Proben x 92 µl
3. Verdünnte Beads gründlich vortexen.
4. 160 µl verdünnte Sample Purification Beads zur Probe geben und durch Auf- und Abpipettieren
gut mischen.
5. 5 min. bei RT inkubieren
6. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
7. 250 µl Überstand (enthält die gewünschte DNA!!!) in ein neues Gefäß überführen.
8. Sample Purification Beads gründlich vortexen.
9. 30 µl unverdünnte Sample Purification Beads zur Probe geben und durch Auf- und Abpipettieren
gut mischen.
10. 5 min. bei RT inkubieren
11. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
Folgende Schritte 12-18 werden im Magnetständer durchgeführt:
12. 276 µl Überstand entfernen ohne die Beads zu lösen.
13. 200 µl 80 % EtOH zugeben ohne die Beads zu lösen und 30 s inkubieren.
14. Überstand entfernen und nochmals mit 200 µl EtOH waschen.
15. Verbleibenden EtOH mit 10 µl Pipette entfernen.
16. 5 min. bei RT im Magnetständer trocknen lassen. (Beads sollten nicht „bröseln“)
17. 20 µl Resuspension Buffer über die Beads laufen lassen, dabei die Beads nicht mit der
Pipettenspitze berühren, da sie sonst an der Spitze hängen bleiben.
18. Probe aus dem Magnetständer nehmen und die Beads durch wiederholtes Auf- und
Abpipettieren resuspendieren.

F1-Laborpraktikum SoSe 2015
20
19. 2 min. bei Raumtemperatur inkubieren.
20. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
21. 17,5 µl klaren Überstand in ein neues Gefäß überführen, dabei die Beads nicht lösen.
Möglichkeit zum Lagern bei -20°C
Adenylierung der 3‘ Enden
1. 12,5 µl A-Tailing Mix zur Probe geben und durch Auf- und Abpipettieren gut mischen.
2. Probe kurz anzentrifugieren.
3. Probe bei folgendem Programm im Thermocycler inkubieren:
37 °C für 30 min. 70 °C für 5 min. 4 °C für 5 min. Hold at 4 °C
4. Probe kurz anzentrifugieren.
Umgehend mit Adapterligation fortfahren!
Adapterligation
1. 2,5 µl Resuspension Buffer zur Probe geben
2. 2,5 µl Ligation Mix 2 zur Probe geben
3. 2,5 µl DNA Adapter Index zur Probe geben (Rücksprache mit dem Betreuer halten! Index
notieren!)
4. Probe durch Auf- und Abpipettieren gut mischen.
5. Probe kurz anzentrifugieren.
6. Probe bei 30 °C für 10 min. im Thermocycler inkubieren.
7. 5 µl Stop Ligation Buffer zur Probe geben und durch Auf- und Abpipettieren gut mischen.
Zweifache Aufreinigung
1. Sample Purification Beads gründlich vortexen.
2. 42,5 µl Sample Purification Beads zur Probe geben und durch Auf- und Abpipettieren gut
mischen.
3. 5 min. bei RT inkubieren
4. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
Folgende Schritte werden im Magnetständer durchgeführt:
5. 80 µl Überstand entfernen ohne die Beads zu lösen.
6. 200 µl 80 % EtOH zugeben ohne die Beads zu lösen und 30 s inkubieren.
7. Überstand entfernen ohne die Beads zu lösen und nochmals mit 200 µl EtOH waschen.
8. Verbleibenden EtOH mit 10 µl Pipette entfernen.
9. 5 min. bei RT im Magnetständer trocknen lassen. (Beads sollten nicht „bröseln“)

F1-Laborpraktikum SoSe 2015
21
10. 52,5 µl Resuspension Buffer über die Beads laufen lassen, dabei die Beads nicht mit der
Pipettenspitze berühren, da sie sonst an der Spitze hängen bleiben.
11. Probe aus dem Magnetständer nehmen und die Beads durch wiederholtes Auf- und
Abpipettieren resuspendieren.
12. 2 min. bei Raumtemperatur inkubieren
13. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
14. 50 µl klaren Überstand in ein neues Gefäß überführen, dabei die Beads nicht lösen.
15. Sample Purification Beads gründlich vortexen.
16. 50 µl Sample Purification Beads zur Probe geben und durch Auf- und Abpipettieren gut mischen.
17. 5 min. bei RT inkubieren
18. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
Folgende Schritte werden im Magnetständer durchgeführt:
19. 95 µl Überstand entfernen ohne die Beads zu lösen.
20. 200 µl 80 % EtOH zugeben ohne die Beads zu lösen und 30 s inkubieren.
21. Überstand entfernen ohne die Beads zu lösen und nochmals mit 200 µl EtOH waschen.
22. Verbleibenden EtOH mit 10 µl Pipette entfernen.
23. 5 min. bei RT im Magnetständer trocknen lassen. (Beads sollten nicht „bröseln“)
24. 27,5 µl Resuspension Buffer über die Beads laufen lassen, dabei die Beads nicht mit der
Pipettenspitze berühren, da sie sonst an der Spitze hängen bleiben.
25. Probe aus dem Magnetständer nehmen und die Beads durch wiederholtes Auf- und
Abpipettieren resuspendieren.
26. 2 min. bei Raumtemperatur inkubieren
27. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
28. 25 µl klaren Überstand in ein neues Gefäß überführen, dabei die Beads nicht lösen.
Möglichkeit zum Lagern bei -20°C
Enrichment
1. 5 µl PCR Primer Cocktail zur Probe geben
2. 20 µl Enhanced PCR Mix zur Probe geben und durch Auf- und Abpipettieren gut mischen.
3. Probe kurz anzentrifugieren.
4. Probe bei folgendem Programm im Thermocycler inkubieren:
95 °C für 3 min.
8 Zyklen: 98 °C für 20 s
60 °C für 15 s
72 °C für 30 s
72 °C für 5 min
Hold at 4 °C
5. Probe kurz anzentrifugieren
Aufreinigung
1. Sample Purification Beads gründlich vortexen.
2. 50 µl Sample Purification Beads zur Probe geben und durch Auf- und Abpipettieren gut mischen.
3. 5 min. bei RT inkubieren

F1-Laborpraktikum SoSe 2015
22
4. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
Folgende Schritte werden im Magnetständer durchgeführt:
5. 95 µl Überstand entfernen ohne die Beads zu lösen.
6. 200 µl 80 % EtOH zugeben ohne die Beads zu lösen und 30 s inkubieren.
7. Überstand entfernen ohne die Beads zu lösen und nochmals mit 200 µl EtOH waschen.
8. Verbleibenden EtOH mit 10 µl Pipette entfernen.
9. 5 min. bei RT im Magnetständer trocknen lassen. (Beads sollten nicht „bröseln“)
10. 32,5 µl Resuspension Buffer über die Beads laufen lassen, dabei die Beads nicht mit der
Pipettenspitze berühren, da sie sonst an der Spitze hängen bleiben.
11. Probe aus dem Magnetständer nehmen und die Beads durch wiederholtes Auf- und
Abpipettieren resuspendieren.
12. 2 min. bei Raumtemperatur inkubieren
13. Probe in den Magnetständer stellen, warten bis die Lösung klar ist.
14. 30 µl klaren Überstand in ein neues Gefäß überführen, dabei die Beads nicht lösen.
Möglichkeit zum Lagern bei -20°C
Quantifizierung der Library mit Qubit HS Assay
Der Qubit ermöglicht eine fluorometrische Konzentrationsbestimmung von Nukleinsäuren.
Verwendet wird das Qubit dsDNA HS Assay Kit, das zur Messung von DNA-Konzentrationen zwischen
10 pg/µl und 100 ng/µl geeignet ist. Ein im Qubit Reangenz enthaltender Farbstoff bindet spezifisch
an doppelsträngige DNA und kann daraufhin mit einem Fluorometer detektiert werden. Die Stärke
des Fluoreszenzsignals ist proportional zur Konzentration der DNA-Probe. Die Verwendung eines
Eichstandards ermöglicht eine exakte Konzentrationsbestimmung.
Lassen Sie alle Lösungen 30 min auf RT equilibrieren. Die Farbstoffe sollten dabei nicht direktem
Sonnenlicht ausgesetzt sein. Die Qubit-Tubes werden nur mit Handschuhen angefasst.
1. Qubit-Tubes auf dem Deckel beschriften: eines je Probe und zwei Standards (S1 und S2)
2. Qubit-Arbeitslösung herstellen:
Volumen der Arbeitslösung = [(Zahl der Proben + 1) x 199 µl] + [2 x 190 µl]
Zusammensetzung der Arbeitslösung: Qubit dsDNA HS Reagent 1:200 verdünnt in Qubit
dsDNA HS buffer
3. In die Qubit-Tubes für die Standards 190 µl Arbeitslösung vorlegen.
4. In die Qubit-Tubes für die Proben 199 µl Arbeitslösung vorlegen.
5. 10 µl der Standards zur vorgelegten Arbeitslösung pipettieren und gut vortexen.
6. 1 µl der Probe zur vorgelegten Arbeitslösung pipettieren und gut vortexen.
7. Proben 2 min. inkubieren und zügig messen.

F1-Laborpraktikum SoSe 2015
23
Qualitätskontrolle, Molekulargewichtsverteilung und Quantifizierung der Library mit Agilent
Bioanalyzer Nano-Assay
Die Messmethode des Bioanalyzers entspricht dem einer Mikrokapillargelelektrophorese. Die Proben
werden durch Anlegen einer geeigneten Spannung elektrophoretisch aufgetrennt, wobei ein sehr
sensitiver, interkalierender Fluoreszenzfarbstoff von einer Kamera detektiert wird. Die gemessene
Fluoreszenz wird gegen die Laufstrecke (in Sekunden) ausgegeben. Durch Verwendung eines
Molekulargewichtsstandards (Leiter) kann dann aus der Laufstrecke das Molekulargewicht der
einzelnen Fragmente bestimmt werden. Wie auch bei normalen Gelelektrophoresen entscheidet die
Konzentration des Gels über die Auftrennungseigenschaften. Mit dem DNA High Sensitivity Kit
können Fragmente zwischen 50 und 7000 Basenpaaren mit Quantitäten von 5-500 pg/µl detektiert
werden.
Richtiges Pipettieren: niemals an den Rand der Wells, sondern immer auf den Boden pipettieren.
Ein Gelaliquot versetzt mit dem Farbstoff (Gel-Dye-Mix) wird Ihnen bereitgestellt. Lassen Sie alle
Lösungen 30 min auf RT equilibrieren. Die Farbstoffe sollten dabei nicht direktem Sonnenlicht
ausgesetzt sein.
1. Nehmen Sie einen neuen DNA High Sensitivity Chip aus der Packung und legen Sie Ihn in die
Vorbereitungsstation.
2. Pipettieren Sie 9 µl Gel-Dye-Mix in das mit gekennzeichnete Well (weißer
Punkt in der unten aufgeführten Grafik). Bitte entnehmen Sie das Gel aus dem
Aliquot knapp unterhalb des Meniskus, da sich am Boden Schwebstoffe absetzen,
die den Lauf beeinträchtigen können.
ACHTUNG: Lesen Sie an dieser Stelle bitte die Punkte 5-7 und bereiten Sie Pipetten und Lösungen
vor, bevor Sie weiterarbeiten
3. Stellen Sie die Spritze auf 1 ml ein, schließen Sie die Vorbereitungsstation und drücken Sie die
Spritze herunter, bis diese vom Clip gehalten wird.
4. Warten Sie exakt 60 Sekunden (Timer) und lösen Sie den Clip.
5. Warten Sie 5 Sekunden und ziehen Sie dann die Spritze LANGSAM wieder in die
Ausgangsposition zurück.
6. Öffnen Sie die Vorbereitungsstation und pipettieren Sie 9 µl Gel-Dye-Mix in die
mit gekennzeichneten Wells.
7. Pipettieren Sie nun 5µl DNA High Sensitivity Marker (grün •) in die verbleibenden
12 Wells.

F1-Laborpraktikum SoSe 2015
24
8. Pipettieren Sie 1 µl der DNA High Sensitivity Ladder (gelb •) in das mit
gekennzeichnete Well.
Pipettieren Sie nun jeweils 1 µl Ihrer Proben in die 11 vorhandenen Wells und
notieren Sie die Reihenfolge. In alle unbenutzten Wells pipettieren Sie 1 µl DNA
High Sensitivity Marker (grün •).
9. Legen Sie den Chip in den beigestellten IKA Vortexer (nur eine Orientierung ist
möglich!) und starten Sie diesen.
HINWEIS: Der IKA Vortexer stoppt automatisch nach einer Minute
10. Legen Sie den Chip in den Bioanalyzer (vermeiden Sie ruckartige Bewegungen) und beginnen Sie
den Lauf innerhalb von 5 min nach Präparation des Chips durch Betätigung des Start-Knopfes
(wird grün wenn der Chip erkannt wurde).

F1-Laborpraktikum SoSe 2015
25
Theorie Teil 2: Bioinformatische Analyse von „Next-Generation Sequencing“-
Daten
Sequenzalignment
In der Bioinformatik wird unter einem Alignment der Vergleich von zwei (paarweises Alignment) oder
mehreren (multiples Alignment, MSA) Zeichenketten oder Sequenzen verstanden. Voraussetzung für
einen Vergleich ist, dass beide Sequenzen derselben Notation (Alphabet) folgen. Beide Sequenzen
werden für das Alignment untereinander gelegt und die einzelnen Stellen der Zeichenkette
miteinander verglichen. Sind beide Stellen identisch, so spricht man von einem match, sind sie
ungleich von einem mismatch. Da auch ganze Abschnitte der einen Sequenz innerhalb der anderen
fehlen können, müssen diese Stellen im Alignment berücksichtigt werden. Diese Lücken (gaps)
stellen eine große Herausforderung in der Bioinformatik dar, da sie nicht direkt erkennbar sind.
Für die Visualisierung von Sequenzalignments gibt es verschiedene
Methoden. Die einfachste ist der sogenannte Dot-Plot (Abbildung 9).
Hier werden in einer Matrix, aufgespannt durch die orthogonal
zueinander liegenden Sequenzen, mögliche matches entsprechend
eingetragen, wodurch sich spezielle Muster ergeben. Diese Muster
deuten auf verschiedene Eigenschaften hin, wie z.B. längere
Abschnitte mit Übereinstimmung (lange Diagonalen),
Wiederholungen der Sequenz (repeats, horizontale oder vertikale
wiederkehrende Diagonalen), oder Poly-Nukleotid-Abschnitte bzw.
sehr kurze Wiederholungen (dunkle Blöcke). Da es für zwei
Sequenzen alleine schon mehrere Möglichkeiten des Alignments gibt, müssen die jeweiligen
Anordnungen einzeln bewertet werden. Hierbei vergibt man für matches, mismatches sowie gaps
Punkte, welche zusammengezählt am Ende zu einem score führen. Dieser stellt ein Maß für die Güte
des Alignments dar. Für die Berechnung des scores werden scoring-Matrizen benutzt. Diese geben
für jedes mögliche Alignment einen Wert aus. Für Nukleotidsequenzen sind diese Matrizen sehr
einfach gehalten. Auf Ebene der Aminosäuren gibt es komplexer aufgebaute Matrizen, da ein
Basenaustausch nicht zwangsläufig auch eine Veränderung der Aminosäuresequenz zufolge hat.
Sogenannte Substitutionsmatrizen spiegeln hier die Wahrscheinlichkeit einer Mutation durch
entsprechend angepasste scoring-Werte wider. (Dayhoff et al. 1978; Henikoff & Henikoff 1992).
Assemblierungsmethoden MSAs bilden die Grundlage der ersten Assemblierungsmethoden. Mit dem Aufkommen der ersten
Sequenziertechniken war es nötig, die Ursprungssequenz(en), welche durch Scherung des
genetischen Materials zerstört wurden, wieder zu rekonstruieren. Das Scheren - ob chemisch oder
mechanisch - bildet bis heute einen Bestandteil der Herstellung von Sequenzbibliotheken, da keine
Technik über eine Leseweite von mehr als einer Kilobase (kb) verfügt und neuere Ansätze,
sogenannte next-generation sequencing - Methoden noch kürzere Sequenzschnipsel (Reads) liefern.
Durch Amplifizierungschritte vor der Scherung entstehen Reads, welche sich in der Ursprungsequenz
überlagern und somit einen Überlapp (overlap) besitzen. Diesen macht man sich im
Assemblierungsprozess zunutze, indem man durch Sequenzalignments der Randregionen größere
Abbildung 9 Dot Plot (http://upload.wikimedia.org/wikipedia/commons/3/33/Zinc-finger-dot-plot.png)

F1-Laborpraktikum SoSe 2015
26
Sequenzstücke produziert. Im optimalen Fall kann man so die Ursprungssequenz rekonstruieren.
Erschwert wird das Ganze durch eine, jeder Technik zugrunde liegende Fehlerrate, welche z.B. zu
falschen Basen innerhalb der Reads führt.
Erste Assemblierungsalgorithmen aus den 70er und 80er Jahren beruhen auf der Tatsache, dass
relativ wenige, lange Reads aus der damals am weitesten verbreiteten Technik, der Sanger-
Sequenzierung vorliegen. Sogenannte greedy-Algorithmen, die schnelle, suboptimale Lösungen
lieferten, waren die ersten Ansätze für die Implementierung solcher Assemblierungsstrategien.
Obwohl die Hard- und Softwareindustrie in den letzten Jahrzehnten große Fortschritte gemacht hat,
konnten die Algorithmen mit den immer riesiger werdenden Datenmengen nicht mithalten, da ihre
Rechenzeit mit dem Wachstum der Daten exponentiell anstieg. Neuere Strategien bedienen sich
Methoden der dynamischen Programmierung, heuristischen Ansätzen sowie mathematischen
Methoden aus den Graphentheorien.
BLAST
Einer der bekanntesten Algorithmen, welcher sich dieser Methoden bedient, ist der BLAST-
Algorithmus („Basic Local Alignment Search Tool“, Altschul et. al. 1990). Er basiert auf der Annahme,
dass Sequenzen mit großer Ähnlichkeit kurze, identische Abschnitte besitzen.
Abbildung 10 BLAST-Algorithmus
Durch das Erstellen einer Liste mit kurzen Teilsequenzen (words) aus der Anfrage- (query) und
Zielsequenz (subject) und durch beidseitiges Verlängern dieser Übereinstimmungen werden so
gezielt kurze, lokale Alignments zwischen nahe verwandten Sequenzen erzeugt. Diese geben
Aufschluss über ihre Funktion oder die Beziehung zueinander. Mithilfe dieser Technik wird die Anzahl
der Sequenzvergleiche deutlich reduziert und somit eine Lösung in endlicher Zeit möglich gemacht.

F1-Laborpraktikum SoSe 2015
27
Obwohl der BLAST-Algorithmus keine Assemblierungsmethode ist, findet man diese Technik in vielen
neueren kommerziellen Assemblierungsprogrammen (DNAStar, SeqMan, NGEN etc.) wieder. So
werden im Vorfeld schon gezielt Sequenzen für einen näheren Vergleich (Alignment) aufgrund
identischer Subsequenzen in Betracht gezogen.
Trotz dieser heuristischen Ansätze sind auch neuere Implementierungen bisweilen mit der Menge
der Daten überfordert, da ein simples Alignment zweier Sequenzen sehr speicherintensiv sein kann.
So wurden in den letzten Jahren verstärkt Ansätze aus den Graphentheorien zu Rate gezogen. Vor
allem Sequenzierungen aus NGS-Projekten erfordern eine veränderte Denkweise für
Assemblierungsstrategien. Da hier mehr qualitativ hochwertigere, kürzere Reads mit einer höheren
Abdeckung (coverage) der Nukleotidpositionen erzeugt werden, rückt der Fokus weg von
rechenintensiven Alignments einzelner Sequenzpaare hin zu speicherentlastenden Vergleichen von
identischen Subsequenzen auf Nukleotidebene. Unterschiede, wie Punktmutationen (SNPs) spiegeln
sich hier in Aufgabelung der Graphen wider, sich wiederholende Abschnitte in Blasen innerhalb des
Graphen. Einer der erfolgreichsten dieser Methode ist der Ansatz des De-Bruijn-Graphen.
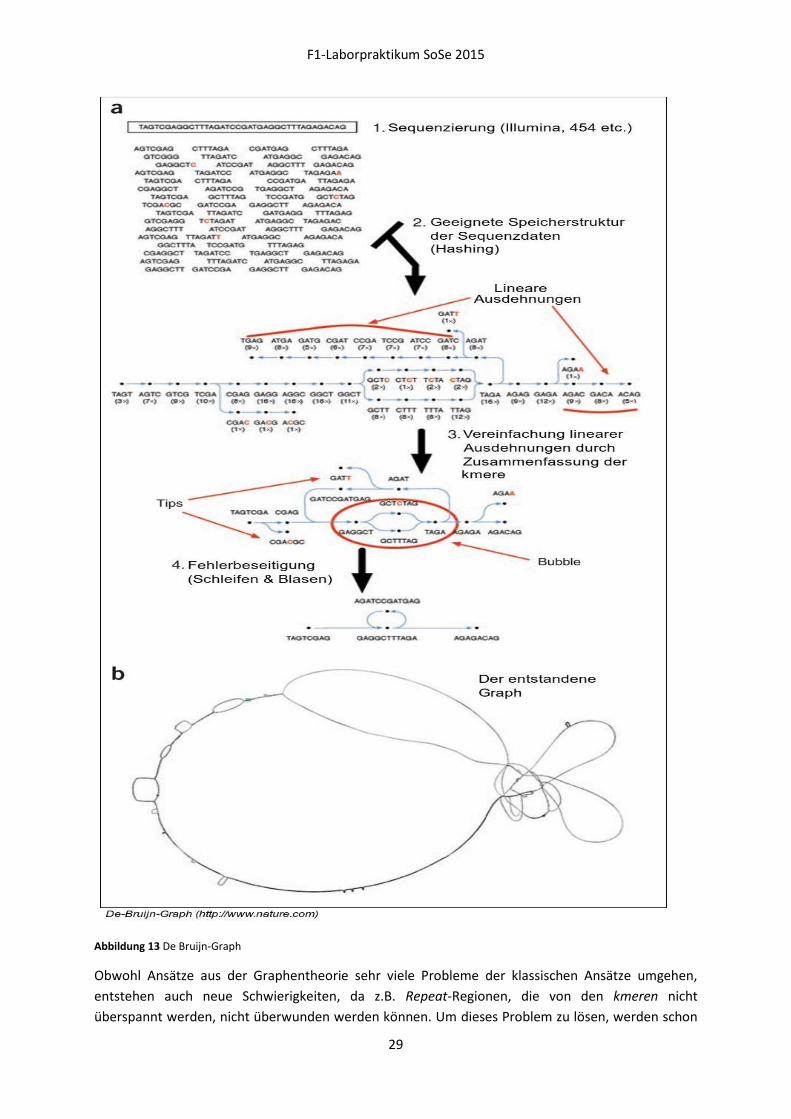
De-Bruijn-Graph (DBG)
Die Grundlage des DBG sind Datensätze, welche:
- Eine hohe Abdeckung der Ursprungssequenz besitzen,
- eindeutig in ihrer Notation sind (kein IUPAC-Code),
- Reads gleicher Länge besitzen (wenn möglich).
Da der DBG keine Alignments im klassischen Sinne durchführt (und somit keine gaps einbaut), wird
der Überlapp durch eine Präprozessierung der Rohdaten gewährleistet. Dieser 'Rückwärtsschritt'
besteht in der Zerlegung der Eingangsdaten in eine Menge von Subsequenzen (kmers). Diese
Zerlegung geschieht in nukleotidlangen Schritten, wodurch sich die kmers eines einzelnen Reads
überlappen und zwar mit der Länge kmer–1. Obwohl dieser Schritt eine Amplifizierung der
Datenmenge bedeutet, kann durch geschickte Implementierung die Menge des Speichers reduziert
werden, da Subsequenzen, die öfters auftreten, nur einmal gespeichert werden müssen.
Der Ablauf des Algorithmus sieht nun wie folgt aus:
1. Beginne bei einem beliebigen kmer.
2. Schneide am 5' Ende ein Nukleotid ab.
3. Erzeuge 4 mögliche Nachbar-kmere, indem am 3' Ende einzeln alle 4 Nukleotide anhängt werden.
4. Wiederhole die Schritte 1-3 mit dem Ursprungs-kmer mit dem 5' und dem 3' Ende vertauscht.

F1-Laborpraktikum SoSe 2015
28
Abbildung 11 Mögliche Nachbar-kmere
Dadurch erhält man für ein einzelnes kmer 8 mögliche Nachbar-kmere (Fehler! Verweisquelle konnte
nicht gefunden werden.). Für jedes Nachbar-kmer, das im Datenpool existiert, wird im Graphen eine
Kante zwischen den beiden kmeren gezogen. Der so entstandene Graph zeichnet sich durch eine
Vielzahl komplexer Strukturen (Blasen, Schleifen) aus, welche auf verschiedene Charakteristiken der
Ursprungssequenz wie SNPs oder repeats hindeuten. Zur Auflösung dieser Strukturen werden Meta-
Informationen über die Abdeckung oder Qualität der Sequenzdaten hinzugezogen. Durch den Ablauf
des Graphen können nun im letzten Schritt die mögliche(n) Ursprungssequenz(en) ermittelt werden.
Abbildung 12 Schematische Darstellung einer Assemblierung mithilfe des De Bruijn Graphen
F1-Laborpraktikum SoSe 2015
29
Abbildung 13 De Bruijn-Graph
Obwohl Ansätze aus der Graphentheorie sehr viele Probleme der klassischen Ansätze umgehen,
entstehen auch neue Schwierigkeiten, da z.B. Repeat-Regionen, die von den kmeren nicht
überspannt werden, nicht überwunden werden können. Um dieses Problem zu lösen, werden schon

F1-Laborpraktikum SoSe 2015
30
während der Sequenzierung neue Techniken angewandt, welche Datensätze mit read-Paaren
erzeugen (paired-end-Reads, mate-pairs). Der Abstand dieser Paare innerhalb der Ursprungssequenz
ist bekannt und kann somit im Assemblierungsprozess berücksichtigt werden.
Arbeitsablauf Sequenzverarbeitung
Aufgrund der immer stärker wachsenden Menge der Daten werden im Institut für Molekulargenetik
gezielt Methoden der parallelen Datenverarbeitung etabliert, sei es durch Nutzung neuer, bereits
parallel implementierter Algorithmen oder durch parallele Verarbeitung auf Ebene der voneinander
unabhängigen Eingangsdaten. Als Beispiel dient hierfür die Parallelisierung des Blast-Prozesses.
Die Blast-Option des NCBI (National Center for Biotechnology Information) bietet nur beschränkte bis
keine Möglichkeiten der Hochdurchsatzdatenverarbeitung, da Sequenzen einzeln eingegeben
werden müssen oder nur kleinere Datensätze hochgeladen werden können. Durch die Verlagerung
der Berechnungen auf lokale Desktop-PCs, wird die Verarbeitung der Daten zwar unabhängig von
Aspekten wie Serverauslastung und Netzwerkverbindungen, jedoch bildet der PC selbst aufgrund der
(z. Zt.) geringen Ausstattung den Flaschenhals des Verarbeitungsprozesses. Des Weiteren müssen für
lokale Blast-Suchen entsprechende Datenbanken zuerst heruntergeladen oder eigene
Sequenzdatenbanken erzeugt werden.
Möglichkeiten der Parallelisierung Es gibt mehrere Ansätze der Parallelisierung der Blast-Suche. Durch die Nutzung von Multicore-
Systemen oder HPC (High-Performance-Cluster, vernetzte Computer mit zentraler Steuerung und
Verwaltung durch entsprechende Software) ist es möglich, voneinander unabhängige Aufgaben
parallel zu verarbeiten, deren Ergebnisse zu speichern und am Ende wieder zusammenzuführen.
Durch gezielte parallel implementierte Algorithmen ist es möglich auf Softwareebene die vorhandene
Hardware maximal auszunutzen und somit eine (im optimalen Falle) lineare Skalierung zu erzielen.
Ein Ansatz für paralleles Arbeiten mit dem Blast-Prozess ist die Aufteilung der Datenbank. Dies
erfordert aber eine intensive Kommunikation zwischen den Prozessen, da dieselbe Sequenz
gleichzeitig in mehreren Datenbankuntereinheiten gesucht wird. Durch Aufteilung der Datenbank
wird darüber hinaus die Berechnung des E-value komplizierter, da dieser von der Datenbankgröße
abhängig ist. Ein anderer Ansatz ist die Aufteilung auf Sequenzebene. Das bedeutet, dass die
Eingangsdaten im Vorfeld in mehrere Datensätze aufgeteilt werden, um diese unabhängig
voneinander auf getrennten Systemen zu verarbeiten. Der Vorteil dieses Prozesses ist, dass die
Statistik nicht verändert wird. Nachteil dieser Art der Prozessierung ist allerdings, dass die Datenbank
für jeden einzelnen Prozess geladen werden muss und somit eine erhöhte Auslastung des
Arbeitsspeichers erfolgt.

F1-Laborpraktikum SoSe 2015
31
Praxis Teil 2: Bioinformatische Analyse von „NGS“-Daten
In diesem Teil des Praktikums analysieren Sie Ihre in Teil 1 gewonnenen Genom-Daten im Hinblick
auf folgende Fragestellungen:
Vergleichen Sie Ihre Daten mit der bekannten geschlechtsbestimmenden Genomregion (SDR)
aus C. thummi bzw. C. piger.
Können neue Gene identifiziert werden?
Stimmt die Genabfolge bzw. deren Orientierung mit den Genen aus der SDR von C. thummi
bzw. C. piger überein?
Identifizieren Sie mögliche strukturelle Variationen bzw. repetitive Sequenzen.
Vergleichen Sie die SDR hinsichtlich des Geschlechts (♀ / ♂) auf Unterschiede.
Vergleichen Sie die SDR aller Arten (C. thummi, C. piger, C. luridus und
C. annularius)miteinander.
Dabei sollen Sie verschiedene Analyseverfahren wie Mapping, „Read Count“, DotPlot, BLAST, „Large
Gap Read Mapping“ und Assembly mit verschiedenen Programmen kennen lernen. Zentrale, aber
nicht ausschließliche, Analysesoftware soll dabei die schon letzte Woche benutzte „CLC Genomics
Workbench“ sein.

Sequenzierung und Basecalling
Da auf der HiSeq Flow Cell lediglich 8 seperate Reaktionsräume, sogenannte Lanes vorhanden sind,
wurde zusätzlich zur eigentlichen Sequenzierung ein Index Read durchgeführt. Schon bei der
Konstruktion der Libraries wurden leicht variierte Adapter eingesetzt. Diese tragen noch vor der
Bindestelle für den Sequenzierprimer eine 6 Basenpaar lange Erkennungssequenz. Im Index Read
werden ausgehend vom revers-komplementären Sequenzierungsprimer diese 6 Nukleotide
ausgelesen. Dies wird notwendig, wenn verschiedene Libraries in einer Lane zusammen sequenziert
werden. Dies kann unterschiedliche Gründe haben. Beispielsweise benötigen viele Projekte nicht die
Masse an Daten einer kompletten Lane, oder - wie in diesem Fall - der Organismus besitzt einen
extremen A/T oder G/C –Gehalt. Nach dem Mischen der Libraries in den gewünschten Verhältnissen,
werden diese auf eine Endkonzentration von ca. 7 pM gebracht. Erfahrungsgemäß sollte diese
Konzentration eine Clusterdichte auf der Flow Cell-Oberfläche von ca. 450.000 bis 500.000 Cluster
pro mm2 ergeben. Mit dieser nahezu optimalen Clusterdichte sind ca. 80-100 Mio. Reads pro Lane
möglich.
Bei der Sequenzierung wird die Flow Cell zunächst in 16 Analysebereiche, sogenannte „Tiles“,
unterteilt. Dies sind zwei 8er Reihen mit Bildabschnitten, die zusammen die gesamte
Abbildung 14 Analysepipeline – vom Bild zur Sequenz

F1-Laborpraktikum SoSe 2015
33
Oberfläche der Flow Cell abdecken. Ausgehend von dem Bild im TIFF-Format jedes einzelnen Tiles
werden die Clusterintensitäten im „Firecrest“-Modul der RTA („Real Time Analysis“ Software)
verarbeitet (Abbildung 13). Die daraus resultierenden CIF-Dateien („cluster intensity files“) werden
dem „Bustard“-Modul zum eigentlichen Basecalling übergeben. Dieses erstellt zunächst binäre
Sequenz-Dateien, die BCL-Dateien („basecall files“). Diese werden separat in das Standardformat für
NGS-Daten, das FASTQ-Format, gebracht.
Datenformat FASTQ
Wichtigstes Dateiformat für NGS-Daten ist das FASTQ-Format. Dieses besteht immer aus 4 Zeilen pro
Sequenz (Abbildung 15). Die durch ein „@“ eingeleitete erste und die mit einem „+“ beginnende
dritte Zeile stellen dabei den Namen der Sequenz dar. In der zweiten Zeile folgt die Basensequenz. Im
Fall von „+“ folgen in der vierten Zeile die Qualitätswerte („ Quality Scores“) für jede einzelne Base
im ASCII-Code.
Abbildung 15 Aufbau des FASTQ-Formats
Im Namen der Sequenz finden sich weitere Informationen kodiert. Diese reichen vom Namen des
Geräts, auf dem sequenziert wurde bis zur Koordinate des Clusters auf der Flow Cell (Abbildung 15).
Abbildung 16 Aufbau des Sequenznamens der ersten und dritten Zeile
F1-Laborpraktikum SoSe 2015
34
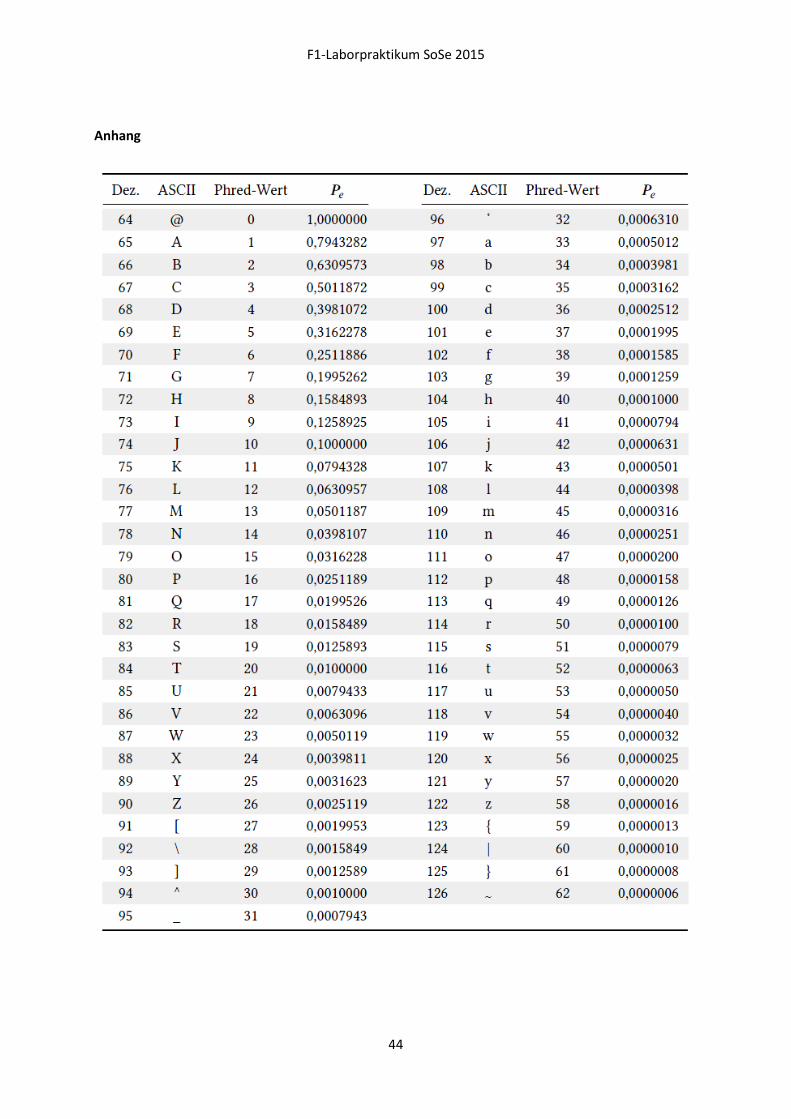
Quality Scores
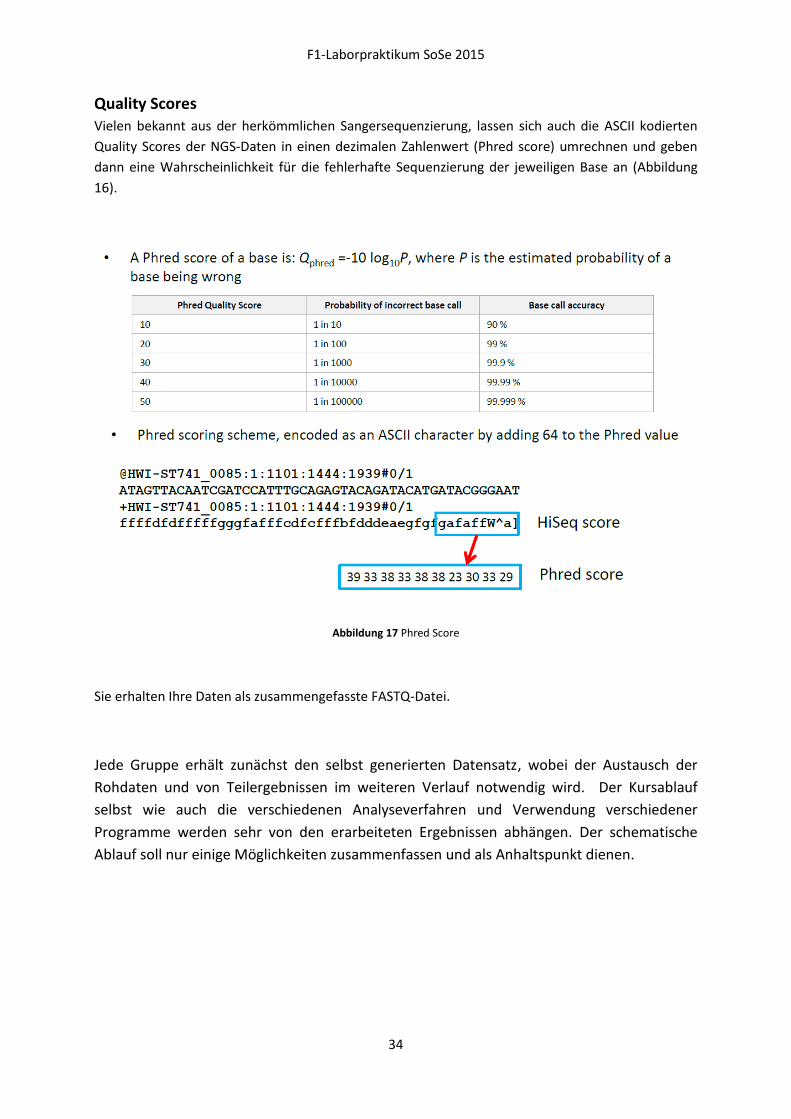
Vielen bekannt aus der herkömmlichen Sangersequenzierung, lassen sich auch die ASCII kodierten
Quality Scores der NGS-Daten in einen dezimalen Zahlenwert (Phred score) umrechnen und geben
dann eine Wahrscheinlichkeit für die fehlerhafte Sequenzierung der jeweiligen Base an (Abbildung
16).
Abbildung 17 Phred Score
Sie erhalten Ihre Daten als zusammengefasste FASTQ-Datei.
Jede Gruppe erhält zunächst den selbst generierten Datensatz, wobei der Austausch der
Rohdaten und von Teilergebnissen im weiteren Verlauf notwendig wird. Der Kursablauf
selbst wie auch die verschiedenen Analyseverfahren und Verwendung verschiedener
Programme werden sehr von den erarbeiteten Ergebnissen abhängen. Der schematische
Ablauf soll nur einige Möglichkeiten zusammenfassen und als Anhaltspunkt dienen.
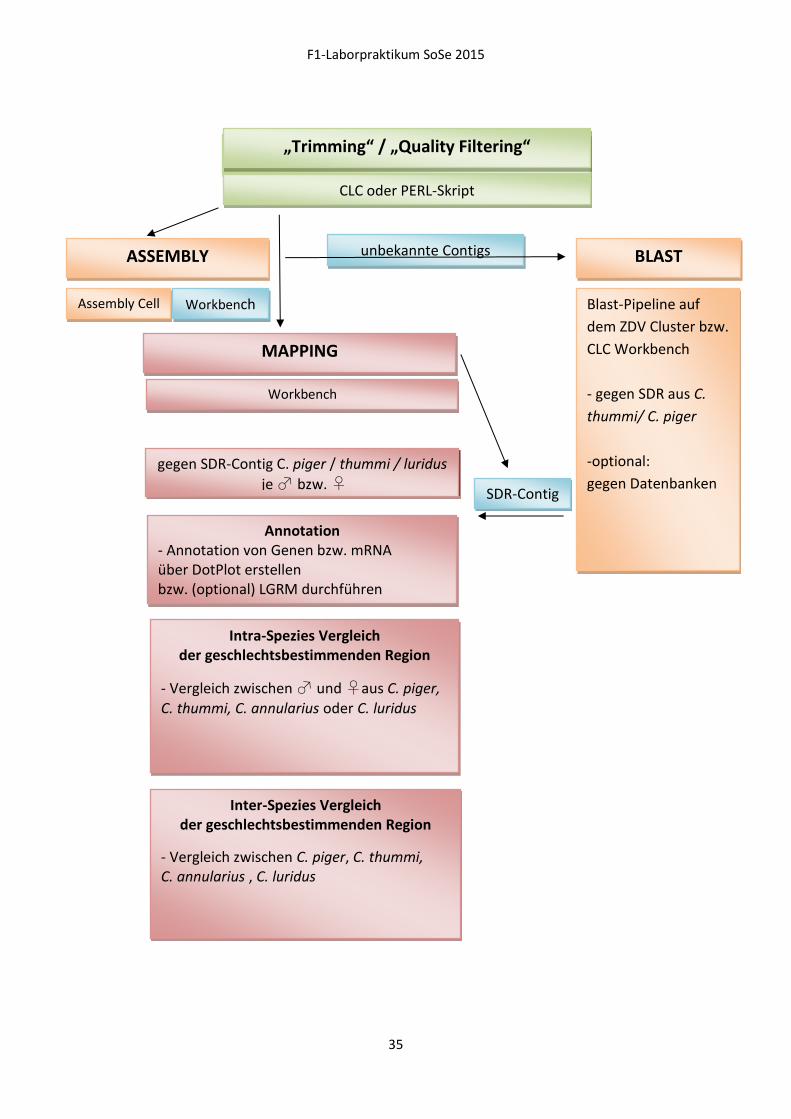
F1-Laborpraktikum SoSe 2015
35
FASTQ „Trimming“ / „Quality Filtering“
CLC oder PERL-Skript
ASSEMBLY
MAPPING
BLAST
Assembly Cell Workbench
Workbench
gegen SDR-Contig C. piger / thummi / luridus
je ♂ bzw. ♀
Annotation - Annotation von Genen bzw. mRNA über DotPlot erstellen bzw. (optional) LGRM durchführen
Intra-Spezies Vergleich der geschlechtsbestimmenden Region
- Vergleich zwischen ♂ und ♀aus C. piger, C. thummi, C. annularius oder C. luridus
Inter-Spezies Vergleich der geschlechtsbestimmenden Region
- Vergleich zwischen C. piger, C. thummi, C. annularius , C. luridus
Blast-Pipeline auf
dem ZDV Cluster bzw.
CLC Workbench
- gegen SDR aus C.
thummi/ C. piger
-optional:
gegen Datenbanken
unbekannte Contigs
SDR-Contig

F1-Laborpraktikum SoSe 2015
36
Bioinformatische Analyse der „Next-Generation Sequencing“ Daten mit Hilfe der
„CLC Genomics Workbench“
Laden Sie sich bitte das ausführliche Manual der „CLC Genomics Workbench“ unter dem folgenden
Link herunter, da das Skript auf verschiedene Abschnitte innerhalb dieser Beschreibung verweisen
wird:
http://www.clcbio.com/files/usermanuals/CLC_Genomics_Workbench_User_Manual.pdf
Die Analyse beinhaltet folgende Schritte:
Import der „Next-Generation Sequencing“-Daten
„Trimming“ der Reads
„Mapping“ der Reads an eine genomische Referenzsequenz
Vergleichende Analyse der „gemappten“ Reads zwischen weiblichen und männlichen
Genomen sowie zwischen den unterschiedlichen Arten (C. piger, C. luridus, C. annularius)
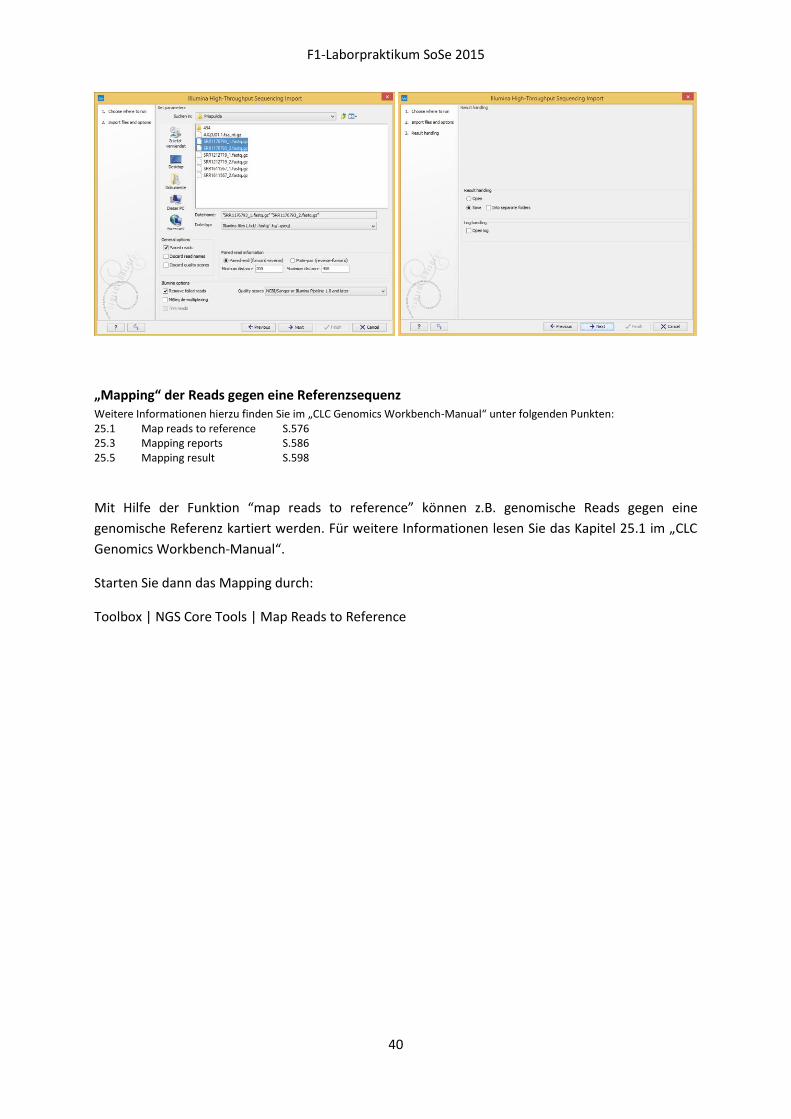
Import der „Next-Generation Sequencing“-Daten Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 1.6 When the program is installed: Getting started S.37 6.1 Standard import S.97 6.3 Import high-throughput sequencing data S.101
Öffnen Sie die „CLC Genomics Workbench 8“ durch die Verknüpfung auf Ihrem Desktop und machen
Sie sich mit der Softwareoberfläche vertraut, die im Wesentlichen aus 3 Bereichen besteht:
Oben links: die „Navigation Area“ zeigt alle importierten Daten an, die der Software zur
Verarbeitung zur Verfügung stehen
Unten links: in der „Toolbox“ befindet sich die Analysesoftware, durch Auswahl des Reiters
„Processes“ ist es später möglich, den Fortschritt einzelner Analysen nachzuverfolgen
Rechts: in der „View Area“ werden Daten durch eine grafische Oberfläche dargestellt
Erstellen Sie sich zunächst einen eigenen Ordner innerhalb der „Navigation Area“, indem Sie auf dem
Lokalen Datenträger (D:) einen Ordner „F1CLC“ erstellen und diesen in der Funktionsleiste der
„Navigation Area“ durch folgende Auswahl hinzufügen:
Add file location
Laden Sie Ihre Illumina-qseq-Dateien über den folgenden Pfad oder den Button „Import“
„Illumina“ der oberen Funktionsleiste in Ihren neu erstellten Ordner ein:
File | Import | Illumina

F1-Laborpraktikum SoSe 2015
37
Beachten Sie hierbei, dass es sich eventuell um Reads ohne „paired-end“-Information handelt.
Verwerfen Sie beim Import die Reads ohne erfolgreiche Sequenzierungsreaktion. Erhalten Sie dabei
jedoch die Informationen über die automatisch ausgelesenen Qualitätsscores sowie die Readnamen.
Die importierten Sequenzen werden jetzt in der „Navigation Area“ in einer Liste zusammengefasst,
die Sie durch einen Doppelklick in der „View area“ öffnen können. Die einzelnen Reads enthalten
außer der eigentlichen Sequenz auch die Information über die Sequenzierungsqualität der einzelnen
Basen („Quality score“), die für die weitere Bearbeitung der Reads genutzt werden kann.
Die Reads enthalten noch Sequenzabschnitte, die bei der Assemblierung zu einem nicht optimalen
oder sogar falschen Ergebnis führen könnten.
Erkennen Sie in der Liste Ihrer Reads solche Abschnitte, und wenn, welche sind dies und
wodurch werden diese verursacht?
Welche Auswirkungen hätten diese Abschnitte auf die Assemblierung?
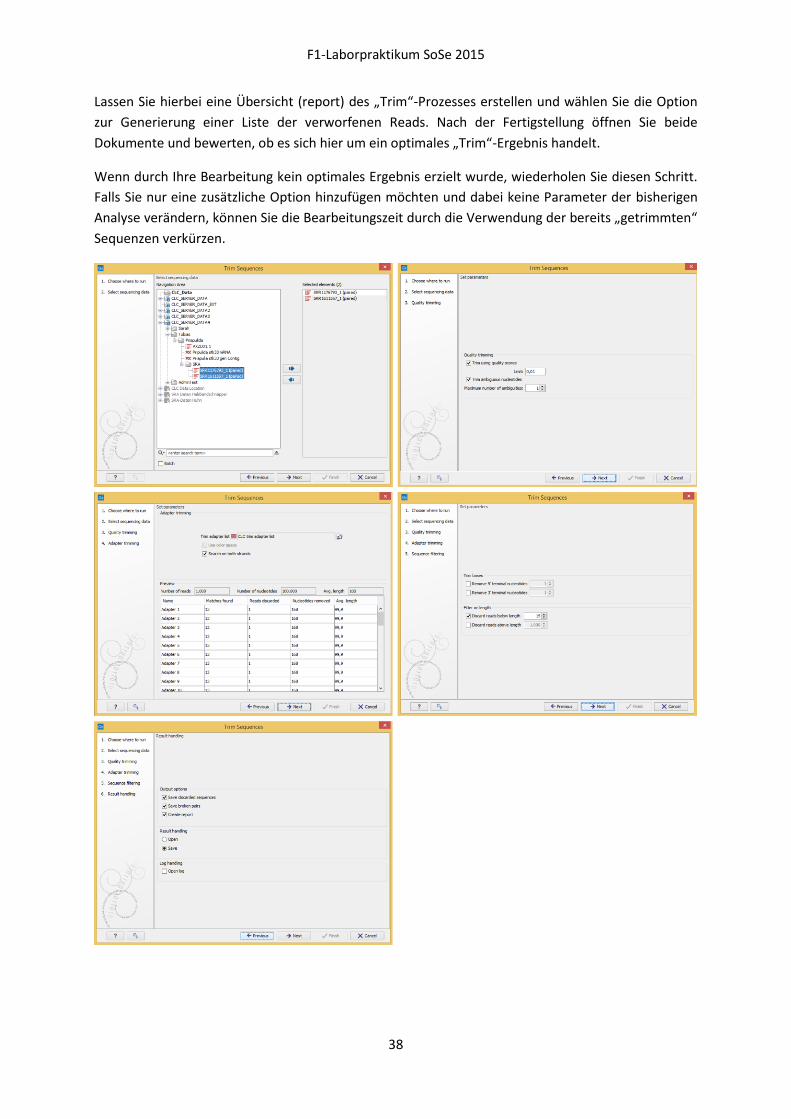
„Trimmen“ der Reads Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 23.1 Trim sequences S.517
Die „CLC Genomics Workbench“ beinhaltet unterschiedliche Möglichkeiten, Reads auf den optimalen
Bereich für eine Weiterverarbeitung in einer Assemblierung zu verkürzen:
Bewertung der Qualitätsangaben einzelner Basen
Erkennung uneindeutiger Basenfolgen
Entfernung der Adaptersequenzen (können variieren)
o UNIV_adapter:
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT
o TAG_adapter:
CAAGCAGAAGACGGCATACGAGAT*6bpINDEX*GTGACTGGAGTTCAGACGTGTGCTCTTCCGATC
Eliminierung einer spezifischen Anzahl von Basen am 5’- bzw. 3’-Ende
Ausschluss einzelner Reads aufgrund von Längenbeschränkungen
Lesen Sie Kapitel 23.1 im „CLC Genomics Workbench-Manual“ und bewerten Sie dabei, welche
Bereiche Ihrer Reads im „Trimm“-Prozess berücksichtigt werden müssen und welche
Parametereinstellungen Sie hierfür verwenden.
Starten Sie dann den „Trim“-Prozess durch:
Toolbox | NGS Core Tools | Trim Sequences
F1-Laborpraktikum SoSe 2015
38
Lassen Sie hierbei eine Übersicht (report) des „Trim“-Prozesses erstellen und wählen Sie die Option
zur Generierung einer Liste der verworfenen Reads. Nach der Fertigstellung öffnen Sie beide
Dokumente und bewerten, ob es sich hier um ein optimales „Trim“-Ergebnis handelt.
Wenn durch Ihre Bearbeitung kein optimales Ergebnis erzielt wurde, wiederholen Sie diesen Schritt.
Falls Sie nur eine zusätzliche Option hinzufügen möchten und dabei keine Parameter der bisherigen
Analyse verändern, können Sie die Bearbeitungszeit durch die Verwendung der bereits „getrimmten“
Sequenzen verkürzen.

F1-Laborpraktikum SoSe 2015
39
Assemblierung der Reads Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 29.1 De novo assembly S.839 25.1 Map reads to reference S.576 25.3 Mapping reports S.586 25.5 Mapping result S.598
Folgende für unsere Fragestellung nutzbare Möglichkeiten der Assemblierung sind im
Programmpaket „CLC Genomics Workbench“ implementiert:
De novo-Assemblierung: Erstellung von „Contigs“ durch Assemblierung aller Reads nach „De
Bruijn Graphen“, die Reads werden danach in den „Contig“-Referenzsequenzen abgebildet
(„gemapped“)
Abbildung/Kartierung der Reads gegen eine genomische Referenzsequenz („Map reads to
reference“).
Abbildung/Kartierung der (transkriptomischen) Reads gegen eine genomische
Referenzsequenz („Large gap read mapping“)
De novo-Assemblierung Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 29.1 De novo assembly S. 839
Das de novo Assembly der Genomdaten wird durch die „CLC Genomics Workbench“ mit Hilfe des „De
Bruijn Graphen“ berechnet. Das Ziel ist dabei, aus den relativ kurzen Reads größere genomische
Contigs zusammenzusetzen.
Durch die BLAST-Analyse gegen Datenbanken ist es möglich, die zusammengesetzten Contigs zu
charakterisieren.
Für eine Einführung in die de novo-Assemblierung führen Sie das Tutorial „De novo assembly und
BLAST“ (siehe „Help“) durch und informieren sich über die verschiedenen Parameter im „CLC
Genomics Workbench-Manual“.
Starten Sie dann den „Trim“-Prozess durch:
Toolbox | DeNovo Sequencing | DeNovo Assembly
F1-Laborpraktikum SoSe 2015
40
„Mapping“ der Reads gegen eine Referenzsequenz Weitere Informationen hierzu finden Sie im „CLC Genomics Workbench-Manual“ unter folgenden Punkten: 25.1 Map reads to reference S.576 25.3 Mapping reports S.586 25.5 Mapping result S.598
Mit Hilfe der Funktion “map reads to reference” können z.B. genomische Reads gegen eine
genomische Referenz kartiert werden. Für weitere Informationen lesen Sie das Kapitel 25.1 im „CLC
Genomics Workbench-Manual“.
Starten Sie dann das Mapping durch:
Toolbox | NGS Core Tools | Map Reads to Reference

F1-Laborpraktikum SoSe 2015
41
„Large Gap Read Mapping“ Weitere Informationen hierzu finden Sie im „CLC User-Manual“ unter folgenden Punkten: Transcript Discovery Plugin 2.0 2. Map reads to reference S.5
Das Large Gap Read Mapping ermöglicht Ihnen das Mappen von transkriptomischen Reads gegen
eine genomische Referenz. Da in Transkriptomdaten keine Informationen über die Intronsequenzen
vorhanden sind, muss der Algorithmus das Einfügen langer Lücken (gaps) gewährleisten, sodass die
Introns überspannt werden können. Das „Large Gap Plug-in“ der „CLC Genomics Workbench“
erreicht dies durch folgende Schritte:
Für jeden Read wird der beste „match“ in der Referenzsequenz ermittelt („seed“ Segment)
Wenn der „match“ den definierten Parametern entspricht, wird die passende Region auf der
Referenzsequenz „gemappt“ und die nicht alignierten Enden werden bei ausreichender
Länge (>17 bp) erneut assembliert („non seed“ Segment)
Dabei sind zwei zusätzliche Parameter zu beachten:
Die maximale Anzahl der Treffer für ein Segment: wenn ein „non seed“ Segment diese
Anzahl überschreitet, wird der Read als „nicht gemappt“ klassifiziert

F1-Laborpraktikum SoSe 2015
42
Maximale Distanz zwischen „seed“ Segment und „non seed“ Segment. Treffer, die eine
größere Distanz haben, werden verworfen.
Alternative Methoden zu dieser „Seed-extend“-Methode (b) nutzen den „Exon-first“-Ansatz (a), der
erst alle perfekten Übereinstimmungen zur Referenzsequenz mappt und dann die Reads untersucht,
die sich nur zum Teil mit der Referenz decken (Abbildung 17). Diese werden dann geteilt und den
einzelnen Exon-Bereichen zugeordnet. Daraus ergibt sich das Problem, dass Reads von Genen mit
einer Pseudogen-Kopie im Genom besser am Pseudogen mappen, wenn diese durch eine mRNA als
Intermediat entstanden ist.
Abbildung 17: Vergleich zwischen Exon-first- und Seed-extend-Ansatz
Sie starten das „Mapping“ durch:
Toolbox | Transcriptomic Analysis | RNA-Seq Analysis | Large Gap Read Mapping

F1-Laborpraktikum SoSe 2015
43
F1-Laborpraktikum SoSe 2015
44
Anhang










![Domestikation kurz.ppt [Kompatibilitätsmodus] · Gen Chromosom Zelle Organ Körper Familie Population Molekulargenetik Zellgenetik Immunogenetik Biochemische Genetik Populationsgenetik](https://img.pdfslide.org/doc/110x75/5d4d126988c993b66c8bacad/domestikation-kurzppt-kompatibilitaetsmodus-gen-chromosom-zelle-organ-koerper.jpg)


















