Embed Size (px)
Citation preview

schlechte verbesserteLebensqualität
changing haemophilia®
PATTARAKON POOLCHAIThailand
Hämophilie A
Die wichtigsten Fragen zu Hämophilie und HemmkörperPatientenbroschüre
Hämophilie verändern. Lebensqualität verbessern.

2
Die wichtigsten Fragen zu Hämophilie und Hämophilie und HemmkörperDiese Broschüre richtet sich an Menschen mit Hämophilie, bei denen das Immunsystem Antikörper (Hemmkörper) gegen die therapeutischen Gerinnungsfaktorkonzentrate gebildet hat.
Für Patienten mit Hämophilie und Hemmkörpern müssen individuelle Behandlungsstrategien entwickelt werden, die in der Akutsituation, aber auch langfristig greifen. Hierbei steht für die Akutbehandlung der schnelle Blutungsstopp und für die Langzeitstrategie die Hemmkörperentfernung im Vordergrund.Diese Broschüre soll helfen, die häufigsten Fragen von Patienten und ihren Angehörigen zur Hämophilie und Hemmkörper zu beantworten.
Darüber hinaus steht Ihnen Ihr behandelnder Arzt gerne zur Verfügung.
Wir danken Frau Dr. med. Ivonne Wieland, Medizinische Hochschule Hannover, Pädiatrische Hämatologie und Onkologie, Carl-Neuberg-Str. 1, 30625 Hannover, unter deren Federführung die vorliegende Broschüre entstanden ist.
Name des Arztes
Krankenhaus
Telefonnummer
Stempel

3
Inhaltsverzeichnis
Angeborene Hämophilie .................................................. 4Wie funktioniert Blutgerinnung? 4Was ist angeborene Hämophilie? 4Was sind die Folgen? 4Wie häufig kommt die Erkrankung vor? 4
Vererbung ........................................................................ 6Wie wird Hämophilie vererbt und warum sind Jungenbetroffen? 6Warum wird von der „Krankheit der Könige“ gesprochen? 7
Diagnose .......................................................................... 8Welche Rolle spielt die Krankheitsvorgeschichte? 8Wie wird die Diagnose bestätigt? 8 Aktivität der Gerinnungsfaktoren bestimmt die Schwereder Symptome 8
Symptome und Schweregrade ....................................... 10Was sind typische Krankheitszeichen? 10Welcher Schweregrad macht welche Blutungsneigung? 10Was ist sonst zu beachten? 13
Therapie ......................................................................... 14Welche Behandlungsmöglichkeiten gibt es? 14Wie sieht die Alltagsbewältigung aus? 14
Hemmkörper .................................................................. 16Was sind Hemmkörper? 16Wann treten Hemmkörper auf? 17 Wie werden Hemmkörper entdeckt? 17Welche Hemmkörper gibt es? 18Wie werden Hemmkörper behandelt? 19
Nützliche Adressen ........................................................ 22 Referenzen ..................................................................... 23

4
Angeborene Hämophilie
Wie funktioniert Blutgerinnung? Wenn man irgendwo eine Wunde hat, blutet es. Damit die Blutung stoppt, ist die Blutgerinnung erforderlich. Zuerst kommen die Blutplättchen (Thrombozyten). Sie lagern sich in der Wunde an und verschließen sie. Dabei werden sie vom von-Willebrand-Faktor miteinander und mit der Gefäßwand verbunden. Durch die Aktivierung der Blutplättchen werden 13 andere Gerinnungsfaktoren aktiviert, wodurch am Ende ein festes Netz um den Plättchenthrombus gebildet wird: Die Blutung steht und die Wunde kann heilen.
Was ist angeborene Hämophilie?Als angeborene Hämophilie wird eine erbliche Form von Blutge rinnungsstörungen bezeichnet, bei der die Gerinnungs-faktoren (mit der Nummer) VIII oder IX fehlen. Durch Veränderun-gen in den Genen, die für die Bildung der Gerinnungsfaktoren verantwortlich sind, wird zu wenig oder gar kein Gerinnungs-faktor gebildet. Die Ursache ist ein Mangel an für die Gerinnung wichtigen Eiweißmolekülen im Blut. Zwei Formen der ange-borenen Hämophilie werden unterschieden: Der Mangel an Gerinnungsfaktor VIII (FVIII, „8“) wird als Hämophilie A, der Mangel an Gerinnungsfaktor IX (FIX, „9“) als Hämophilie B bezeichnet.1,2
Was sind die Folgen?Da die Faktoren VIII und IX eine wichtige Rolle in der oben beschriebenen Gerinnungskaskade spielen, kommt es bei ihrer Verminderung oder wenn sie sogar fehlen, zu einer eingeschränkten oder stark eingeschränkten Blutgerinnung und einer erhöhten Blutungsneigung. Auftretende Wunden schließen sich nicht vollständig oder nicht schnell genug, sodass es zu schweren Blutungen kommen kann.
Da die Hämophilie durch eine Veränderung in den Genen verursacht wird, besteht sie lebenslang und verschwindet nicht von selbst.
Wie häufig kommt die Erkrankung vor?Hämophilie ist selten. Die Wahrscheinlichkeit, dass ein Junge mit Hämophilie geboren wird, beträgt Schätzungen zufolge 1 zu 5.000 bis 1 zu 10.000. Dabei ist die Hämophilie A etwa fünfmal häufiger als die Hämophilie B.
In Deutschland sind etwa 5.000 bis 6.000 Fälle von Hämophilie A und B bekannt.2-4

5
KURZ UND KNAPP
Bei angeborener Hämophilie ist die Blutgerinnung ohne Therapie lebenslang gestört.
Die Ursache ist ein Mangel an für die Gerinnung wichtigen Eiweißmolekülen im Blut. Bei Hämophilie A ist zu wenig Faktor VIII vorhanden, bei Hämophilie B zu wenig Faktor IX.
In Deutschland sind 5.000 bis 6.000 Fälle von Hämophilie A oder B bekannt.
!
NATHAN GRAYKanada
Hämophilie A

6
Vererbung
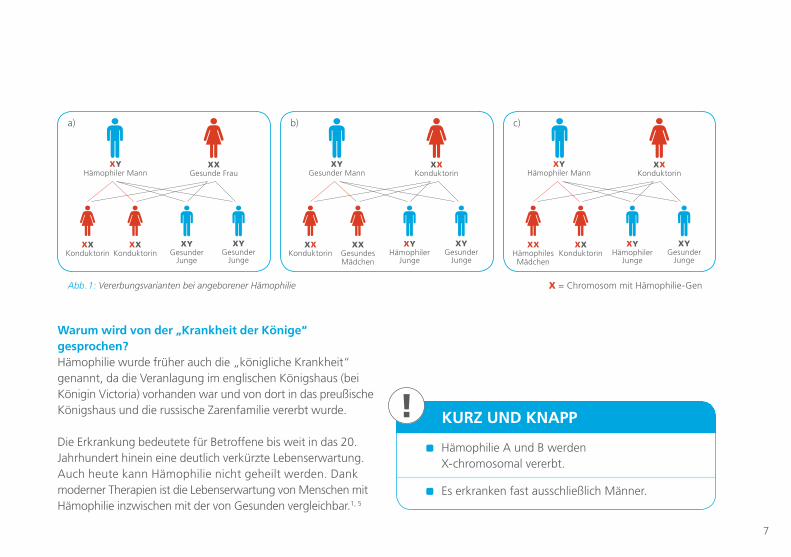
Wie wird Hämophilie vererbt und warum sind Jungen betroffen? Mädchen/Frauen haben zwei X-Chromosomen (XX), Jungen/Männer haben ein X- und ein Y-Chromosom (XY). Da die Mutter zwei X-Chromosomen hat, erhalten Kinder von der Mutter ein X-Chromosom. Vom Vater können sie ein X- oder ein Y-Chromo-som bekommen. Gibt der Vater das Y-Chromosom weiter, wird ein Junge (XY) geboren. Vererbt er das X-Chromosom, wird ein Mädchen (XX) geboren.2
Da Mädchen zwei X-Chromosomen haben, sind sie nicht von Hämophilie betroffen, wenn bei ihnen das Gen zur Herstellung von Gerinnungsfaktoren auf mindestens einem der beiden X-Chromosomen intakt ist und es das aktive Chromosom ist (Abb. 1a). Sie sind Merkmalsträgerinnen (Konduktorinnen), die das X-Chromosom mit dem fehlerhaften Gen mit einer Wahrscheinlichkeit von 50 % an ihre Kinder weitergeben.Jungen, die den Fehler im Gen auf dem X-Chromosom von ihrer Mutter erben, erkranken, weil sie die Störung nicht mit ihrem zweiten Geschlechtschromosom (dem Y-Chromosom) „ausgleichen“ können (Abb. 1b).
Mädchen erkranken extrem selten an einer Hämophilie, und zwar nur dann, wenn sie von beiden Elternteilen ein merk-maltragendes X-Chromosom (mit fehlerhaftem Gen) erhalten (Abb. 1c). Allerdings weiß man heutzutage, dass auch Kon-duktorinnen eine erhöhte Blutungsneigung haben können.
Bei etwa jedem dritten Patienten werden in der Familie keine früheren Fälle von Hämophilie gefunden. Hier entsteht die Erkrankung wahrscheinlich als Folge einer spontanen Verände-rung (Mutation; „Neumutation“) im Faktor VIII- oder IX-Gen.2
Faktor VIII und IX: Der genetische Bauplan liegt auf dem X-Chromosom Die angeborene Hämophilie betrifft fast ausschließlich Jungen. Der Grund dafür ist, dass die Anlage – die Gene – für die Gerinnungsfaktoren VIII und IX nur auf dem X- (Geschlechts-) Chromosom liegt. Das bedeutet, die Anlage, an Hämophilie zu erkranken, wird über das X- (Geschlechts-) Chromosom vererbt.2
7
Warum wird von der „Krankheit der Könige“ gesprochen?Hämophilie wurde früher auch die „königliche Krankheit“ genannt, da die Veranlagung im englischen Königshaus (bei Königin Victoria) vorhanden war und von dort in das preußische Königshaus und die russische Zarenfamilie vererbt wurde.
Die Erkrankung bedeutete für Betroffene bis weit in das 20. Jahrhundert hinein eine deutlich verkürzte Lebenserwartung. Auch heute kann Hämophilie nicht geheilt werden. Dank moderner Therapien ist die Lebenserwartung von Menschen mit Hämophilie inzwischen mit der von Gesunden vergleichbar.1, 5
KURZ UND KNAPP
Hämophilie A und B werden X-chromosomal vererbt.
Es erkranken fast ausschließlich Männer.
!
Abb. 1: Vererbungsvarianten bei angeborener Hämophilie X = Chromosom mit Hämophilie-Gen
XX Gesunde Frau
XYHämophiler Mann
XXKonduktorin
XXKonduktorin
XYGesunder
Junge
XYGesunder
Junge
a)
XX Konduktorin
XYGesunder Mann
XXKonduktorin
XXGesundes Mädchen
XYHämophiler
Junge
XYGesunder
Junge
b)
XX Konduktorin
XYHämophiler Mann
XXHämophiles Mädchen
XXKonduktorin
XYHämophiler
Junge
XYGesunder
Junge
c)

8
Diagnose
Welche Rolle spielt die Krankheitsvorgeschichte?Bei Verdacht auf Hämophilie wird der Arzt zunächst nach einer möglichen Krankheitsvorgeschichte beim Betroffenen selbst oder in dessen Familie fragen (Eigen- bzw. Familienanamnese). Hier sollte man möglichst genaue Angaben machen, da aus Informationen zu früheren Blutungsereignissen wichtige Schlüsse für die Diagnose gezogen werden können.
Wie wird die Diagnose bestätigt?Die Gerinnungsanalyse weist den Weg zur Therapie: Da beide Faktoren für eine funktionierende Gerinnung wichtig sind, sind die Krankheitszeichen und -folgen bei Hämophilie A und B praktisch identisch. Die Kenntnis der jeweils vorliegenden Variante ist jedoch Voraussetzung für die erfolgreiche Behandlung. Daher muss bei Verdacht auf Hämophilie eine Gerinnungs-analyse erfolgen.
Weiteren Aufschluss geben Labortests zur Gerinnung. Typisch für die Hämophilie (A und B) ist eine verlängerte aktivierte partielle Thromboplastinzeit, kurz aPTT.2
Krankheitsvorgeschichte ist aufschlussreich Eine Hämophilie A oder B wird häufig diagnostiziert, wenn die Erkrankung in einer Familie bereits bekannt ist und bei Kindern gezielt danach gesucht wird. Es kommt auch vor, dass ein betroffenes Kind zuerst durch Symptome auffällt und anschließend Familienmitglieder diagnostiziert werden, die bis dahin nichts von ihrer Erkrankung wussten, weil sie keine oder nur geringe Symptome hatten.2

9
Aktivität der Gerinnungsfaktoren bestimmt die Schwere der Symptome Die Diagnose wird weiter erhärtet, wenn bei einem Betroffenen eine verminderte Aktivität von FVIII oder FIX nachgewiesen werden kann. Wenn die Erkrankung in der Familie bekannt ist, kann die Bestimmung der FVIII bzw. FIX-Aktivität bereits aus Nabelschnurblut direkt nach der Entbindung erfolgen.
Achtung: Bei einer Verminderung des FVIII muss auch ein von-Willebrand-Syndrom ausgeschlossen werden, da auch in diesem Fall der FVIII vermindert sein kann.
Das Ergebnis zur Aktivität von Gerinnungsfaktor VIII bzw. IX ist zudem für die Schweregradeinteilung der Erkrankung bedeutsam:
• 5 – 40 % vom Normwert = milde (leichte) Hämophilie • 1 – 5 % vom Normwert = mittelschwere Hämophilie • < 1 % vom Normwert = schwere Hämophilie (betrifft
ca. 50 % der männlichen Hämophiliepatienten).6
Zur Diagnostik gehört auch eine Mutationsanalyse. Die Art der Genveränderung kann insbesondere bei der Hämophilie A einen Hinweis auf das Hemmkörperrisiko geben. Sie wird z. B. auch benötigt, um zweifelsfrei zu klären, ob die Mutter oder Schwestern eines Jungen mit Hämophilie Konduktorinnen sind.
KURZ UND KNAPP
Eigen- und Familienanamnese spielen bei der Erkennung der angeborenen Hämophilie eine wichtige Rolle.
Typisch für Hämophilie A und B ist eine verlängerte aktivierte partielle Thromboplastinzeit.
Je weniger aktiver Gerinnungsfaktor vom Körper hergestellt werden kann, desto ausgeprägter sind die Krankheitszeichen einer angeborenen Hämophilie.
!

10
Symptome und SchweregradeWas sind typische Krankheitszeichen?Die Krankheitssymptome sind abhängig vom Schweregrad der Hämophilie. Während bei der schweren Hämophilie (Restaktivität <1%) Spontanblutungen (Blutungen ohne vorherige Verletzung) typisch sind, starke Blutungen schon bei leichten Verletzungen auftreten und Wunden fast nicht aufhören zu bluten, können Patienten mit einer milden Hämophilie (Restaktivität >5%) im alltäglichen Leben fast komplett unauffällig sein, haben eventuell mehr blaue Flecken. Bei Menschen mit mittelschwerer aber auch milder Hämophilie (Restaktivität 1-5%) sind spontane Blutungen schon eher selten, allerdings können sie nach Operationen und Unfällen eine sehr starke Blutungsneigung haben.
MASSIMO SERAFINIItalien
Hämophilie A

11
Welcher Schweregrad macht welche Blutungsneigung?
• Schwere Hämophilie Typische Blutungen bei Patienten mit einer schweren Hämophilie sind Muskel-, Weichteil und Gelenkblutungen, die auch spontan ohne vorherigen Unfall auftreten.
Typisch sind zudem Blutungen im Körperinneren, die ohne erkennbaren Anlass auftreten. Solche spontanen Blutungen können in der Unterhaut stattfinden, wo sie als großflächige blaue Flecken (Hämatome) sichtbar werden. Häufig kommt es zu Blutungen in belastete Gelenke wie Knie (Abb. 2), Ellenbogen und Sprunggelenk sowie zu Blutungen in die Muskulatur. Das allererste Symptom kann auch ein großes Hämatom mit „Beule“ nach einer Impfung oder eine an-haltende Nach blutung nach der Impfung sein. Erste Symptome einer schweren Hämophilie treten meist ab der zweiten Hälfte des ersten Lebensjahres auf, wenn das Baby aktiver wird. Dies sind häufig auffallend viele Blutergüsse (Hämatome), viel mehr als sie andere Kinder in dem Alter haben. Auch erste Gelenkblutungen, die durch eine Schwellung, Überwärmung oder auch nur Schonhaltung auffallen, können dann auftreten. Nach harmlosen Verletzungen kann es zu anhaltenden Blutungen kommen.
Anhaltende Blutungen sind auch nach scheinbar harmlosen äußeren Verletzungen möglich. So kann es passieren, dass Betroffene z. B. wegen einer leichten Schnittwunde ärztlich versorgt werden müssen.
Abb. 2: Zielgelenkblutung (Quelle: Dr. Morio Arai, Department of Laboratory Medicine, Tokyo Medical University, Japan)
Anhaltende Blutungen sind typisch für die schwere Hämophilie Das wichtigste Krankheitszeichen bei Hämophilie A und B sind anhaltende Blutungen. Diese können nach äußeren Verletzungen und nach Verletzungen im Körperinneren auftreten.6 Spontane Blutungen im Kopf und der inneren Organe sind zum Glück sehr selten. Sie können aber nach Unfällen auftreten und sind dann sehr gefährlich.

12
• Problem hämophile ArthropathieKommt es zu wiederholten Einblutungen in das gleiche Gelenk, wird das als Zielgelenk bezeichnet. Es kommt zunächst zu einer schmerzhaften Entzündung (Synovitis) und die Muskulatur bildet sich zurück.
Dadurch wird diese besser durchblutet und es kommt immer wieder und noch leichter zu spontanen Einblutungen. Als Folge geht der Knorpel und schließlich das ganze Gelenk kaputt. Es wird steif und tut immer weh.
Beim Hämophiliepatienten müssen diese Blutungsereignisse nach Möglichkeit vermieden werden, um eventuelle schwere Schäden zu verhindern.6
• Mittelschwere HämophilieBei der mittelschweren Hämophilie (Restaktivität 1-5%) ist die Blutungsneigung nicht so ausgeprägt wie bei der schweren Hämophilie. Spontane Blutungen sind schon eher selten, allerdings können sie nach Operationen und Unfällen eine sehr starke Blutungsneigung haben. Auch hier können anhaltende Blutungen auftreten.
• Leichte Hämophilie kann unbemerkt bleibenBei minimaler Aktivität von Gerinnungsfaktoren sind die typischen Krankheitszeichen einer schweren Hämophilie im frühen Kindesalter sichtbar. Liegt eine leichte Hämophilie vor, sind die Symptome schwächer ausgeprägt und Spontanblutungen selten. Hier muss in bestimmten Lebenssituationen (etwa vor einer Operation) der erniedrigte Gerinnungsfaktor ersetzt werden, um eine Blutstillung zu gewährleisten. Bei Betroffenen mit geringen Symptomen kann eine leichte Hämophilie unbemerkt bleiben.6 Allerdings können auch bei der milden Hämophilie traumatische Gelenkblutungen auftreten. Da diese bei der milden Hämophilie aufgrund der sonst milden Symptomatik nicht erwartet werden, werden diese häufig nicht oder nicht rechtzeitig erkannt und behandelt, so dass sich auch hier bleibende Schäden entwickeln können, wie eine hämophile Arthropathie.

13
Was ist sonst zu beachten?Bei allen unklaren Symptomen sollte zunächst an eine Blutung gedacht werden. Zum Beispiel könnte eine Ursache für Bauch-schmerzen eine Psoasblutung (Muskel zwischen Wirbelsäule und Oberschenkelknochen) sein.
Impfungen sollten immer subkutan (unter die Haut), nicht wie üblich in den Muskel erfolgen.
Medikamente, die zusätzlich die Blutgerinnung stören, sollten vermieden werden. Fragen Sie immer ihren Arzt, ob ein Medikament unbedenklich ist.
KURZ UND KNAPP
Anhaltende Blutungen sind ein Krankheitszeichen bei Hämophilie A und B.
Häufige Orte von Blutungen sind Unterhaut (blaue Flecken), Gelen ke und Mus ku latur.
Seltener kommt es zu Blutungen in in ne ren Organen und im Zentral ner ven system.
!
NATHAN GRAYKanadaHämophilie A

14
Therapie
Welche Behandlungsmöglichkeiten gibt es?Bei der medikamentösen Behandlung der Hämophilie A und B werden die fehlenden Gerinnungsfaktoren VIII und IX durch Injektionen von Gerinnungsfaktorkonzentraten ersetzt (Faktor-ersatztherapie). Leider gibt es keine Tablette und eine Injektion unter die Haut geht auch nicht. Der Gerinnungsfaktor muss in die Vene gespritzt werden. Die Ersatzstoffe (Gerinnungs-faktorkonzentrate) werden aus Blutplasma gewonnen oder rekombinant (biotechnologisch) hergestellt.6 Man unterscheidet eine Behandlung bei Bedarf („on demand“), d. h. man spritzt nur, wenn eine Blutung bereits aufgetreten ist, von einer vorbeugenden Behandlung („Prophylaxe“), d. h. man spritzt regelmäßig, um Blutungen zu vermeiden. Das Problem einer „on Demand“ Behandlung ist, dass eine Blutung schon aufgetreten ist, z. B. sich Blut im Gelenk befindet, und schon allein dadurch Schäden verursachen kann. In Deutschland ist die prophylaktische Behandlung mit Gerinnungsfaktor der Standard, um Blutungen und Folge-schäden primär zu vermeiden. Die Häufigkeit und Dosis hängt unter anderem von Blutungsneigung und körperlicher Aktivität ab. Sollten dennoch Blutungen, z. B. durch Verletzungen oder Unfälle auftreten, muss meist noch zusätzlich gespritzt werden. Wichtig ist, dass das so schnell wie möglich erfolgt. Bei einer
größeren Blutung reicht häufig eine Injektion nicht aus. Dann muss so lange behandelt werden, bis die Blutung vollständig abgebaut ist.
Wie sieht die Alltagsbewältigung aus? Bei gutem Ansprechen auf die Behandlung haben Hämophilie-patienten eine vergleichbare Lebensqualität und Lebens-erwartung wie gesunde Menschen. Die Patienten können einen normalen Alltag haben und berufstätig sein. Sportliche Aktivitäten für Patienten mit Hämophilie werden empfohlen, da eine gute Muskulatur und Geschicklichkeit einen Schutz vor Blutungen darstellen.
Heimselbstbehandlung ist Standard Am Anfang wird das Spritzen des Gerinnungsfaktors der Arzt übernehmen. Wenn das Kind etwas älter ist (meist >2-3 Jahre), können die Eltern das Spritzen erlernen und etwa ab dem 8. Lebensjahr dann der Patient selbst. Dies nennt man Heim-selbstbehandlung. Sie erfordert vom Patienten hohe Mitver-antwortung. Bei der heute üblichen Heimselbstbe handlung erlaubt die vorbeugende (prophylaktische) Injektion eine dauerhafte Versorgung mit Gerinnungsfaktoren.

15
Einschränkungen gibt es bei Aktivitäten mit offensichtlichem Verletzungsrisiko, etwa bei Kampf- oder Kontaktsportarten (Boxen, Fußballspielen im Verein). Dagegen werden z. B. Schwimmen und Radfahren im Hinblick auf den Aufbau einer gelenkschützenden Muskulatur und einer guten Geschicklich-keit empfohlen. Empfehlungen, welche Sportart gut und welche weniger geeignet ist, kann der behandelnde Arzt geben.
Generell wird empfohlen, dass an Tagen mit Schulsport „Prophylaxetage“ sein sollten.
KURZ UND KNAPP
Angeborene Hämophilie wird durch Injektionen von Gerinnungsfaktorkonzentraten behandelt (Heimselbstbehandlung).
Therapieziele: Blutungen und ihre Folgeschäden sollten komplett vermieden und eine gute Lebensqualität mit einem normalen sozialen Leben angestrebt werden.
Für Hämophiliepatienten werden verschiedene sportliche Aktivitäten empfohlen. Risikosportarten sollten gemieden werden.
!
WUSSTEN SIE …
... wie die Dosierung von zugeführten Gerinnungs-faktoren berechnet werden kann?
Die Faustregel lautet:
Die Injektion einer Internationalen Einheit (IE) Ge rin nungs faktor pro Kilogramm Körpergewicht erhöht die Aktivität des Gerinnungsfaktors im Körper um 1 bis 2 %.
?

16
Hemmkörper
Was sind Hemmkörper?Das Problem der Hemmkörperbildung ist, dass das Gerinnungs-faktorkonzentrat nicht mehr wirkt und dadurch trotz Sub-stitution Blutungen nicht verhindert oder behandelt werden können.
Hemmkörper werden bei Hämophilie A deutlich häufiger beobachtet (15 bis 30 %) als bei Hämophilie B (zu etwa 3 bis 5 %). Bislang ist nicht vollständig geklärt, welche Ursachen zur Bildung von Hemmkörpern führen. Es sind einige Faktoren bekannt, die das Risiko dafür erhöhen. Die Hemmkörper-bildung tritt z. B. häufiger bei schwerer Hämophilie
und bei bestimmten genetischen Fehlern (bestimmten „Mutationstypen“ im Faktor VIII- oder IX-Gen) auf. Weitere Ursachen werden im Immunsystem selbst oder eher in der „Umwelt“ (sehr frühe Behandlung einer schweren Blutung mit hohen Faktordosierungen, „Gefahrensignale“) vermutet. Wenn bereits ein Familienmitglied einen Hemmkörper hatte, ist das Risiko erhöht. Menschen mit verschiedenen ethnischen Zugehörig keiten haben ein unterschiedliches Risiko.
Auch die Art der Behandlung von Patienten mit Hämophilie und Hemmkörpern kann sich möglicherweise auf die weitere Entwicklung der Hemmkörperspiegel auswirken. Zum Beispiel reicht es nicht aus bei Patienten mit starker Hemmkörper-bildung, den Faktor abzusetzen: Nach dem Absetzen des Gerinnungsfaktorkonzentrats kann eine spontane Rückbildung der Hemmkörper im Blut erreicht werden. Allerdings ist dieser nicht verschwunden, sondern steigt wieder an, wenn erneut ein entsprechendes Faktor VIII- oder IX-Präparat verabreicht wird.2-3, 7-8
Hemmkörper verkomplizieren die Therapie Bei einem Teil der behandelten Patienten bildet das Immun-system Antikörper (Abwehrstoffe) gegen die therapeutischen Gerinnungsfaktorkonzentrate. Insbesondere bei der schweren Hämophilie, bei der der Körper selbst keinen Faktor bildet, wird der substituierte Faktor als Fremdprotein erkannt und der Köper bildet „Abwehrstoffe“- Hemmkörper. Diese sogenannten Hemmkörper bauen die verabreichten Gerinnungsfaktoren rasch (teilweise sofort) ab, so dass diese nicht mehr oder nur noch eingeschränkt wirken können.2-3

17
Wann treten Hemmkörper auf?Wenn Hemmkörper auftreten, bilden sie sich oft am Anfang einer (Gerinnungs-) Faktorersatztherapie. Das größte Risiko besteht nach den ersten 9 – 50 Verabreichungstagen. Nach 100 Verabreichungstagen mit dem Gerinnungsfaktor ist das Risiko einer Hemmkörperentwicklung äußerst selten.7-8 Daher erfolgen in dieser Zeit regelmäßige Laborkontrollen zur Hemmkörperbestimmung.
Wie werden Hemmkörper entdeckt?Hemmkörper fallen entweder durch Unwirksamkeit der Behandlung („hochtitrige Hemmkörper“) oder durch die routine-mäßigen Laborkontrollen auf. Im Labor kann man die Höhe des Hemmkörpers messen. Außerdem ist die Halbwertszeit vermindert (d. h. der Faktor wird schneller abgebaut) und die Aktivität steigt nach der Faktorgabe nicht in erwartetem Maße an. Das daraus folgende Therapieversagen macht sich häufig in den ersten Wochen bemerkbar, wenn trotz Therapie anhaltende oder spontane Blutungen auftreten.7-8
KURZ UND KNAPP
An Hemmkörper sollte gedacht werden, wenn trotz Therapie anhaltende oder spontane Blutungen auftreten.
Das Risiko für eine Hemmkörperbildung ist in den ersten Wochen einer Faktor ersatz therapie am höchsten.
!
18
Welche Hemmkörper gibt es?Als Maß für die Hemmkörperbildung wurde die sogenannte Bethesda-Einheit (BE) definiert. Werte oberhalb von 5 BE pro Milliliter Plasma bezeichnen eine starke Hemmkörperbildung („hochtitriger Inhibitor/Hemmkörper“). Bei Werten von weniger als 5 BE pro Milliliter Plasma spricht man von einer schwachen Hemmkörperbildung („niedrigtitriger Inhibitor/Hemmkörper“). Je höher der Hemmkörper ist, umso mehr und schneller wird der Faktor abgebaut. Bei Patienten mit einem niedrigtitrigen Hemmkörper kann in den meisten Fällen die Weiterbehandlung mit dem entsprechenden Faktorpräparat fortgesetzt werden, ggf. sind höhere Dosierungen erforderlich. Die Weiterbehandlung der Patienten mit einem hohen Hemm-körpertiter ist in der Regel schwieriger und mit einem höheren Therapieaufwand verbunden, da das entsprechende Faktor-präparat nicht mehr wirkt.9 Erneute Faktorgaben können sogar dazu führen, dass der Hemmkörper weiter ansteigt.
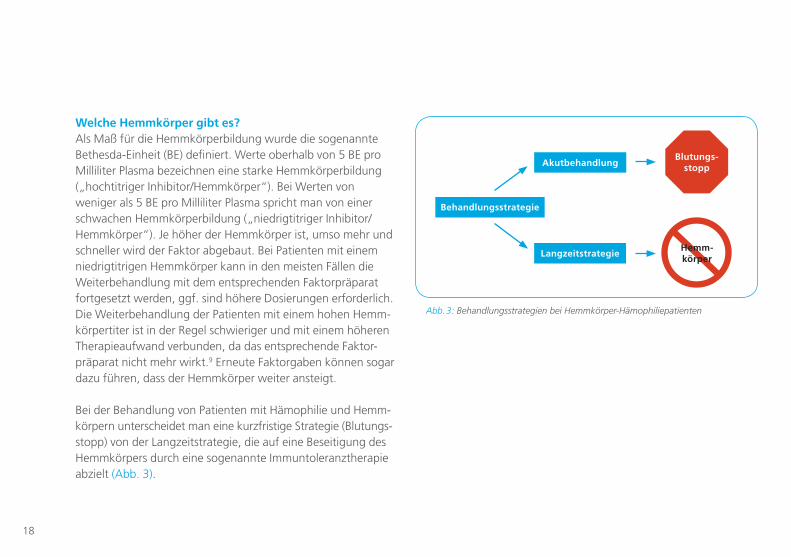
Bei der Behandlung von Patienten mit Hämophilie und Hemm-körpern unterscheidet man eine kurzfristige Strategie (Blutungs-stopp) von der Langzeitstrategie, die auf eine Beseitigung des Hemmkörpers durch eine sogenannte Immuntoleranztherapie abzielt (Abb. 3).
Blutungs-stopp
Behandlungsstrategie
Akutbehandlung
LangzeitstrategieHemm-körper
Abb. 3: Behandlungsstrategien bei Hemmkörper-Hämophiliepatienten

19
Wie werden Hemmkörper behandelt?
• Akutbehandlung: BlutungsstoppBei niedrigtitrigen Hemmkörpern (<5 BE) können die Patienten die Faktorenkonzentrate zur Stillung akuter Blutungen meist weiter verwenden und sich auch Operationen unterziehen. In der Regel heißt das, dass man einfach mit der Prophylaxe weitermacht, ggf. mit einer höheren Dosis. Auch bei Blutungen müssen die Faktorkonzentrate ggf. höher dosiert werden als bei Patienten ohne Hemmkörper.
Bei hochtitrigen Hemmkörpern (>5 BE) benötigen die Patienten in der Akutsituation andere Behandlungsstrategien, da die entsprechenden Faktorkonzentrate nicht wirken. Hier kommen sogenannte Bypass-Präparate zum Einsatz, diese umgehen die Faktor VIII- oder IX-Wirkung im Gerinnungssystem und führen dadurch zu einer Blutstillung.
In Deutschland stehen als Bypass-Präparate rekombinanter aktivierter Faktor VII (rFVIIa) und aktiviertes Prothrombin-komplexkonzentrat (aPCC) zur Verfügung. aPCC wird aus menschlichen Blutspenden hergestellt, ist also plasmatischen Ursprungs. In ihm sind neben aktiviertem Faktor II, VII, IX und X auch noch geringe Mengen Faktor VIII enthalten. rFVIIa ist gentechnologisch hergestellt (rekombinant) und ist frei von Bestandteilen aus menschlichem Spenderblut. Was das optimale Präparat für den Patienten ist, wird individuell entschieden. Eine konsequente Therapie ist zur Vermeidung von Spät- und Folgeschäden unbedingt erforderlich.
• Langzeitstrategie: ImmuntoleranztherapieMit der Immuntoleranztherapie (ITT) soll die Hemmkörper-bildung dauerhaft überwunden werden. Die Wahl des Behandlungsschemas erfolgt in Abhängigkeit von der Höhe des Antikörpertiters und des Hämophilietyps (A oder B). Bei niedrigtitrigen Hemmkörpern reicht es in der Regel aus, die Prophylaxe fortzusetzten. Die ITT bei hochtitrigen Hemmkörpern gegen FV III basiert auf der hoch dosierten Gabe von Faktorkonzentraten bis zum Erreichen der Hemm-körperelimination. Kinder und Erwachsene injizieren zweimal täglich hoch dosiert FVIII.9 Für die Hämophilie B sind oft andere Therapie schemata erforderlich. Die ITT bewirkt, dass sich das
Individuelle Behandlungsstrategie erforderlich Bei Patienten mit Hämophilie und Hemmkörpern muss eine individuelle Behandlungs strategie entwickelt werden, die in der Akutsituation, aber auch langfristig greift. Das Vorgehen hängt unter anderem vom Alter des Patienten und vom Aus maß der Hemmkörper bildung ab.

20
Immunsystem an den ersetzten Faktor gewöhnt und nicht mehr mit der Bildung von Hemmkörpern reagiert.
Der Faktor VIII dient in diesem Fall nicht der Verhinderung oder Stillung von Blutungen, sondern dem Erreichen einer Immun-toleranz des Körpers gegenüber dem Faktor VIII. Zur Blutungs-prophylaxe oder -behandlung müssen während dieser Zeit in der Regel Bypass-Präparate eingesetzt werden. Die ITT kann Monate oder bis zu zwei Jahre dauern.
Da Hemmkörper meist bei kleinen Kindern mit „noch schlechten Venen“ auftreten und je nach Behandlungsschema zweimal am Tag der hochdosierte FVIII und ggf. zusätzlich noch ein Bypass-Präparat gespritzt werden muss, ist meist ein zentralvenöser Katheter (port a cath- oder Broviak-Katheter) erforderlich. Auch das wird Ihr behandelnder Arzt mit Ihnen ausführlich besprechen.
Bei der ITT handelt es sich häufig um eine sehr langwierige Therapie, da in der Regel erst nach vielen Monaten ein Ansprechen gesehen werden kann und es am Anfang sogar noch zu einem Anstieg des Hemmkörpers kommen kann. Dies erfordert für alle Beteiligten viel Geduld und Disziplin. Da die Voraussetzung für einen Therapieerfolg die konsequente Durchführung ohne Unterbrechung ist.
In der Mehrzahl der Patienten ist diese Art der ITT erfolgreich. Wenn nicht, können noch andere Medikamente zur Immun-suppression zum Einsatz kommen.
KURZ UND KNAPP
Je nach Ausmaß der Hemmkörperbildung unterscheidet man hoch- und niedrigtitrige Hemmkörper.
Bei hochtitrigen Hemmkörpern können akute Blutungen mit rekombinantem Faktor VIIa oder aktiviertem Prothrombinkomplex-Konzentrat behandelt werden.
Die langfristige Strategie zur Überwindung von Hemmkörpern ist die Immuntoleranz therapie.
!

21
ANIL ÖZCANTürkei
Hämophilie A
HALIL ÖZCANTürkeiHämophilie A und Hemmkörper

22
Nützliche Adressen
Deutsche Hämophiliegesellschaft (DHG)Neumann-Reichardt-Straße 34 22041 Hamburg Telefon: +49 (0) 40 672 29 70 Fax: +49 (0) 40 672 49 44 E-Mail: [email protected] Internet: www.dhg.de
Interessengemeinschaft Hämophiler e.V. (IGH)Remmingsheimer Str. 3, 72108 Rottenburg am Neckar Telefon: +49 (0) 7472 226 48 E-Mail: [email protected] Internet: www.igh.info
Bündnis zur Förderung der Sicherheit von Hämophilen e.V. (BFSH)Marktstraße 50 99084 Erfurt Telefon: +49 (0) 361 663 82 60 Fax: +49 (0) 361 663 82 70 E-Mail: [email protected] Internet: www.bfsh.info
Gesellschaft für Thrombose- und Hämostaseforschung e.V. (GTH)Feodor-Lynen-Str. 5 30625 Hannover Telefon: +49 (0) 511 532 84 88 Fax: +49 (0) 511 532-16 84 88 E-Mail: [email protected] Internet: www.gth-online.org

23
Referenzen 1. Ingram GIC, J Clin Path 1976; 29 (6): 469 – 479. 2. von Depka Prondzinski M et al. (eds), UNI-MED-Verlag AG
Bremen, 2002: 36 – 60.3. Biggs R, Br J Haematol 1977; 35 (4): 487 – 504.4. World Federation of Hemophilia, WFH Annual Global Survey
on Hemophilia 2008; 34 pp.5. Otto JC, Med Repos 1803; 6: 1 – 4.6. Forbes CD et al. (eds), Chapman and Hall London,
1997: 380 pp.7. McMillan CW et al., Blood 1988; 71 (2): 344 – 348.8. Allain JP, Frommel D, Blood 1976; 47 (6): 973 – 982.9. Bundesärztekammer (BÄK). Querschnitts-Leitlinien zur
Therapie mit Blutkomponenten und Plasmaderivaten. 4. Auflage, 2009.
BENJAMIN GRAYKanada
Hämophilie A

Hämophilie verändern. Lebensqualität verbessern.
Physiotherapie
TrainingSchmerz
Reisen
Ernährung Novo Nordisk hat HaemCare™ ins Leben gerufen, um Menschen mit Hämophilie, deren Familien sowie behandelnde Ärzte, Betreuer und Therapeuten zu unterstützen.
HaemCare™ ist Bestandteil der inter nationalen Changing Haemophilia® Initiative.
Mit HaemCare™ bieten wir ein umfassendes Angebot an Service leistungen und stehen bei Fragen oder Anregungen jederzeit zur Verfügung.
www.novonordisk.de
Novo Nordisk Pharma GmbH, Brucknerstraße 1, 55127 Mainz Tel.: 06131-9030, Fax: 06131-9031370, www.novonordisk.de
Changing Haemophilia® ist eine eingetragene Marke der Novo Nordisk Health Care AG und der Apis-Stier ist eine eingetragene Marke von Novo Nordisk A/S. © 2017 Novo Nordisk Healthcare AG, Zurich, Switzerland. D
E/C
H/1
116/
0201
Art
.-Nr.:
703
861
Stan
d 01
/201
7








![Rep Päd Teil1.ppt [Kompatibilitätsmodus] · Besonderheiten des kindlichen Skeletts Knochen weicher, dehnbarer → bricht anders Knorpel höhere Elastizität, Wachstumsfugen Periost](https://img.pdfslide.org/doc/110x75/5d539e0588c993007d8b4fac/rep-paed-teil1ppt-kompatibilitaetsmodus-besonderheiten-des-kindlichen-skeletts.jpg)




















