Embed Size (px)
Citation preview

64 GEI~KARD PFLEIDERER :
M~glichkeiten und Grenzen in der Anwendung moderner biochemischer Analysenverfahren GERHA.RD I~FLEIDERER
Aus dem Institut fiir Biochemie im Institut fiir Organisehe Chemic der J. W. Goethe- Universit~t, Frankfurt a. M.
Eingegangen am 1. April 1965
Summary. The advantages and disadvantages of the various methods of separation used in modern biochemical research are described. Without going into too many details, distribution, adsorption and ion-exchange chromatography, electrophoresis with and without supporting media, and gel filtration were briefly treated. Using examples taken from widely different disciplines, it is shown that each method has its own limits for the analysis and the preparative separation of mixtures of low and high molecular substances. The combination of more than one separation system could usually achieve the desired result.
Einleitung Die stiirmische Entwicklung der biochemischen Forschung mit all ihren Grenzgebieten hat zwangsl~ufig eine Bliitezeit der Methodik mit sich gebracht. Dabei mul~ jedoch einsehrs festgestellt werden, da$ die Mehrzahl der heutigen Trennverfahren auf wenige altbekannte Prinzipien wie Verteilung einer Substanz zwisehen zwei nieht misehbaren LSsungs- mitteln, Adsorption an eine feste Oberflgehe oder Wanderung im elek- trisehen Feld zuriiekgefiihrt werden kann, die laufend verbessert und vor allem aufMikroteehnik umgestellt wurden. In Forsehung und Praxis Mrd nur der optimal erfo]greich sein, der die ~ehrhei t der modernen ~ethodik fiberschaut und beherrscht. Da eine exakte Analytik im weitesten Sinn nieht ohne vorherige Reindarstellung einer Substanz mSglich ist, mSchte ich mir erlauben, keine scharfe Trennlinie zwischen analytischen und prs Trennverfahren zu ziehen, zumal beide das Kandwerkszeug des mit Naturstoffen arbeitenden Wissenschaftlers sind. Sinn dieses Vortrags vor einem breit gestreuten tISrerkreis aus Chemic, Medizin und Biologie soll es ja sein, dem 5~ichtfaehmann oder dem, der sieh auf be- stimmte Methoden speziaHsiert hat, unhand eines ~berblieks in kritischer Weise die heutigen MSgliehkeiten und Grenzen der ~e thodik aufzuzeigen.
Mikro- und Ultramikromethoden Die auffallendste Entwicklung der letzten Jahre geht wohl dahin, alle Methoden auf Mikrotechnik oder sogar Ultramikrotechnik auszurichten. Man verlangt heute von einem analytischen Verfahren, dal~ es bis herab zu #g-Mengen und darunter noch reelle und gut reproduzierbare Werte liefert. Vielfach ist die unterste Erfassungsgrenze nur dureh die Empfind- liehkeit der Mel~ger~te oder aber schon durch St6rungen in Form yon

Anwendung moderner biochemischer Analysenverfahren 65
Verunreinigungen in den LSsungsmitteln, Reagentien oder Tr/iger- substanzen der Chromatographic (Papierleerwert) bedingt. So ist es heute m6glieh, mit weniger als 0,5 mg Substanz eine ehemisehe Elementaranalyse auf Kohlenstoff-, Wasserstoff- und Stiekstoffgehalt und mit weniger als 50 #g auf Sehwefelgehalt durehzufiihren. Ffir eine Aminos/tureanalyse benStigt man heute nur noeh je Aminosgure etwa 0,1 #Mol. EDST~6M 9 beschrieb 1952 ein Ultramikroverfahren auf der Basis der Papierehromatographie, mit dessen Hilfe man aus dem Hydro- lysat yon 1,5--5 #g Ribonueleinsgure eine Basenanalyse dutch Kontakt- photographie bei 257 und 275 nm durehf/ihren konnte, oder der B/iehersehe Arbeitskreis hat mit 1,4/~Mol Gesamtphosphat auf einer Mikro- ionenaustausehersgule 32 Fraktionen eines Leberpunktates abtrennen, grSl?tentefls identifizieren und quantitativ bestimmen kSnnen 31. Ebenso empfindlich ist die Gasehromatographie mit Erfassungsgrenzen yon 1 #g und darunter. Die enzymatisehen Bestimmungsmethoden benStigen ebenfalls in der Regel Bruehteile yon ,uMol Substanz, bzw. k6nnen bei katalytisehen Messungen mit Coenzymen noeh wesentlieh darunter liegen, t t o n z ~ u. Mitarb. a2 publizierten kfirzlieh eine Pyri- doxalphosphatbestimmung mit tIilfe yon Hefe-Transaminase, die herab bis zu 10 -4 #g Coenzym noeh brauehbare Werte liefert. Die hoeh spezi- fisehe ATP-Bestimmung mit Lueiferase aus Leuehtks erlaubt noeh gut die Bestimmung yon 0,01 #g ATP/ml aufgrund der charakteristisehen Lmnineseenz, nieht zu reden yon den mikrobiologisehen Wuehsstoff- messungen, deren Erfassungsgrenze z.B. bei der Vitamin B12-Bestim- mung in der Gegend yon 10-1~g liegt. Grunds~tzlieh kann bei allen optisehen Verfahren dutch Verwendung yon Mikroktivetten der Sub- stanzbedarf je Bestimmung wesentlieh gesenkt werden. Die hohe Empfindliehkeit der Isotopenteehnik kann als bekannt vorausgesetzt werden. I)iese Auswahl soll stellvertretend zeigen, in welehen I)imensio- nen sieh die heutige Analytik bewegt.
Wahl der Trennverfahren
Wie wh'd nun in einem gegebenen Fall die Auswahl der geeignetsten Trennmethoden, falls diese ftir eine Analyse tiberhaupt notwendig sind, zu treffen sein? Verh/~ltnismgBig einfaeh ist die Situation, wenn die Struktur der zu bestimmenden Substanz bekannt ist. Aus der Konstitution lassen sieh ehemische und physikalisehe Eigensehaften ableiten, die sowohl fiir die Trennteehnik, als aueh fiir die naehfolgende Naehweismethode wiehtig sind. Es sei an dieser Stelle sehon darauf hingewiesen, dal3 unter Umstgnden eine f~r die Analytik entwiekelte Trennteehnik sieh nieht ohne weiteres dureh einfaehe Erweiterung der apparativen I)imensionen auf prgparative Zweeke -- insbesondere im g-Magstab -- umwandeln 1/tBt. Folgende Eigenscha/ten der zu trennenden Substanzen sind zu beriiek-
Z. analyt. Chem., Bd. 212 5

66 ~ERHARD PFLEIDERER :
siehtigen: Ladung, LSslichkeit, Adsorbierbarkeit, Flfichtigkeit und neuerdings MolekfilgrSl~e. Handelt es sich um eine sehr komplexe Misehung aus biologisehem Material, so ist in vielen F~llen eine grobe Vortrennung ratsam. Man denke an die Extraktion lipophiler 8ubstanzen wie z.B. Steroide durch organische LSsungsmittel, die Ausfgllung yon Proteinen durch Hitze- oder S~turebehandlung oder die Abtrennung nieder- molekularer Substanzen durch Dialyse, Ultrafiltration oder Gelfiltration. Unentbehrlich far viele Zwecke ist nach wie vor die Adsorption aroma~i- seher nnd heteroeyeliseher Naturstoffe an I-Iolzkohle und ihre ansehlie- ~ende Elution, ein Proze~, der naeh geeigneter Vorbehandlung des Adsorbens aueh im Mikroma~stab vielfaeh mit 100% Ausbeute ver- l/~nft. Allein der Anreicherungs- und Entsalzungseffekt ist hier uniiber- troffen. ])as wohl theoretiseh iibersichtlichste Trennverfahrenist die yon L.C.CI~AIO eingefiihrte Gegenstromverteiluny, bei der sieh bekanntlieh die gel6sten Stoffe zwisehen 2 nicht oder nur begrenzt mischbaren LSsungsmitteln im Idealfall in einem konstanten Verhaltnis verteilen. Obwohl dieses Ver- fahren ausgezeiehnet geeignet ist fiir die Analytik bzw. Reinheitspriifung, aber aueh dureh die Kombhlation versehiedenartigster L6sungsmittel- gemisehe fiir die Trennung fast aller Stoffarten brauchbar, hat es erstaun- lieherweise verh~Itnism~l~ig wenig Eingang in die Forsehung gefunden. ])abei ist wirklieh hier unter bestirnmten Voraussetzungen eine mathe- matisehe Voransbereehnung des gesamten Verteilungsvorganges m6glieh. Abweiohungen der experimentenen Verteilnngskurve yon der bereehne- ten lassen auf Verunreinigungen schlieBen. Ist der Verteilungskoeffizient sehr ungiinstig (sehr grol3 oder klein, z.B. Phosphors~ureester), so kann er durch Zusatz eines Salzbildners mit enspreehenden Substituenten im gewiinschten Sinn verbessert werden. So setzten KA~AU U. ZAOHAV ~s bei der Verteilung Aminos~nre-spezifischer Transfer-l~NS diese zuvor in das Tri-n-Butylammoniumsalz urn. Ein grol3er Vorteil des Verfahrens, das heute weitgehend automatisiert ist (bis zu 1000faehe Verteilung), beruht auf der einfachen EntnahmemSglichkeit im Verlauf und am Ende des Verfahrens. 1Xeben der Trennung yon I-Iormonen und Antibiotiea werden neuerdings ausgezeiehnete Erfolge bei der eben erwahnten Trennung versehiedener Transfer-R1NS erzieltlS, is. StSrungen treten dann ein, wenn die gelSsten Substanzen die Tendenz zur Assoziation aufweisen. ])ureh geeignete LSsungsmittelwahl kann dieser Effekt kompensiert werden, zumal dieser sehr stark yore p}I und der Ionenst~rke abh~ngt. Bei sehr unreinen Systemen treten manchmal zu Anfang unangenehme Emulsio- nen auf, die meist durch Zentrifugation aufgehoben werden. Breite Anwendung gefnnden hat die Verteilungschromatographie zwischen 2 Phasen, yon denen meist die wal~rige (stationare) Phase dureh einen unlSslichen Tr~ger festgehalten wird. MA~mI~ u. SY~G~ 22 benutzten zu

Anwendung moderner biochemischer Analysenverfahren 67
Anfang in Saulen geftilltes Siliealgel zur Bindung der stationaren Phase und entwickelten mit einem mit Wasser nieht misehbaren organisehen LSsungsmittel (mobile Phase). Die hier und auch auf anderen Trgger- stoffen wie Cellulose oder Stgrke hintereinandergesehalteten zahllosen Verteilungsprozesse sind Ursaehe der hervorragenden Trennung selbst ehemiseh sehr ahnlieher Verbindungen. Eine erste KrSnung erfuhr die Verteilungsehromatographie dutch Einf/ihrung yon Papier als Trager der stationaren Phase dureh Co~sD~, GO~DON u. 7~/[AI~TIN 6. Auf die alte Streitfrage, ob die tibliche Papierehromatographie mehr als Verteflungs-, Adsorptions- oder Ionenaustauseh-Chromatographie aufzufassen ist, soll hier nieht mehr eingegangen werden. In den meisten Fallen diirfte der Verteilungskoeffizient zwisehen mobiler und stationarer Phase haupt- verantwortlieh ftir den Rf-Wert sein. Bekanntlieh kann man die Phasen dureh Impragnieren des Tragers mit hydrophoben Stoffen umkehren. Die Analytilc und milcropr~iparative Anwendung des Papierchromatographie ist so universell und allgemein bekannt, dab sich viele Worte hierfiber er- iibrigen. Wichtige Grundiage ist die weitgehende Unabhangigkeit der Rf-Werte yon der Menge und Anzahl der Begleitstoffe, abgesehen yon anorganischen Salzen, die bei hoher Konzentration betr/~chtliehe Ver- zerrungen und Verz6gerungen der Substanzflecke verursachen. I)a die R~-Werte auBerordentlich stark yon den auBeren Reaktionsbedingungen und dem Material abhangen, ist die Verwendung absoluter t~f-Werte nicht sehr sinnvoll, sondern as wird hier -- wie auch in der Diinnschicht- Chromatographie oder I-Iochspannungselektrophorese -- immer empfeh- lenswert sein, bei der entscheidenden Identifizierung eine Vergleiehs- substanz mit heranzuziehen. Die y o n BATE-SMITII U. WESTALL 2, sparer yon I~EIO~L 2v oder SO,AVOn u. BvLrascg 3~ abgeleitete Abhangigkeit der
t~f-Werte yon der ehemisehen Konstitution Rz~ = log ~ [
bei homologen Reihen in allerdings relativ engen Bereiehen zur Aufklarung unbekannter Konstitution heranziehen, wobei sieh der Rlu-Wert aus versehiedenen experimentell ermittelten Gruppenkonstanten erreehnen laBt. Xhnliehe Beziehungen wurden aueh in der Gas-Chromatographie aufgefunden. Da S/turen und Basen als ionisierte und nieht-ionisierte Verbindungen ehromatographiert werden k6nnen, kann dureh Vergleieh mehrerer Substanzen in versehiedenen L6sungsmitteln mit versehiedenem pt{ zndem auf Art und Zahl polarer Gruppen gesehlossen werden. Die Einfiihrung der Diinnschichtchromatographie dureh STAI{L 37 hut in neuerer Zeit die Papierehromatographie etwas in den Kintergrund gedrangt. /-[ier handelt es sieh praktisch um offene Chromatographie- saulen mit 250 # Dicke, die durch Aufstreiehen oder Sprfihen yon mit Gips als Bindemittel durchmisehten Tragern wie Kieselgel G entstehen. Je naeh Auswahl des Tragers und LSsungsmittels fiberwiegt aueh hier die
5*

68 ~EI~IIARD PFLEIDEI~EI~ :
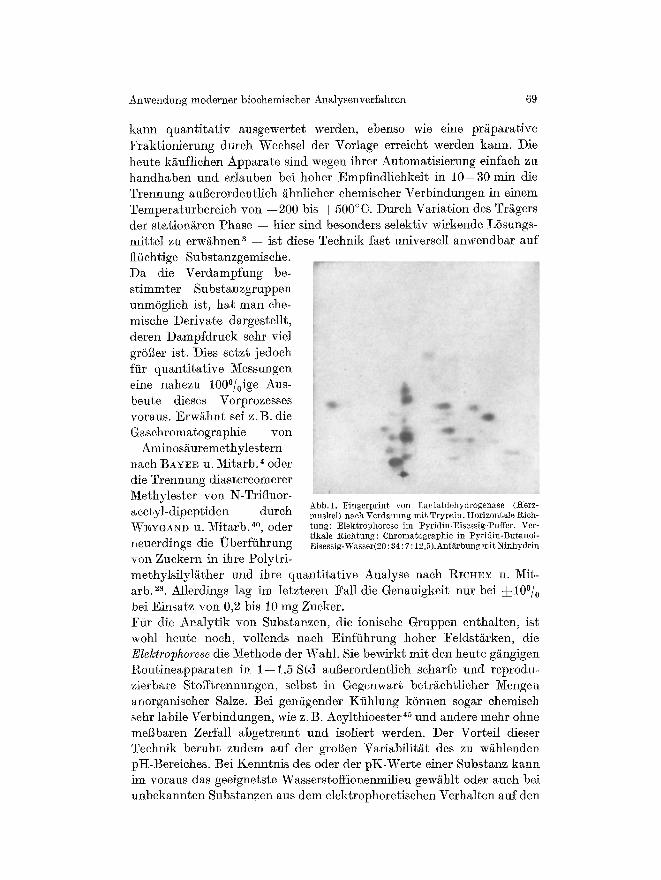
Adsorptions-, Verteilungs- oder Ionenaustausehehromatographie. Die VorteJle gegeniiber dem Papierverfahren liegen vor allem bei der idealen Trennung lipophiler Substanzen, sehr viel kiirzeren Laufzeiten (0,5 his 3 Std gegeniiber 12--18) und hoher Trennseh~rfe bei groger Naehweis- empfindliehkeit. Sehlieglieh lassen sieh auf diinnen Sehiehten im Gegen- satz zu Papier sehr drastisehe Naehweismethoden wie Einwirkung kon- zentrierter S/~ure und hohe Temperatur durehf/ihren. Eine groBe Zahl unl6slieher Trggerstoffe ist bisher mit und ohne Bindemittel verwendet worden, zuletzt aneh Cellulose, Celluloseaustauseher, Sephadex und Sephadexaustbuseher. Bei kleinen I~tWerten hilft man sich mit mehr- faeher Entwieklung auf derselben Platte. Die offene S~ulenteehnik wird bueh bnalytiseh erfolgreieh fiir Proteine, insbesondere bei Anwendung einer Gradiententeehnik benutzt a3. Ahnlieh wie bei der Pbpierehrombto- graphie sind die siehtbar gemaehten Substbnzfleeken nbeh Elution, die hier besonders einfbeh ist, oder in der Durehsieht liehtelektriseh bus- wertbbr mit einem tSehlerbereieh yon durehschnittlieh 5--10~ Aueh die Autorbdiographie naeh Verwendung radioaktiv markierter Substan- zen bereitet keine Sehwierigkeiten. Besonders giinstig erweist sieh die erstmals bei der Papierteehnik angewbndte Kombination yon Chromato- grbphie und Elektrophorese auf Diinnsehiehtplatten, wie sie z.B. bei der Fingerprintmethode iiblieh ist. WIELASD u. Gs.O~GO~OUI, OS ~s haben kiirzlieh ftir die Trennung yon Peptidgemisehen bus reinen Proteinen nacb enzymatiseher Verdaunng sehr gut reproduzierbare Bedingungen besehrie- ben, die Mr lbufend mit bestem Erfolg bei der Identifizierung radioaktiv- markierter Wirkgruppenpeptide yon Enzymen bnwenden (Abb. 1). Selbstverst/indlieh kann die Diinnsehiehtehrombtographie auf ]angen Glasplatten preparative Anwendung finden. In den Laboratorien der Fa. Merck, Darmstadt, werden sehon lange auf 1 m langen, 20 am hohen Dfinnsehiehtplatten und bei 2 mm Sehiehtdieke Chromatogramme im g-Magstab durehgeffihrt (z.B. IIALPAAP13). Eine Verteilungschromatographie zwischen gas/6rmiger und ]li~ssiger Phase ist das Prinzip der Gasehromatographie, wobei die station/~re Phase ver- k6rpert wird dureh einen mit L6sungen niederen Dampfdrueks getr/~nkten inerten Tr/tger (Celit, Diatomeen-Erde, Schamottesteine) und die mobile Phase durch ein inertes Gas, das die station~re Phase durchstr6mt und in das das zu anMysierende Substanzgemiseh unter Verdampfung ein- getragen wird. Der Dbmpfdruek der Komponenten darf bei der Arbeits- temperatur yon h6ehstens 500~ nieht zu gering sein. Je naeh Vertei- lungsgleiehgewieht zwisehen gasf6rmiger und fltissiger Phase wird ein Stoff beim Durehwbndern der Sgule mehr oder weniger stark verz6gert und dbnn naeh Verlassen der Sgule dureh einen Detektor registriert (Zeit oder Volumen vom Auftragen bis zum Erseheinen im Detektor bezeiehnet man als Retentionszeit bzw. I~etentionsvolumen). Die Registrierung
Anwen4ung moderner bioohemiseher Ana,lysenverf~hren 69
kann quantitativ ausgewertet werden, ebenso wie eine prgparative Fraktionierung dutch Weehsel der Vorlage erreicht werden kann. Die heute kgufliehen Apparate sind wegen ihrer Automatisierung einfaeh zu handhaben and erlauben bei hoher Empfindliehkeit in 10--30 min die Trennung augerordentlieh ghnlieher ehemiseher Verbindungen in einem Temperaturbereieh yon --200 bis q-500~ Dureh Variation des Trggers der stationgren Phase -- bier sind besonders selektiv wirkende L6sungs- mittel zu erwghnen a -- ist diese Teehnik fast universell anwendbar auf flfiehtige Substanzgemisehe. Da die Verdampfung be- stimmter Substanzgruppen nnm6glieh ist, hat man ehe- misehe Derivate dargestellt, deren Dampfdruek sehr viel gr6ger ist. Dies setzt jedoeh fiir quantitative Messungen eine nahezu 1000/0ige Aus- beute dieses Vorprozesses voraus. Erw/ihnt sei z. B. die Gasehromatographie von
Aminos~uremethylestern naeh BAY~ u. Mitarb. 4 oder die Trennung diastereomerer Methylester yon N-Trifluor-
Abb. 1. Fingerprint yon Laetatdehydrogenase (Herz- aeetyl-dipeptiden dureh muskeI) naeh Verda.uung tait Trypsin. I{orizongale I%ieh- W E Y G A N D U. Mitarb. 40, oder tung: Elektrophorese im Pyridin-Eisessig-Puffer. Ver-
tikale Riehtung: Chromatographie in Pyriclin-?3utanol- neuerdings die Uberf/ihrung Eisessig-Wasser(20: 34 : 7 : 12,5).Anffirbungmit Ninhydrin
yon Zuekern in ihre Polytri- methylsilyl/~ther and ihre quantitative Analyse naeh RICI{Eu u. ~V[it- arb. 2s. Allerdings lag im letzteren Fall die Genauigkeit nur bei •176 bei Einsatz yon 0,2 his 10 mg Zueker. Fiir die Analytik yon Substanzen, die ionisehe Gruppen enthalten, ist wohl heute noeh, vollends naeh Einfiihrung hoher Feldstgrken, die Elelctrophorese die Methode der Wahl. Sie bewirkt mit den heute g/tngigen Routineapparaten in 1--1,5 Std augerordentlieh seharfe und reprodu- zierbare Stoff~rennungen, selbst in Gegenwart betrs Nengen anorganiseher Salze. Bei gen/igender Kiihlung k6nnen sogar ehemiseh sehr labile Verbindungen, wie z. B. Aeylthioester 45 und andere mehr ohne meBbaren Zerfall abgetrennt und isoliert werden. Der Vorteil dieser Teehnik beruht zudem auf der grogen Variabilit~t des zu wghlenden pI-I-Bereiehes. Bei Kenntnis des oder der pK-Werte einer Substanz kann im voraus das geeignetste Wasserstoffionenmflieu gew/thlt oder aueh bei unbekannten Substanzen aus dem elektrophoretisehen Verhalten auf den

70 GERHARD PFLEIDERER:
pK-Wert geschlossen werden. Giinstig kann sich auch die Verwendung yon ptt-Gradienten auswirken. Fiir die hohe Reproduzierbarkeit dieser Methode, im Gegensatz zu den teilweise stark schwankenden R~-Werten der Papierchromatographie, sprieht das konstante Verhaltnis der Wanderungsstrecken verschiedener ionischer Verbindungen zu der einer Bezugssubstanz unter identisehen Bedingungen. Ftir verschiedene Aminosguren lagen die Abweiehungen bei pit 1,9 gegenfiber Alanin bei •176 ~. Erstaunlicherweise hat sich dieses niitzliehe Verfahren noeh wenig in der pr/~parativen organisehen Chemie durehsetzen k6nnen. Kostspielige kgufliehe Wirkstoffe oder radioaktiv-markierte Produkte lassen sieh raseh und mit hoher Ausbeute auf diese Weise im NikromaBstab rein darstellen. Unbrauehbar ist die Verwendung yon Papier als Trgger ffir die Hoch- spannungselektrophorese hoehmolekularer Substanzen. ]tier sind besser geeignet Folien aus aeetylierter Cellulose (Membranfolien der Fa. Sehl. & Seh.), auf denen der bei Papier immer beobaehtete starke Ad- sorptionseffekt ffir Proteine nahezu vSllig ausgesehaltet ist. ]gin sehr seh6nes Beispiel hierffir ist die yon uns mehrfaeh publizierte Trennung der 5 Isozyme der Laetatdehydrogenase aus S/~ugetierorganen 24,43. Die naeh l~berspr/ihen mit Substrat- und Coenzyml6sung unter der UV-Lampe deutlieh siehtbaren Einzelkomponenten k6nnen naeh dem Aussehneiden der Zonen in wenigen Sekunden in einer MeBkfivette eluiert und enzyma- tiseh getestet warden bei bis zu 100% Gesamtausbeute an urspriinglieh eingesetzter Enzymaktivit~t. Die mikropr/~parative Anwendung auf dieken PapierbSgen oder im St~rkebloek geht bis in die Gr6Benordnung yon 100--500 mg Substanz ]e Trennung. In grSgeren Mengen lassen sieh Substanzen auf Cellulose- und Stgrkes~ulen elektrophoretiseh trennen. lq'tir kontinuierliehe Trennungen bieten sieh die Vertlkalelel~trophorese au[ Papiervorhiingen naeh der Besehreibung yon G~assMa~z~ u. I~ANNIG 12 und neuerdings die trggerfreie Elektrophorese an. So sind Gergte besehrieben, bei denen zwisehen zwei geneigten Glasplatten fortlaufend ein Pufferfflm bewegt wird, der gleiehzeitig dutch ein GMehspannungsfeld beeinflugt wird. Nit diesen Get,ten kann z.B. in 24 Std die Auftrennung von etwa 1,5 g Serum erreieht werden. I-IA~Z~IG 14 konnte sehHeBlieh sogar Zell- elemente, wie Nitoehondrien, Nikrosomen und Zellkerne bei ausgezeieh- neter ttomogenitgt der einzeinen t~raktionen absondern. Wohl die seh/~rfste Auftrennung eines Proteingemisehes gewghrleistet die Elektrophorese in Gelen, wobei die yon S?aIT~I~S 34 eingef~hrteVerwendung partiell hydrolysierter Stgrke und die Polyaerylamidteehnik hervor- zuheben sind. Bis zu 16 Komponenten kSnnen naeh der Elektrophorese yon Serum identifiziert werden. Man denke an die sehSnen genetisehen Studien mit mensehliehen I-Iaptoglobinen 2~. Es gibt wohl kein besseres Reinheitskriterium ftir den Enzymehemiker als die Einheitliehkeit eines

Anwendung moderner biochemischer AnMysenverfahren 71
Enzympr/~parates in der Gelelektrophorcse, wobei Amidoschwarzanf~r- bung und enzymatischer Aktivit/~tsnachweis fibereinstimmen mfissen. Die grof~e TrennschKrfe wird mitbegiinstigt durch den bier beobachteten 5/folekularsiebeffekt, der bei der Wanderung hochmolekularer Teilchen durch die Gelmatrix zustande kommt und sich an der unterschiedlichen Wanderung yon fl-Lipoproteid, Transferrin und H~moglobin bei pI-I 8,55 im St/irkegel gegeniiber einer gleichartigen Wanderung auf Papier ab- zeichnet. Das Ausmal~ der Retardierung h~ngt dabei von der St&rke- konzentration im Gel ab und ist anscheinend relativ nur yon der GrSBe des Molekfils 35, nicht aber yon der GrSl~e der Ladung abh/ingig. Nachteile der Eiweii3gelelektrophorese liegen in der schwierigen quantitativen Auswertung der angef~rbten Banden oder der pr/iparativen Isolierung hinterher. Nut in beschrs Mal~e lassen sich Proteine nach Gefrieren und Auftauen der Gelsegmcnte eluieren.
Isolierung von einzelnen Stoffen
Die bisher genannten Methoden finden ihre hauptsttchliche Anwendung in der Analytik, also bei der Auftrennung kleinerer Substanzgemische. Will man jedoch zu grSl~eren ~engen iibergehen und niedermolekulare Stoffe frei yon allen Verunreinigungen in Substanz isolieren, so wird es immer vorteilhaft sein, zur Siiulentechnik mit wasserunl6slichen Tr/igern ~berzugehen. Je nach Problemstellung sind hier Kuns~harzaustauscher, Cellulosepulver oder Sephadexgele mit und ohne ionische Gruppen und natiirlich die klassischen Tr~ger der Adsorptions- und Verteilungs- chromatographie zu nennen. Die Technik der Kunstharzaustauscher- chromatographie mit Gradientenelution (Ionenst/~rke oder pI-I) ist heute perfektioniert in Verbindung mit automatisch registrierenden Photo- zellen und Fraktionssammlern. Die bekanntesten Beispiele sind die Eiweil~- und Peptidanalytik sowie die Verwendung in der Nucleotid- chemie. Vielfach ist jedoch l~stig das Einengen und EntsMzen der hoch- verdiinnt anfMlenden Fraktionen. da nich~ in jedem Fall flfichtige Puffer als Elutionsmittel verwendet werden k6nnen. Die automatische I%egi- strierung der Elutionsfltissigkeiten beschr/inkte sich bisher fast ausschliel~- ]ich auf die t tg-Bande bei 253 nm (Uvicord der Fa. LKB, Stockholm). I-Iier bringt die Mikrodurchflul~kiivette der Fa. Zeiss, Oberkochen, in Verbindung mit einem ~onochromator neue Anwendungsm6glichkeiten. Neuerdings entwickelten STou~Fn~ u. Mitarb. as einen modifizierten Flammeniolxisationsdetektor, wie er im Prinzip in der Gaschromato- graphie wirksam ist, um auch bei nicht-absorbierenden und schwierig nachzuweisenden hochmolekularen Substanzen ffir die Flfissigkeits- chromatographie eine Nachweismcthode bei h6chster Empfindlichkeit in I-I&nden zu haben.

72 ~:ERHARD PFLEIDERER :
Naehteilig wirken sieh bei den Kunstharzaustauschern die Diffusions- effekte in die Poren derK6rner aus oder die dureh Anhgufung aromatiseher Ringe verursaehten Adsorptionseffekte fiir aromatisehe und hetero- eyelisehe Verbindungen, wie sieh/iberhaupt dieser Austausehertyp fiir die Trennung hochmolekularer Substanzen nieht als geeignet erwiesen hat. Diesen Tr/~gern erw/~ehst groge Konkurrenz in Gestalt der Cellulose- und Sephadexaustauseher. Erstere wurden 1956 yon SOBE~ U. P~TER- SO~ ~6 eingefiihrt. Im Untersehied zu den Kunstharzen ist die Cellulose- matrix hydrophil, dazu fiihrt die Fasereigensehaft tier Cellulose zur Anhaufung der austausch-aktiven Gruppen nahe an der Oberflaehe, so dag groBe Molektile leieht adsorbiert werden. Trotz der an sieh kleineren Gesamtaustausehkapazit/tt besitzen daher diese Tr/~ger eine hShere Kapa- z i t i t fiir Proteine. Zum Tell noeh/ibertroffen werden sie yon den dutch PO~ATI~ u. F~oDrs 26 1959 entdeekten Sephadexionenaustausehern, die dureh gezielte Vernetzung noeh zusgtzlieh einen Gelfiltrationseffekt je naeh Molekiilgr61~e beMrken k6nnen. Die Kapazit/~t beziiglieh hoeh- molekularer Substanzen ist bei den letzteren noeh gr6ger bei hoher Trennseh/~rfe und hoher DurehfluBgesehwindigkeit infolge der KSrnung des Materials. In der t~egel ist die Verwendung eines Koehsalzgradienten bei der Elution besonders wirksam. Es mul3 allerdings darauf hingewiesen werden, dab meist erst dureh die Reehromatographie einer Fraktion deren Einheitliehkeit gew/~hrleistet ist. Erwghnenswert ist, dab wir in versehiedenen F~llen bei der Trennung yon Enzymen auf Carboxymethyl-Cellulose oder -Sephadex unter Ver- wendung eines ansteigenden ptt-Gradienten (pit 5--8) zwar sehr sehSne Fraktionierungen erreiehten, aber im Eluat naehtrgglieh eine kontinuier- liehe Inaktivierung beobaehteten, die am besten dutch Struktur/~nde- rungen beim Adsorptionsvorgang und nieht dureh einen pH-Effekt erkl/~rt werden kann. Aueh bei der Reinigung niedermolekularer ioniseher Verbindungen hat sieh in letzter Zeit die Sephadex- und Cellulose-Ionenaustauseher- ehromatographie ausgezeiehnet bewghrt (z. B. die Trennung yon Peptid- gemisehen auf Phosphoeellulose 5). Wie sehon vorhin erwghnt, kann die Konzentrierung der hoehverdiinnt vorliegenden Fraktionen gewisse Sehwierigkeiten mit sieh bringen. Insbesondere ist nieht in allen Fallen die Ultrafiltration, die Gefriertroeknung oder Ausf/tllung mit Neutral- salzen bei der Konzentrierung der Proteine erfolgreieh. Dagegen ist das Entsalzungsproblem in idealer Weise heute ffir hoeh- molekulare Substanzen gelSst dutch Einffihrung der Gelfiltration, die im folgenden kurz zu bespreehen sein wird 7. Sephadex ist bekanntlieh die Bezeiehnung ffir rgumlieh vernetzte I)extrane, also wasserunl6sliehe, aber doeh hydrophfle Tr/~gersubstanzen 26. Vom Vernetzungsgrad des Dextrans und der NolekfilgrSBe der zu analysierenden Substanz h~ngt es

Anwendung moderner biochemischer Analysenverf~hren 73
nun ab, ob letztere in die Poren der Gelk6rner eindringen kann oder nieht. Bei den k/~uflichen Typen liegen die oberen Ausschlul~grenzen fiir G 25 bei einem Molgewicht yon 5000, fiir G 50 bei 10000, ffir G 75 bei 50000, fiir G 100 bei 100000 und fiir G 200 bei 200000. Bei ~olekular- gewiehten oberhalb der Ausschlul3grenzen ist die Verteilung innerhalb und au•erhalb der Gelk6rner wie 0:1, Kd-Wert = 0, bei vollstandigem Durchdringungsverm6gen ist dieVerteilung 1 : 1 (K d = 1). Zwischen diesen Grenzwerten miil]ten alle Ka-Werte liegen, wenn keine zusgtzlichen Einflfisse auf die gel6sten Stoffe beim Durehwandern der Gels~ule ein- treten. Dies ist jedoch in weitem Mal~ nicht der Fall. Der Kd-Wert wird ermittelt nach folgendem Ansatz :
Ve- V0 , wobei Ve das Volumen ist Kd -- V~
vom Aufbringen der L6sung auf die S~ule bis zum Erscheinen des Maxi- mums der Konzentrat ion im Eluat. V0 ist das Ausschlul3volumen, das aui~erhalb der Gelk6rner ist und mit einem sehr hochmolekularen Stoff Ka = 0 geeieht wird. Vi ist das innere Volumen in den GelkSrnern, das ermittelt wh'd durch )/[essung der Wasseraufnahme beim Quellen der trockenen K6rner. Aus den yon GELOT~E 11 ver6ffentlichten ~e~da ten ist zu entnehmen, dal3 z.B. bei der Gelfiltration yon Aminosi~uren durch eine Sephadex G-25-S~ule mit reinem Wasser die Ka-Werte der meisten neu- tralen, aliphatischen Aminos~uren bei 0,8 liegen. Tryptophan hat da- gegen einen Ka-Wert yon 1,9, Asparagins~ure yon 0,2, Glutamins/~ure yon 0,2, Lysin yon 3,3 und Arginin yon 13. Man sieht, dal~ sowohl ein Ioneneffekt durch im Gel lokalisierte COOI-LGruppen als auch ein Ad- sorptionseffekt gegenfiber aromatisehen Verbindungen festzustellen ist. Oer Ioneneffekt kann weitgehend ausgeschaltet werden durch Ver- wendung yon Elektrolyten, z.B. Jst K a bei 0,05 m NaC1 ffir Arginin 1,0, ffir Asparagins~ure 0,8. ~hnliche Effekte treten bei den Ribonucleotiden auf. Wit verwenden in unserem Arbeitskreis laufend diesen Ioneneffekt zur Vorreinigung yon komplexen Peptidgemischen. So lassen sich bei pI-I 6,5 anodisch wandernde Peptide in einem Arbeitsgang yon s~mt- lichen neutralen und basischen trennen. Dagegen gelingt in vielen F~tllen nicht die Entsalzung niedermolekularer Substanzen nach ihrer chromatographischen Auftrennung mit den fiblichen Sephadexgelen, da die Molgewichtsunterschiede zwischen Elutionspuffer und z.B. Nucleotiden durch den Adsorptionseffekt der Keterocyclen ausgeglichen werden. Wir haben das auf Polyacrylamid- basis hergestellte Biogel untersucht, yon dem behauptet wird, dal~ es weder Ionen- noch Adsorptionswirkung zeigt. Naeh ersten Uberschlags- messungen scheint trier der Kd-Wert aueh yon der Ladung, nieht aber yon aromatischen Bestandteilen abzuh~ngen.

74 GERhArD PFLEIDEKER :
Die Hauptanwendung der Gelfiltration liegt in der Entsalzung hoch- molekularer Substanzen, so da~ sich in Znkunft eine erseh6pfende Dialyse erfibrigt. Dies is~ vor allem wiehtig bei Benutzung der Ionen- austauscherchromatographie, weft die Proteine meist auf m6gliehst salz- freies ~il ieu vor Beginn der Chromatographie umgestellt werden mfissen. Ganz hervorragend geeignet ist diese Technik bei radioaktiven Markie- rungsversuchen mit Proteinen, mit denen wir uns zur Zeit sehr stark beseh~ftigen. Es gelingt in nahezu allen Fallen, in einem Arbeitsgang das fiberschfissige radioaktive l~eagens quantitativ von dem t~eaktionspro- dukt des Proteins abzutrennen. Ganz besonders deutlich wurde das sicht- bar, als wir mit radioaktiv-markierter diazotierter Su]fanilsaure Proteine kuppe]ten, wie das in der Immunchemie vie]faeh gemaeht wird. Bekannt- lieh ist eine tagelange Dialyse notwendig, um das gekuppelte Protein yon dem fiberschiissigen Reagens zu trennen, ws wir in einem Dutch- flul~ dies erwirken konnten. Weiterhin ist die Gelfiltration in breitem Mal~e anwendbar, nm Komplexe zwischen nieder- und hochmolekularen Stoffen zu verfolgen. Wir haben das DPNtiLBindungsvermSgen ver- sehiedener Dehydrogenasen in der Weise gemessen, dal~ wir eine Sephadex G 50-Ss znerst mit einer bestimmten DPNI~I-Konzentration vorbe- handelten, dann das Enzym mit der gleichen Coenzymmenge auftrugen und wieder mit DPNK-L6sung nachspfilten. Aus der Elutionsab- sorptionskurve sieht man, dab zuerst das Enzym austritt, dann folgt ein Tal, das der DPNtLMenge entspricht, die yon dem Enzym aufgenommen wurde ~s. Vor allem ist diese Teehnik erfolgreich bei der Messung des DPNH-BindungsvermSgens yon partiell inaktivierten Enzymen. Eine Reinigung des Proteine aufgrund der Molekfilgr613e ist ebenfalls mittels der zuletzt geschflderten Teehnik m6glich. Wie verschiedene Autoren in letzter Zeit gezeigt haben, besteht ein ]inearer Zusammen- hang zwisehen der 3. Wurzel aus dem K,-Wert eines Proteins und der 2. Wnrzel aus dam Molgewieht. Wit haben die von WI~I,A~D, DVESBERG u. D~TE~MA~S a~ entwickelte Molgewiehtsbestimmung mit Sephadex G 100 oder G 200 an mehreren unreinen Enzymsystemen fiberprfift bei Verfolgung der Enzymaktivits im Eluat nnd ausgezeiehnete Resultate in ~bereinstimmung mit ~nderen physikalisehen Methoden erzielt 1. Man kann anf diese Weise mit einem Minimum yon wenigen #g Protein eine verhs exakte Bestimmung durchffihren. Schlie~lieh kann -- am besten am Ende der iibliehen l~einigungsmethoden -- eine weitere Anreicherung eines Proteins dutch Gelfiltration fiber G 100 oder 200 erwirk~ werden. Besonders vielverspreehend scheint die yon PO~ATg beschriebene Recycling-Chromatographie zu sein, wobei in einem mehr- stufigen Kreisprozel~ dieselbe Sephadexsaule durehstr6mt wurde as. tIier- bei trennen sich auch Snbstanzen mit s Molgewieht nach mehr-

Anwendung moderner biochemiseher Analysenverfahren 75
fachen Umlgufen. t t J s ~ ~ hat /~hnliehe Molekularsiebehromatogra- phien an vernetztem Polyaerylamidgel und an Agarose mit gutem Erfolg durchf/ihren k6nnen.
Sehlul~betraehtung Zum SchluB m6chte ich nochmals kurz auf die quantitative Auswer~ung yon Chromatogrammen und Pherogrammen eingehen, die sowohl auf der Trennschicht selbst als auch nach der Elution auf lichtelektrischem Wege erfolgen kann. Bei dem ersten Verfahren migt man haupts~ehlieh densitometrisch ira durchfallenden Lieht, wobei die Art des Tr~gers bei geringen Sehichtdieken keine entscheidende Rolle spielt. Die Messung kann also sowohl auf Diinnschichtplatten als aueh auf Papier, St~rkegel, Polyaery]amidgel oder ~{embranfolien durchgefiihrt werden, jedoeh ist naeh vorgesehaltetem Trennverfahren meist keine abso]ut gleichm~[~ige Farbverteilung der Substanzflecken vorhanden, so dat3 bei den heute fibliehen Meggers noeh keine optimale Genauigkeit zu erwarten ist. Dazu verlguft in vielen l~llen die ehemisehe Anf~rbereaktion nur un- vollstgndig auf dem Tr~ger ab. Als Beispiel sei die Anf~rbung yon Aminos~uren naeh Elektrophorese oder Chromatographie auf Papier dureh Behandlung mit 2,4,6-Trinitrobenzol-l-sulfos~ure erw~hnt, die erst naeh E]ution der TNB-Verbindungen und naeh Zugabe frischen Reagen- ses in der L6sung vollst/~ndig wird, d~nn aber quantitativ mit j:3~ Genauigkeit erfaBt werden kann a3. Die fr/iher/ibliehe Imprggnierung der Papiere, um sic transparent zu maehen, dureh Paraffin61-Brom- naphthalin, Benzylalkohol oder Glycerin kann in vielen F/~llen unter- bleiben. Die exaktesten Werte seheint die Messung des reflektierten Liehtes zu geben, wie sie K o R ~ u. Mitarb. 19 z.B. mit Hflfe einer Ulbrieht- Kugel und eines selbstregistrierenden Spektralphotometers aufPapier mit Substanzmengen yon 0,1--5 #g beschrieben haben. I~GL~ u. MXNSI~ALL ~v untersuchten die relative Abweiehung der l~IeBwerte bei Trans- missions- und Reflexionsmessungen mit transparent gemaehtem und niehtbehandeltem Papier. Die besten Werte wurden mit troekenem Papier erzielt.
Die obigen Verfahren erlauben ohne den groBen apparativen Aufwand dcr Stein- und Moore-Teehnik Aminosgurenanalysen naeh Dosv.S oder R6WE, F E ~ R u. FISCItE~ 29 in Proteinhydrolysaten mit 5--10% Fehlerbreite. WALL~r~LS U. A ~ s a~ ermittelten nach Umsetzung eines Proteinhydrolysats mit Dflfitrofluorbenzol und ansehlieBender papierchromatographiseher Auftrennung des Gemisehes die Amino- s/~urenzusammensetzung der Alkoholdehydrogenase. Sehlieftlich lassen sieh -- wic zu Anfang erw~hnt -- auf diese Weise Bausteinanalysen fiir RNS und DNS durehf/ihren. Meine Ansf/ihrungen sollten zeigen, dag

76 G. PFLEIDERER : Anwendung moderner biochemischer Analysenverfahren
j ede M e t h o d e ffir b e s t i m m t e Z w e c k e o p t i m a l se in k a n n , es a b e r k e i n e
u n i v e r s e l l a n w e n d b a r e T e e h n i k g ib t , s o n d e r n d e r w a h r e E r f o l g i n d e r
g e s c h i c k t e n K o m b i n a t i o n d e r v e r s e h i e d e n e n V e r f a h r e n l iegt .
Zusammenfassung O h n e d ie in d e r m o d e r n e n b i o c h e m i s c h e n F o r s c h u n g b e n f i t z t e n ~ e t h o d e n
a u s f f i h r l i e h zu e r l s w i r d die A n w e n d u n g s b r e i t e d e r V e r t e i l u n g s - ,
A d s o r p t i o n s - u n d I o n e n a u s t a u s c h c h r o m a t o g r a p h i e , d e r E l e k t r o p h o r e s e
m i t u n d o h n e T r a g e r u n d d e r G e l f i l t r a t i o n n a c h n e u e s t e n E r k e n n t n i s s e n
ge sch f lde r t . E s w i r d a n we i r g e s t r e u t e n B e i s p i e l e n geze ig t , da[~ j ede
M e t h o d e ffir s i ch a l l e in i h r e G r e n z e n be i d e r A n a l y s e u n d p r s
T r e n n u n g n i ede r - u n d h o c h m o l e k u l a r e r S t o f f g e m i s c h e h a t u n d l e t z t ] i c h
d ie K o m b i n a t i o n m e h r e r e r V e r f a h r e n d e n b e s t e n E r f o l g gewi~hr le i s te t .
Literatur 1 A v ~ i c c ~ o , F., u. C. B. BRuNI: Biochem. Z. 840, 321 (1964). -- 2 BA_TE-SIVIITIt, E. C., and R. G. WESTz%LL: Biochem. biophysica Acta 4, 427 (1950). - - 3 BAYIng, E. : Angew. Chem. 71, 299 (1959). - - 4 BArley, E., K. H. REVT~ER U. F. BORN : Angew. Chem. 69, 640 ( 1 9 5 7 ) . - 5 CANFIELD, R. E., and C. B. ANFINSEN: J. biol. Chemistry 288, 2684 (1963). - - ~ CONS])EN, R., A. H. GORDON, and A . J . P . MARTIN: Bio- chemic. J . 88, 224 (1944). - - ~ ])]~,TER~ANN, I t . : Angew. Chem. 76, 635 (1964). - - s DosE, K. : Biochem. Z. 329, 416 (1957). - - 9 E])sT~5~[, J. E. : Biochim. biophysica Acta 9, 528 (1952). - - lo GEDIN, H. I., u. J . PoR~T~: Biochim. biophysica Acta 26, 159 (1957). -- 21 GELOTTE, B.: J. Chromatogr. (Amsterdam) 8, 330 (1960). - - 22 GRASS~ANN, W., u. K. I-I)_NNm: Naturwissenschaften 87, 397 (1950). - - Hoppe- Sey]ers Z. physiol. Chem. 292, 32 (i953). - - la H~-Lr~r , H. : Chem.-Ing.-Techn. 35, 488 (1963). - - 24 ~tANR~[G, K. : Hoppe-Seylers Z. physiol. Chem. 388,211 (1964) . - 15 HZERT~N, S. : Arch. Biochem. Biophysics, Suppl. 1,147 (1962). - - ~6 HOLLEY, R. W., J . ArGA~, B. P. DOCTOR, J . FARROW, IV[. A. MARINI, and S. I t . MERRILL : J. biol. Chem- istry 236, 200 (1961). - - ~7 INGLE, 1%. B., and E. MINS~L~: J. Chromatogr. (Amsterdam) 8, 369 (1962). -- ~s K ~ v , W., u. H. G. Z~c~kv: Biochim. biophysica Acta 91,549 (1964). -- ~KoRTE, F., u. H. W E ~ M P : Angew. Chem. 70,434 (1958). - - ~0 KU~KEL, I t . G., in D. GL~C~: Meth. of Biol. Anal., Vol. I, p. 156. New York: Inter- sci. Publ. 1954. -- ~1LA~-GE, V. : Biochem. Z. 388, 503 (1961). - - ~e M A ~ , A. J. P., and 1~. L. SYNGE : Biochemic. J. 35, 1358 (1941). - - ea PFLE~D]~RE~, G., u. F. Au~C- c ~ o : Biochem. Biophys. Res. Comm. 16, 53 (1964). -- e~ PE~E~])~ER, G., u. E. D. W ~ c ~ s ~ v ~ : Bioehem. Z. 334,185 (1961). -- ~ PO~kT~, J. , and H. BENN~Cl~: Arch. Biochem. Biophysics., Suppl. 1,152 (1962). - - "~a P O ~ T ~ , J . , and P. FLOD~N: Nature (London) 183, 1657 (1959). - - e~ RE~C~L, R. : Mh. Chem. S6, 69 (1955). - - ~s RIc~EY, J. M., H. G.]~C]ZEY, and 1~. Sc~a~ER: Analyt. Biochemistry9, 272 (i964). -- ~ RSWE, K., E. FlY,BEn U. H. FISC~]~R: Hoppe-Seylers Z. physiol. Chem. 818, 174 (1958). - - ~0 ScHuyler, I t . K., u. 1%. BVL~SC~: Z. Naturs 10b, 683 (1955). - - a~ SChNIT- ZEl, H., K. P~r~NBE~G, E. G~NSE, R. CZOK, T. B i i c ~ u. H. AD~I : Biochem. Z. 382, 167 (1959). - - ae SCn~aE~B]~R, G., M. EEKS~E~N, A. 0SE~ u. H. HOLZE~: Biochem. Z. 340, 35 (1964). -- 3a S~INODA, T., and K. SkTAK]~: J. Biochemistry 50, 293 (1961). - - a~ S ~ T ~ S , 0 . : Biochemic. J. 61, 629 (1955); 71,585 (1959). -- a~ S~I~H~ES, O. : Advances Prote in Chemistry 14, 65 (1959). - - a~ SOBEr, H. A., and E. A. PE~]~RSON: J. Amer. chem. Soe. 76, 1711 (1954). -- aT S T y , E. : Pharmazie 11, 633 (1956). - - as STOUFFER, J. E., T. E. KERSTEN, and P. IV[. K~VEGE~: Bio-

BERG~EYER: Grundlagen und Fortschritte der enzymatischen Analyse 77
ehim. biophysica Acta 93, 191 (1964). -- a9 WALLE:NFELS, K., u. A. ARENS: Biochem. Z. 832, 217 (1960). -- ~0 W~YGA~D, F., A. PI~OX, L. SC~nl)~AM~ER U. W. K6~IG: Angew. Chem. 75, 282 (1963). -- ~1 WIELA~*]), TI~., P. DUES]3EI~G U. H. D~TER~IA~I: Biochem. Z. 387, 303 (1963). -- 42 WIELA~D, T~., u. D. GEOI~GO- rOVLOS: ]~iochem. Z. 840, 476 (1964). -- ha V~TIELA~D, TH., U. H. DETEICMAN~: Experientia 18, 431 (1962). -- ~4 WIELA~D, TH., G. PFLEIDElCER, I. HAUPT U. W. W61~EI~: Biochem. Z. 832, 1 (1959). -- a~ WI~LAxI), T~., B. SA~D~A~ - U. G. P~L]~ID~RnR: Biochem. Z. 328, 239 (1956).
Prof. Dr. G. P]~L]~ID]~RI~I%, Inst i tut fiir Biochemie im Inst i tut ffir organische Chemie, J. W. Goethe-Universits 6 Frankfur t /M, Robert Mayer-StraBe 7--9
Grundlagen und Fortsehritte der enzymatisehen Analyse HANS ULRICH BERGMEYER
Aus der Biochemischen Abteilung Tutzing der C. F. Boehringer & Soehne GmbH, Mannheim
Eingegangen am 19. M~rz 1965
Summary. A review of enzymatic analysis is given and the newer developments are shown. Some examples are discussed.
Einleitung Ich fiirchte, das Thema ein wenig zu verfehlen, wenn ich fiber Grundlagen und Fort- schritte der enzymatischen Analyse berichten soll. Die Grund]agen sind weitgehend bekannt und die Fortschritte sind nicht iiberw~ltigend, nachdem Mitre der dreiBiger Jahre WAI~BUI~G 86 bereits ein so umfassendes Konzept gab fiber die M6glichkeiten, mit Enzymen Analysen durchzuffihren, dab schwerlich grundlegend Neues hinzu- kommen konnte. Nur klarere Begriffe lassen uns das Gebiet heute vielleicht besser fibersehen und jetzt verfiigbare reinste Enzyme lassen auch komplizierte Mei~- anordnungen gelingen.
A n a l y s e m i t H i l f e y o n E n z y m e n u n d die B e s t i m m u n g der A k t i v i t s v o n
E n z y m e n als M e r k m a l des f u n k t i o n e l l e n Z u s t a n d e s ihres Trs das i s t e n z y m a t i s c h e A n a l y s e s.
Zweifellos sind Fortschritte in den apparativen !Viel3anordnungen zu verzeichnen. Ich denke an das Zweistrahlphotometer (double beam)la, an die vibrierende oder rotierende Platinelektrode 2~, an den pH-Stat 14 zur Registrierung yon Hydrolase- reaktionen. Fortsehritte sind auch in den Ergebnissen mit Hilfe der enzymatischen Analyse erzielt worden: die Entdeekung der Isoenzyme 20, ihr Nachweis mit der ,,Sprfihteehnik ''as nach elektrophoretischer Trennung. Und die Auffindung der ,,proportionskonstanten Enzymgruppen" in verschiedenen Organen 1~
A b e r m e i n T h e m a g e h t y o n d e n G r u n d l a g e n aus u n d soll die F o r t s c h r i t t e
in d e n e h e m i s c h e n P r i n z i p i e n de r e n z y m a t i s e h e n A n a l y s e aufze igen .





























