Embed Size (px)
Citation preview

This work has been digitalized and published in 2013 by Verlag Zeitschrift für Naturforschung in cooperation with the Max Planck Society for the Advancement of Science under a Creative Commons Attribution4.0 International License.
Dieses Werk wurde im Jahr 2013 vom Verlag Zeitschrift für Naturforschungin Zusammenarbeit mit der Max-Planck-Gesellschaft zur Förderung derWissenschaften e.V. digitalisiert und unter folgender Lizenz veröffentlicht:Creative Commons Namensnennung 4.0 Lizenz.
Activated Nitriles in Heterocyclic Synthesis: The Reaction of Cinnamonitrile Derivatives with Active Methylene Reagents
Galal Eldin Hamza Elgemeie*. Samier Anwar Elees, Ibraheim Elsakka, and Mohamed Hilmy Elnagdi Chemistry Department, Faculty of Science, Minia University, Minia, A . R . E g y p t
Z. Naturforsch. 38b, 639-642 (1983); received November 2, 1982
Heterocyclic Synthesis, Cinnamonitrile Derivatives
The reaction of substituted cinnamonitrile derivatives with cyanoacetanilide and ethyl acetoacetate is reported. Several new polyfunctional pyridine and pyran derivatives could be synthesised. The structures of the prepared compounds and the mechanisms of their formation is reported.
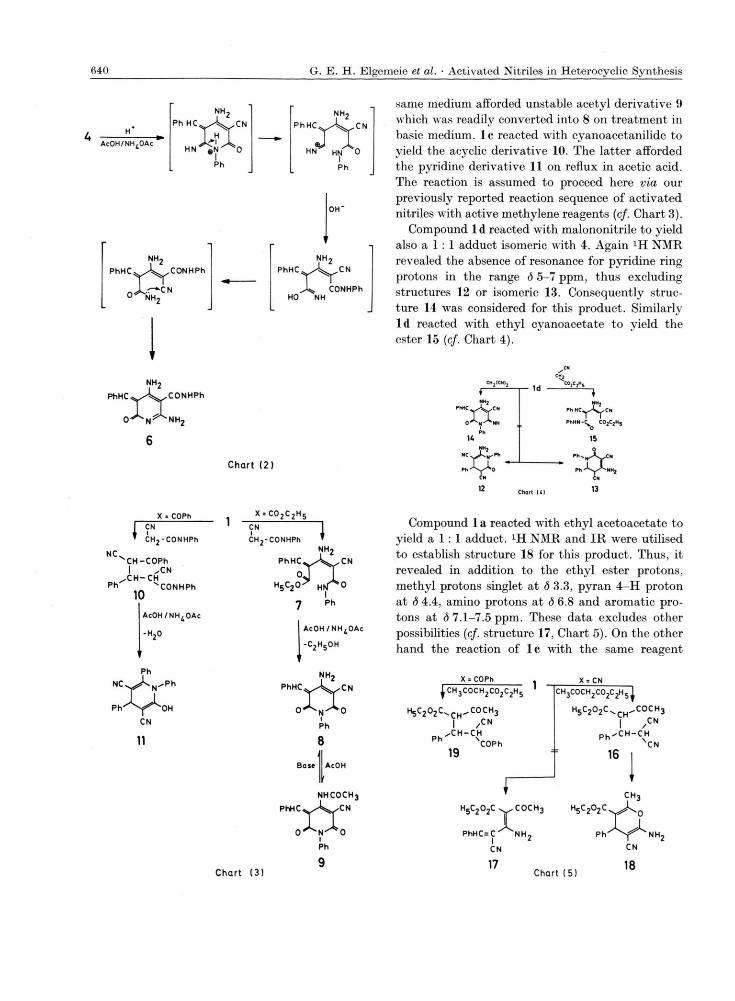
In the last decade we have been involved in a program aiming to explore the synthetic potential, scope and limitations of a,ß-unsaturated nitrile derivatives in heterocyclic synthesis [1,2]. Several new approaches for synthesis of five, six and fused heterocyclic derivatives could be achieved during this work [3-6]. As a part of this program the reaction of the cinnamonitrile derivatives la - c with cyanoacetanilide and with ethyl acetoacetate was investigated. Thus, it has been found that 1 a reacts with cyanoacetanilide to yield a 1 : 1 adduct. Several theoretically possible isomeric structures were con-sidered (cf. structures 2-5, Chart 1). Although struc-tures 2 and 3 seemed more likely based on analogy to the well established behaviour of l a toward active methylene reagents [7], both structures were ruled out based on XH NMR which revealed the absence of any proton resonance between <5 4-7 ppm for pyridine 4-H proton in either 2 or 3. XH NMR also ruled out structure 5 as it revealed also the absence of pyrimidine-H resonance. Thus, structure 4 was considered for the reaction product. The for-mation of 4 from the reaction of l a and cyano-acetanilide may be assumed to proceed via addition of the amide nitrogen to the cyano group in 1 a and subsequent cyclisation into 4. Alternately addition of the methylene group to the cyano group in 1 a followed by cyclisation by addition of the amide nitrogen to the other cyano group would also lead to the formation of 4.
When 4 was heated with acetic acid in the pre-sence of aqueous ammonium acetate solution it rearranged into the amide 6. The formation of 6 is
* Reprint requests to Dr. G. E. Hamza Elgemeie. 0340-5087/83/0500-0639/$ 01.00/0
assumed to proceed via acid catalysed ring opening of 4 and recyclisation (cf. Chart 2).
CN /
Ph C H = C
X 1a: X*CN l b : X=CO2C2H5
1c: x =coPh 1d: X = CONHPH
H 2 n
5
Ph
CN
C H P h
I NL.
NC
/
NH PhHC* ^
NC COCH 2 CN
CN
PhHC
I
Ph
* \
CH,-CONHPh
NH2
P h H C ^ A ^ C N
NC CONHPh
Ph' J s Sj^NH 2
CN
t P h - N - C - C H , C N
I 2
P h - C H - C H ( C N ) ,
l a
CN I CH,-CONHPh
P h - N H - C - C H - C N I
P h H C - C H ( C N ) 2
1 1/ Ph
N H ,
Chart (1)
CN
2
Compound l b reacted with cyanoacetanilide in ethanol and in presence of triethylamine to yield the acyclic adduct 7. The latter cyclised into 8 on reflux in acetic acid for 5h. Long reflux of 7 in the
640 G. E. H. Elgemeie et al. • Activated Nitriles in Heterocyclic Synthesis
AcOH/NH OAc
P h H C
P h HC CN
HN
P h H C CN
HfT HN 0
C O N H P h P h H C ^ ^ C N
^ CONHPh HO NH
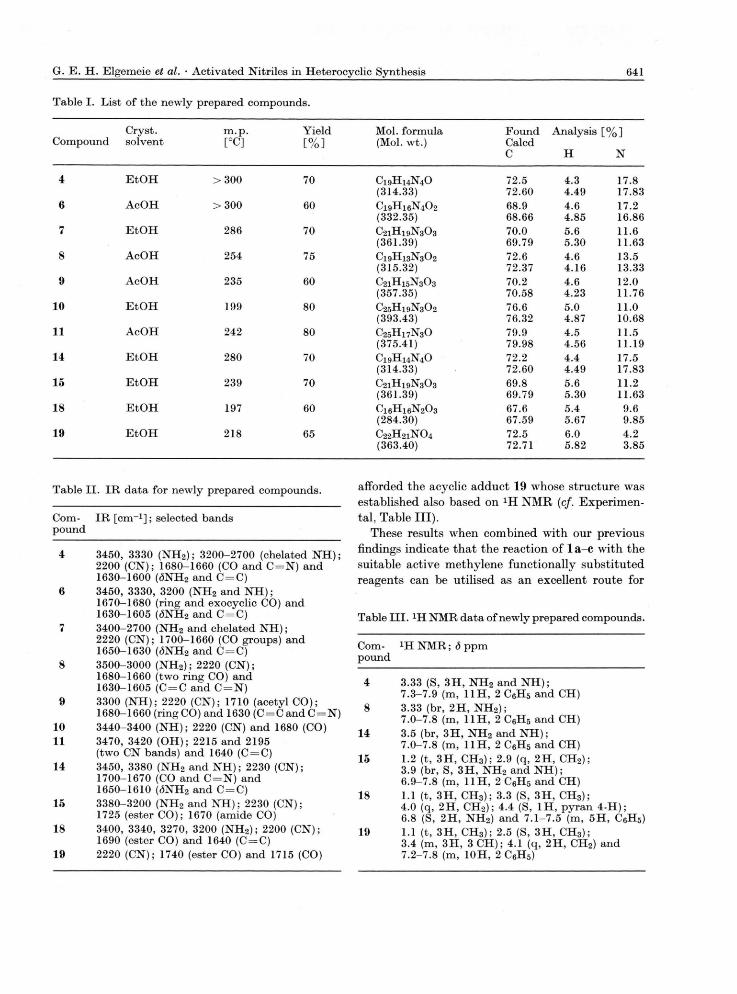
same medium afforded unstable acetyl derivative 9 which was readily converted into 8 on treatment in basic medium, l c reacted with cyanoacetanilide to yield the acyclic derivative 10. The latter afforded the pyridine derivative 11 on reflux in acetic acid. The reaction is assumed to proceed here via our previously reported reaction sequence of activated nitriles with active methylene reagents (cf. Chart 3).
Compound 1 d reacted with malononitrile to yield also a 1 : 1 adduct isomeric with 4. Again *H NMR revealed the absence of resonance for pyridine ring protons in the range <5 5-7 ppm, thus excluding structures 12 or isomeric 13. Consequently struc-ture 14 was considered for this product. Similarly ld reacted with ethyl cyanoacetate to yield the ester 15 [cf. Chart 4).
NH,
PhHC^xW^CONHPh
Chart (2)
* |— Id
NHj -V-CN
N NH Ph
JÜ2 Ph
* NH2
PhHC- V CN PhHN-t C02C2H5 0
15
PhV|(yJL,CN Ph
13
X = COPh
1 T CHj-CONHPh
N C . C H - C 0 P h I CN
/CH-CH P h ^ C O N H P h
10
A c O H /NH 4 O A c
-H ,0
P h
N C ^ s k ^ P h
P h - ^ y ^ O H CN
11
X = C O 2 C 2 H 5
TS 1 CH 2 -C0NHPH »
NH,
PhHC<^A^CN
H 5 C 2 0 ^ HKI 0
ACOH/NH 4OAC
- C 2 H 5 O H
NH 2
O ^ n ^ - O i P h
8 Base AcOH
N H C O C H J
PhHC A ^ / C N
Compound 1 a reacted with ethyl acetoacetate to yield a 1 : 1 adduct. XH NMR and IR were utilised to establish structure 18 for this product. Thus, it revealed in addition to the ethyl ester protons, methyl protons singlet at d 3.3, pyran 4-H proton at d 4.4, amino protons at d 6.8 and aromatic pro-tons at <3 7.1-7.5 ppm. These data excludes other possibilities (cf. structure 17, Chart 5). On the other hand the reaction of l c with the same reagent
^ C H 3 C O C H 2 C O 2 C 2 H 5
HSC 2 0 2 C C O C H 3
, C N
1 X = CN
P h
I /v
. C H - C H
19 COPh
CH 3 C0CH 2 C0 2 C 2 H 5 |
H 5 C 2 ° 2 C - C H - C O C H 3
I , C N
p h . C H - C H XCN
16
r H 5 C 2 O 2 C _ C O C H 3
C H ,
Chart (3)
P h H C = C ' ~NH I CN
17
H 5 C 2 O 2 C
P h
Chart (5)
CN
18
N H ,
G. E. H. Elgemeie et al. • Activated Nitriles in Heterocyclic Synthesis 641
Table I. List of the newly prepared compounds.
Compound Cry st. solvent
m.p. ra
Yield [ % ]
Mol. formula (Mol. wt.)
Found Calcd C
Analysis [ % ]
H N
4 E t O H >300 70 C19H14N4O 72.5 4.3 17.8 (314.33) 72.60 4.49 17.83
6 A c O H >300 60 C19H16N4O2 68.9 4.6 17.2 (332.35) 68.66 4.85 16.86
7 E t O H 286 70 C2IH19N303 70.0 5.6 11.6 (361.39) 69.79 5.30 11.63
8 A c O H 254 75 CI9HI3N302 72.6 4.6 13.5 (315.32) 72.37 4.16 13.33
9 A c O H 235 60 C2IH 1 5N303 70.2 4.6 12.0 (357.35) 70.58 4.23 11.76
10 E t O H 199 80 C2 5HI9N302 76.6 5.0 11.0 (393.43) 76.32 4.87 10.68
11 A c O H 242 80 C25H17N3O 79.9 4.5 11.5 (375.41) 79.98 4.56 11.19
14 E t O H 280 70 C19H14N4O 72.2 4.4 17.5 (314.33) 72.60 4.49 17.83
15 E t O H 239 70 C2IHI9N303 69.8 5.6 11.2 (361.39) 69.79 5.30 11.63
18 E t O H 197 60 CI6HI6N203 67.6 5.4 9.6 (284.30) 67.59 5.67 9.85
19 E t O H 218 65 C22H2IN04 72.5 6.0 4.2 (363.40) 72.71 5.82 3.85
Table II . I R data for newly prepared compounds.
Com- I R [cm - 1 ] ; selected bands pound
4 3450, 3330 (NH2); 3200-2700 (chelated N H ) ; 2200 (CN); 1680-1660 (CO and C = N) and 1630-1600 (<5NH2 and C = C)
6 3450, 3330, 3200 (NH2 and N H ) ; 1670-1680 (ring and exocyclic CO) and 1630-1605 (<5NH2 and C = C)
7 3400-2700 (NH2 and chelated NH); 2220 (CN); 1700-1660 (CO groups) and 1650-1630 (<5NH2 and C = C)
8 3500-3000 (NH2); 2220 (CN); 1680-1660 (two ring CO) and 1630-1605 (C = C and C = N)
9 3300 (NH); 2220 (CN); 1710 (acetyl CO); 1680-1660 (ring CO) and 1630 (C = C and C = N)
10 3440-3400 (NH); 2220 (CN) and 1680 (CO) 11 3470, 3420 (OH); 2215 and 2195
(two CN bands) and 1640 (C = C) 14 3450, 3380 (NH2 and N H ) ; 2230 (CN);
1700-1670 (CO and C = N ) and 1650-1610 (<5NH2 and C = C)
15 3380-3200 (NH2 and N H ) ; 2230 (CN); 1725 (ester CO); 1670 (amide CO)
18 3400, 3340, 3270, 3200 (NH2); 2200 (CN); 1690 (ester CO) and 1640 (C = C)
19 2220 (CN); 1740 (ester CO) and 1715 (CO)
afforded the acyclic adduct 19 whose structure was established also based on !H NMR (cf. Experimen-tal, Table III).
These results when combined with our previous findings indicate that the reaction of 1 a-c with the suitable active methylene functionally substituted reagents can be utilised as an excellent route for
Table III . 1 H N M R data of newly prepared compounds.
Com- i H N M R ; ö ppm pound
4 3.33 (S, 3H, N H 2 and N H ) ; 7.3-7.9 (m, 1 1 H , 2 C 6 H 5 and CH)
8 3.33 (br, 2H, NH 2 ) ; 7.0-7.8 (m, 1 1 H , 2 C 6 H 5 and CH)
14 3.5 (br, 3H, N H 2 and N H ) ; 7.0-7.8 (m, 1 1 H , 2 C 6 H 5 and CH)
15 1.2 (t, 3H, CH 3 ); 2.9 (q, 2H, CH2); 3.9 (br, S, 3 H , N H 2 and N H ) ; 6.9-7.8 (m, 1 1 H , 2 C 6 H 5 and CH)
18 1.1 (t, 3H, CH 3 ) ; 3.3 (S, 3H, CH3); 4.0 (q, 2H, CH2); 4.4 (S, I H , pyran 4-H); 6.8 (S, 2H, NH 2 ) and 7.1-7.5 (m, 5 H , C6H5)
19 1.1 (t, 3H, CH 3 ); 2.5 (S, 3H, CH3); 3.4 (m, 3H, 3 CH); 4.1 (q, 2H, CH2) and 7.2-7.8 (m, 10H, 2 C6H5)

642 G. E. H. Elgemeie et al. • Activated Nitriles in Heterocyclic Synthesis 642
synthesis of several, otherwise difficulty accessible, heterocyclic derivatives in most cases via initial Michael addition. However, several exceptions to this generalisation were observed. Thus, cautious should be made on suggesting structures for reac-tion products of similar reactions based on analogy to our results.
Experimental
All melting points are uncorrected. IR spectra were obtained (KBr) on a Pye Unicam Sp-1000 spectrophotometer or a Shimadzu IR 200. *H NMR were measured on a Varian EM-390-90 MHz in DMSO using TMS as internal standard and chemical shifts are expressed as d ppm. Analytical data were obtained from the analytical data unit at Cairo University. Compounds 1 a-d were prepared follow-ing literature procedures [8-10].
The reaction of l a - c with cyanoacetanilide and with ethyl acetoacetate
General procedure: To a suspension of each of 1 a-c (0.01 mole) in ethanol (30 ml) and cyanoacetanilide or ethyl acetoacetate (0.01 mole) and one ml of triethylamine were added. The reaction mixture was refluxed for 6 h then cooled and poured into water. The solid product, so formed, was collected
by filtration and crystallised from the proper solvent {cf. Table I).
Reaction of 4, 7 and 10 with acetic acid -ammonium acetate mixture
General procedure: To a solution of each of 4, 7 and 10 (0.01 mole) in acetic acid (30 ml), ammonium acetate (0.03 mole) was added. The reaction mixture was boiled under reflux for 6 h. The solid product, so formed, was collected by filtration and crystallised from the proper solvent (cf. Table I).
Reaction of 8 with acetic acid
A solution of 8 (0.01 mole; 3.03 g) in acetic acid (20 ml) was refluxed for 12 h. The solvent was then evaporated to half its original volume. The solid product, so formed, was collected by filtration and crystallised from the proper solvent (cf. Table I).
Reaction of 1 with malononitrile and ethyl cyanoacetate
A solution of 1 (0.01 mole; 2.5 g) in ethanol (30 ml), malononitrile or ethyl cyanoacetate (0.01 mole) and 1 ml of triethylamine, the reaction mixture was refluxed for 5 h, then colled and poured into water. The solid product, so formed, was collected by filtration and crystallised from the proper solvent (cf. Table I).
[1] M. H. Elnagdi, H. A. Elfahham, M. R . H. Elmog-hayar, K . U. Sadek, and G. E . H . Elgemeie, J. Chem. Soc. Perkin Trans. I 1982, 989.
[2] M. H. Elnagdi, Tetrahedron 30, 2791 (1974). [3] S. M. Fahmy, N. M. Abed, R . M. Mohareb, and
M. H. Elnagdi, Synthesis 1982, 490. [4] M. H. Elnagdi and H. Wamhoff, J. Heterocycl.
Chem. 18, 1287 (1981). [5] M. H. Elnagdi, H. A . Elfahham, S. A . S. Ghozlan,
and G. E . H. Elgemeie, J. Chem. Soc. Perkin Trans. I, 2667 (1982).
[6] H. A . E . Daboun, S. E . Abdou, M. M. Hussein, and M. H. Elnagdi, Synthesis 1982, 502.
[7] M. R. H. Elmoghayar, M. K . A. Ibraheim, M. A. Khalifa, and M. H.^Elnagdi, Monatsh. Chem. 113, 57 (1982).
[8] G. N. Sausen, V. A . Engelhardt, and W . J. Middleton, J. Am. Chem. Soc. 80, 2815 (1958).
[9] E. Campaigne, G. F . Bulbenko, W . E . Kreigh-baum, and N. R. Maulding, J. Org. Chem. 27, 4482 (1962).
[10] H. Kauffmann, Ber. Dtsch. Chem. Ges. 50, 527 (1917).


![Gehinderte Ligandbewegungen in Übergangsmetall-Komplexen ...zfn.mpdl.mpg.de/data/Reihe_B/38/ZNB-1983-38b-1424.pdf4.0 International License. ... (CO)2(C4H6)2 ergibt [15], ist dieser](https://img.pdfslide.org/doc/110x75/5cebc78188c993031a8bfeb6/gehinderte-ligandbewegungen-in-uebergangsmetall-komplexen-zfnmpdlmpgdedatareiheb38znb-1983-38b-1424pdf40.jpg)




![Über das [C H Fe (CO) ]--Anion (C H = Cycloheptatrienyl)zfn.mpdl.mpg.de/data/Reihe_B/38/ZNB-1983-38b-1446.pdf · Z. Naturforsch. 38b, 1446-1453 (1983); eingegangen am 7. ... von](https://img.pdfslide.org/doc/110x75/5a7894a97f8b9aa2448e0c3a/ber-das-c-h-fe-co-anion-c-h-cycloheptatrienylzfnmpdlmpgdedatareiheb38znb-1983-38b-1446pdfz.jpg)








![NaCoS - ein Thiocobaltat mit isolierten CoS -Anionenzfn.mpdl.mpg.de/data/Reihe_B/38/ZNB-1983-38b-0012.pdf · manganat, Na2Mn2S3, darstellen und seine Struktur bestimmen konnten [5],](https://img.pdfslide.org/doc/110x75/5e5b6cb5db395d6f74163149/nacos-ein-thiocobaltat-mit-isolierten-cos-manganat-na2mn2s3-darstellen-und.jpg)




![Asymmetrische Katalysen, 13 [1] Chelat-Liganden und ihre ...zfn.mpdl.mpg.de/data/Reihe_B/38/ZNB-1983-38b-1332.pdf · Hydrosilylierung von Acetophenon mit Diphenyl-silan katalysieren](https://img.pdfslide.org/doc/110x75/5e5e18a05cbcc1776a3c8b22/asymmetrische-katalysen-13-1-chelat-liganden-und-ihre-zfnmpdlmpgdedatareiheb38znb-1983-38b-1332pdf.jpg)





