Embed Size (px)
Citation preview

Herstellung von Kaliumbromat
Laborbericht
von
Simon Wyss
Martin Rüegg
Gruppe 17
Horgen, den 23. November 2009
Dozent:
Dr. Christian Hinderling

Laborbericht Seite 2
23. November 2009
I Zusammenfassung Kaliumbromat ist ein mutagener Stoff, welcher trotz dieser Eigenschaft in der Backindustrie
eingesetzt wird, um den Teig luftiger zu machen. Der Stoff ist in der Lebensmittelbranche
bekannt unter der Bezeichnung E924, ist aber unterdessen in den meisten Ländern verbo-
ten, da sich das Kaliumbromat, bei zu hoher Konzentration im Mehl, beim Backvorgang nicht
vollständig zersetzt.
In diesem Bericht wird gezeigt wie Kaliumbromat über die Disproportionierung von Brom mit
Kalilauge hergestellt werden kann. Die Identität des dabei erhaltenen Kaliumbromats wurde
mittels Infrarotspektroskopie nachgewiesen. Das erhaltene Spektrum ist mit dem Referenz-
spektrum des „National Institute of Standards and Technology“ (NIST) vergleichbar.
Die Gehaltsbestimmung der hierbei erhaltenen Kristallisate erfolgte mittels einer Redox-
Titration, genauer gesagt mittels Iodeometrie. Der Gehalt des Kaliumbromats betrug 97.3
g/100g (± 2.2 g/100g).

Laborbericht Seite 3
23. November 2009
Inhaltsverzeichnis I Zusammenfassung .............................................................................................................. 2
Inhaltsverzeichnis .................................................................................................................. 3
1 Zielsetzung / Aufgabenstellung ........................................................................................... 4
2 Theoretischer Teil ............................................................................................................... 4
2.1 Kaliumbromat ............................................................................................................... 4
2.2 Sauerstoffsäuren der Halogene ................................................................................... 4
2.2.1 Disproportionierung ................................................................................................... 5
2.3 Analysentechniken ....................................................................................................... 5
2.3.1 Spektroskopische Analysentechniken.................................................................... 5
2.3.2 Iodometrie ............................................................................................................. 6
3 Geräte und Materialien ....................................................................................................... 8
3.1 Chemikalien und Materialien ........................................................................................ 8
3.2 IR ................................................................................................................................. 8
4 Praktischer Teil ................................................................................................................... 9
4.1 Herstellung von Kaliumbromat ..................................................................................... 9
4.1.1 Erste Stufe: Herstellung des Eisenkomplexes ....................................................... 9
4.1.2 Umkristallisation .................................................................................................... 9
4.1.3 Vernichtung von überschüssigem Brom ................................................................ 9
4.2 Analytik .......................................................................................................................10
4.2.1 IR-Spektroskopie ..................................................................................................10
4.2.2 Iodometrie ............................................................................................................10
5 Ergebnisse ........................................................................................................................13
5.1 Synthese und Umkristallisation ...................................................................................13
5.2 Analytik .......................................................................................................................13
5.2.1 IR .........................................................................................................................13
5.2.2 Iodometrie ............................................................................................................16
5.3 Ausbeute ....................................................................................................................17
6 Diskussion .........................................................................................................................18
7 Literaturverzeichnis ...........................................................................................................19
8 Verzeichnis der Abbildungen .............................................................................................20
Laborbericht Seite 4
23. November 2009
1 Zielsetzung / Aufgabenstellung Im Rahmen des allgemeinen chemischen Grundpraktikums soll Kaliumbromat aus Brom
hergestellt werden. Das dabei erhaltene Produkt soll mittels Infrarotspektroskopie charakteri-
siert und anschliessend der Gehalt mittels Iodometrie bestimmt werden.
Anhand dieses Versuchs soll ein Einblick in die Chemie der Sauerstoffsäure und die Dispro-
portionierungsreaktionen gegeben werden.
2 Theoretischer Teil
2.1 Kaliumbromat
Kaliumbromat ist bei Raumtemperatur ein weisses, kristallines Pulver. Kaliumbromat ist giftig
und brandfördernd und besitzt mutagene Eigenschaften. Heutzutage wird Kaliumbromat
noch als Mehlzusatz verwendet (E924), um den Teig beim Backvorgang zu vergrössern.
Wenn nicht zu viel eingesetzt wird, zersetzt sich das Kaliumbromat im Ofen komplett und das
Brot ist ungiftig.
Als Nahrungsmittelzusatz ist Kaliumbromat unterdessen jedoch in den meisten Ländern nicht
mehr zugelassen. (1)
2.2 Sauerstoffsäuren der Halogene
Von den Halogenen gibt es verschiedene Verbindungen mit Sauerstoff und Wasserstoff,
welche alle Säurecharakter besitzen. Von Chlor, Brom und Iod sind Oxosäuren mit einem
oder mehreren Sauerstoff-Atomen bekannt, vom Fluor jedoch nur die HOF-Verbindung
(Hypofluorige Säure), welche sehr instabil ist. Die Oxidationszahlen der Halogene reichen
dabei von +I bis +VII, wobei nur ungerade Oxidationszahlen anzutreffen sind.
Tabelle 1: Bekannte Oxosäuren der Halogene
Oxidationszahl des
Halogens
Zusammensetzung Name der Säure Name des Anions
+I HOF, HOCl, HOBr,
HOI
Hypohalogenige
Säure
Hypohalogenit
+III HClO2 Halogenige Säure Halogenit
+V HClO3, HBrO3, HIO3 Halogensäure Halogenat
+VII HClO4, HBrO4, HIO4 Perhalogensäure Perhalogenat
H4I2O9, H5IO6
Die Halogen-Sauerstoff-Bindungen sind kurze, stark polare Bindungen die oftmals auch mit
Doppelbindungen beschrieben werden. Die Oxosäuren der Halogene sind starke Oxidati-

Laborbericht Seite 5
23. November 2009
onsmittel, so dass einige der Säuren theoretisch auch Wasser bzw. OH- Ionen oxidieren
können, jedoch läuft diese Reaktion so langsam ab, dass die Oxohalogen-Verbindungen in
wässrigen Lösungen dennoch haltbar sind. (2)
2.2.1 Disproportionierung
Bei der Disproportionierung (auch Dismutation) handelt es sich um eine Redox-Reaktion, bei
welcher ein Molekül zugleich oxidiert und reduziert wird, dabei entstehen aus einer Verbin-
dung zwei Produkte. Beispielsweise disproportioniert Brom (Br2), bei der Anwesenheit von
Kaliumhydroxid (KOH), zu Bromid (Br-) und Bromat (BrO3-). Bei diesem Beispiel ist es jedoch
so, dass nur jedes sechste Brom-Atom in eine Bromat-Verbindung disproportioniert. Der
Grund dafür ist, dass die Oxidationszahl des Broms nach Bromid nur auf –I springt, bei der-
jenigen zum Bromat jedoch auf +V.
3 Br2 + OH-
5 Br-
BrO3-+ + 3 H2O
Bei den Salzen der Halogene sind die Halogenate und die Perhalogenate immer instabiler
als die Halogenide.
Das Gegensteil der Disproportionierung nennt man Komproportionierung oder Syn-
proportionierungt. Bei dieser Reaktion wird analog zur Disproportionierung durch gleichzeiti-
ge Reduktion und Oxidation, zweier Atome des gleichen Elements, welche jeweils eine hohe
und eine niedrige Oxidationsstufe aufweisen, in eine mittlere Oxidationsstufe umgewandelt.
(2) (3)
4 OH-
2 MnO4-
3 Mn2+
5 MnO2 2 H2O+ + +
2.3 Analysentechniken
2.3.1 Spektroskopische Analysentechniken
Spektroskopie ist ein Verfahren bei welchem anhand des Spektrums, die Wechselwirkungen
zwischen elektromagnetischer Strahlung und Materie untersucht wird. Jedes Spektrum,
gleich welcher Art, entsteht durch eine Wechselwirkung der zu untersuchenden Materie mit
elektromagnetischer Strahlung. Diese Materie kann zum Beispiel Strahlung aufnehmen
(Strahlungsabsorption) oder abgeben (Strahlungsemission), wobei sich ihr eigener energeti-
scher Zustand ändert. Moleküle können verschiedene Energieniveaus einnehmen. Es kön-
nen sich zum Beispiel Bindungen dehnen, biegen oder verdrehen, Elektronen können sich
von Orbital zu Orbital bewegen oder der Kernspin kann mittels elektromagnetischer Strah-
lung ausgelenkt werden. Alle diese Änderungen können nur mit bestimmten Frequenzen der
elektromagnetischen Strahlung angeregt werden. Das heisst, nur bestimmte Frequenzen

Laborbericht Seite 6
23. November 2009
können Bindungen in Molekülen dehnen, biegen oder verdrehen und Elektronen können nur
zwischen Orbitalen mit definierter Energiedifferenz wechseln. Die Beobachtung bzw. Mes-
sung von Strahlung, die in Wechselwirkung mit der Materie steht, erlaubt Rückschlüsse auf
die Zustandsänderung der Materie und daraus letztendlich auch ihre Identifizierung oder
Quantifizierung (4).
2.3.1.1 IR-Spektroskopie
Neben den oben dargestellten Übergängen der Elektronen eines Atoms haben Moleküle
auch die Möglichkeit, durch Änderung der Atomabstände, zu vibrieren oder um verschiedene
Achsen zu rotieren.
Prinzip der Infrarotspektroskopie (IR) ist die Messung von Wellenlängen und Intensitäten der
Absorption infraroter Strahlung durch eine Probe. Der IR-Bereich der elektromagnetischen
Strahlung befindet sich zwischen 700 nm und 1‘000‘000 nm (14‘300 cm -1 - 10 cm -1). Der IR-
Bereich lässt sich noch in drei kleinere Bereiche einteilen; das nahe IR (14‘300 – 4‘000 cm-1),
das mittlere IR (4‘000 – 200 cm -1) und das ferne IR (200 – 10 cm -1) (5).
Wird ein Molekül mit Licht aus dem IR-Bereich bestrahlt, so stellt man bei einer Transmissi-
onsmessung fest, dass bei bestimmten Wellenlängen Licht absorbiert wird. Diese fehlende
Energie hat demnach Schwingungen im Molekül angeregt.
Bei Molekülen mit mehr als zwei Atomen sind immer mehrere Bindungen von einem Schwin-
gungsvorgang betroffen. Die beobachteten Schwingungen lassen sich auf Streck- und Bie-
geschwingungen zurückführen. Streckschwingungen, welche auch als Valenzschwingungen
bezeichnet werden, können dabei sowohl symmetrisch als auch asymmetrisch sein. Biege-
schwingungen (Deformationsschwingungen) beinhalten jedoch eine Änderung des Bin-
dungswinkels und lassen sich in vier Arten einteilen. Twisting, Wagging, Scissoring und Ro-
cking (6). Damit durch die Absorption eines Photons eine Vibration angeregt werden kann,
muss die betreffende Atomgruppe ein permanentes Dipolmoment besitzen dessen Betrag
durch die anzuregende Schwingung variiert wird.
2.3.2 Iodometrie
Die Iodometrie ist eine vielseitige Redoxtitration, da sie sowohl als Oxidations- als auch als
Reduktionsverfahren eingesetzt werden kann. Die Iodometrie beruht dabei auf der oxidie-
renden Wirkung von elementarem Iod (I2) sowie auf der reduzierenden Wirkung von Iodid (I-).
Die Reaktion beruht dabei auf folgender Gleichung:
2 I- I2 2 e
-+
Reduzierende Stoffe können dabei direkt mit einer Iodmasslösung titriert werden. Oxidieren-
de Stoffe oxidieren jedoch Iodid zu Iod. Das erhaltene Iod wird anschliessend mit Natrium-
thiosulfatlösung titriert. (7)

Laborbericht Seite 7
23. November 2009
Bei der Iodometrie handelt es sich um eine Absolutmethode. Bei diesen Absolutmethoden
lässt sich der Gehalt der zu bestimmenden Probe direkt über den Verbrauch an der
Iodmasslösung, oder bei einer indirekten Methode, durch den Verbrauch des Reduktionsmit-
tels bestimmen.
In jedem Fall muss vor der Gehaltsbestimmung der Probe, der Titer der Iodmasslösung, oder
bei einer Rücktitration der Natriumthiosulfatmasslösung mittels eines Urtiters bestimmt wer-
den.
Je nach der Art des Analyten (oxidieren oder reduzierend), entsteht am sogenannten
Äquivalenzpunkt Iod oder das letzte noch vorhandene Iod wird reduziert. Obwohl Iod in ge-
löster Form eine gelbliche Färbung hat, ist es nicht möglich auf Grund der Entstehung oder
dem Verschwinden der gelben Farbe, den exakten Endpunkt zu erkennen. Daher wird kurz
vor Erreichen des Äquivalenzpunktes ein wenig Stärkelösung zu der Titrationsprobe zuge-
geben, was in einer intensiven Blaufärbung der Probe resultiert. Die auftretende intensive
Blaufärbung beruht dabei auf der Bildung einer Einschlussverbindung von Iod und Stärke,
genauer gesagt mit Amylose. Amylose besteht aus schraubenförmig angeordneten Molekül-
ketten. In den Hohlräumen dieser Ketten entstehen lineare Polyiodidketten. Diese intensive
Blaufärbung erlaubt nun die exakte Bestimmung des Äquivalenzpunktes. (8)

Laborbericht Seite 8
23. November 2009
3 Geräte und Materialien
3.1 Chemikalien und Materialien
Name Hersteller Artikelnummer
Stärke VWR 1.01252.0250
Natriumthiosulfat-pentahydrat Sigma-Aldrich 72050
Wasser deion. In house -
Brom Sigma-Aldrich 30202
Kaliumhydroxid-Plätzchen Sigma-Aldrich 60370
Kaliumiodid Sigma-Aldrich 60405
Salzsäure 37 % Sigma-Aldrich 320331
Kaliumiodat Urtitersubstanz VWR 1.02404.0100
Rundfilter LS 14; diam. 55 mm Schleicher & Schüll 120416695067
3.2 IR
Gerät Hersteller
IR-Spektrometer; Spectrum 1000 Perkin Elmer
Golden Gate ATR Golden Gate
Software Spectrum One Perkin Elmer

Laborbericht Seite 9
23. November 2009
4 Praktischer Teil
4.1 Herstellung von Kaliumbromat
4.1.1 Erste Stufe: Herstellung des Eisenkomplexes
Br2 + KOH KBr KOBr+
3 KOBr 2 KBr KBrO3+
62 g Kaliumhydroxid-Plätzchen wurden in einem 250 ml Sulfierkolben vorgelegt und in 62 ml
Wasser deion. gelöst. Dabei kam es zu einer erheblichen Temperaturerhöhung (ca. 80°C).
Anschliessend wurden 26 ml Brom langsam unter rühren zugetropft. Beim zutropfen kam es
zu einer Gelbfärbung der Lösung und danach zur Kristallisation von Kaliumbromat und Kali-
umbromid.
Nach der kompletten Zugabe des Broms wurde die Reaktionslösung langsam unter rühren
auf Raumtemperatur abgekühlt. Dabei fiel weiteres Kaliumbromat und Kaliumbromid aus.
Die ausgefallenen Kristalle wurden über einen Rundfilter abgenutscht. Dieser Nutschkuchen
wurde mit ca. 10 ml eiskaltem Wasser deion. nachgewaschen.
4.1.2 Umkristallisation
Das bei der Synthese erhaltene Kristallisat wurde in einem 250 ml Rundkolben vorgelegt und
mit 130 ml Wasser deion. versetzt. Die Lösung wurde zum sieden erhitzt bis sich das Kristal-
lisat komplett löste. Anschliessend wurde die Lösung unter Rühren langsam auf Raumtem-
peratur abgekühlt, wodurch es zur Kristallisation des Kaliumbromats kam. Das Kristallisat
wurde über einen Rundfilter abgenutscht und mit ca. 10 ml eiskaltem Wasser deion. nach-
gewaschen (Kristallisat 1.). Das dabei erhaltene Filtrat wurde unter rühren, im Eisbad, lang-
sam auf ca. drei bis vier Grad abgekühlt. Das noch auskristallisierte Kaliumbromat wurde
ebenfalls abgenutscht und mit ca. 10 ml eiskaltem Wasser deion. nachgewaschen (Kristalli-
sat 2.). Die Mutterlauge wurde anschliessend am Rotavap bei ca. 50 °C unter reduziertem
Druck zur Trockene eingeengt.
4.1.3 Vernichtung von überschüssigem Brom
Die Glasapparaturen wurden in einem Plastikbecken in ca. 1 mol/l Natriumthiosulfatlösung
eingelegt um das überschüssige Brom zu vernichten.

Laborbericht Seite 10
23. November 2009
4.2 Analytik
4.2.1 IR-Spektroskopie
Sowohl von Kristallisaten als auch von der Mutterlauge wurde jeweils ein Spektrum im IR-
Bereich zwischen 4000 cm-1 und 500 cm-1, mittels ATR, aufgenommen.
4.2.2 Iodometrie
4.2.2.1 Herstellung Reagenzien und Masslösung
Natrumthiosulfatmasslösung (1 mol/l)
25.008 g Natriumthiosulfat-pentahydrat wurden in einen 1000 ml Messkolben eingewogen, in
Wasser deion. gelöst und zur Marke gestellt.
Stärkelösung
2 g Stärke wurden in 200 ml kochendem Wasser deion. gelöst.
Salzsäure 2 mol/l
165 ml Salzsäure 37 % wurden mit Wasser deion. zu 1000 ml verdünnt.
4.2.2.3 Titerbestimmung
Ca. 70.0 mg Kaliumiodat (genau gewogen) wurden in einen 300 ml Erlenmeyer eingewogen,
mit 100 ml Wasser deion. und 20 ml Salzsäure (2 mol/l) versetzt. Anschliessend wurde der
Lösung 1 g Kaliumiodid zugegeben. Diese Lösung wurde mittels Natriumthiosulfatlösung (1
mol/l) bis kurz vor den Umschlagspunkt titriert. Nun wurden ca. 2 ml Stärkelösung zugesetzt
und bis zum Umschlagspunkt, von Violett zu klar, titriert.
Von der Titerbestimmung wurde eine Dreifachbestimmung durchgeführt.
Tabelle 2: Titerbestimmung
Einwaage [mg]
Kaliumiodat
Verbrauch [ml]
Natriumthiosulfatlsg.
73.4 20.40
72.7 20.30
71.5 19.90

Laborbericht Seite 11
23. November 2009
Einwaage Verbrauch Verbrauch Theor. Verbrauch Titer
mg ml mol ml Praxis/Theorie 73.4 3.430E-04 20.40 20.58 0.9913 72.7 3.397E-04 20.30 20.38 0.9959 71.5 3.341E-04 19.90 20.05 0.9927
0.9933 Mittelwert [g/100g]
0.0024 Stdev
0.24 RSD [%]
4.2.2.4 Analyse der Proben
BrO3- + 6 I
-
+ 6 H3O+
3 I2 + Br-
9 H2O+
I2 2 S2O3 2-
2 I-
S4O6 2-+ +
Ca. 55.0 mg (genau gewogen) der beiden Kristallisate beziehungsweise ca. 900.0 mg der
Mutterlauge wurden in einen 300 ml Erlenmeyer eingewogen und in 100 ml Wasser deion.
gelöst. Anschliessend wurde die Lösung mit 1 g Kaliumiodid und 40 ml Salzsäure (2 mol/l)
versetzt. Diese Lösung wurde während ca. 20 Minuten unter Lichtausschluss stehengelas-
sen.
Danach wurde die Lösung mit Natriumthiosulfatlösung (1 mol/l) von Braun bis Hellgelb titriert.
Dieser Lösung wurde nun ca. 2 ml Stärkelösung zugegeben, wodurch es zu einem Farbum-
schlag nach Violett kam. Diese violette Lösung wurde mit der Natriumthiosulfatlösung bis
zum Umschlagspunkt, von Violett zu klar, titriert.
Tabelle 3: Kristallisat 1
Einwaage [mg]
Kristallisat 1
Verbrauch [ml]
Natriumthiosulfatlsg.
56.3 19.80
56.8 19.95
56.2 19.85
Tabelle 4: Kristallisat 2
Einwaage [mg]
Kristallisat 2
Verbrauch [ml]
Natriumthiosulfatlsg.
55.8 19.60
56.0 19.80
56.1 20.05
Laborbericht Seite 12
23. November 2009
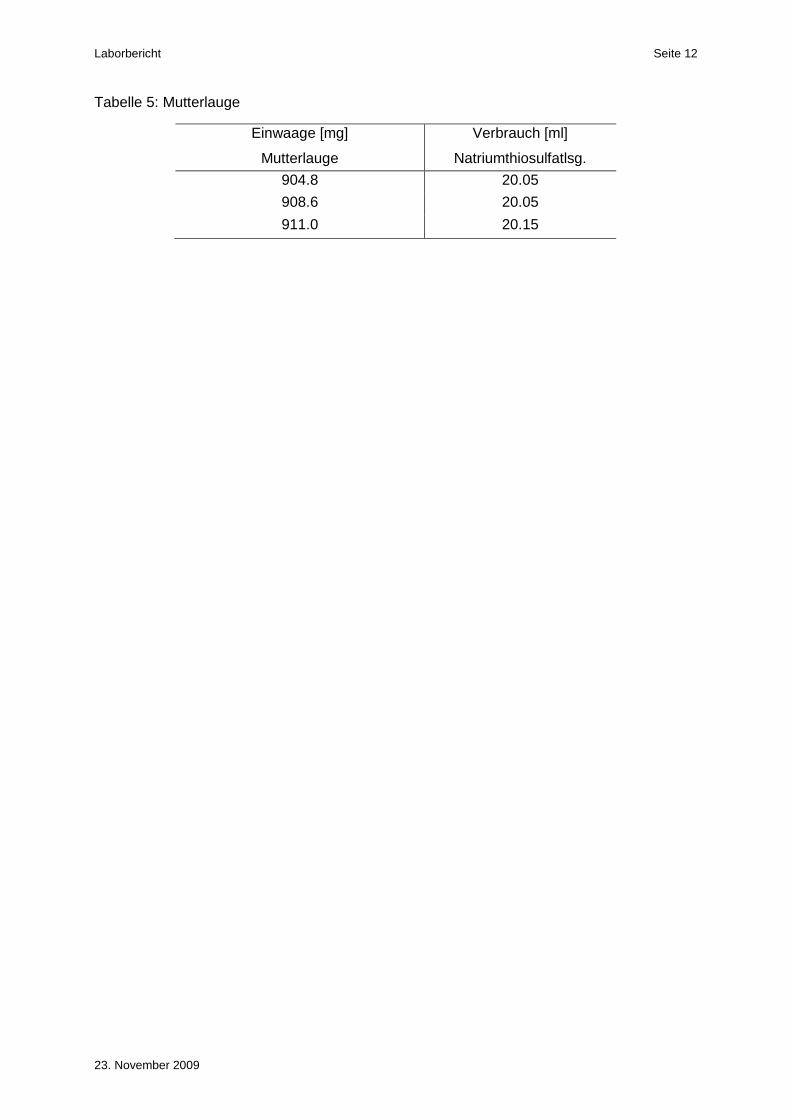
Tabelle 5: Mutterlauge
Einwaage [mg]
Mutterlauge
Verbrauch [ml]
Natriumthiosulfatlsg.
904.8 20.05
908.6 20.05
911.0 20.15

Laborbericht Seite 13
23. November 2009
5 Ergebnisse
5.1 Synthese und Umkristallisation
Die Herstellung des Kaliumbromats mittels des oben beschriebenen Verfahrens verlief ein-
wandfrei. Bei der Zugabe von Brom in die Reaktionslösung kam es zuerst zu einem gelben
Farbumschlag und nach einiger Zeit zu einer Ausfällung von Kaliumbromat bzw. von Kalium-
bromid als weisse Kristalle. Dieser Niederschlag konnte anschliessend problemlos abge-
nutscht werden.
Die Umkristallisation, welche im Anschluss an die Synthese des Kaliumbromats durchgeführt
wurde, gelang ebenfalls einwandfrei. Wie in Kapitel 5.2 ersichtlich, konnte das Kaliumbromat
mittels der hier durchgeführten Umkristallisation in sehr reiner Form erhalten werden. Der
Verlust durch diesen Aufreinigungsschritt belief sich auf 9.8 %. Dieser etwas erhöhte Verlust
liesse sich vermutlich durch ein weiteres Abkühlen der Mutterlauge ebenfalls noch kristalli-
sieren. Nichtsdestotrotz konnte wie oben erwähnt sehr reines Kaliumbromat erhalten wer-
den.
5.2 Analytik
5.2.1 IR
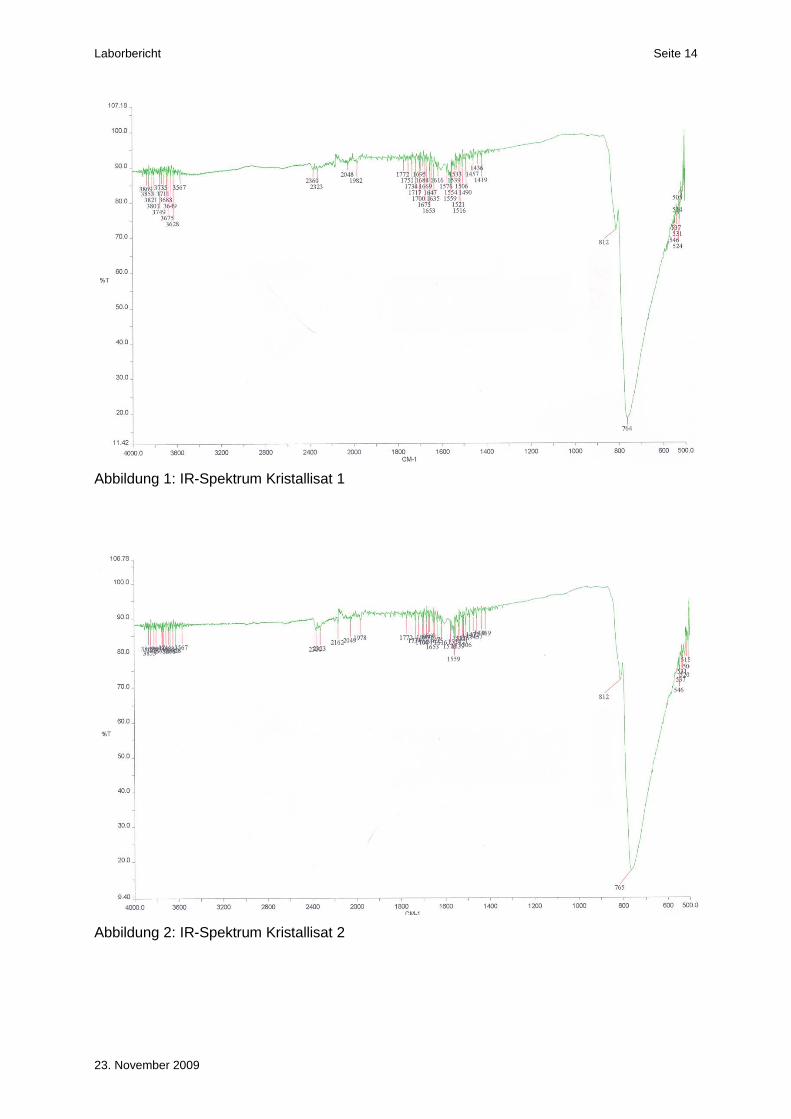
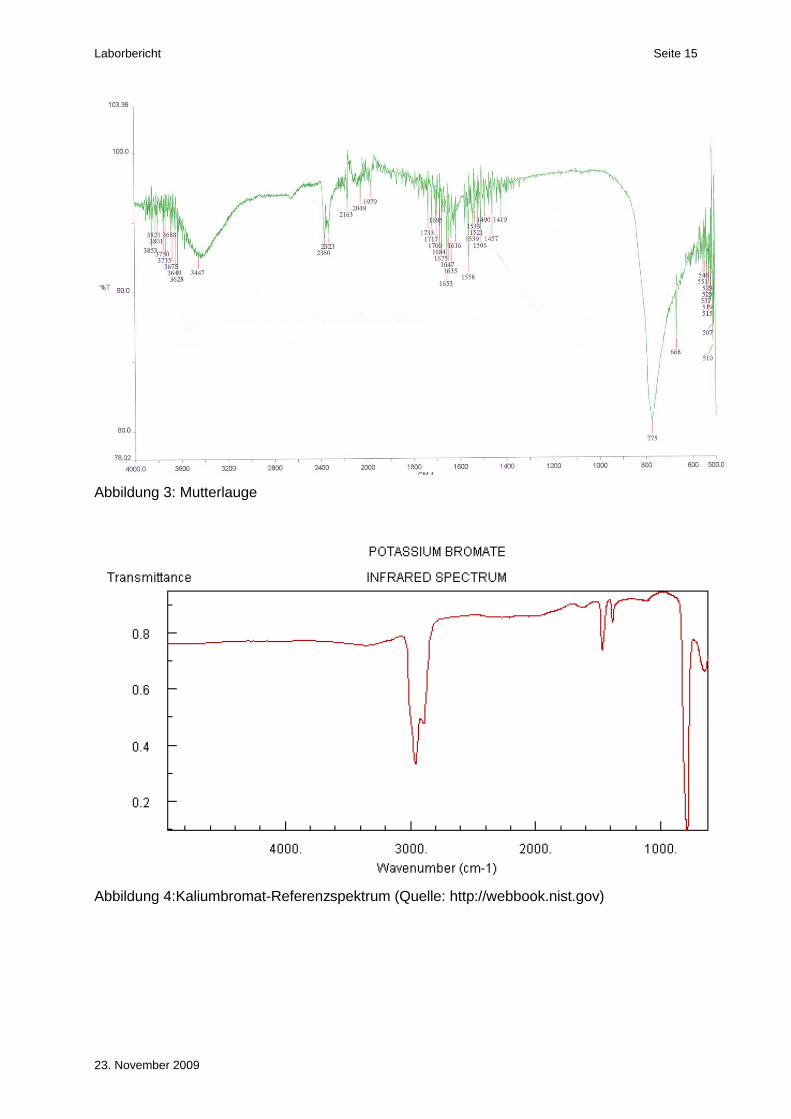
Sowohl im Spektrum des ersten als auch des zweiten Kristallisats (Abbildung 1 und Abbil-
dung 2) konnte die markante Bande bei ca. 760 cm-1 nachgewiesen werden. Diese Bande ist
auch im Spektrum des „National Institute of Standards and Technology“ (NIST) gut ersicht-
lich (Abbildung 4). Die weiteren drei Banden im IR-Spektrum der NIST, bei ca. 2900, 1450
und 1380 cm-1 sind auf Störbanden des Nujols zurückzuführen.
Im Spektrum der Mutterlauge ist ebenfalls noch die Bande bei ca. 760 cm-1 ersichtlich. Die
Bande ist jedoch nicht so intensiv wie bei den beiden Kristallisaten. Somit sind in der Mutter-
lauge noch Spuren von Kaliumbromat enthalten.
Damit konnte die Identität der beiden Kristallisate eindeutig bewiesen werden.
Laborbericht Seite 14
23. November 2009
Abbildung 1: IR-Spektrum Kristallisat 1
Abbildung 2: IR-Spektrum Kristallisat 2
Laborbericht Seite 15
23. November 2009
Abbildung 3: Mutterlauge
Abbildung 4:Kaliumbromat-Referenzspektrum (Quelle: http://webbook.nist.gov)
Laborbericht Seite 16
23. November 2009
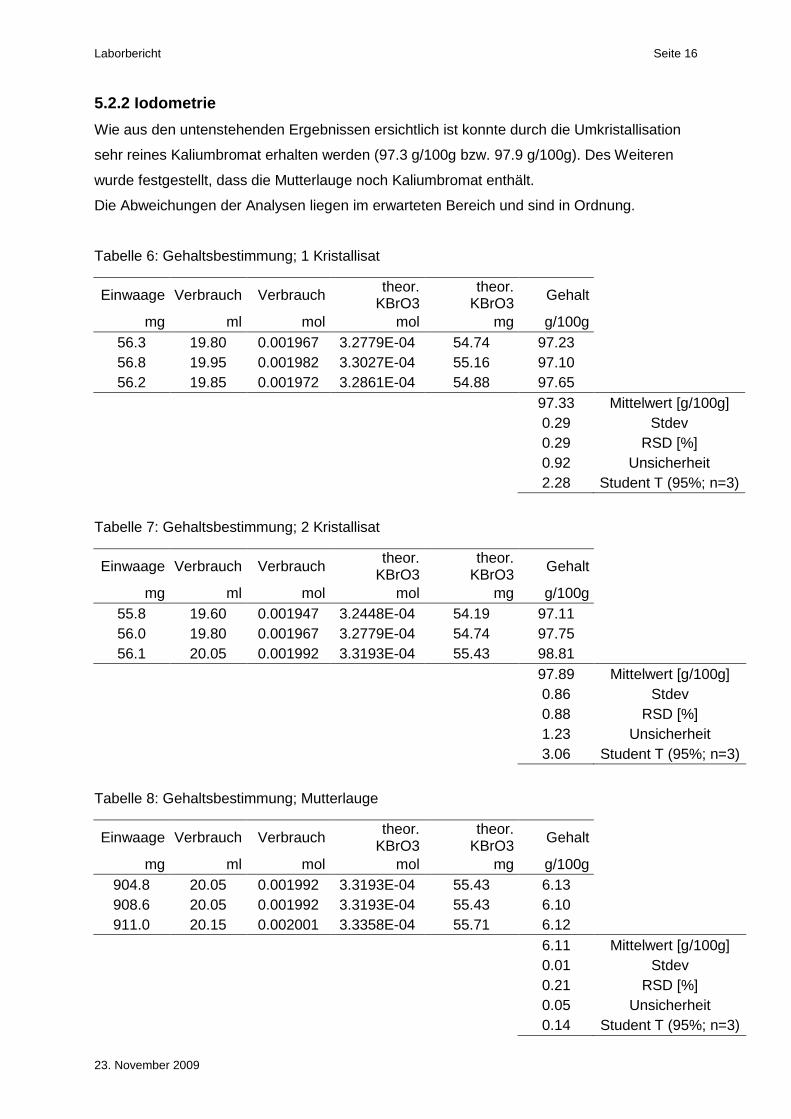
5.2.2 Iodometrie
Wie aus den untenstehenden Ergebnissen ersichtlich ist konnte durch die Umkristallisation
sehr reines Kaliumbromat erhalten werden (97.3 g/100g bzw. 97.9 g/100g). Des Weiteren
wurde festgestellt, dass die Mutterlauge noch Kaliumbromat enthält.
Die Abweichungen der Analysen liegen im erwarteten Bereich und sind in Ordnung.
Tabelle 6: Gehaltsbestimmung; 1 Kristallisat
Einwaage Verbrauch Verbrauch theor. KBrO3
theor. KBrO3 Gehalt
mg ml mol mol mg g/100g 56.3 19.80 0.001967 3.2779E-04 54.74 97.23 56.8 19.95 0.001982 3.3027E-04 55.16 97.10 56.2 19.85 0.001972 3.2861E-04 54.88 97.65
97.33 Mittelwert [g/100g]
0.29 Stdev
0.29 RSD [%] 0.92 Unsicherheit 2.28 Student T (95%; n=3)
Tabelle 7: Gehaltsbestimmung; 2 Kristallisat
Einwaage Verbrauch Verbrauch theor. KBrO3
theor. KBrO3 Gehalt
mg ml mol mol mg g/100g 55.8 19.60 0.001947 3.2448E-04 54.19 97.11 56.0 19.80 0.001967 3.2779E-04 54.74 97.75 56.1 20.05 0.001992 3.3193E-04 55.43 98.81
97.89 Mittelwert [g/100g]
0.86 Stdev
0.88 RSD [%] 1.23 Unsicherheit 3.06 Student T (95%; n=3)
Tabelle 8: Gehaltsbestimmung; Mutterlauge
Einwaage Verbrauch Verbrauch theor. KBrO3
theor. KBrO3 Gehalt
mg ml mol mol mg g/100g 904.8 20.05 0.001992 3.3193E-04 55.43 6.13 908.6 20.05 0.001992 3.3193E-04 55.43 6.10 911.0 20.15 0.002001 3.3358E-04 55.71 6.12
6.11 Mittelwert [g/100g]
0.01 Stdev
0.21 RSD [%] 0.05 Unsicherheit 0.14 Student T (95%; n=3)

Laborbericht Seite 17
23. November 2009
5.2.2.2 Unsicherheitsfortpflanzung
Für die Unsicherheitsfortpflanzung wurde die Standardabweichung der Bürette, der Waage,
der Titerbestimmung und der jeweiligen Gehaltsbestimmung mit einbezogen. Die Unsicher-
heitsfortpflanzung wurde nach untenstehender Formel berechnet.
�����ℎ��ℎ�� = ��� ���������� + � ��������ℎ���� + � ��ü�������� !���� + ��"##$�%������ ∙ ��ℎ�������
Die Standardabweichung der Bürette musste vor deren Verwendung noch nach dieser For-
mel umgerechnet werden. ��ü����� = (.*+,√* = 0.17 !� Die Standardabweichung der Waage betrug gemäss beiliegendem Datenblatt 0.1 mg.
Die jeweiligen berechneten Unsicherheitsfortpflanzungen sind in den Tabellen 5 bis 7 er-
sichtlich.
Um die zusätzliche Unsicherheit durch die relativ kleine Mehrfachbestimmung ebenfalls ein-
zubeziehen wurde der Student-T-Faktor herangezogen. Der Stichprobenumfang betrug bei
den jeweiligen Bestimmungen drei, somit ergibt sich ein Freiheitsgrad von zwei. Der Vertrau-
ensbereich wurde auf 95 % festgelegt.
5.3 Ausbeute
Die Ausbeute betrug wie aus Tabelle 9 ersichtlich ist 72.4 %. Diese Ausbeute ist vergleichs-
weise tief. Jedoch sind weitere 7.9 % an Kaliumbromat in der Mutterlauge enthalten. Diese
liessen sich durch weiteres abkühlen, bzw. Kristallisieren noch gewinnen. Dies ändert jedoch
nichts daran das ca. 20 % nicht in Kaliumbromat umgesetzt werden konnte bzw. in der Reak-
tionslösung zurückblieben. Vermutlich ist in der Mutterlauge der Reaktionslösung noch Kali-
umbromat vorhanden. Des Weiteren verlief die Reaktion vermutlich nicht vollständig.
Tabelle 9:Ausbeute
Name Ausbeute in % Ausbeute in % mit Gehaltskorrektur
1 Kristallisat 64.9 63.2
2 Kristallisat 9.5 9.3
Summe 74.4 72.4

Laborbericht Seite 18
23. November 2009
6 Diskussion Wie aus den Ergebnissen ersichtlich ist, konnte sehr reines Kaliumbromat erhalten werden.
Die Synthese des Kaliumbromats und die anschliessend durchgeführte Umkristallisation ver-
liefen problemlos. Als negative Punkte sind jedoch die relativ tiefe Ausbeute und die ca. 7.9
% Verlust an Kaliumbromat bei der Umkristallisation zu werten. Der Verlust liesse sich durch
tieferes abkühlen der Mutterlauge zu einem grossen Teil noch kristallisieren.
Durch die Kristallisation des Kaliumbromats, welches noch in der Mutterlauge enthalten ist
liesse sich die Ausbeute auf 80 % erhöhen. Die restlichen 20 % Verlust sind vermutlich dar-
auf zurückzuführen das die Mutterlauge der Reaktionslösung noch Kaliumbromat enthält,
welches nicht vollständig ausfiel. Des Weiteren verlief die Reaktion wahrscheinlich nicht
komplett.
Nichtsdestotrotz, konnte sehr reines Kaliumbromat erhalten werden. Wie aus der Durchge-
führten Gehaltsbestimmung ersichtlich ist, konnte durch die Umkristallisation das Kalium-
bromat einwandfrei vom Kaliumbromid abgetrennt werden. Weiter konnte die Identität des
Kaliumbromats durch die Infrarotspektroskopie bestätigt werden. Wie in Kapitel 5.2.1 ersicht-
lich, sind die markanten Banden von Kaliumbromat welche von der NIST beschrieben wer-
den in den hier aufgenommenen Spektren wiedergefunden worden.
.

Laborbericht Seite 19
23. November 2009
7 Literaturverzeichnis 1. US Food and Drug Administration. [Online] 1 April 2005. [Cited: 11 November 2009.]
http://www.fda.gov/.
2. Mortimer, Charles E. and Müller, Ulrich. Chemie. Stuttgard : Georg Thieme Verlag,
2003.
3. Holleman, Arnold F. and Wiberg, Egon. Lehrbuch der Anorganischen Chemie. Berlin :
de Gruyter, 2007.
4. Cammann, Karl, [ed.]. Instrumentelle Analytische Chemie. Heidelberg : Spektrum
Akademischer Verlag GmbH Heidelberg, 2001.
5. HFP Modul F3; Überblick grundlegende Analysenmethoden. Mansardo, Dr. G. Zürich :
s.n., 2006.
6. Spektroskopie – Praxis Infrarot-Spektroskopie. Geiger. Dübendorf : s.n., 2008.
7. Kunze, Udo R. and Schwedt, Georg. Grundlagen der quantitativen Analyse. Weinheim :
WILEY-VCH Verlag GmbH & Co. KGaA, 2009.
8. Joachim Strähle, Eberhard Schweda. Einführung in das anorganisch-chemische
Praktikum. Stuttgard : Hirzel S. Verlag, 2005.

Laborbericht Seite 20
23. November 2009
8 Verzeichnis der Abbildungen Abbildung 1: IR-Spektrum Kristallisat 1 ................................................................................14
Abbildung 2: IR-Spektrum Kristallisat 2 ................................................................................14
Abbildung 3: Mutterlauge ......................................................................................................15
Abbildung 4:Kaliumbromat-Referenzspektrum (Quelle: http://webbook.nist.gov) ..................15





![Smog - molekuelwald.square7.chmolekuelwald.square7.ch/biblio/%d6kologie/Oeko_2013_Theorie_Weiterf... · Web view[1] Mit virtuellem Wasser ist die Wassermenge bezeichnet, die nach](https://img.pdfslide.org/doc/110x75/5d51da7a88c993e2358b7527/smog-d6kologieoeko2013theorieweiterf-web-view1-mit-virtuellem-wasser.jpg)
















![10 ISO IEC 17025 [Kompatibilitätsmodus]molekuelwald.square7.ch/biblio/QualyManagement/QM_ISO17025.pdf · ISO 17025:1999 2005 Die grundsätzlichen Änderungen • Anpassung der ISO/IEC](https://img.pdfslide.org/doc/110x75/5e137e60187d536f565b1c44/10-iso-iec-17025-kompatibilittsmodus-iso-170251999-2005-die-grundstzlichen.jpg)





![Herstellung von Na[Co(EDTA)]molekuelwald.square7.ch/biblio/Allgemeine Chemie Praktikum/AllgCP_2009... · Herstellung von Na[Co(EDTA)] Aufgabenstellung / Abstract ZHAW CH 09 – 3](https://img.pdfslide.org/doc/110x75/5e5a5175001ebf13462111ba/herstellung-von-nacoedta-chemie-praktikumallgcp2009-herstellung-von-nacoedta.jpg)